
Method Article
Inmunofluorescencia múltiple combinada con análisis de imágenes espaciales para la evaluación clínica y biológica del microambiente tumoral
En este artículo
Resumen
En este artículo, se describe un protocolo para la inmunofluorescencia múltiple (mIF) de amplificación manual de señal de tiramida (TSA) combinada con análisis de imágenes y análisis espacial. Este protocolo se puede utilizar con secciones fijadas en parafina con formalina (FFPE) para la tinción de dos a seis antígenos por portaobjetos, dependiendo del escáner de portaobjetos disponible en el laboratorio.
Resumen
El microambiente tumoral (TME) se compone de una gran cantidad de diferentes tipos de células, como las células inmunes citotóxicas y las células inmunomoduladoras. Dependiendo de su composición y de las interacciones entre las células cancerosas y las células peritumorales, el TME puede afectar la progresión del cáncer. La caracterización de los tumores y su complejo microambiente podría mejorar la comprensión de las enfermedades del cáncer y puede ayudar a los científicos y médicos a descubrir nuevos biomarcadores.
Recientemente desarrollamos varios paneles de inmunofluorescencia multiplex (mIF) basados en la amplificación de señal de tiramida (TSA) para la caracterización del TME en cáncer colorrectal, carcinoma de células escamosas de cabeza y cuello, melanoma y cáncer de pulmón. Una vez finalizada la tinción y el escaneo de los paneles correspondientes, las muestras se analizan en un software de análisis de imágenes. La posición espacial y la tinción de cada célula se exportan desde este software de cuantificación a R. Desarrollamos scripts R que nos permiten no solo analizar la densidad de cada tipo de célula en varios compartimentos tumorales (por ejemplo, el centro del tumor, el margen del tumor y el estroma), sino también realizar análisis basados en la distancia entre diferentes tipos de células.
Este flujo de trabajo en particular agrega una dimensión espacial al análisis de densidad clásico que ya se realiza rutinariamente para varios marcadores. El análisis de mIF podría permitir a los científicos tener una mejor comprensión de la compleja interacción entre las células cancerosas y el TME y descubrir nuevos biomarcadores predictivos de respuesta a los tratamientos, como los inhibidores del punto de control inmunitario y las terapias dirigidas.
Introducción
Con el desarrollo de terapias dirigidas e inhibidores de puntos de control inmunitario, se ha vuelto de suma importancia caracterizar mejor las interacciones entre las células cancerosas y su microambiente tumoral, y este es actualmente un campo importante de investigación traslacional. El TME está compuesto por una plétora de diferentes tipos de células, con un equilibrio de células citotóxicas inmunes dirigidas a las células cancerosas y células inmunomoduladoras que podrían favorecer el crecimiento tumoral y la invasividad 1,2,3,4. La caracterización de este entorno complejo podría mejorar la comprensión de las enfermedades oncológicas y puede ayudar a los científicos y clínicos a descubrir nuevos biomarcadores predictivos y pronósticos para seleccionar mejor a los pacientes para el tratamiento futuro 5,6. Por ejemplo, Galon y su equipo desarrollaron el Immunoscore, que es un método de puntuación reproducible que se puede utilizar como un biomarcador predictivo. El Immunoscore se calcula utilizando la densidad de linfocitos T CD3+ y CD8+ en el margen invasivo y en el centro del tumor 7,8.
En las últimas décadas, se han desarrollado soluciones comerciales para mIF, pero a menudo son costosas y están diseñadas para paneles específicos de antígenos. Para superar la necesidad de paneles específicos de antígenos en la investigación académica y traslacional, desarrollamos un método rentable para realizar mIF en secciones tumorales FFPE, permitiendo la tinción de dos a seis antígenos agregados a la contratinción de núcleos celulares en muestras humanas y de ratón.
Una vez que todas las secciones de tejido se tiñen y escanean con un escáner de portaobjetos de fluorescencia, las muestras pueden ser analizadas por varios programas de análisis de imágenes que admiten grandes conjuntos de datos piramidales. Finalmente, los datos sin procesar se pueden utilizar en un entorno para computación estadística y gráficos como el software R (v.4.0.2) para realizar análisis de densidad y espaciales.
En este manuscrito se presenta un protocolo optimizado para la tinción de cinco marcadores, así como trucos y consejos para optimizar nuevos paneles. Además, se explican los pasos detallados del análisis de imágenes y las funciones R utilizadas para el análisis estadístico y espacial.
Protocolo
Todas las muestras utilizadas en el presente protocolo provienen de un estudio aprobado por los comités de ética locales y autorizado por la autoridad competente. Todos los participantes en el estudio dieron su consentimiento informado por escrito. El ensayo está registrado con ClinicalTrials.gov (NCT03608046).
1. Inmunofluorescencia múltiple
- Seccionamiento FFPE
- Fije el tejido en paraformaldehído al 4% e incruste el tejido fijo en parafina.
- Corte secciones de 5 μm y colóquelas en portaobjetos de microscopio adhesivo.
- Seque los portaobjetos durante la noche a temperatura ambiente (RT).
- Desparafinización e inhibición de peroxidasas endógenas
- Desparafinar los tejidos sumergiendo los portaobjetos en tolueno (3x durante 5 minutos cada uno) y metanol (3x durante 5 minutos cada uno) bajo una campana extractora.
- Inhibir las peroxidasas endógenas sumergiendo los portaobjetos en peróxido de hidrógeno al 3% diluido en metanol durante 20 minutos bajo una campana extractora.
- Enjuagar las diapositivas en destilado (d)H2O(1x durante 3 min).
- Tinción de inmunofluorescencia multiplex
- Sumerja los portaobjetos en un frasco de tinción de 300 ml que contenga 10 mM de citrato (pH 6) o EDTA (pH 9) complementado con TritonX-100 al 0,1%.
NOTA: El tampón utilizado (pH 6 o pH 9) depende del antígeno teñido (ver Tabla 1). - Coloque el frasco de tinción con la tapa cerrada en un microondas durante 3-5 minutos a la potencia máxima (por ejemplo, 900 W) hasta que el tampón comience a hervir.
NOTA: El momento óptimo para hervir depende del microondas y del volumen del tampón. Los ajustes pueden ser necesarios para encontrar el momento perfecto. Para algunos antígenos frágiles o especímenes frágiles y menos adherentes (por ejemplo, organoides y esferoides), la ebullición por microondas puede ser demasiado dura. En este caso, se puede usar una olla a presión en su lugar. - Mantenga el tampón a una temperatura cercana a la ebullición colocando el frasco de tinción cerrado en el microondas a baja potencia (por ejemplo, 90 W) durante 15 minutos.
- Realice el último paso de calentamiento poniendo el microondas a máxima potencia durante 90 s.
- Retire el frasco del microondas y deje que el tampón se enfríe durante 15 minutos en RT.
- Enjuague las diapositivas 3 veces durante 5 minutos cada una en dH2O y 1 vez durante 5 minutos en solución salina tamponada con tris que contenga 0,1% de Tween 20 (TBS-T).
- Retire el TBS-T secando las diapositivas en una toalla de papel
- Coloque los portaobjetos (planos) en una bandeja de la cámara de tinción o en una caja portaobjetos para microscopio (consulte la Tabla de materiales).
- Rodee el tejido con un bolígrafo hidrófobo.
- Bloquear los sitios de unión no específicos cubriendo el tejido con albúmina sérica bovina (BSA) al 5% disuelta en TBS-T durante 30 min.
- Retire el amortiguador de bloqueo secando las diapositivas en una toalla de papel.
NOTA: No enjuague las diapositivas después del paso de bloqueo. - Incubar el tejido durante 60 min con el anticuerpo primario (ver Tabla 1) diluido en BSA TBS-T al 1% cubriendo el tejido con alrededor de 300 μL de la solución.
- Enjuague las diapositivas 3 veces durante 3 minutos cada una con TBS-T.
- Incubar el tejido durante 40 min con el anticuerpo secundario poli-HRP (ver Tabla 1) cubriendo el tejido con alrededor de 300 μL de la solución.
- Enjuague las diapositivas 3 veces durante 3 minutos con TBS-T.
- Incubar el tejido durante 10 min con reactivo de fluorocromo-tiramida (ver Tabla 1) diluido 200 veces en tampón de borato (borato 0,1 M, pH 7,8, 3 M NaCl) suplementado extemporáneamente con 0,003% deH2O2cubriendo el tejido con alrededor de 300 μL de la solución.
- Enjuague las diapositivas 3 veces durante 3 minutos con TBS-T.
- Repita los pasos 1.3.1-1.3.16 hasta que se haya realizado toda la tinción de TSA.
- Incubar el tejido durante la noche a 4 °C con el último anticuerpo primario (ver Tabla 1) diluido en BSA TBS-T al 1%.
NOTA: Dado que la incubación es durante la noche, es importante cubrir la bandeja de la cámara de tinción o la caja de portaobjetos del microscopio y agregar dH2O en una toalla de papel en el fondo de la caja (debajo de los portaobjetos) para asegurarse de que los tejidos no se sequen durante la incubación. - Enjuague el pañuelo 3 veces durante 5 minutos cada una con TBS-T.
- Incubar el tejido durante 120 min con el anticuerpo secundario (acoplado directamente con fluorocromo) diluido 200 veces en BSA TBS-T al 1%.
- Enjuague el pañuelo 3 veces durante 5 minutos cada una con TBS-T.
- Tinción de los núcleos incubando el tejido durante 5 min en bisbenzimida (20 mM) diluida 1.000 veces en BSA TBS-T al 10%.
NOTA: La bisbenzimida puede ser reemplazada por DAPI, pero este último es más tóxico y debe manejarse con cuidado bajo una campana extractora. - Enjuagar el pañuelo 3 veces durante 3 min cada una en dH2O.
- Monte las guías utilizando un medio de montaje de fluorescencia y vidrios de cubierta de borosilicato.
- Sumerja los portaobjetos en un frasco de tinción de 300 ml que contenga 10 mM de citrato (pH 6) o EDTA (pH 9) complementado con TritonX-100 al 0,1%.
2. Escaneo de diapositivas
- Digitalice las diapositivas escaneándolas en un escáner de diapositivas de fluorescencia con un aumento de 20x (los detalles del escáner de diapositivas se proporcionan en la Tabla de materiales).
NOTA: En la Figura 1 se muestra un escaneo representativo de un múltiplex óptimo.
3. Análisis de imágenes
- Importe los escaneos a un software de análisis de imágenes (File > Open Image).
- Vaya a la pestaña Clasificadores y seleccione el complemento DenseNet AI V2 .
- Entrene el complemento DenseNet AI V2 para reconocer núcleos rodeando alrededor de 500 núcleos en una imagen.
- Entrene a la IA en varias otras diapositivas del mismo lote y diferentes lotes de tinción mIF rodeando varios núcleos (50) en varias diapositivas (alrededor de 10).
NOTA: Las instrucciones detalladas sobre cómo usar el complemento AI se pueden encontrar en el manual del software. El uso de IA para la detección de núcleos es opcional. Otros métodos para detectar núcleos están disponibles dependiendo del software de análisis de imágenes utilizado. - Guarde la IA entrenada (Acciones clasificadoras > Guardar).
- Vaya a la pestaña Anotaciones y cree una anotación para cada región de interés (ROI), como el centro del tumor y el margen del tumor, mediante la herramienta Anotación de pluma.
- Si es necesario, elimine las regiones con pliegues y las regiones que aparecen borrosas con la herramienta Anotación de exclusión.
NOTA: La tinción con hematoxilina-eosina de una sección adyacente a la utilizada para mIF se puede realizar antes de la tinción mIF para asegurar que las células tumorales estén presentes en la muestra y para ayudar a los anatomopatólogos a determinar el ROI. - Vaya a la pestaña Análisis y seleccione el algoritmo HighPlex FL (Acciones de configuración > Cargar > HighPlex FL).
- Seleccione la pestaña Selección de tinte y seleccione el tinte que le interese.
- En la pestaña Detección nuclear, vaya a Tipo de segmentación nuclear y seleccione AI custom.
- En Clasificador de segmentación nuclear, seleccione la IA guardada en el paso 3.5.
- En la pestaña Detección de membrana y citoplasma, elija el radio máximo de citoplasma (en este estudio, se utilizó 1,5) y el número de colorantes de membrana .
- Para cada colorante, seleccione el umbral positivo del núcleo, el umbral positivo del citoplasma y el umbral positivo de la membrana.
NOTA: El umbral es diferente para cada tinción y debe ajustarse para cada lote de portaobjetos y cada antígeno teñido. El uso de la herramienta Configuración de vista (Ver > Ver configuración) puede ayudar a seleccionar un umbral adecuado utilizando el valor de intensidad al final del pico de intensidad (a la derecha). - Para cada colorante, seleccione el núcleo, la membrana y el porcentaje de citoplasma de los valores de completitud.
NOTA: Este parámetro es importante para evitar la detección de falsos positivos cuando dos células con tinción diferente están cerca una de la otra (Figura 2). - Guarde el algoritmo (Acciones de configuración > Guardar).
- Analice los ROI (Analyze > Annotation Layer).
- Vaya a la pestaña Resultados y seleccione todos los datos en Datos de objeto (ctrl + A).
- Exporte los datos en formato .csv (haga clic con el botón derecho > Exportar datos de > objeto . Csv).
NOTA: Esta tabla contiene la posición (Xmin, Xmax; Ymin, Ymax) y la positividad de cada marcador de cada célula analizada.
4. Bioinformática usando R
NOTA: Un script R que proporciona más detalles sobre los siguientes pasos está disponible en GitHub (benidovskaya/Ring: Pipeline for the analysis of multiplex immunofluorescence stainings. [github.com])
- Usando la tabla exportada, primero defina los diferentes tipos de celdas en función de las tinciones de colocalización. Por ejemplo, defina las células T citotóxicas mediante células CD3+/CD8+ doblemente positivas.
- A continuación, reconstruya una imagen simplificada de la diapositiva en un diagrama de puntos utilizando las coordenadas exportadas desde el software de análisis de imágenes y ggplot2 (Figura 3). Utilizando estos datos, se pueden realizar varios tipos de análisis:
- Análisis de densidad
NOTA: El análisis más simple es un análisis de densidad.- Realice un análisis de densidad para todos los tipos de células utilizando todo el portaobjetos para biopsias o algún área específica del tejido. Por ejemplo, calcule la densidad de las células T CD3+ y CD8+ en el centro del tumor y el margen del tumor (Figura 4A-C).
- Para calcular esas densidades, utilice un software de análisis de imágenes para producir un marco de datos específico por muestra con el fenotipo y las coordenadas de cada célula. A través de una función de agrupamiento (k-vecino más cercano) en R, cree un objeto poligonal utilizando los bordes de la biopsia estudiada y calcule la densidad de los tipos celulares de interés dentro de ella.
NOTA: Esto permite comparar las densidades de diferentes tipos de células entre diferentes condiciones (como diferentes puntos de tiempo, tipos de tratamiento, tipos de tejido y respuesta al tratamiento) y localizaciones (centro del tumor, margen del tumor, fibrosis del estroma y área de necrosis) dependiendo de la hipótesis biológica. Debido a la alta proximidad entre las células cancerosas, las células peritumorales y las células infiltrantes de tumores, el software de análisis de imágenes puede detectar células doblemente positivas como células inmunes y cancerosas al mismo tiempo. En ese caso, uno necesita corregir bioinformáticamente este problema mencionando cuáles son estas células doblemente positivas. En este caso, las células CD3+CD8+citoqueratina+ se marcaron como células citotóxicas porque la positividad de la citoqueratina se debía a las células tumorales que rodean los linfocitos infiltrantes.
- Mapas de calor
- Usando la densidad de cada tipo de celda de diferentes paneles y aplicando una normalización (por ejemplo, centrado de escala), dibuje mapas de calor (Figura 5) que representen la abundancia celular en la población de muestras.
- Mediante el uso de agrupamiento jerárquico no supervisado basado en la densidad celular, agrupe a los pacientes con composiciones similares de EMT y correlacione estos grupos con parámetros clínicos como la respuesta al tratamiento y la supervivencia.
NOTA: Los mapas de calor y la agrupación en clústeres se pueden realizar fácilmente con el paquete R ComplexHeatmap9.
- Distribución celular espacial
- Calcular bioinformáticamente las distancias entre las células (por ejemplo, células inmunes y tumorales; Figura 6A, B) basadas en las coordenadas de celda proporcionadas por el análisis de imágenes. Utilice las distancias medianas y medias entre los tipos de células de interés para comparar la proximidad celular en todas las muestras de una cohorte.
- Funciones descriptivas espaciales
- Utilice la función G-cross del vecino más cercano de tipo cruz, disponible a través del paquete R spatstat10, para determinar la probabilidad de que una célula de interés, X (por ejemplo, una célula tumoral), se encuentre con la célula más cercana, Y (por ejemplo, una célula T), dentro de un cierto radio alrededor de la célula X.
- Calcular el área bajo la curva empírica para obtener un valor numérico que represente la infiltración tumoral de células T CD3+ alrededor de las células tumorales11 (Figura 6C). Utilice otras funciones descriptivas espaciales como la función F o la función J12.
- Análisis Immunoscore
- Calcular el Immunoscore (I), desarrollado por el equipo de Galon7,8, utilizando la densidad de células T CD3+ y CD8+ en el centro del tumor y el margen invasivo del tumor.
NOTA: La puntuación oscila entre I0 e I4. Una baja densidad de células T CD3+ y CD8+ en el centro y el margen del tumor se asocia con una puntuación de I0, mientras que una alta densidad de células T CD3+ y CD8+ en ambas regiones se asocia con una puntuación de I4. Recientemente, el impacto pronóstico del Immunocore fue validado en un estudio con muestras de 2.681 pacientes con cáncer de colon en estadio I-III de 14 centros en 13 países7. Sin embargo, para ser calculado, Immunoscore requiere una muestra resecada quirúrgicamente que contenga tanto el centro como el margen del tumor. Para las biopsias, que generalmente carecen de margen, recientemente se ha desarrollado un Immunoscore adaptado a la biopsia13. - Para calcular el Immunoscore adaptado a la biopsia, convierta el valor de la densidad de células T CD3+ y CD8+ en un percentil y, a continuación, utilice el percentil medio de células T CD3+ y CD8+ para la puntuación en una de tres categorías (es decir, baja, intermedia y alta)13.
- Calcular el Immunoscore (I), desarrollado por el equipo de Galon7,8, utilizando la densidad de células T CD3+ y CD8+ en el centro del tumor y el margen invasivo del tumor.
- Análisis de puntos calientes
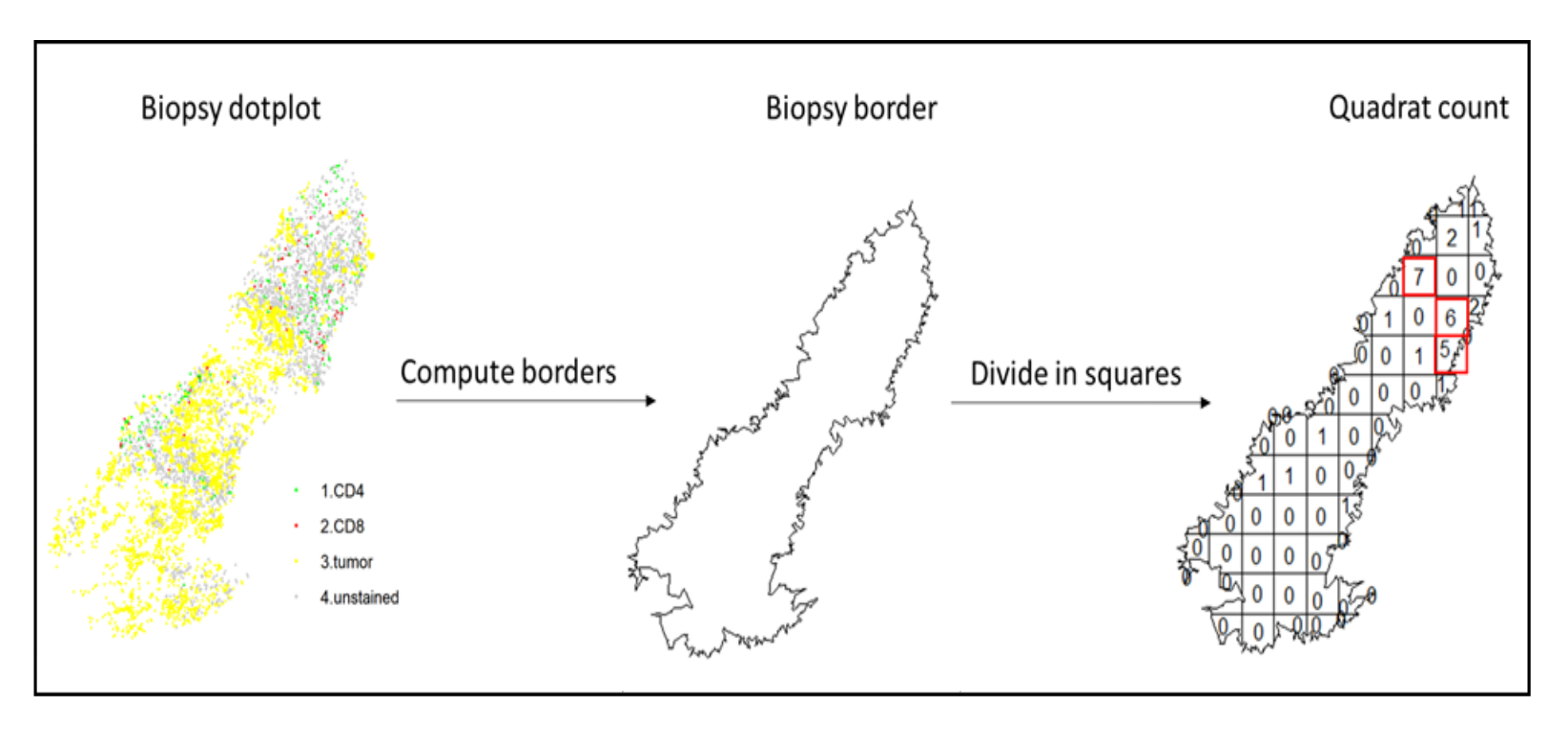
- Utilice el análisis de puntos calientes, utilizando la función quadratcount (spatstat)10, para comparar las densidades de diferentes tipos de células en el área más infiltrada del tejido. Por ejemplo, es posible calcular una puntuación "similar a Immunoscore" utilizando el valor de la densidad de células T CD3 y CD8 de los cuadrados más infiltrados del tejido (Figura 7). Aplicar este método para el análisis de cualquier tipo de célula con una distribución no homogénea a través del tejido.
- Análisis de densidad
Resultados
Siguiendo este protocolo, se deben investigar varios parámetros para garantizar que el tejido se tiñe correctamente. En primer lugar, la tinción TSA debe mostrar un buen rango dinámico cuando se utilizan tiempos de exposición bajos (generalmente 2-100 ms) durante el proceso de escaneo. Un tiempo de exposición bajo implica que la amplificación se ha realizado correctamente durante la reacción con HRP. Para los antígenos teñidos con el anticuerpo secundario directamente acoplado con el fluorocromo, el tiempo de exposición podría ser mucho más largo, lo que podría conducir a un fotoblanqueo (una disminución en la intensidad de la señal debido a un largo tiempo de exposición). En segundo lugar, es importante verificar que cada tinción muestre una SNR alta. Una señal de fondo alta con una señal de antígeno baja puede ser una indicación de que el anticuerpo primario no es lo suficientemente específico, que las peroxidasas endógenas no se inactivaron correctamente o que un paso del protocolo no se realizó adecuadamente. En tercer lugar, dependiendo del escáner de diapositivas y los conjuntos de filtros utilizados para el escaneo, es posible ver superposiciones entre dos colores (por ejemplo, AF555, AF594 y AF647). La elección de los conjuntos de filtros correctos en el escáner y la dilución de anticuerpos primarios correctos son cruciales para evitar posibles detecciones cruzadas. El control de calidad consiste en la detección de células teñidas individuales para cada marcador en el archivo escaneado. Por último, también es importante añadir un control positivo y negativo para cada lote de tinción. Para las células inmunes, la amígdala es un buen control positivo. Un resultado representativo de la tinción óptima se muestra en la Figura 1.

Figura 1: Cáncer rectal localmente avanzado teñido por inmunofluorescencia múltiple. Abreviaturas: PD-1 = proteína de muerte celular programada 1; PD-L1 = ligando de muerte programada 1; ROR-γ = receptor huérfano gamma relacionado con RAR; CD3 = grupo de diferenciación 3; hPanCK = pancitoqueratina humana. Cada tinción de antígeno se escanea en escala de grises, y los colores presentados en la figura son pseudocolores. Barra de escala de bajo aumento: 200 μm; Barra de escala de gran aumento: 100 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Detección de núcleos y tinción de un cáncer rectal localmente avanzado utilizando un software de análisis de imágenes. Sin el parámetro de porcentaje de completitud configurado correctamente, el software detecta dos células CD8 + (círculo verde) porque están cerca una de la otra, pero solo una celda está teñida. El uso de un 70% de integridad ayuda a evitar esta detección de falsos positivos. Verde = hPanCK; amarillo = CD3; Naranja = CD8. Barra de escala: 100 μm Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Análisis de imagen y reconstitución del diagrama de puntos R de una metástasis de cáncer colorrectal de hígado. En la tinción múltiple (izquierda), la pancitoqueratina humana está en amarillo, CD3 está en verde, CD8 está en azul claro y IDO está en naranja. En el diagrama de puntos (derecha), las células pancitoqueratina + humanas están en amarillo, las células CD3 + CD8 − están en verde, las células CD3 + CD8 + están en azul y las células IDO + están en naranja. Haga clic aquí para ver una versión más grande de esta figura.
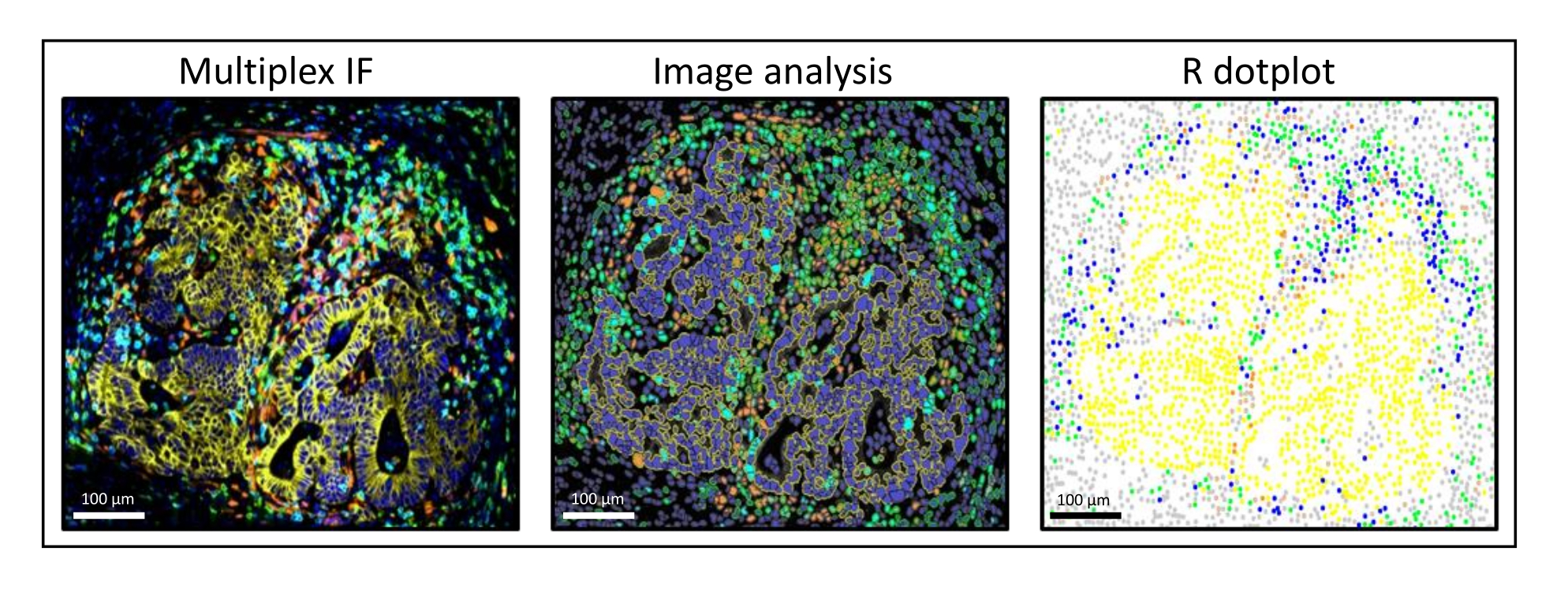{kind=link}

Figura 4: Análisis de una sección quirúrgica de un HNSCC. (A) Una sección quirúrgica de un HNSCC. Las células cancerosas son visibles en verde. Las células peritumorales se visualizan alrededor de los islotes tumorales (CD3 en amarillo y CD8 en púrpura). (B) El centro del tumor (en amarillo con un borde negro) se calcula bioinformáticamente mediante el algoritmo k-nearest-neighbor basado en la distancia entre las islas tumorales de una sola área. Alrededor de esta área, se calcula un margen invasivo (amarillo claro con un borde gris) sobre una base arbitraria de 500 μm. (C) Las células T invasivas se resaltan con puntos negros en el centro del tumor y puntos grises en el margen invasivo. Otras células T se resaltan en puntos verdes claros. Barra de escala: 1 mm. Haga clic aquí para ver una versión más grande de esta figura.
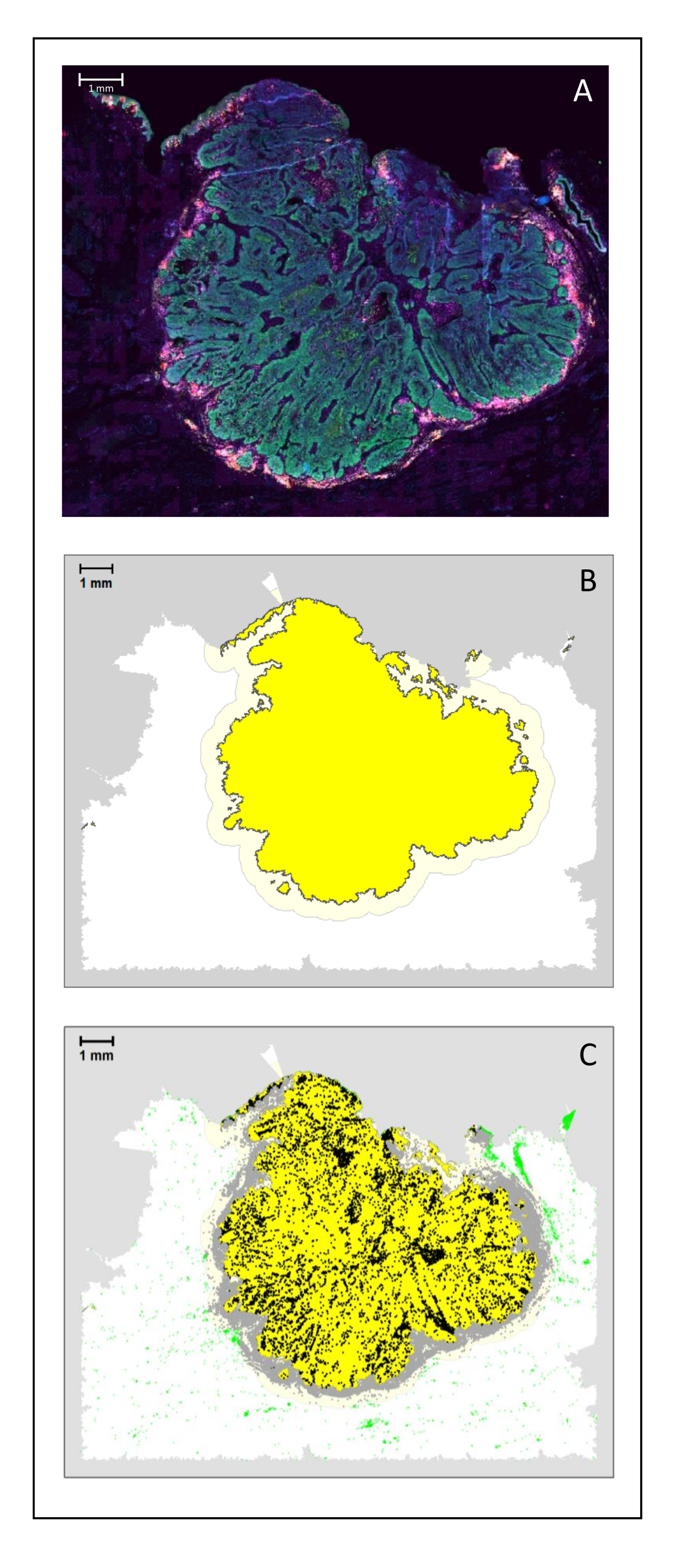{kind=link}

Figura 5: Mapa de calor de la densidad de diferentes tipos de células de biopsias de cáncer de recto localmente avanzadas. El mapa de calor se dibujó utilizando la agrupación no supervisada de las densidades de diferentes tipos de células de diferentes paneles multiplex con el paquete ComplexHeatmap. El escalado y el centrado se utilizaron para la normalización. Haga clic aquí para ver una versión más grande de esta figura.
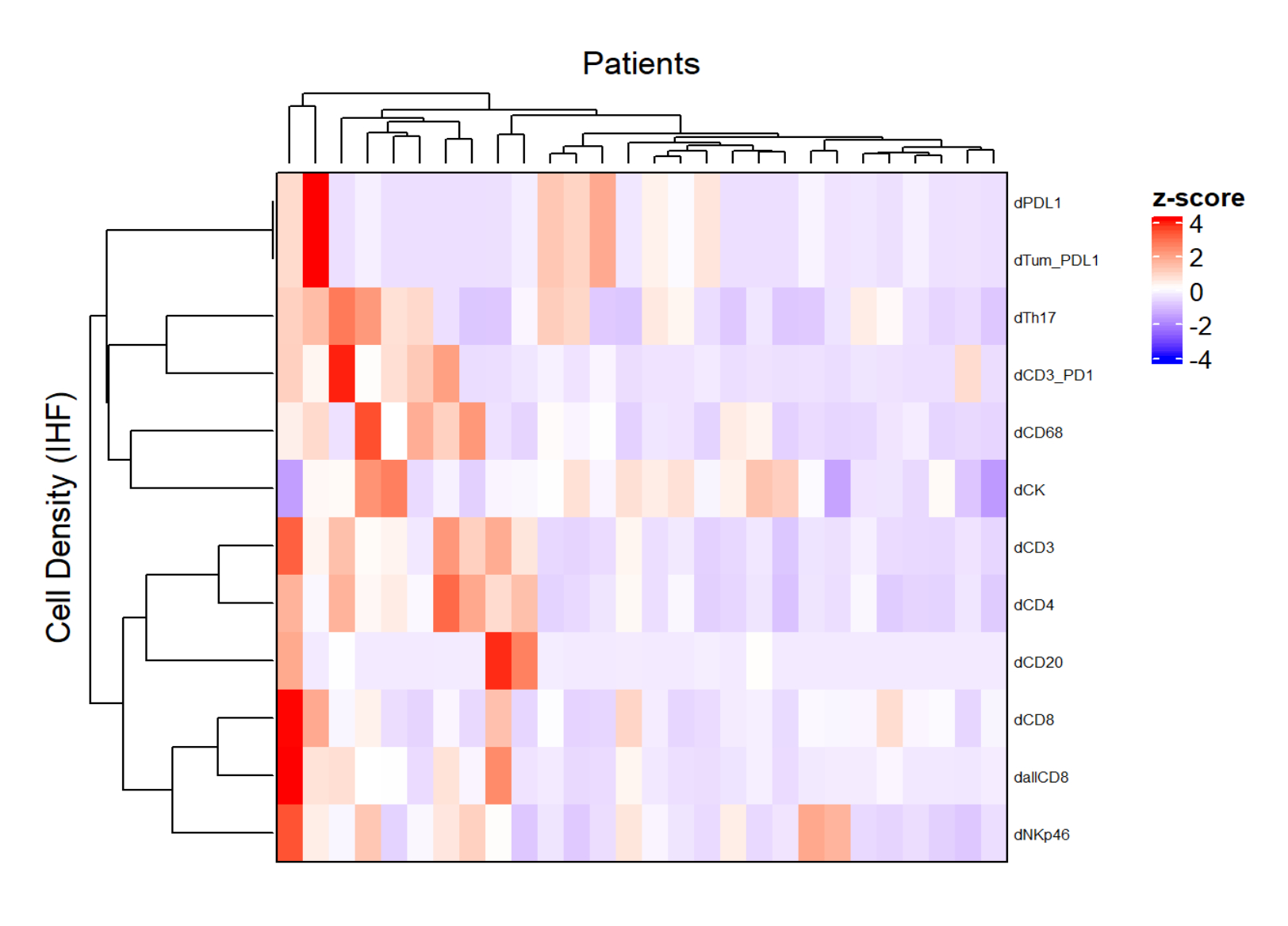{kind=link}

Figura 6: Distancias de las células CD4+ y CD8+ a cada célula IDO+ o tumoral. Las células pancitoqueratina + humanas están en amarillo, las células CD3 + CD8 − están en verde, las células CD3 + CD8 + están en azul y las células IDO + están en naranja. (A) La distancia más cercana entre las células tumorales y cada célula T CD8 +. (B) Gráficos de barras de las distancias entre las células IDO+ y cada célula T CD8+ (azul) o linfocitos T CD4+ (verde). (C) Ejemplo de una muestra analizada por la función G-cross. El eje y muestra la probabilidad de que una célula tumoral encuentre un linfocito CD3+ en un radio que oscila entre 0 y 200 μm alrededor de la célula tumoral. Se muestran tres curvas; la curva teórica está en verde punteado (distribución de Poisson), la curva empírica corregida con corrección km está en negro y la curva empírica corregida con corrección de borde está en rojo punteado. Haga clic aquí para ver una versión más grande de esta figura.
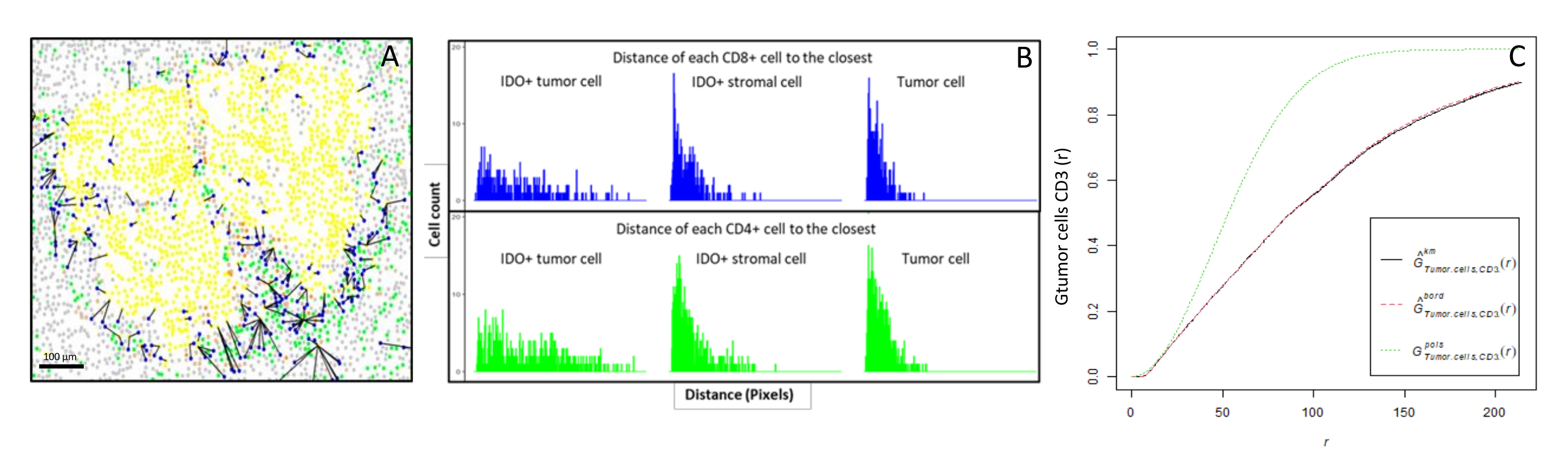{kind=link}

Figura 7: Ilustración de un cuadrado. El cálculo del borde y el recuento de cuadrantes se realizaron utilizando el paquete spatstats. Los cuadrados más infiltrados (hotspots) se pueden usar para estadísticas posteriores. CD4 está en verde, CD8 está en rojo y las células tumorales están en amarillo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

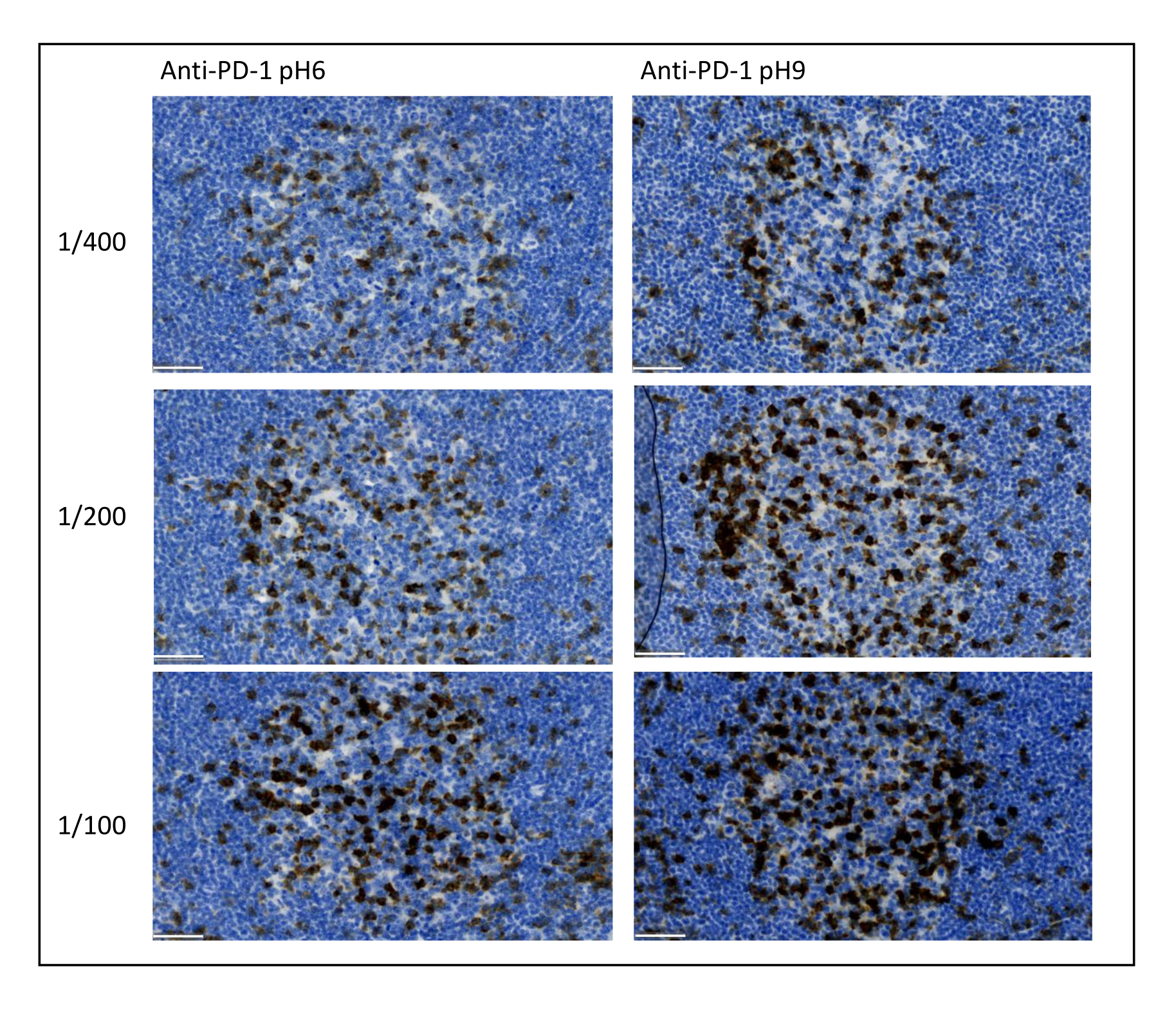
Figura 8: Dilución de anticuerpos y optimización de la recuperación de antígenos. Detección cromogénica de PD-1 utilizando tres diluciones diferentes y dos soluciones diferentes de recuperación de antígenos del anticuerpo primario (Citrato pH 6 y EDTA pH 9). Barra de escala: 50 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Anticuerpo primario | Dilución | Recuperación de antígenos | Anticuerpo secundario | Fluorocromo | Posición |
| PD-1 | 1/100 | EDTA (pH 9) | Anti-conejo | AF647 | 1 |
| PD-L1 | 1/1000 | EDTA (pH 9) | Anti-conejo | AF488 | 2 |
| ROR-γ | 1/200 | EDTA (pH 9) | Anti-ratón | ATT0-425 | 3 |
| CD3 | 1/100 | Citrato (pH 6) | Anti-conejo | AF555 | 4 |
| hPanCK | 1/50 | Citrato (pH 6) | Anti-ratón acoplado con AF750 | 5 | |
Tabla 1: Ejemplo de un panel multiplex optimizado. Abreviaturas: PD-1 = proteína de muerte celular programada 1; PD-L1 = ligando de muerte programada 1; ROR-γ = receptor huérfano gamma relacionado con RAR; CD3 = grupo de diferenciación 3; hPanCK = pancitoqueratina humana; AF = AlexaFluor; EDTA = ácido etilendiaminotetraacético. CD3 se utiliza para detectar linfocitos T; PD-1 se utiliza para detectar linfocitos agotados; ROR- γ se utiliza para detectar Th-17; y hPanCK se utiliza para detectar células tumorales. La columna de posición indica el orden en que se debe realizar el múltiplex secuencial.
Discusión
Los parámetros más importantes a tener en cuenta para optimizar la tinción múltiple son la dilución, la especificidad y la recuperación de antígenos utilizados para cada anticuerpo primario. Antes de comenzar un protocolo multiplex, la dilución óptima de cada anticuerpo primario y la recuperación óptima del epítopo (pH 6 o pH 9) deben probarse mediante tinción cromogénica (DAB). Aconsejamos probar tres diluciones para cada tampón de recuperación de antígeno: la dilución que generalmente especifica la marca que comercializa el anticuerpo, la misma dilución dividida dos veces y la misma dilución multiplicada por dos (Figura 8). Elegir la dilución correcta es un paso muy importante para verificar la especificidad del anticuerpo y optimizar la relación señal-ruido (SNR) de la tinción. Después de elegir la dilución correcta en el DAB, se debe probar la misma dilución para cada anticuerpo primario utilizando TSA uniplex. Una vez que se seleccionan el tampón de dilución y recuperación de epítopos para cada tinción de antígeno, también es importante configurar correctamente la secuencia del múltiplex; Específicamente, algunos antígenos se tiñen mejor en la primera posición y otros en la última posición. Aconsejamos probar el etiquetado múltiplex utilizando todas las permutaciones de orden posibles para elegir qué tinción de antígenos debe venir primero, segundo, etc. Este es también un paso muy importante porque algunos antígenos frágiles pueden degradarse después de varias rondas de recuperación de epítopos, y algunos antígenos se tiñen mejor después de varias rondas de recuperación de epítopos. Por ejemplo, la SNR siempre es más alta en la última posición para CD3 y en la primera posición para la tinción de PD-1. Además, la tinción de varios antígenos colocalizados puede verse obstaculizada por un efecto paraguas (la saturación de los sitios reactivos de tiramida). Esto puede atenuarse disminuyendo la concentración de tiramida. Cuando la expresión de un antígeno está condicionada por la expresión de otro (CD8 solo presente en células T que expresan CD3), aconsejamos teñir el antígeno con la expresión más amplia (CD3 en este caso) después del otro. Finalmente, elegir el fluorocromo correcto para cada tinción de antígeno de acuerdo con las especificidades del escáner también es un paso importante para evitar la detección cruzada.
Las principales ventajas de esta técnica son la amplificación y la relación señal-ruido obtenida. Sin embargo, esta técnica viene con una limitación, que es que la tinción es secuencial, y los fluorocromos están unidos covalentemente al tejido. Sin embargo, después de realizar todas las rondas de amplificación de la señal de tiramida, también es posible agregar una última tinción con un anticuerpo secundario directamente acoplado con un fluorocromo (sin TSA). En algunos paneles, utilizamos este método para agregar tinción en el canal 750. Esto era necesario porque no había tiramida-AF750 disponible comercialmente en ese momento. Cabe destacar que el tiempo de exposición (durante la exploración) del antígeno teñido con AF750 será mucho más largo que para los otros antígenos teñidos con TSA. En ese caso, aconsejamos teñir una proteína altamente expresada como la citoqueratina o aumentar la concentración del anticuerpo primario. Al hacer eso, es posible teñir un máximo de cinco a seis antígenos por portaobjetos en un lote dependiendo del escáner de fluorescencia.
En oposición, varias técnicas disponibles comercialmente utilizan la tinción en serie con varias rondas de tinción, escaneo y extracción o fotoblanqueo para mejorar el número de antígenos que se pueden teñir en una sola sección de tejido. Sin embargo, estas técnicas a menudo consumen mucho tiempo, son costosas, no tienen amplificación de señal, requieren pasos computacionales avanzados para fusionar los escaneos en serie correctamente y, en nuestra experiencia, pueden inducir daños irreversibles en los tejidos debido a los numerosos pasos del procedimiento. Sin embargo, se ha descrito que hasta 30 antígenos podrían ser teñidos en un solo tejido utilizando este método14.
En conclusión, nuestro método es una técnica de inmunohistofluorescencia robusta, reproducible, fácil de usar y rentable que se puede usar en cualquier laboratorio que posea un escáner de diapositivas de fluorescencia. Se puede usar cualquier anticuerpo primario comercializado adecuado para IHQ, y los paneles no son específicos para ningún kit comercial. El análisis de imágenes se puede realizar en varios programas diferentes, incluidos programas de código abierto como QuPath y R. Sin embargo, creemos que este método podría incluso mejorarse en el futuro para paneles de antígenos grandes, permitiendo realizar tinción / escaneo en serie de la misma diapositiva con diferentes paneles de antígenos y con la ventaja de la amplificación de la señal.
Divulgaciones
Los autores no tienen conflictos de intereses que declarar.
Agradecimientos
Los autores desean agradecer a la Dra. Derouane F por su ayuda y apoyo. Nicolas Huyghe es investigador apoyado por una beca del Fondo Nacional Belga para la Investigación Científica (Télévie/FNRS 7460918F).
Materiales
| Name | Company | Catalog Number | Comments |
| anti-CD3 primary antibody | Abcam | ab16669 | rabbit monocolonal |
| anti-CD8 primary antibody | DAKO | M710301 | mouse monoclonal |
| anti-hPanCK primary antibody | DAKO | M3515 | mouse monoclonal |
| anti-PD-1 primary antibody | Cell Signalling | D4W2J | rabbit monocolonal |
| anti-PD-L1 primary antibody | Cell Signalling | 13684 | rabbit monocolonal |
| anti-RORC primary antibody | Sigma | MABF81 | mouse monoclonal |
| ATTO-425 | ATTOtec | ||
| Axioscan Z1 | Zeiss | Light source: Colibri 7 (385, 430, 475, 555, 590, 630, 735 nm) Filtersets: Excitation 379/34 – beam splitter 409 – emission 440/40; Excitation 438/24 – beam splitter 458 – emission 483/32; Excitation 490/20 – beam splitter 505 – emission 525/20; Excitation 546/10 – beam splitter 556 – emission 572/23; Excitation 592/21 – beam splitter 610 – emission 630/30; Excitation 635/18 – beam splitter 652 – emission 680/42; Excitation 735/40 – beam splitter QBS 405 + 493 + 611 + 762 - emission QBP 425/30 + 524/51 + 634/38 + 785/38; Objective: Plan-Apochromat 20x/0.8; Camera : Orca Flash 4.0 V3 | |
| Borosilicate Cover Glass | VWR | 631-0146 | |
| Envision+ anti-mouse | DAKO | K4001 | |
| Envision+ anti-rabbit | DAKO | K4003 | |
| Fluorescence mounting medium | DAKO | S3023 | |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 750 | ThermoFischer | A-21037 | |
| HALO software | Indicalabs | ||
| Hoescht | Sigma | 14533 | |
| Superfrost plus microscope slides | Fisherscientific/Epredia | 10149870 | |
| Tyramide-AF488 | ThermoFischer | B40953 | |
| Tyramide-AF555 | ThermoFischer | B04955 | |
| Tyramide-AF647 | ThermoFischer | B04958 |
Referencias
- Ge, P., et al. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomedicine & Pharmacotherapy. 118, 109228(2019).
- Fridman, W. H. The immune microenvironment as a guide for cancer therapies. Oncoimmunology. 1 (3), 261-262 (2012).
- Fridman, W. H., Pages, F., Sautes-Fridman, C., Galon, J. The immune contexture in human tumours: Impact on clinical outcome. in Nature Reviews. Cancer. 12 (4), 298-306 (2012).
- Hanahan, D., Weinberg, R. A. Hallmarks of cancer: The next generation. Cell. 144 (5), 646-674 (2011).
- Calu, V., et al. Key biomarkers within the colorectal cancer related inflammatory microenvironment. Scientific Reports. 11 (1), 7940(2021).
- Havel, J. J., Chowell, D., Chan, T. A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nature Reviews. Cancer. 19 (3), 133-150 (2019).
- Pages, F., et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 391 (10135), 2128-2139 (2018).
- Mlecnik, B., et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 44 (3), 698-711 (2016).
- Gu, Z., Eils, R., Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 32 (18), 2847-2849 (2016).
- Baddeley, A., Rubak, E., Turner, R. Spatial Point Patterns: Methodology and Applications with R. , CRC Press. Boca Raton, FL. (2022).
- Barua, S., et al. Spatial interaction of tumor cells and regulatory T cells correlates with survival in non-small cell lung cancer. Lung Cancer. 117, 73-79 (2018).
- Parra, E. R. Methods to determine and analyze the cellular spatial distribution extracted from multiplex immunofluorescence data to understand the tumor microenvironment. Frontiers in Molecular Biosciences. 8, 668340(2021).
- El Sissy, C., et al. A diagnostic biopsy-adapted immunoscore predicts response to neoadjuvant treatment and selects patients with rectal cancer eligible for a watch-and-wait strategy. Clinical Cancer Research. 26 (19), 5198-5207 (2020).
- Bolognesi, M. M., et al. Multiplex staining by sequential immunostaining and antibody removal on routine tissue sections. The Journal of Histochemistry and Cytochemistry. 65 (8), 431-444 (2017).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados