
Method Article
Un ensayo validable de reacción digital de la polimerasa en cadena de gotas para la detección de vectores virales adenoasociados en estudios de bioderramamiento de lágrimas
En este artículo
Resumen
Aquí, presentamos un protocolo para el desarrollo y validación de buenas prácticas de laboratorio en la detección compatible de vectores virales adenoasociados en lágrimas humanas por reacción en cadena de la polimerasa digital de gotitas en apoyo del desarrollo clínico de vectores de terapia génica.
Resumen
El uso de vectores virales para tratar enfermedades genéticas ha aumentado sustancialmente en los últimos años, con más de 2.000 estudios registrados hasta la fecha. Los vectores virales adenoasociados (AAV) han tenido un éxito particular en el tratamiento de enfermedades relacionadas con los ojos, como lo ejemplifica la aprobación del voretigene neparvovec-rzyl. Para llevar nuevas terapias al mercado, las agencias reguladoras generalmente solicitan estudios de biodiseminación calificados o validados para evaluar la liberación del vector en el medio ambiente. Sin embargo, la Administración de Alimentos y Medicamentos de los Estados Unidos no ha publicado directrices oficiales para el desarrollo de ensayos moleculares para respaldar tales estudios de desprendimiento, lo que deja a los desarrolladores determinar las mejores prácticas por sí mismos. El propósito de este protocolo es presentar un protocolo validable para la detección de vectores AAV en lágrimas humanas por reacción en cadena de la polimerasa digital de gotitas (ddPCR) en apoyo de estudios clínicos de bioderramamiento. Este manuscrito analiza los enfoques actuales de la industria para la validación de ensayos moleculares y demuestra que el método excede los criterios de aceptación del ensayo objetivo actualmente propuestos en los libros blancos. Finalmente, se discuten los pasos críticos en el desempeño de cualquier ensayo de ddPCR, independientemente de la aplicación.
Introducción
Las definiciones de terapia génica varían, pero generalmente inducen una alternancia permanente intencional y a menudo esperada de una secuencia específica de ADN del genoma celular para modificar o manipular la expresión de un gen o alterar las propiedades biológicas de una célula viva con un propósito clínico 1,2. Los vectores virales se utilizan cada vez más como vehículos para la terapia génica debido a su eficiencia de transducción, con un informe que sugiere que más del 70% de los ensayos clínicos actuales de terapia génica utilizan vectores virales3. El interés en los vectores virales para la terapia génica ha ido ganando constantemente. El Informe de datos trimestrales del cuarto trimestre de 2022 sobre el panorama de la terapia génica, celular y de ARN de la Sociedad Americana de Terapia Génica y Celular informó que en 2022, la línea de terapia de genes, células y ARN desde la preclínica hasta el registro previo creció un 7%, lo que elevó el número total de terapias en desarrollo a 3,726, de las cuales 2,053 (55%) fueron terapias génicas4. La Administración de Alimentos y Medicamentos de los Estados Unidos (EE.UU. FDA) ha aprobado actualmente 27 terapias celulares y genéticas para uso clínico en humanos, cinco de las cuales utilizan específicamente vectores virales5.
Los virus adenoasociados (AAV) han ganado un interés específico como vehículos para la terapia génica. Un metaanálisis reciente reveló que ha habido aproximadamente 136 ensayos clínicos que investigan el uso de AAV en las últimas dos décadas6. Además, tres de las cinco terapias génicas aprobadas por la FDA de EE.UU. están basadas en AAV. Esto se debe a su naturaleza altamente editable, amplia gama de huéspedes que puede ajustarse en función del uso de vectores naturales específicos o artificialmente diseñados, baja patogenicidad y toxicidad en humanos y, en general, baja inmunogenicidad 7,8. Los AAV también se han utilizado con éxito para tratar enfermedades oculares en un entorno clínico aprobado. Voretigene neparvovec-rzyl es una terapia basada en AAV2 que fue aprobada por la FDA de EE.UU. en 2017 y por la Agencia Europea de Medicamentos (EMA) en 2018 para tratar la distrofia retiniana asociada a la mutación bialélica RPE65 9.
Con el creciente interés en el desarrollo de terapias basadas en AAV viene la necesidad de orientación regulatoria sobre ensayos. La detección y cuantificación precisas de cualquier vector viral es una parte integral de las fases de descubrimiento, fabricación y pruebas preclínicas / clínicas del desarrollo del producto. La FDA de los Estados Unidos ha comenzado a emitir algunas directrices para las terapias génicas, incluidas las aplicaciones de nuevos fármacos en investigación de la química, la fabricación y el control de la terapia génica humana 10, el seguimiento a largo plazo después de la administración de la terapia génica11, las pruebas de retrovirus competentes en replicación 12 y las recomendaciones para los vectores microbianos utilizados en las terapias génicas 13. La EMA también ha publicado una serie de directrices sobre el desarrollo de productos de terapia génica que generalmente se alinean con las recomendaciones de la FDA, aunque existen algunas diferencias14. Es importante tener en cuenta que, si bien estas directrices no establecen responsabilidades legalmente exigibles, excepto cuando se hace referencia a regulaciones específicas, proporcionan claridad sobre el pensamiento actual de las agencias reguladoras sobre el tema y sus expectativas para los ensayos requeridos para la presentación de medicamentos y la aprobación regulatoria.
La FDA recomienda específicamente que se realicen estudios para evaluar la distribución, persistencia y eliminación de un vector desde el sitio de administración hasta los tejidos oculares y no oculares, los fluidos intraoculares y la sangre15. Estos toman la forma de estudios de biodistribución y desprendimiento. Los estudios de biodistribución evalúan la exposición investigando cómo se propaga un producto por todo el cuerpo de un paciente desde el sitio de administración. El derramamiento evalúa específicamente la liberación del producto del paciente al medio ambiente y plantea la posibilidad de transmisión del vector a individuos no tratados16. La FDA hace recomendaciones para el diseño de estudios de biodistribución y derramamiento con respecto a la frecuencia de recolección de muestras, la duración de la recolección de muestras, los tipos de muestras recolectadas y las condiciones de almacenamiento.
Además, la FDA recomienda el uso de la reacción en cadena de la polimerasa cuantitativa (qPCR o PCR en tiempo real) para la detección cuantitativa de genomas de vectores debido a su facilidad de rendimiento, formato de alto rendimiento, tiempos de respuesta rápidos y sensibilidad al ensayo. Sin embargo, hay una relativa falta de recomendaciones para el diseño y la evaluación del rendimiento de los métodos moleculares en comparación con los que existen para moléculas pequeñas y grandes. Muchas de las directrices para tales estudios son difíciles de aplicar a los métodos moleculares debido al diseño único y complejo tanto de los productos como de los propios ensayos, lo que plantea dudas sobre la idoneidad de las plataformas disponibles para las evaluaciones recomendadas y los métodos apropiados para la validación de ensayos. Hasta la fecha, la FDA no ha requerido la validación formal de los ensayos basados en PCR, aunque la EMA ha impuesto este requisito17. A la luz de este vacío, diferentes grupos y talleres han emitido libros blancos y recomendaciones que los fabricantes y las organizaciones de investigación por contrato han tratado de seguir 18,19,20,21,22,23,24,25. La mayoría de estas recomendaciones están escritas específicamente teniendo en cuenta los ensayos de qPCR, con sugerencias o alteraciones para plataformas emergentes, como la PCR digital de gotas (ddPCR), incluidas solo cuando se consideran relevantes. Las recomendaciones más recientes se han centrado en las consideraciones para los ensayos de ddPCR, pero se han centrado en gran medida en sus aplicaciones a la cuantificación del genoma vectorial en un entorno de fabricación en lugar de en las complejas matrices biológicas encontradas en los estudios de biodesprendimiento.
Dependiendo de la aplicación clínica y los objetivos, se puede preferir la ddPCR a la qPCR en apoyo de los estudios de biodistribución y eliminación debido a la mayor sensibilidad y capacidad de la ddPCR para manejar la interferencia de la matriz en comparación con la qPCR. Además, debido a la partición de muestras en aproximadamente 20.000 gotas, se puede lograr una cuantificación precisa del número de copias sin el uso de una curva estándar utilizando estadísticas de Poisson, simplificando el desarrollo y la validación del método. El objetivo de este protocolo es describir un enfoque estandarizado para el desarrollo y validación de un método basado en ddPCR para la detección de vectores AAV en lágrimas recogidas de la superficie ocular en apoyo de estudios clínicos de biodesprendimiento.
Protocolo
1. Preparación de un fragmento de ADN sintético
- Diseñar y ordenar un fragmento de ADN sintético que contenga la región de amplificación objetivo para su uso como control de calidad.
- Asegúrese de que la secuencia contiene toda la secuencia de amplicón desde el cebador directo hasta el cebador inverso del gen objetivo de interés, con una extensión de cuatro a seis pares de bases de secuencia en los extremos 5' de cada secuencia de unión al cebador.
- Evite los homopolímeros de adenina y timina mayores de 12 pares de bases o los pares de bases de guanina y citosina mayores de ocho pares de bases, ya que los homopolímeros pueden interferir con la síntesis del fragmento del gen.
NOTA: Si los amplicones contienen tales secuencias, se pueden hacer sustituciones de bases siempre que se mantengan los sitios de recocido para los cebadores y sondas. - Alternativamente, prepare un plásmido linealizado que contenga el amplicón utilizando estrategias típicas de clonación.
- Centrifugar el tubo que contiene el fragmento de ADN sintético en una microcentrífuga durante ~10 s para asegurarse de que el material se recoge en la parte inferior del tubo.
- Resuspender el fragmento de ADN sintético utilizando tampón tris-EDTA (TE) hasta una concentración de 1,0 ×10 10 copias/μL, o según corresponda, según el intervalo de ensayo objetivo.
- Vórtice brevemente, luego incubar a 50 °C durante 20 ± 5 min. Enfriar sobre hielo.
- Preparar alícuotas múltiples, idealmente de un solo uso, y almacenar a -70 a -90 °C hasta su uso.
NOTA: Los fragmentos de ADN sintético preparados de esta manera suelen ser estables durante al menos 24 meses a partir de la fecha de resuspensión. - Si lo desea, determine la concentración exacta del stock de ADN sintético preparado antes de usarlo como control de calidad, o estime la concentración nominal en función de la resuspensión utilizada.
2. Preparación de cebadores y sonda
- Diseñar y ordenar cebadores y una sonda de hidrólisis para apuntar a la región de amplificación deseada utilizando estrategias de diseño típicas26,27.
- Utilice un tinte reportero fluorescente de 5' (por ejemplo, FAM) y un enfriador de 3' (por ejemplo, Iowa Black dark quencher) compatible con el sistema ddPCR.
NOTA: Existen numerosos paquetes de software de diseño de ensayos de PCR, y cualquiera puede ser utilizado. Por ejemplo, Primer-BLAST del Centro Nacional de Información Biotecnológica28 se usa ampliamente debido a las opciones robustas para el diseño de ensayos y la facilidad con la que se puede evaluar bioinformáticamente la especificidad para identificar posibles efectos fuera del objetivo. Cabe señalar que la preparación de cebadores y sondas puede variar de los pasos enumerados aquí dependiendo del formato en el que se suministran.
- Utilice un tinte reportero fluorescente de 5' (por ejemplo, FAM) y un enfriador de 3' (por ejemplo, Iowa Black dark quencher) compatible con el sistema ddPCR.
- Centrifugar los tubos que contienen el cebador directo, el cebador inverso y la sonda en una microcentrífuga durante ~10 s para granular el material hasta el fondo del tubo.
- Resuspender los cebadores a 20 μM utilizando tampón TE. Vórtice brevemente.
- Vuelva a suspender la sonda a 10 μM utilizando el búfer TE. Vórtice brevemente.
- Preparar múltiples alícuotas, idealmente de un solo uso, y almacenar a un mínimo de -20 °C hasta su uso.
NOTA: Los cebadores y sondas preparados de esta manera suelen ser estables durante al menos 24 meses a partir de la fecha de resuspensión.
3. Preparación del tampón de dilución de la muestra
- Descongelar el tampón de PCR y cortar el ADN del esperma de salmón a temperatura ambiente. Vórtice a fondo para mezclar.
- Prepare un tampón de dilución de muestra, según la Tabla 1.
- Vórtice a fondo. Conservar a 2-8 °C hasta 1 mes después de la preparación.
Tabla 1: Preparación del tampón de dilución de la muestra. Haga clic aquí para descargar esta tabla.
4. Preparación de la mezcla maestra
- Descongele la mezcla maestra de ddPCR para sondas, cebador directo, cebador inverso y sonda a temperatura ambiente y deje calentar durante al menos 10 minutos después de la descongelación antes de su uso. Guarde este producto a temperatura ambiente hasta su uso.
NOTA: Estos reactivos deben llevarse completamente a temperatura ambiente para garantizar una formación eficiente de gotas. No sostenga los reactivos en hielo durante la preparación.- Vortex centrifugar completa y brevemente en una mini centrífuga antes de su uso.
NOTA: Las enzimas de restricción generalmente se suministran en glicerol y deben retirarse del almacenamiento inmediatamente antes de su uso. Mezclar suavemente. No vorágice.
- Vortex centrifugar completa y brevemente en una mini centrífuga antes de su uso.
- Prepare una mezcla maestra de PCR para cada objetivo de amplificación. Consulte la Tabla 2 para obtener una composición de mezcla maestra de PCR sugerida y modificar las concentraciones de cebadores y sondas según sea necesario.
- Vórtice a fondo y centrifugar brevemente antes de la adición de la enzima de restricción. Añadir la enzima de restricción e invertir para mezclar.
NOTA: En este paso, se requieren 22 μL de reacción de PCR para obtener un volumen final de 40 μL de reacción de PCR después de la formación de gotitas (que consiste en 15 μL de mezcla maestra de PCR, 5.0 μL de plantilla y 20 μL de aceite de generación de gotas).
- Vórtice a fondo y centrifugar brevemente antes de la adición de la enzima de restricción. Añadir la enzima de restricción e invertir para mezclar.
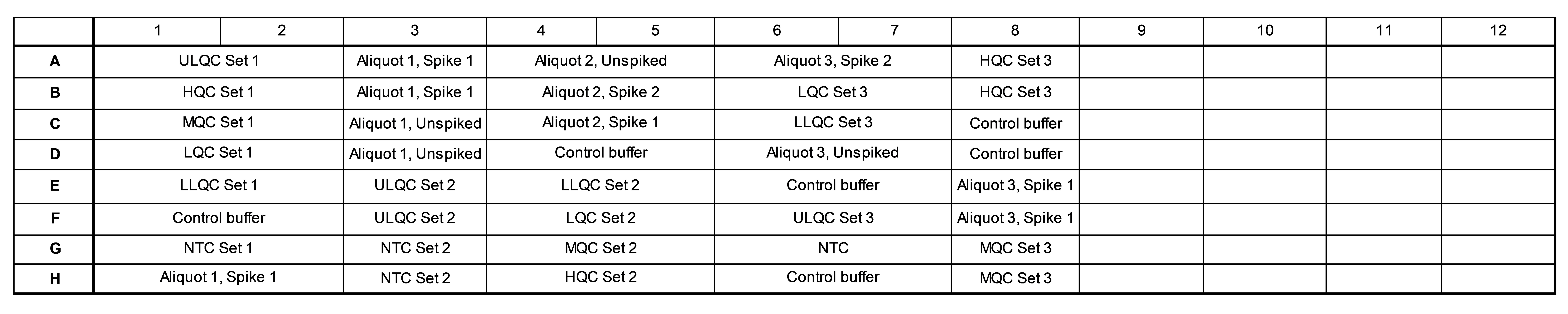
- Agregue 16.5 μL de mezcla maestra a cada pocillo de acuerdo con el mapa de placas. Consulte la figura 1 para ver un mapa de placas de ejemplo para obtener una ejecución de precisión y exactitud de validación.
- Asegúrese de que una placa contenga tres preparaciones independientes de la serie de control de calidad (QC), tres alícuotas de lágrimas endógenas probadas independientemente, clavadas a un nivel alto y bajo y sin púas, y tres controles independientes sin plantilla (NTC).
- Varíe el diseño de estos pocillos a través de la placa, donde el Conjunto 1 se carga en orden decreciente de concentración, el Conjunto 2 se carga en orden creciente de concentración y el Conjunto 3 se carga en un orden aleatorio para evaluar si hay algún efecto específico de la ubicación de la placa.
- Organice las muestras para llenar la mayor cantidad posible de una columna y llene los pozos no utilizados dentro de una columna con búfer de control. Incluya múltiples lotes de control endógenos (por ejemplo, más charcos de lágrimas o lágrimas recolectadas de individuos) en los pozos restantes, si lo desea.
- Selle la placa con una película adhesiva transparente. Mantenga la placa a temperatura ambiente durante la preparación de la plantilla. Alternativamente, sostenga la placa durante un máximo de 4 h a 2-8 °C, pero vuelva a ponerla a temperatura ambiente durante al menos 10 minutos antes de agregar la plantilla.
Tabla 2: Ejemplo de preparación de la mezcla maestra de PCR. Haga clic aquí para descargar esta tabla.

Figura 1: Ejemplo de mapa de placas para la validación de la exactitud y la ejecución de precisión. Abreviaturas: ULQC = control de calidad del límite superior; HQC = control de alta calidad; MQC = control de calidad medio; LQC = control de baja calidad; LLQC = control de calidad de límite inferior; NTC = sin control de plantilla. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
5. Preparación de QCs
- Descongele los fragmentos de ADN sintético o los plásmidos linealizados a temperatura ambiente y deje calentar durante al menos 10 minutos después de la descongelación antes de su uso. Lleve las plantillas a temperatura ambiente para garantizar una formación eficiente de gotas.
- Guarde este producto a temperatura ambiente hasta su uso. Vortex centrifugar completa y brevemente en una mini centrífuga antes de su uso.
- Prepare diluciones de control de calidad utilizando el tampón de dilución de la muestra como diluyente. En la Tabla 3 se presenta un ejemplo de las concentraciones recomendadas para prepararse para una validación de exactitud y precisión.
NOTA: Después de completar con éxito las ejecuciones de exactitud y precisión, solo se deben ejecutar en cada placa el control de alta calidad (HQC), el control de calidad media (MQC) y el control de baja calidad (LQC). Para la exactitud y las ejecuciones de precisión, se incluyen al menos tres diluciones independientes de los QC para la evaluación de la exactitud y precisión intraensayo. Después de las ejecuciones de exactitud y precisión, solo se debe incluir una serie de dilución. - Después de la preparación, guarde las diluciones a temperatura ambiente hasta que se agreguen al plato.
- Conservar las diluciones en hielo o a 2-8 °C si es necesario. Antes del uso posterior, deje que las diluciones se calienten a temperatura ambiente durante al menos 10 minutos antes de su uso. Deseche los QC al final del día.
Tabla 3: Ejemplo de preparación de control de calidad (QC) utilizando fragmentos sintéticos de ADN bicatenario. Abreviaturas: ULQC = control de calidad del límite superior; HQC = control de alta calidad; MQC = control de calidad medio; LQC = control de baja calidad; LLQC = control de calidad de límite inferior; NTC = sin control de plantilla. Haga clic aquí para descargar esta tabla.
6. Preparación de muestras
- Descongele las muestras de lágrimas recogidas de un ensayo clínico a temperatura ambiente hasta que se descongelen y deje calentar durante al menos 10 minutos después de la descongelación antes de su uso.
- Guarde este producto a temperatura ambiente hasta su uso. Centrifugar minuciosa y brevemente en una microcentrífuga antes de su uso.
- Diluya las muestras de lágrimas 1:10 (o más) utilizando tampón de dilución de la muestra como diluyente en tubos de PCR de 0,2 ml o tiras de PCR de 8 pocillos. Selle los tubos.
NOTA: Dependiendo de la concentración esperada de objetivo en lágrimas, puede ser necesario diluir aún más las muestras o probar múltiples diluciones de cada muestra. - Calentar las muestras en un termociclador a 95 °C durante 10 min, seguido de mantener a 4 °C durante al menos 5 minutos para que se enfríen. Utilice una velocidad de rampa de 3 °C/s.
NOTA: Las muestras pueden permanecer en el termociclador a 4 °C hasta su uso el mismo día o pueden congelarse a -70 a -90 °C para un almacenamiento más prolongado. Este paso sirve para desnaturalizar la cápside vectorial, liberando el genoma. Como los fragmentos de ADN sintético de control de calidad o los plásmidos linealizados son bicatenarios, no deben someterse a este paso de calentamiento. - Devuelva las muestras después del enfriamiento a temperatura ambiente (o si están congeladas, descongele a temperatura ambiente) y deje calentar durante al menos 10 minutos.
NOTA: Las muestras deben llevarse completamente a temperatura ambiente para garantizar la formación eficiente de gotas.
7. Adición de plantillas
- Recupere la placa ddPCR que contiene la mezcla maestra. Vórtice cada muestra o tubo de dilución de control de calidad a fondo y centrifugar brevemente para recoger el material.
- Retire la película adhesiva y agregue 5.5 μL de QC o muestras a los pocillos apropiados de la placa de 96 pocillos, según el mapa de placas.
NOTA: Consulte el paso 4.2.1 para obtener una explicación de los volúmenes requeridos - Agregue 5.5 μL de tampón de dilución de muestra a los pocillos NTC.
- La generación de gotitas requiere que todos los pocillos de una columna tengan una reacción o control de amortiguación. Si alguno de los pocillos de una columna no contiene reacciones de muestra, diluir el control tampón 2x ddPCR 1:2 utilizando agua libre de nucleasa. Agregue 22 μL de control de tampón ddPCR 1x a cualquier pocillo vacío de una columna.
NOTA: Si no se utiliza una columna completa, no es necesario agregar control de búfer a estos pozos. - Agregue un sello de aluminio perforable a la placa. Colocar la placa en el sellador de placas y sellar durante 5 s a 180 °C.
- Alternativamente, selle la placa de acuerdo con las recomendaciones del fabricante del sistema ddPCR.
- Realice el vórtice de la placa a la velocidad máxima durante al menos 30 s (utilizando el ajuste de vórtice continuo; no utilice el vórtice táctil) y centrifugar brevemente en un girador de placas.
NOTA: La mezcla completa y completa de la placa en este paso es fundamental para la partición adecuada de la reacción de PCR en gotas. Asegúrese de que no haya burbujas visibles en los pozos. Si es necesario, la placa se puede mantener a 2-8 °C antes de la generación de gotas durante un máximo de 4 h. Si se sostiene, deje que la placa alcance la temperatura ambiente durante un mínimo de 10 minutos antes de la generación de gotas.
8. Generación automatizada de gotas, ciclos térmicos y lectura de gotas
- Genere gotas en el generador de gotas automatizado de la siguiente manera.
- En la pantalla táctil, seleccione las columnas del mapa de placas que contienen muestras. La plataforma del instrumento se iluminará para indicar qué consumibles (cartuchos DG32, puntas, contenedor de residuos, aceite de generación de gotas) se requieren. Las luces amarillas indican que es necesario agregar un consumible, mientras que las luces verdes indican que hay suficientes consumibles disponibles.
- Cargue el generador de gotas de atrás hacia adelante.
- Para las sondas de hidrólisis, asegúrese de que el aceite de generación de gotas para sondas esté instalado y que quede suficiente aceite para el número de pozos. Si se utilizan productos químicos de PCR alternativos, asegúrese de que se instale un aceite de generación de gotas compatible.
- Coloque un bloque frío en el soporte de la placa de gotas. Asegúrese de que el bloque sea completamente de color azul y que no se vea ningún rosa. Coloque una nueva placa de ddPCR de 96 pocillos en el bloque frío.
- Coloque la placa de PCR preparada en el soporte de la placa de muestra. Cierre la tapa de la máquina. Presione inicio para la generación de gotas.
- Después de la formación de gotitas, un total de 40 μL por reacción se transfiere automáticamente a la nueva placa de PCR.
- Dentro de los 30 minutos posteriores a la finalización de la generación de gotas, retire la placa que contiene las gotas del bloque frío. Trabaje suavemente ya que las gotitas son más frágiles en esta etapa.
- Agregue un sello de aluminio perforable a la placa. Colocar la placa en el sellador de placas y sellar durante 5 s a 180 °C.
- Alternativamente, selle la placa de acuerdo con las recomendaciones del fabricante del sistema ddPCR.
- Coloque la placa en un termociclador compatible. Introduzca las condiciones de ciclismo (consulte la Tabla 4).
- Después del final del ciclo térmico, sostenga la placa en el termociclador, transfiera a 2-8 ° C o léala inmediatamente.
NOTA: Mantener la placa durante 12 h a 4-12 °C puede mejorar el recuento de gotitas, pero esto no es necesario. Se deben obtener suficientes gotas sin la retención. - Cargue la placa en el lector de gotas, asegurándose de que quede suficiente aceite del lector y que el contenedor de residuos tenga suficiente espacio. Lea las gotas. Realice la lectura de gotas dentro de las 24 h posteriores al inicio del ciclo térmico.
Tabla 4: Condiciones típicas de ciclos térmicos. Haga clic aquí para descargar esta tabla.
9. Análisis de datos
NOTA: Se necesita un mínimo de 10.000 gotas por pocillo para el cálculo adecuado de la concentración utilizando las estadísticas de Poisson. No intente analizar ningún pozo con menos de 10,000 gotas.
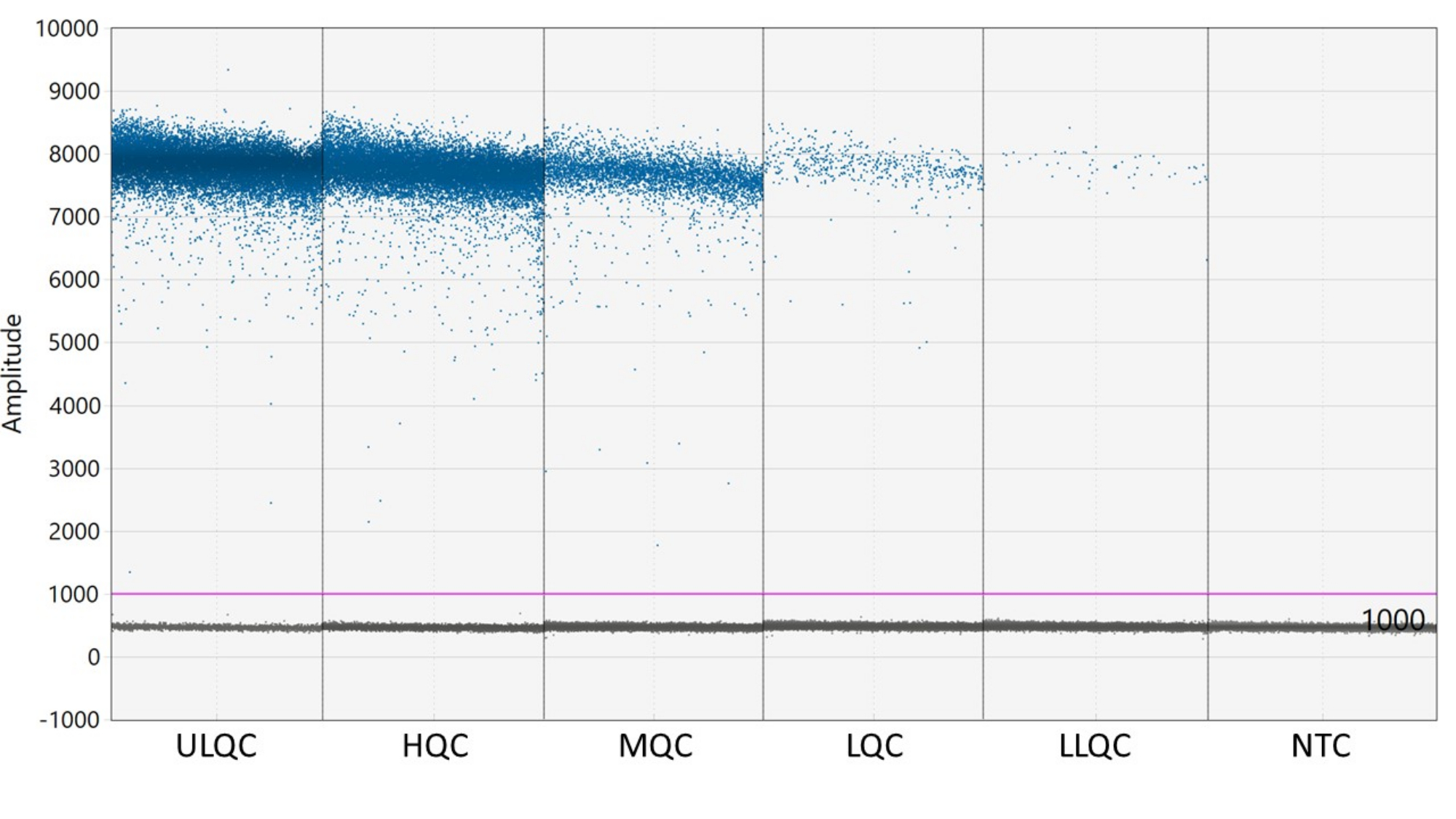
- Se requiere un umbral para definir las gotitas como positivas o negativas. El software de análisis ddPCR aplica automáticamente un umbral que puede variar entre pozos. Sin embargo, establezca manualmente un umbral para todos los pocillos de la placa ligeramente por encima de la intensidad fluorescente de los pocillos NTC para obtener resultados más consistentes, exactos y precisos.
NOTA: La colocación adecuada del umbral puede requerir una optimización dependiendo de la separación de las gotas positivas y negativas y de la cantidad de lluvia de gotas que existe (véase la figura 2). En este ejemplo, el gráfico de amplitud de gotas muestra pozos de ejemplo en cada nivel de control de calidad y el NTC. La línea púrpura indica un umbral de 1.000, establecido ligeramente por encima de la población de gotas negativas. - El modelado estadístico de Poisson requiere al menos tres gotas positivas para calcular la concentración con un 95% de confianza. Considere que todos los pozos que contienen cero, una o dos gotas positivas son negativos y se establecen en una concentración de cero27.
- Calcule el número de copias en cada muestra de lágrimas.
- La concentración, en copias/μL, se proporciona en el informe de datos. Utilice este valor para determinar la concentración en copias/μL de la muestra original (es decir, en la muestra de lágrimas).
- Para calcular la dilución de la reacción de ddPCR, divida el volumen de reacción inicial de PCR antes de la formación de gotitas por el volumen de plantilla agregado. Cuando se utilizan los volúmenes presentados en este método, esto produce un valor de 4.

- Determinar el factor de dilución en serie a partir de la muestra original (paso 6.2).
- Para determinar las copias/μL en la muestra, multiplique las copias/μL por la dilución de la reacción ddPCR y, a continuación, por el factor de dilución en serie. Por ejemplo, la concentración en copias/μl generada en el informe de datos fue de 966; Se agregaron 5,5 μL de plantilla por cada 22 μL de reacción. Se utilizó una dilución seriada de 1:50.000 de la muestra.


- Si se probaron múltiples diluciones de la misma muestra, analice todas las diluciones válidas dentro del rango y calcule la media.
- Para cada CC, calcular las copias esperadas/μL de reacción de PCR dividiendo la concentración de la dilución de CC dada (en copias/μL) por el volumen de reacción de ddPCR (20 μL). Esto permite la comparación directa de este valor nominal con el valor de copias/μL proporcionado en el informe de datos sin más cálculos.
NOTA: Este enfoque también se utilizó para el análisis de las muestras de lágrimas con púas utilizadas en los resultados representativos. - Determine el valor medio, la desviación estándar, el coeficiente de variación (%CV) y el porcentaje de error relativo a la concentración nominal (%RE) de la muestra o el valor de control de calidad utilizando los pocillos replicados (incluya diluciones múltiples si corresponde).
- Para la evaluación de la precisión entre pozos, determine esto para cada uno de los duplicados de pozos, si se incluye.
- Para la evaluación de la exactitud y precisión intraensayo, determinar esto para cada serie de dilución o alícuota utilizada dentro de un lote.
- Para la evaluación de la exactitud y precisión entre ensayos, determinar esto utilizando los medios intraensayo de cada uno de los lotes incluidos.

Figura 2: Ejemplo de establecimiento de umbral. Abreviaturas: ULQC = control de calidad del límite superior; HQC = control de alta calidad; MQC = control de calidad medio; LQC = control de baja calidad; LLQC = control de calidad de límite inferior; NTC = sin control de plantilla. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
10. Criterios de aceptación del ensayo
- Utilice las siguientes especificaciones para los datos calculados para cada lote para determinar si el lote es aceptable. Si no se cumplen estas condiciones, invalide y repita el lote.
NOTA: Estos criterios se determinaron como un consenso a partir de los libros blancos publicados sobre la validación de ensayos basados en PCR 18,19,20,21,22,23,24,25. Puede ser necesario modificar los criterios objetivo según sea apropiado para la aplicación clínica. - Sin control de plantilla (NTC)
- Asegúrese de que cada pozo NTC tenga al menos 10.000 gotas.
- Asegúrese de que cada pocillo NTC tenga menos de 3 gotas positivas.
- Control de calidad y rango de ensayo
- Asegúrese de que cada pozo de control de calidad tenga al menos 10,000 gotas.
- Asegúrese de que la precisión de los pozos replicados de una concentración de control de calidad sea de ≤25.0% CV, excepto en los límites superior e inferior de cuantificación, donde ≤30.0% es aceptable. Evalúe esto de forma independiente para cada conjunto de control de calidad y nivel de concentración.
- Asegúrese de que el error relativo de la concentración calculada hacia atrás en cada nivel medio de CC esté dentro de ±25,0% RE de la concentración nominal (copias/reacción PCR), excepto en los límites superior e inferior de cuantificación, donde ±30,0% RE es aceptable. Evalúe esto de forma independiente para cada conjunto de control de calidad y nivel de concentración.
- Asegúrese de que al menos 2/3 de las muestras de control de calidad (por ejemplo, cuatro de seis resultados) y el 50% de las muestras de control de calidad en cada nivel (bajo, medio, alto) cumplan con estas pautas.
- Muestras
- Asegúrese de que los pozos de muestra a analizar tengan al menos 10,000 gotas.
- Asegúrese de que la precisión de los pocillos replicados de una dilución de muestra a analizar sea de ≤25.0% CV.
- Asegúrese de que al menos una dilución incluida de la muestra dada esté dentro del rango de cuantificación definido del ensayo, como se definió anteriormente en función de los QC límite superior e inferior.
- Si todas las diluciones incluidas producen resultados superiores al límite superior definido de cuantificación, y si queda un volumen de muestra suficiente, repita el ensayo utilizando una dilución más alta de la muestra.
- Si todas las diluciones incluidas producen un resultado inferior al límite inferior de cuantificación, y si queda un volumen de muestra suficiente, repita el ensayo utilizando una dilución inferior de la muestra.
NOTA: Las muestras que contienen más de tres gotas positivas, pero que tienen una concentración por debajo del límite inferior de cuantificación, pueden describirse como detectables, pero no cuantificables.
Resultados
Con fines demostrativos, se desarrolló un ensayo diseñado para detectar un vector AAV2 que expresa la proteína fluorescente verde mejorada (eGFP) disponible comercialmente, con un fragmento de ADN bicatenario sintético que contiene eGFP como control de calidad. Actualmente, existe un debate en curso sobre si el vector en sí mismo o un fragmento de ADN sintético o plásmido linealizado es el más apropiado para su uso como QC. Generalmente, se puede usar un fragmento de ADN sintético o un plásmido linealizado si se demuestra la equivalencia con el vector en el desarrollo del método (datos no mostrados). Se diseñaron y optimizaron cebadores y sondas para detectar el transgén eGFP. Consulte la Tabla Suplementaria S1 para las secuencias utilizadas en este trabajo. La concentración del stock de fragmentos de CC se determinó empíricamente mediante ddPCR. Todos los ensayos se realizaron utilizando las concentraciones y las condiciones de PCR dadas como ejemplos en la sección de protocolo.
Para los ensayos de qPCR, se recomienda evaluar la linealidad, sensibilidad, rango dinámico, exactitud y precisión de la curva estándar. Dado que la ddPCR no se basa en una curva estándar para la cuantificación del objetivo, estas recomendaciones deben modificarse. En cambio, se utilizaron QC que consisten en fragmentos sintéticos de ADN bicatenario diluidos a varias concentraciones para abarcar el rango cuantificable esperado de una reacción ddPCR basada en el modelo estadístico de Poisson 29,30,31,32 para definir el rango dinámico y la sensibilidad y para evaluar la exactitud y la precisión. La elección de las concentraciones de control de calidad se basó principalmente en la proporción esperada de gotas positivas a totales dentro de un pocillo a una concentración dada. Matemáticamente, la ddPCR es teóricamente más precisa cuando aproximadamente el 80% de las particiones se amplifican positivamente. A medida que la relación de gotas positivas a totales aumenta por encima de 0,8, la precisión disminuye debido a la saturación de las particiones, y la cuantificación no es posible una vez que el 100% de las gotas son positivas. En el extremo inferior, teóricamente, se puede detectar y cuantificar tan solo una gota positiva, aunque la precisión es menor y el ensayo está sujeto a falsos positivos de bajo nivel. Por lo general, al menos tres gotitas deben ser positivas para que un resultado se calcule con un 95% de confianza, que es el umbral para calcular una concentración que usamos aquí.
Se preparó una serie de cinco concentraciones de CC diferentes, y se espera que el número de copias objetivo/μL de volúmenes de reacción de PCR calculados produzca relaciones positivas a gotas totales que abarcan el rango cuantificable de ddPCR, como se muestra en la Tabla 5. Estos se utilizaron para evaluar la exactitud y precisión del ensayo. En la evaluación aquí, los límites superior e inferior de cuantificación no se llevaron al máximo teórico posible en ddPCR. La cuantificación precisa puede ser posible a niveles más altos y más bajos que los demostrados aquí. La gama debe desarrollarse en consonancia con las aplicaciones posteriores de este método.
Se prepararon un total de tres series de dilución preparadas independientemente de estos CC en tampón de dilución de muestra para cada lote para evaluar la exactitud y precisión intraensayo. Se incluyeron pocillos duplicados de cada dilución de control de calidad. Para simular un protocolo de validación real, varios analistas realizaron un total de seis lotes de exactitud y precisión durante varios días. Los resultados de estos seis lotes se analizaron para definir la exactitud intraensayo e interensayo y la precisión del método y para definir el rango dinámico del ensayo.
El rendimiento intraensayo se evaluó para cada lote en cada nivel de control de calidad. Esperábamos que todos los pozos de control de calidad y NTC tuvieran al menos 10.000 gotas. Esto se cumplió en 216 de los 216 pozos probados en los seis lotes, con un recuento promedio de gotas de 19,748 gotas / pocillo (Tabla 6). A continuación, se esperaba que el %CV entre pozos de cada conjunto de pozos duplicados de cada QC fuera de ≤25,0%, excepto para el límite superior e inferior de QC, donde se esperaba un ≤30,0%. Esto se cumplió en conjuntos de 90 de los 90 pozos probados en los seis lotes para los QC, con un promedio de %CV entre pozos de 3.9% en todos los niveles de QC (Tabla 7). Todos los QC produjeron relaciones medias positivas a gotas totales dentro de los rangos esperados descritos anteriormente (Tabla 6).
Dentro de cada lote, se calcularon la media intraensayo y la desviación estándar para cada uno de los puntos de la serie de dilución preparados de forma independiente, y se utilizaron para calcular una media intraensayo para cada concentración en cada ensayo. Esto se utilizó para evaluar la exactitud y precisión del ensayo (Tabla 8). La precisión se refiere a la variabilidad en los datos de las réplicas de la misma muestra homogénea en condiciones normales de ensayo y se evalúa calculando el %CV de las múltiples alícuotas incluidas. Se esperaba que las tres alícuotas probadas dentro de cada lote produjeran un %CV intraensayo ≤25,0%, excepto para el límite superior e inferior de CC, donde se esperaba un ≤30,0%. Esto se cumplió para los cinco niveles de control de calidad en cada uno de los 60 lotes (30 de 30 actuaciones totales). En general, se pudo lograr una mayor precisión intraensayo que los criterios objetivo, con un %CV intraensayo medio del 7,7% en todos los niveles de control de calidad. La precisión se refiere a la cercanía de acuerdo entre el valor determinado experimentalmente y el valor nominal. Esto se evalúa calculando el porcentaje de error relativo (%RE o %Bias) entre las concentraciones calculadas de cada QC y sus concentraciones nominales teóricamente esperadas. Se esperaba que la media intraensayo de las tres alícuotas fuera de ±25,0% RE de la concentración nominal, excepto para el límite superior e inferior de CC, donde se esperaba un ±30,0%. Esto se cumplió para los cinco niveles de control de calidad en cada uno de los 60 lotes (30 de 30 actuaciones totales). En general, se pudo lograr una mayor precisión intraensayo que nuestro objetivo, con un % de ER intraensayo absoluto medio del 4,2% en todos los niveles de control de calidad. En todas las actuaciones del NTC (30 en total), no se detectaron gotitas positivas.
La exactitud y la precisión entre ensayos también se calcularon utilizando la media intraensayo de cada nivel de control de calidad dentro de cada lote. Se esperaba que la precisión entre ensayos fuera de ≤25,0% CV, excepto para el límite superior e inferior de control de calidad, donde se esperaba ≤30,0%. Asimismo, para la precisión entre ensayos, se esperaba una RE de ±25,0%, excepto para el límite superior e inferior de CC donde se esperaba ±30,0%. Se observó una exactitud y precisión entre ensayos significativamente mayor que estos objetivos (Tabla 9), con una precisión entre ensayos que varió de 4,0% a 8,5% y una exactitud absoluta entre ensayos que varió de 1,0% a 3,2%. En conjunto, estos resultados demuestran que este método puede lograr suficiente exactitud y precisión dentro y entre ensayos dentro de los objetivos actuales de la industria. Se puede definir un rango dinámico de este ensayo de 2.500-2,5 copias por μL de reacción de PCR en función de estos resultados, con una sensibilidad general del ensayo de 2,5 copias por μL de reacción de PCR. Como se mencionó anteriormente, puede ser posible validar rangos dinámicos más amplios.
A continuación, fue necesario evaluar la exactitud y la precisión del ensayo dentro de la matriz objetivo, en este caso, las lágrimas. Por lo general, los ensayos se validan antes del inicio de los estudios clínicos, lo que significa que es poco probable que las lágrimas recogidas de pacientes tratados con vectores estén disponibles para fines de validación. Esto se puede crear artificialmente clavando el vector AAV objetivo en lágrimas recolectadas de donantes voluntarios para crear QC con púas de matriz. Las lágrimas humanas agrupadas fueron recolectadas por un tercero (BioIVT). Como prueba de principio, se utilizó un vector AAV2 que expresa eGFP adquirido de una fuente comercial. La concentración del stock vectorial AAV2 se determinó empíricamente mediante ddPCR, sin el uso de un paso de aislamiento de ADN, como se describe en este protocolo. En cada ejecución, el AAV2 se introdujo de forma independiente en las tres alícuotas lagrimales a un nivel alto (reacción esperada de PCR de 1,41 x 103 copias/μL) y bajo (reacción de PCR de 28,2 copias/μL). Se incluyeron alícuotas sin púas como control para demostrar la especificidad del método.
El rendimiento intraensayo se evaluó para cada lote en cada nivel de pico. Se esperaba que todas las muestras de lágrimas tuvieran al menos 10.000 gotas. Esto se cumplió en 108 de los 108 pozos probados en los seis lotes, con un número total medio de gotas de 20.208 gotas / pocillo (Tabla 10). A continuación, se esperaba que el %CV entre pozos de cada conjunto de pozos duplicados de cada QC fuera de ≤25.0% para los niveles de pico alto y bajo. Esto se cumplió en 36 de los 36 conjuntos de pozos probados en los seis lotes para los QC, con un promedio de %CV entre pozos de 3.2% (Tabla 11).
Dentro de cada lote, se calcularon la media intraensayo y la desviación estándar para cada uno de los picos lagrimales preparados de forma independiente, y se utilizaron para calcular una media intraensayo para cada concentración en cada ensayo. Esto se utilizó para evaluar la exactitud y precisión del ensayo en matriz (Tabla 12). Se esperaba que el %CV intraensayo fuera del ≤25,0% y niveles de pico altos y bajos. Esto se cumplió en seis de los seis lotes para cada nivel. En general, se pudo lograr una mayor precisión intraensayo en matriz que el objetivo, con un %CV intraensayo medio del 3,7% en el nivel alto y del 12,2% en el nivel bajo (8,0% global). También se esperaba que el %ER intraensayo fuera del ±25,0% en ambos niveles de pico. Esto se cumplió en seis de los seis lotes para cada nivel. Del mismo modo, se encontró en general que se podía lograr una mayor precisión intraensayo en la matriz que el objetivo, con un %ER absoluto medio intraensayo del 8,1% en el nivel bajo y del 11,3% en el nivel alto (9,7% global). Para el control sin picos, no se detectó ninguna señal de eGFP en ninguna de las alícuotas (Tabla 12), lo que demuestra la especificidad del método en la matriz lagrimal humana.
La exactitud entre ensayos y la precisión en la matriz lagrimal también se calcularon utilizando la media intraensayo de cada nivel de pico dentro de cada lote. Se esperaba que la precisión entre ensayos fuera ≤25,0% CV, y para la exactitud entre ensayos, se esperaba ±25,0% RE. Se observó una exactitud y precisión entre ensayos significativamente mayor que estos objetivos (Tabla 13), con una precisión entre ensayos del 5,5% en el nivel alto y del 7,1% en el nivel bajo, y con una exactitud absoluta entre ensayos del 11,3% en el nivel alto y del 8,1% en el nivel bajo. En conjunto, estos resultados demuestran la exactitud, precisión y especificidad del método en la matriz de lágrimas.
Tabla 5: Controles de calidad utilizados para definir el rango dinámico del ensayo. Abreviaturas: ULQC = control de calidad del límite superior; HQC = control de alta calidad; MQC = control de calidad medio; LQC = control de baja calidad; LLQC = control de calidad de límite inferior; NTC = sin control de plantilla. Haga clic aquí para descargar esta tabla.
Tabla 6: Recuentos totales de gotitas y proporciones positivas a totales de gotas de control de calidad de ADN bicatenario sintético y NTC. Abreviaturas: ULQC = control de calidad del límite superior; HQC = control de alta calidad; MQC = control de calidad medio; LQC = control de baja calidad; LLQC = control de calidad de límite inferior; NTC = sin control de plantilla. Haga clic aquí para descargar esta tabla.
Tabla 7: Estadísticas de QC entre pozos (objetivos de copia/reacción de PCR μL). Abreviaturas: ULQC = control de calidad del límite superior; HQC = control de alta calidad; MQC = control de calidad medio; LQC = control de baja calidad; LLQC = control de calidad de límite inferior; NTC = sin control de plantilla. Haga clic aquí para descargar esta tabla.
Tabla 8: Exactitud intraensayo y precisión de los QC (objetivos de copia/reacción de PCR de μL). Abreviaturas: ULQC = control de calidad del límite superior; HQC = control de alta calidad; MQC = control de calidad medio; LQC = control de baja calidad; LLQC = control de calidad de límite inferior; NTC = sin control de plantilla. Haga clic aquí para descargar esta tabla.
Tabla 9: Exactitud entre ensayos y precisión de los QC (objetivos de copia/reacción de PCR de μL). Abreviaturas: ULQC = control de calidad del límite superior; HQC = control de alta calidad; MQC = control de calidad medio; LQC = control de baja calidad; LLQC = control de calidad de límite inferior; NTC = sin control de plantilla. Haga clic aquí para descargar esta tabla.
Tabla 10: Recuentos totales de gotitas de muestras de lágrimas. Haga clic aquí para descargar esta tabla.
Tabla 11: Estadísticas de la muestra de lágrimas entre pocillos (objetivos de copia/reacción de PCR μL). Haga clic aquí para descargar esta tabla.
Tabla 12: Exactitud intraensayo y precisión de muestras de lágrimas (objetivos de copia/reacción de PCR μL). Haga clic aquí para descargar esta tabla.
Tabla 13: Exactitud entre ensayos y precisión de muestras de lágrimas (objetivos de copia/reacción de PCR μL). Haga clic aquí para descargar esta tabla.
Tabla suplementaria S1: Secuencias de cebadores, sondas y control de calidad de ADN bicatenario sintético utilizados en este estudio. Haga clic aquí para descargar esta tabla.
Discusión
Hay varios pasos del protocolo ddPCR que son críticos para el correcto desempeño del ensayo. El primer paso crítico es el diseño y la optimización de los cebadores y la sonda. En general, se recomienda el uso de química basada en sonda de hidrólisis sobre química basada en colorantes (por ejemplo, SYBR Green) en un entorno preclínico o clínico debido a su especificidad superior. Además, la elección del objetivo de amplificación es crítica. Típicamente, el transgén de interés del vector es el objetivo. Sin embargo, en etapas preclínicas más tempranas o en vectores donde puede no ser posible distinguir el transgén vectorial frente al ADN genómico, puede ser apropiado utilizar dianas vectoriales estandarizadas. Por ejemplo, uno podría apuntar a la región de repetición terminal invertida, promotor, cola poli-A o las uniones entre segmentos entre estos componentes vectoriales. La elección del objetivo variará según el diseño vectorial. Las estrategias y el software tradicionales de diseño de sondas y cebadores qPCR suelen ser apropiados para ddPCR. Los parámetros de diseño que se espera que produzcan una temperatura de recocido constante (por ejemplo, 60 °C) deben seleccionarse para reducir la cantidad de optimización requerida. También se ha recomendado diseñar, ordenar y evaluar al menos tres conjuntos diferentes para cada objetivo. A continuación, se debe seleccionar el conjunto que muestre la mayor especificidad (sin amplificación en el pozo de control negativo o en una matriz de ADN objetivo relacionado) y sensibilidad (es decir, límite de detección)20.
Si es ventajoso poder hacer la transición del ensayo entre qPCR y ddPCR, se recomienda optimizar primero las condiciones del ensayo utilizando qPCR e identificar condiciones para el conjunto seleccionado que resulten en eficiencias de amplificación del 90% -110% con un R2 ≥ 0.98. Sin embargo, la ddPCR como método de criterio de valoración suele ser menos sensible que la qPCR debido a las variaciones en las eficiencias de amplificación. Como mínimo, se recomienda ejecutar un gradiente de temperatura térmica en la etapa de recocido/extensión para cubrir temperaturas por encima y por debajo de las temperaturas de recocido esperadas y para evaluar la separación de amplitud de lluvia y fluorescente entre los grupos de gotas negativas y positivas en función de la temperatura. Si el espacio de trabajo lo permite, se recomienda tener estaciones de trabajo dedicadas individuales para la preparación de la mezcla maestra, la adición de plantillas y la amplificación. Siempre que sea posible, estos deben ser separados físicamente por un flujo de trabajo unidireccional con controles de ingeniería incorporados, como acceso controlado y presiones de aire diferenciales, para reducir el riesgo de contaminación cruzada y falsos positivos. Si esto no es posible, se debe tener extrema precaución para evitar la contaminación cruzada.
Hay dos pasos en este protocolo que pueden parecer inusuales para aquellos que están más acostumbrados al desarrollo de ensayos de qPCR. El primero es la inclusión de una enzima de restricción en la mezcla maestra de PCR. Durante la amplificación por ddPCR, cada gota se termocicla hasta el punto final. En un ensayo correctamente optimizado, esto da como resultado dos poblaciones de gotas, un conjunto que muestra un nivel consistentemente alto de señales fluorescentes, los positivos, y otro que muestra consistentemente un bajo nivel de señal fluorescente, los negativos. Si se produce interferencia de PCR, puede desincronizar el inicio de la amplificación por PCR, lo que hace que la gota no alcance una meseta de amplificación y, por lo tanto, puntos finales fluorescentes inconsistentes. En este caso, las gotitas se distribuirán entre los negativos y los positivos, lo que resulta en un fenómeno llamado lluvia ddPCR. Esto puede dar lugar a una cuantificación inexacta del objetivo y a umbrales aplicados de manera incoherente y subjetiva. Nuestra recomendación es establecer el umbral ligeramente por encima de la señal del NTC, lo que debería minimizar los efectos de la lluvia en la cuantificación final, ya que todas las gotas aún se consideran positivas, incluso si no se ciclan completamente hasta el punto final. Los AAV tienen una estructura secundaria altamente compleja que, dependiendo del objetivo de amplificación, puede reducir la accesibilidad a los cebadores y sondas, lo que resulta en interferencia de PCR y, por lo tanto, lluvia. La inclusión de la enzima de restricción en la mezcla maestra escinde esta estructura secundaria para aumentar el acceso de los cebadores y sondas, reduciendo la lluvia, lo que puede mejorar la precisión del ensayo. Los efectos de la inclusión de una enzima de restricción en la reacción ddPCR han sido descritos previamente25,32. Se puede usar cualquier enzima de restricción, siempre y cuando se confirme que no corta dentro de la región de amplificación objetivo. No se requieren pasos de predigestión o composiciones tampón alternativas.
El segundo paso inusual es la preparación de la muestra de lágrimas que contiene AAV. En este protocolo, se utilizó una proporción de lágrimas de 1:10 (o mayor) y posteriormente se calentó la muestra. Por lo general, cuando las lágrimas se recogen a través de un tubo capilar, que es un método de recolección ampliamente utilizado, en promedio se pueden recolectar aproximadamente 10.0 μL33. La dilución ayuda a abordar el volumen limitado de la muestra y proporciona suficiente material para la prueba de pozos duplicados. Si bien esto reduce el límite teórico de detección, la robusta sensibilidad de ddPCR aún debería resultar en la detección de todas las partículas vectoriales, excepto de muy pocas. Este enfoque también crea un pozo de "copia de seguridad" si uno fallara inesperadamente. En este caso, o en casos de volumen de muestra insuficiente para ejecutar dos pozos, el error de Poisson podría usarse para evaluar la precisión. Además, en los casos en que la concentración está por debajo del límite de detección, crea una oportunidad para combinar datos de pozos para determinar una concentración. Es necesario liberar los vectores AAV de las cápsides virales para la detección de ddPCR. Algunos métodos para la cuantificación de AAV han incluido un paso de digestión de proteinasa K para eliminar la cápside viral34,35,36. Todos los serotipos AAV naturales tienen temperaturas de fusión iguales o inferiores a aproximadamente 90 °C, y la mayoría cae por debajo de 80 °C; por lo tanto, esto parece ser una inclusión innecesaria37. El calentamiento por sí solo parece ser suficiente para liberar el ADN vectorial.
Además, la ddPCR es generalmente menos susceptible a los inhibidores de PCR que pueden estar presentes en una muestra que puede afectar a un ensayo de qPCR. Si se incluye un paso específico de aislamiento de ADN, esto también requeriría una validación específica, que se evita en este protocolo. Las muestras se diluyen antes del calentamiento debido a la cinética de difusión de los genomas vectoriales en un líquido. Durante el proceso de calentamiento y enfriamiento posterior, las hebras sensoriales positivas y negativas del genoma de ADN monocatenario pueden unirse para producir un intermedio bicatenario si las concentraciones son lo suficientemente altas. La dilución antes del calentamiento reduce las concentraciones y hace matemáticamente improbable que se formen suficientes productos intermedios bicatenarios para tener un efecto adverso en la precisión de la cuantificación. Cabe señalar que los fragmentos de ADN sintético o plásmidos linealizados utilizados como controles de calidad no deben someterse a este paso de calentamiento. Como estos son de doble cadena, el calentamiento daría como resultado la conversión a intermedios de una sola cadena. Después de la partición independiente de estos QC monocatenarios en gotas, se esperaría que esto resultara en un aumento de dos veces en la concentración de QC en relación con la concentración nominal. Alternativamente, si los CC deben calentarse para estandarizar el método, esto debe tenerse en cuenta en la reconstitución y asignación de una concentración nominal.
Finalmente, con respecto a la preparación de la muestra, muchos protocolos también recomiendan la inclusión de un paso de tratamiento de DNasa para eliminar cualquier ADN vector no encapsidado. Este paso es crítico en los casos en que no se desea cuantificar el ADN libre asociado con la preparación del vector (como durante la cuantificación para fines de dosificación). Sin embargo, en el contexto de los estudios de biodistribución y biodesprendimiento, uno típicamente desea saber dónde ha viajado cualquier ADN vectorial, independientemente de si está encapsidado o no. Por lo tanto, se sugiere típicamente no realizar un paso de tratamiento con DNasa durante dichos estudios. Si se determina que es necesario incluir un paso de DNasa, este paso debe ser previo a las diluciones y el calentamiento.
En este documento, se presentan datos representativos del enfoque para la evaluación del rango dinámico, la sensibilidad, la exactitud y la precisión del método en el contexto de una validación adecuada para el propósito y conforme a las buenas prácticas de laboratorio. La falta actual de orientación sobre este tema deja a los laboratorios de validación para determinar los criterios de ensayo objetivo por sí mismos, en línea con el pensamiento actual de la industria. Diferentes grupos han planteado criterios objetivo más altos y más bajos que los utilizados en este estudio 19,20,21,22,23,24,25. Los criterios del ensayo objetivo, hasta que se definan de forma más rígida, deben seleccionarse antes de la validación en función de las aplicaciones clínicas previstas del método. Dependiendo de las decisiones posteriores que se tomen sobre la base de los datos, pueden ser necesarios niveles más altos de exactitud y precisión. Por el contrario, un simple resultado positivo versus negativo puede ser suficiente.
El enfoque también abordó recomendaciones para la evaluación de la especificidad y un efecto matricial. Un conjunto de lágrimas recogidas de individuos no tratados no produjo un resultado positivo en este ensayo, mientras que el objetivo pudo detectarse cuando el vector se clavó en las lágrimas a una concentración alta y baja dentro de las tasas de recuperación recomendadas. Idealmente, la matriz que contiene vector endógeno (por ejemplo, recolectado después del tratamiento con vector viral) también se incluiría en estas evaluaciones. Sin embargo, es poco probable que tales muestras estén disponibles para su uso en una validación. Para aumentar la solidez de la validación, se pudieron evaluar múltiples grupos de lágrimas, o lágrimas recolectadas de una variedad de individuos, para determinar si se produce un efecto de matriz específico del paciente. Finalmente, se recomienda evaluar la estabilidad. En flujos de trabajo donde la extracción de ADN ocurre fuera de la matriz biológica, puede ser necesario evaluar la estabilidad tanto de la muestra como del ADN extraído. En este flujo de trabajo, la muestra se prueba directamente en el ensayo sin necesidad de extracción de ADN. Por lo tanto, en consideración de la evaluación de la estabilidad para este método, se debe evaluar la estabilidad de las muestras de lágrimas. Por lo general, se recomiendan evaluaciones de sobremesa, refrigerador, congelación / descongelación y estabilidad a largo plazo. Estos no se realizaron como parte de este estudio, pero los métodos desarrollados aquí pueden ser utilizados en esta evaluación, después de manipulaciones a las muestras de entrada.
En general, se ha demostrado que este método es un ensayo robusto, repetible y validable para detectar vectores basados en AAV en muestras de lágrimas. Puede servir como plataforma para adaptarse a vectores específicos para apoyar los ensayos clínicos y proporciona una base para la validación de un ensayo coherente con las buenas prácticas de laboratorio.
Divulgaciones
Todos los autores son empleados de KCAS Bioanalytical and Biomarker Services, una organización de investigación por contrato que proporciona servicios integrales de desarrollo que cumplen con las Buenas Prácticas de Laboratorio / Buenas Prácticas Clínicas desde el soporte de descubrimiento temprano hasta el registro del producto, incluido el soporte de ensayos basados en AAV, como se describe en este protocolo. KCAS financió este proyecto y la publicación de este protocolo. Si bien no existen pautas actuales de la FDA para el diseño y la validación de los experimentos presentados en este documento, los expertos y líderes de opinión que desarrollan dichos estudios en KCAS han adoptado este enfoque como el enfoque de referencia. Los criterios y parámetros adicionales se discuten proyecto por proyecto y pueden incluirse en función del uso previsto.
Agradecimientos
Nos gustaría agradecer a Nick Russell y Brandon McKethan de Bio-Rad por sus útiles discusiones durante el desarrollo de este método.
Materiales
| Name | Company | Catalog Number | Comments |
| AAV-eGFP Vector | Charles River Laboratories | RS-AAV2-FL | Lot AAV2-0720-FL, used as a proof of principle vector |
| AutoDG Droplet Digital PCR system | Bio-Rad | QX200 | Alternative ddPCR system may be used following manufacturer’s protocol. |
| AutoDG Oil for Probes | Bio-Rad | 1864110 | Or use material compatible with ddPCR system. |
| ddPCR Buffer Control for Probes | Bio-Rad | 1863052 | Or use material compatible with ddPCR system and PCR chemistry. |
| ddPCR Droplet Reader Oil | Bio-Rad | 1863004 | Or use material compatible with ddPCR system. |
| ddPCR Piercable Foil Seals | Bio-Rad | 1814040 | Or use material compatible with ddPCR system. |
| ddPCR Plates 96-Well, Semi-Skirted | Bio-Rad | 12001925 | Or use material compatible with ddPCR system. |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023, 1863024, or 1863025 | Use master mix compatible with primers/probes and ddPCR system. |
| DG32 AutoDG Cartidges | Bio-Rad | 1864108 | Or use material compatible with ddPCR system. |
| Droplet Reader | Bio-Rad | QX200 | Alternative ddPCR system may be used following manufacturer’s protocol. |
| GeneAmp PCR Buffer | Applied Biosystems | N8080129 | N/A |
| Nuclease-Free Water | Ambion | AM9906 | N/A |
| PCR Plate Sealer | Bio-Rad | PX1 | Or use material compatible with ddPCR system. |
| Pipet Tips for AutoDG | Bio-Rad | 1864120 | Or use material compatible with ddPCR system. |
| Pluronic F-68 Non-ionic Surfactant | Gibco | 24040 | N/A |
| Primer and Hydrolysis Probes | Various | Various | Design based on target sequence using general approaches for primer/probe design. Select fluorphores and quenchers compatible with ddPCR system. |
| Restriction Enzyme | Various | Various | Varies with target amplification sequence. Use restriction enzyme that does not cut in the amplified sequence |
| Sheared salmon sperm DNA | ThermoFisher | AM9680 | N/A |
| Synthetic DNA gene fragment or linearized plasmid | Various | Various | Design a synthetic DNA fragment containing the target amplification region for use as a quality control |
| TE Buffer | Teknova | T0224 | Ensure prepared or purchases nuclease free. 10 mM Tris-HCl, 1.0 mM EDTA, pH=8.0 |
| Touch Thermal Cycler | Bio-Rad | C1000 | Or use material compatible with ddPCR system. |
Referencias
- Sherkow, J. S., Zettler, P. J., Greeley, H. T. Is it 'gene therapy. Journal of Law and the Biosciences. 5 (3), 786-793 (2018).
- Ginn, S. L., Amaya, A. K., Alexander, I. E., Edelstein, M., Abedi, M. R. Gene therapy clinical trials worldwide to 2017: an update. The Journal of Gene Medicine. 20 (5), 3015 (2018).
- Ghosh, S., Brown, A. M., Jenkins, C., Campbell, K. Viral vector systems for gene therapy: a comprehensive literature review of progress and biosafety challenges. Applied Biosafety. 25 (1), 7-18 (2020).
- Gene, Cell, & RNA therapy landscape, Q4 2022 quarterly data report. American Society for Gene & Cell Therapy Available from: https://asgct.org/global/documents/asgct_citeline-q4-2022-report_final.aspx (2022)
- Approved Cellular and Gene Therapy Products. United States Food and Drug Administration Available from: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (2022)
- Au, H. K. E., Isalan, M., Meilcarek, M. Gene therapy advances: a meta-analysis of AAV usage in clinical settings. Frontiers in Medicine. 8, 809118 (2022).
- Lundstrom, K. Viral vectors in gene therapy. Diseases. 6 (2), 42 (2018).
- Naso, M. F., Tomkowicz, B., Perry, W. L., Strohl, W. R. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 31, 317-334 (2017).
- Russell, S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomized, controlled, open-label, phase 3 trial. Lancet. 390 (10097), 849-860 (2017).
- Chemistry, manufacturing, and control (CMC) information for human gene therapy investigational new drugs (INDs). United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/chemistry-manufacturing-and-control-cmc-information-human-gene-therapy-investigationsal-new-drug (2020)
- Long term follow-up after administration of human gene therapy products. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/long-term-follow-after-adminstration-human-gene-therapy-products (2020)
- Testing of retroviral vector-based human gene therapy products for replication competent retrovirus during product manufacture and patient follow-up. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/testing-retroviral-vector-based-human-gene-therapy-products-replication-competent-retrovirus-during (2020)
- Recommendations for microbial vectors used in gene therapy. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/recommendations-microbial-vectors-used-gene-therapy (2016)
- Multidisciplinary: gene therapy. European Medicines Agency Available from: https://www.europa.eu/en/human-regulatory-development/scientific-guidelines/multidisciplinary/multidisciplinary-gene-therapy (2023)
- Human gene therapy for retinal disorders. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-retinal-disorders (2020)
- Design and analysis of shedding studies for virus or bacterial-based gene therapy and oncolytic products. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-and-analysis-shedding-studies-virus-or-bacteria-based-gene-therapy-and-oncolytic-products (2015)
- Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products. European Medicines Agency Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-clinical-aspects-gene-therapy-medicinal-products_en.pdf (2018)
- Kaur, S. white paper on recent issues in bioanalysis: mass spec of proteins, extracellular vesicles, CRISPR, chiral assays, oligos; nanomedicines bioanalysis; ICH M10 section 7.1; non-liquid & rare matrices; regulatory inputs (part 1A - recommendations on endogenous compounds, small molecules, complex methods, regulated mass spec of large molecules, small molecule, PoC & part 1B - regulatory agencies' inputs on bioanalysis, biomarkers, immunogenicity, gene & cell therapy and vaccine). Bioanalysis. 14 (9), 505-580 (2022).
- Hays, A., Islam, R., Matys, K., Williams, D. Best practices in qPCR and qPCR validation in regulated bio analytical laboratories. The AAPS Journal. 24 (2), 36 (2022).
- Ma, H., Bell, K. N., Loker, R. N. qPCR and qRT-PCR analysis: Regulatory points to consider when conducting biodistribution and vector shedding studies. Molecular Therapy. Methods & Clinical Development. 17 (20), 152-168 (2020).
- Wissel, M. Recommendations on qPCR/ddPCR assay validation by GCC. Bioanalysis. 14 (12), 853-863 (2022).
- Expectations for biodistribution (BD) assessments for gene therapy (GT) products. International Pharmaceutical Regulators Programme Available from: https://admin.iprp.global/sites/default/files/2018-09/IPRP_GTWG_ReflectionPaper_BD_Final_2018_0713.pdf (2018)
- Pinheiro, L., Emslie, K. R. Basic concepts and validation of digital PCR measurements. Methods in Molecular Biology. 1768, 11-24 (2018).
- Tzonev, S. Fundamentals of counting statistics in digital PCR: I measured two target copies-what does it mean. Methods in Molecular Biology. 1768, 25-43 (2018).
- Prantner, A., Marr, D. Genome concentration, characterization, and integrity analysis of recombinant adeno-associated viral vectors using droplet digital PCR. PLoS One. 18, 0280242 (2023).
- Primer-BLAST. National Library of Medicine, National Center for Biotechnology Information Available from: https://www.ncbi.nim.nih.gov/tools/primer-blast/ (2023)
- Koressaar, T., Remm, M. Enhancements and modifications of primer design program Primer 3. Bioinformatics. 23 (10), 1289-1291 (2007).
- Ye, J. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 13, 134 (2012).
- Droplet Digital PCR Application Guide. Bio-Rad Available from: https://www.bio-rad.com/webroot/we/pdf/lsr/literature/Bulletin_6407.pdf (2023)
- Qian, P. L., Sauzade, M., Brouzes, E. dPCR: A technology review. Sensors. 18 (4), 1271 (2018).
- Basu, A. Digital assays part I: portioning statistics and digital PCR. SLAS Technology. 22 (4), 369-386 (2017).
- Sanmiguel, J., Gao, G., Vandeberghe, L. H. Quantitative and digital droplet-based AAV genome titration. Methods in Molecular Biology. 1950, 51-83 (2019).
- Bachhuber, F., Huss, A., Senel, M., Tumani, H. Diagnostic biomarkers in tear fluid: from sampling to preanalytical processing. Scientific Reports. 11, 10064 (2021).
- Martinez-Fernandez de la Camara, C., McClements, M. E., MacLaren, R. E. Accurate quantification of AAV vector genomes by quantitative PCR. Genes. 12 (4), 601 (2021).
- Ai, J., Ibraheim, R., Tai, P. W. L., Gao, G. A scalable and accurate method for quantifying vector genomes of recombinant adeno-associated viruses in crude lysate. Human Gene TherapyMethods. 28 (3), 139-147 (2017).
- Dobnik, D., et al. Accurate quantification and characterization of adeno-associated viral vectors. Frontiers in Microbiology. 10, 1570 (2019).
- Bennett, A., et al. Thermal stability as a determinant of AAV serotype identity. Molecular Therapy Methods and Clinical Development. 6, 171-182 (2017).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados