
Method Article
Ein validierbarer digitaler Tröpfchen-Polymerase-Kettenreaktions-Assay für den Nachweis von Adeno-assoziierten viralen Vektoren in Bioshedding-Studien an Tränen
In diesem Artikel
Zusammenfassung
In dieser Arbeit stellen wir ein Protokoll für die Entwicklung und Validierung guter Laborpraktiken bei der konformen Detektion von Adeno-assoziierten viralen Vektoren in menschlichen Tränen durch digitale Polymerase-Tröpfchenkettenreaktion zur Unterstützung der klinischen Entwicklung von Gentherapievektoren vor.
Zusammenfassung
Der Einsatz von viralen Vektoren zur Behandlung genetischer Krankheiten hat in den letzten Jahren erheblich zugenommen, mit bisher über 2.000 registrierten Studien. Adeno-assoziierte virale (AAV) Vektoren haben besondere Erfolge bei der Behandlung von Augenerkrankungen erzielt, wie die Zulassung von Voretigene Neparvovec-rzyl zeigt. Um neue Therapien auf den Markt zu bringen, verlangen die Zulassungsbehörden in der Regel qualifizierte oder validierte Bioshedding-Studien, um die Freisetzung des Vektors in die Umwelt zu bewerten. Die United States Food and Drug Administration hat jedoch keine offiziellen Richtlinien für die Entwicklung molekularer Assays zur Unterstützung solcher Shedding-Studien veröffentlicht, so dass die Entwickler die Best Practices für sich selbst bestimmen müssen. Ziel dieses Protokolls ist es, ein validierbares Protokoll für den Nachweis von AAV-Vektoren in menschlichen Tränen mittels digitaler Polymerase-Kettenreaktion (ddPCR) zur Unterstützung klinischer Bioshedding-Studien zu präsentieren. Dieses Manuskript diskutiert aktuelle Industrieansätze zur Validierung molekularer Assays und zeigt, dass die Methode die derzeit in White Papers vorgeschlagenen Akzeptanzkriterien für Zielassays übertrifft. Abschließend werden Schritte erörtert, die für die Durchführung eines ddPCR-Assays entscheidend sind, unabhängig von der Anwendung.
Einleitung
Die Definitionen der Gentherapie variieren, induzieren aber im Allgemeinen eine absichtliche und oft erwartete dauerhafte Veränderung einer bestimmten DNA-Sequenz des zellulären Genoms, um die Expression eines Gens zu modifizieren oder zu manipulieren oder die biologischen Eigenschaften einer lebenden Zelle für einen klinischen Zweck zu verändern 1,2. Virale Vektoren werden aufgrund ihrer Transduktionseffizienz zunehmend als Vehikel für die Gentherapie verwendet, wobei ein Bericht darauf hindeutet, dass über 70 % der aktuellen klinischen Studien zur Gentherapie virale Vektoren verwenden3. Das Interesse an viralen Vektoren für die Gentherapie nimmt stetig zu. Der vierteljährliche Datenbericht der American Society of Gene and Cell Therapy für das 4. Quartal 2022 über die Gen-, Zell- und RNA-Therapielandschaft berichtet, dass im Jahr 2022 die Gen-, Zell- und RNA-Therapie-Pipeline von der Präklinik bis zur Vorregistrierung um 7 % gewachsen ist, womit sich die Gesamtzahl der in der Entwicklung befindlichen Therapien auf 3.726 erhöht, von denen 2.053 (55 %) Gentherapien waren4. Die US-amerikanische Food and Drug Administration (FDA) hat derzeit 27 Zell- und Gentherapien für die klinische Anwendung beim Menschen zugelassen, von denen fünf speziell virale Vektoren verwenden5.
Adeno-assoziierte Viren (AAVs) haben als Vehikel für die Gentherapie besonderes Interesse geweckt. Eine kürzlich durchgeführte Metaanalyse ergab, dass es in den letzten zwei Jahrzehnten etwa 136 klinische Studien gab, in denen die Verwendung von AAVs untersucht wurde6. Darüber hinaus basieren drei der fünf von der FDA in den USA zugelassenen Gentherapien auf AAV-Basis. Dies liegt an ihrer hochgradig editierbaren Natur, ihrem breiten Wirtsspektrum, das auf der Grundlage der Verwendung spezifischer natürlich vorkommender oder künstlich hergestellter Vektoren abgestimmt werden kann, ihrer geringen Pathogenität und Toxizität beim Menschen und ihrer allgemein geringen Immunogenität 7,8. AAVs wurden auch erfolgreich zur Behandlung von Augenerkrankungen in einem zugelassenen klinischen Umfeld eingesetzt. Voretigene Neparvovec-rzyl ist eine AAV2-basierte Therapie, die 2017 von der US-amerikanischen FDA und 2018 von der Europäischen Arzneimittel-Agentur (EMA) zur Behandlung der mit der ballelischen RPE65-Mutation assoziierten Netzhautdystrophie zugelassen wurde9.
Mit dem zunehmenden Interesse an der Entwicklung von AAV-basierten Therapien steigt auch der Bedarf an regulatorischen Leitlinien für Assays. Der genaue Nachweis und die Quantifizierung eines viralen Vektors ist ein integraler Bestandteil der Entdeckungs-, Herstellungs- und präklinischen/klinischen Testphasen der Produktentwicklung. Die US-amerikanische FDA hat damit begonnen, einige Leitlinien für Gentherapien herauszugeben, darunter zur Chemie, Herstellung und Kontrolle von Anträgen auf neue Arzneimittel für die menschliche Gentherapie10, zur Langzeitnachsorge nach Verabreichung einer Gentherapie 11, zu replikationskompetenten Retrovirustests 12 und zu Empfehlungen für mikrobielle Vektoren, die in Gentherapien verwendet werden 13. Die EMA hat auch eine Reihe von Richtlinien für die Entwicklung von Gentherapieprodukten veröffentlicht, die im Allgemeinen mit den Empfehlungen der FDA übereinstimmen, obwohl einige Unterschiede bestehen14. Es ist wichtig zu beachten, dass diese Leitlinien zwar keine rechtlich durchsetzbaren Verantwortlichkeiten festlegen, es sei denn, es wird auf spezifische Vorschriften verwiesen, aber sie schaffen Klarheit über die aktuelle Denkweise der Aufsichtsbehörden zu diesem Thema und ihre Erwartungen an Assays, die für Arzneimittelanträge und behördliche Zulassungen erforderlich sind.
Die FDA empfiehlt ausdrücklich, dass Studien durchgeführt werden sollten, um die Verteilung, Persistenz und Clearance eines Vektors vom Verabreichungsort auf okuläres und nicht-okuläres Gewebe, intraokulare Flüssigkeiten und Blut zu bewerten15. Diese finden in Form von Bioverteilungs- und Häutungsstudien statt. Biodistributionsstudien bewerten die Exposition, indem sie untersuchen, wie sich ein Produkt vom Verabreichungsort aus im Körper eines Patienten verteilt. Bei der Ausscheidung wird speziell die Freisetzung des Produkts vom Patienten in die Umwelt bewertet und die Möglichkeit einer Übertragung des Vektors auf unbehandelte Personen erhöht16. Die FDA gibt Empfehlungen für das Design von Bioverteilungs- und Shedding-Studien in Bezug auf die Häufigkeit der Probenentnahme, die Dauer der Probenentnahme, die Art der entnommenen Proben und die Lagerbedingungen.
Darüber hinaus empfiehlt die FDA die Verwendung der quantitativen Polymerase-Kettenreaktion (qPCR oder Real-Time-PCR) für den quantitativen Nachweis von Vektorgenomen aufgrund ihrer einfachen Leistung, ihres Hochdurchsatzformats, ihrer schnellen Durchlaufzeiten und ihrer Assay-Sensitivität. Es gibt jedoch einen relativen Mangel an Empfehlungen für das Design und die Leistungsbewertung molekularer Methoden im Vergleich zu denen, die für kleine und große Moleküle existieren. Viele der Leitlinien für solche Studien lassen sich aufgrund des einzigartigen und komplexen Designs sowohl der Produkte als auch der Assays selbst nur schwer auf molekulare Methoden anwenden, was die Frage nach der Eignung der verfügbaren Plattformen für die empfohlenen Bewertungen und geeigneten Methoden für die Assay-Validierung aufwirft. Bisher hat die FDA keine formale Validierung von PCR-basierten Assays verlangt, obwohl die EMA diese Anforderung auferlegt hat17. Angesichts dieser Lücke haben verschiedene Gruppen und Workshops Whitepaper und Empfehlungen herausgegeben, die Hersteller und Auftragsforschungsinstitute zu befolgen versucht haben 18,19,20,21,22,23,24,25. Die meisten dieser Empfehlungen wurden speziell für qPCR-Assays verfasst, wobei Vorschläge oder Änderungen für neue Plattformen, wie z. B. die digitale Tröpfchen-PCR (ddPCR), nur dann aufgenommen werden, wenn dies als relevant erachtet wird. Neuere Empfehlungen konzentrierten sich auf Überlegungen zu ddPCR-Assays, konzentrierten sich jedoch weitgehend auf deren Anwendungen zur Quantifizierung des Vektorgenoms in einer Produktionsumgebung und nicht auf die komplexen biologischen Matrices, die in Bioshedding-Studien angetroffen werden.
Abhängig von der klinischen Anwendung und den Zielen kann die ddPCR der qPCR zur Unterstützung von Bioverteilungs- und Shedding-Studien vorgezogen werden, da die ddPCR im Vergleich zur qPCR eine höhere Sensitivität und Fähigkeit zur Behandlung von Matrixinterferenzen aufweist. Darüber hinaus kann durch die Aufteilung der Proben in ca. 20.000 Tröpfchen eine genaue Quantifizierung der Kopienzahl ohne die Verwendung einer Standardkurve unter Verwendung der Poisson-Statistik erreicht werden, was die Methodenentwicklung und -validierung vereinfacht. Das Ziel dieses Protokolls ist es, einen standardisierten Ansatz für die Entwicklung und Validierung einer ddPCR-basierten Methode zum Nachweis von AAV-Vektoren in Tränen zu beschreiben, die von der Augenoberfläche gesammelt wurden, um klinische Bioshedding-Studien zu unterstützen.
Protokoll
1. Herstellung eines synthetischen DNA-Fragments
- Entwerfen und bestellen Sie ein synthetisches DNA-Fragment, das die Zielamplifikationsregion enthält, um es als Qualitätskontrolle zu verwenden.
- Stellen Sie sicher, dass die Sequenz die gesamte Amplikonsequenz vom Forward-Primer bis zum Reverse-Primer des Zielgens von Interesse enthält, mit einer Erweiterung von vier bis sechs Basenpaaren der Sequenz an den 5'-Enden jeder Primer-Bindungssequenz.
- Vermeiden Sie Homopolymere von Adenin und Thymin mit mehr als 12 Basenpaaren oder Guanin- und Cytosinbasenpaaren mit mehr als acht Basenpaaren, da Homopolymere die Synthese des Genfragments beeinträchtigen können.
ANMERKUNG: Wenn Amplikone solche Sequenzen enthalten, können Basensubstitutionen vorgenommen werden, solange die Glühstellen für die Primer und Sonden erhalten bleiben. - Alternativ können Sie ein linearisiertes Plasmid herstellen, das das Amplikon enthält, indem Sie typische Klonierungsstrategien verwenden.
- Zentrifugieren Sie das Röhrchen mit dem synthetischen DNA-Fragment ~10 s lang in einer Mikrozentrifuge, um sicherzustellen, dass das Material am Boden des Röhrchens gesammelt wird.
- Resuspendieren Sie das synthetische DNA-Fragment unter Verwendung von Tris-EDTA (TE)-Puffer auf eine Konzentration von 1,0 ×10 10 Kopien/μl oder je nach Bedarf basierend auf dem Ziel-Assay-Bereich.
- Kurz vortexen, dann bei 50 °C 20 ± 5 min inkubieren. Auf Eis abkühlen lassen.
- Bereiten Sie mehrere, idealerweise Einweg-Aliquots vor und lagern Sie sie bis zur Verwendung bei -70 bis -90 °C.
HINWEIS: Auf diese Weise hergestellte synthetische DNA-Fragmente sind in der Regel mindestens 24 Monate ab dem Datum der Resuspension stabil. - Falls gewünscht, bestimmen Sie die genaue Konzentration des präparierten synthetischen DNA-Materials, bevor Sie es als Qualitätskontrolle verwenden, oder schätzen Sie die Nennkonzentration auf der Grundlage der verwendeten Resuspension.
2. Vorbereitung von Primern und Sonde
- Entwerfen und bestellen Sie Primer und eine Hydrolysesonde, um den gewünschten Amplifikationsbereich unter Verwendung typischer Designstrategien anzuvisieren26,27.
- Verwenden Sie einen 5'-Fluoreszenz-Reporterfarbstoff (z. B. FAM) und einen 3'-Quencher (z. B. Iowa Black Dark Quencher), die mit dem ddPCR-System kompatibel sind.
HINWEIS: Es gibt zahlreiche Softwarepakete für das PCR-Assay-Design, und jedes kann verwendet werden. Zum Beispiel wird Primer-BLAST vom National Center for Biotechnology Information28 aufgrund der robusten Optionen für das Assay-Design und der Leichtigkeit, mit der die Spezifität bioinformatisch bewertet werden kann, um mögliche Off-Target-Effekte zu identifizieren, weit verbreitet eingesetzt. Es ist zu beachten, dass die Herstellung von Primern und Sonden je nach Format, in dem sie geliefert werden, von den hier aufgeführten Schritten abweichen kann.
- Verwenden Sie einen 5'-Fluoreszenz-Reporterfarbstoff (z. B. FAM) und einen 3'-Quencher (z. B. Iowa Black Dark Quencher), die mit dem ddPCR-System kompatibel sind.
- Zentrifugieren Sie die Röhrchen mit dem vorderen Primer, dem umgekehrten Primer und der Sonde in einer Mikrozentrifuge für ~10 s, um das Pelletmaterial auf den Boden des Röhrchens zu bringen.
- Resuspendieren Sie die Primer mit TE-Puffer auf 20 μM. Kurz vortexen.
- Resuspendieren Sie die Sonde mit TE-Puffer auf 10 μM. Kurz vortexen.
- Bereiten Sie mehrere, idealerweise Einweg-Aliquots vor und lagern Sie sie bis zur Verwendung bei mindestens -20 °C.
HINWEIS: Auf diese Weise hergestellte Primer und Sonden sind in der Regel mindestens 24 Monate ab dem Datum der Resuspension stabil.
3. Herstellung des Probenverdünnungspuffers
- PCR-Puffer und geschorene Lachsspermien-DNA bei Raumtemperatur auftauen. Zum Mischen gründlich vortexen.
- Bereiten Sie einen Probenverdünnungspuffer gemäß Tabelle 1 vor.
- Gründlich vortexen. Nach der Zubereitung bei 2-8 °C bis zu 1 Monat lagern.
Tabelle 1: Herstellung des Probenverdünnungspuffers. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
4. Herstellung des Mastermixes
- Tauen Sie den ddPCR-Mastermix für Sonden, Vorwärtsprimer, Rückwärtsprimer und Sonde bei Raumtemperatur auf und lassen Sie ihn vor der Verwendung mindestens 10 Minuten nach dem Auftauen erwärmen. Bis zur Verwendung bei Raumtemperatur lagern.
Anmerkungen: Diese Reagenzien müssen vollständig auf Raumtemperatur gebracht werden, um eine effiziente Tröpfchenbildung zu gewährleisten. Halten Sie die Reagenzien während der Zubereitung nicht auf Eis.- Vor Gebrauch gründlich und kurz in einer Minizentrifuge zentrifugieren.
Anmerkungen: Restriktionsenzyme werden in der Regel in Glycerin geliefert und sollten unmittelbar vor der Verwendung aus der Lagerung genommen werden. Vorsichtig mischen. Nicht vorwirbeln.
- Vor Gebrauch gründlich und kurz in einer Minizentrifuge zentrifugieren.
- Bereiten Sie für jedes Amplifikationsziel einen PCR-Mastermix vor. In Tabelle 2 finden Sie eine empfohlene Zusammensetzung des PCR-Mastermixes und ändern Sie die Konzentrationen von Primern und Sonden nach Bedarf.
- Gründlich wirbeln und kurz zentrifugieren, bevor das Restriktionsenzym hinzugefügt wird. Fügen Sie das Restriktionsenzym hinzu und drehen Sie es um, um es zu mischen.
HINWEIS: In diesem Schritt sind 22 μl PCR-Reaktion erforderlich, um ein endgültiges Volumen von 40 μl PCR-Reaktion nach Tröpfchenbildung zu erhalten (bestehend aus 15 μl PCR-Mastermix, 5,0 μl Template und 20 μl Tröpfchenerzeugungsöl).
- Gründlich wirbeln und kurz zentrifugieren, bevor das Restriktionsenzym hinzugefügt wird. Fügen Sie das Restriktionsenzym hinzu und drehen Sie es um, um es zu mischen.
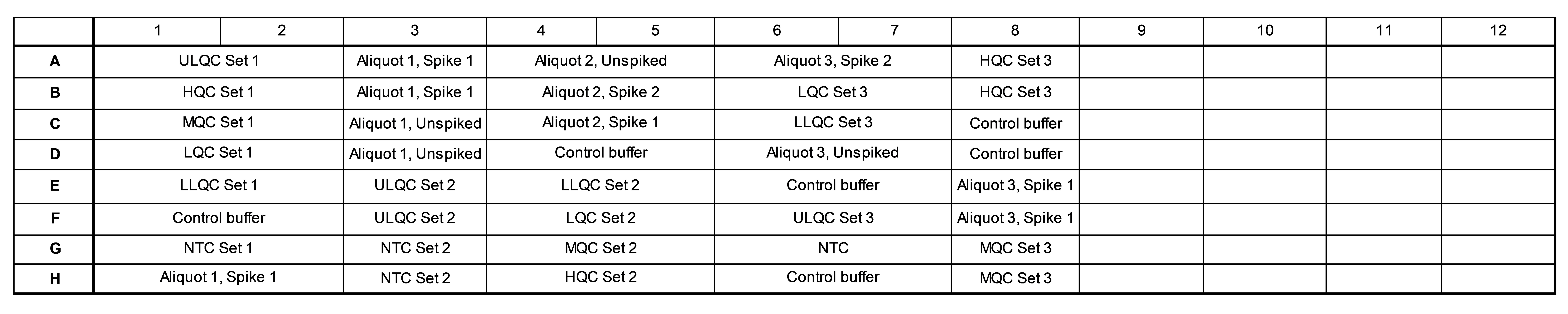
- Geben Sie 16,5 μl Mastermix gemäß der Plattenkarte in jede Vertiefung. In Abbildung 1 finden Sie ein Beispiel für eine Plattenkarte für eine Validierungsgenauigkeit und einen Präzisionslauf.
- Stellen Sie sicher, dass eine Platte drei unabhängige Präparate der Qualitätskontrollreihe (QC) enthält, drei unabhängig getestete endogene Tränenaliquote, die auf ein hohes und niedriges Niveau gespikt und ohne Spikes versehen sind, und drei unabhängige Kontrollen ohne Matrizen (NTCs).
- Variieren Sie das Layout dieser Vertiefungen auf der Platte, wobei Satz 1 in der Reihenfolge der abnehmenden Konzentration, Satz 2 in der Reihenfolge der zunehmenden Konzentration und Satz 3 in zufälliger Reihenfolge geladen wird, um zu bewerten, ob es plattenpositionsspezifische Effekte gibt.
- Ordnen Sie die Proben so an, dass sie so viel wie möglich von einer Säule füllen, und füllen Sie ungenutzte Vertiefungen innerhalb einer Säule mit Kontrollpuffer. Fügen Sie bei Bedarf mehrere endogene Kontrollpartien (z. B. mehr Tränenlachen oder Tränen, die von Einzelpersonen gesammelt wurden) in die verbleibenden Vertiefungen ein.
- Versiegeln Sie die Platte mit einer durchsichtigen Klebefolie. Halten Sie die Platte während der Schablonenvorbereitung bei Raumtemperatur. Alternativ können Sie die Platte bis zu 4 h bei 2-8 °C halten, aber vor dem Hinzufügen der Schablone mindestens 10 Minuten wieder auf Raumtemperatur bringen.
Tabelle 2: Beispiel für die Herstellung eines PCR-Mastermixes. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.

Abbildung 1: Beispiel für eine Plattenkarte für Validierungsgenauigkeit und Präzisionslauf. Abkürzungen: ULQC = obere Grenze der Qualitätskontrolle; HQC = hohe Qualitätskontrolle; MQC = mittlere Qualitätskontrolle; LQC = niedrige Qualitätskontrolle; LLQC = untere Grenze der Qualitätskontrolle; NTC = keine Vorlagensteuerung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
5. Vorbereiten von QCs
- Synthetische DNA-Fragmente oder linearisierte Plasmide bei Raumtemperatur auftauen und vor Gebrauch mindestens 10 Minuten nach dem Auftauen erwärmen lassen. Bringen Sie die Schablonen auf Raumtemperatur, um eine effiziente Tröpfchenbildung zu gewährleisten.
- Bis zur Verwendung bei Raumtemperatur lagern. Vor Gebrauch gründlich und kurz in einer Minizentrifuge zentrifugieren.
- Bereiten Sie QC-Verdünnungen vor, indem Sie den Probenverdünnungspuffer als Verdünnungsmittel verwenden. Ein Beispiel für die empfohlenen Konzentrationen zur Vorbereitung auf eine Validierungsgenauigkeit und -genauigkeit ist in Tabelle 3 dargestellt.
HINWEIS: Nach dem erfolgreichen Abschluss der Genauigkeits- und Präzisionsläufe müssen auf jeder Platte nur die hohe Qualitätskontrolle (HQC), die mittlere Qualitätskontrolle (MQC) und die niedrige Qualitätskontrolle (LQC) durchgeführt werden. Für Genauigkeits- und Präzisionsläufe sind mindestens drei unabhängige Verdünnungen der QCs enthalten, um die Genauigkeit und Präzision innerhalb des Assays zu beurteilen. Nach Genauigkeits- und Präzisionsläufen muss nur eine Verdünnungsserie enthalten sein. - Bewahren Sie die Verdünnungen nach der Zubereitung bei Raumtemperatur auf, bis sie auf die Platte gegeben werden.
- Lagern Sie die Verdünnungen auf Eis oder bei Bedarf bei 2-8 °C. Lassen Sie die Verdünnungen vor der späteren Verwendung mindestens 10 Minuten lang auf Raumtemperatur erwärmen, bevor Sie sie verwenden. Verwerfen Sie die QCs am Ende des Tages.
Tabelle 3: Beispiel für eine Herstellung der Qualitätskontrolle (QC) unter Verwendung synthetischer doppelsträngiger DNA-Fragmente. Abkürzungen: ULQC = obere Grenze der Qualitätskontrolle; HQC = hohe Qualitätskontrolle; MQC = mittlere Qualitätskontrolle; LQC = niedrige Qualitätskontrolle; LLQC = untere Grenze der Qualitätskontrolle; NTC = keine Vorlagensteuerung. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
6. Vorbereitung der Proben
- Tränenproben, die aus einer klinischen Studie entnommen wurden, bis sie aufgetaut sind, bei Raumtemperatur auftauen und vor der Verwendung mindestens 10 Minuten nach dem Auftauen erwärmen lassen.
- Bis zur Verwendung bei Raumtemperatur lagern. Vor Gebrauch gründlich und kurz in einer Mikrozentrifuge zentrifugieren.
- Verdünnen Sie Tränenproben im Verhältnis 1:10 (oder größer) mit Probenverdünnungspuffer als Verdünnungsmittel in 0,2-ml-PCR-Röhrchen oder 8-Well-PCR-Streifen. Verschließen Sie die Röhrchen.
HINWEIS: Abhängig von der erwarteten Konzentration des Ziels in Tränen kann es erforderlich sein, die Proben weiter zu verdünnen oder mehrere Verdünnungen jeder Probe zu testen. - Erhitzen Sie die Proben in einem Thermocycler 10 Minuten lang bei 95 °C und halten Sie sie anschließend mindestens 5 Minuten lang bei 4 °C zum Abkühlen. Verwenden Sie eine Rampenrate von 3 °C/s.
Anmerkungen: Die Proben können bis zur Verwendung am selben Tag bei 4 °C im Thermocycler verbleiben oder für eine längere Lagerung bei -70 bis -90 °C eingefroren werden. Dieser Schritt dient dazu, das Vektorkapsid zu denaturieren und das Genom freizusetzen. Da synthetische QC-DNA-Fragmente oder linearisierte Plasmide doppelsträngig sind, sollten sie diesem Erhitzungsschritt nicht unterzogen werden. - Bringen Sie die Proben nach dem Abkühlen wieder auf Raumtemperatur zurück (oder tauen Sie sie bei Raumtemperatur auf) und lassen Sie sie mindestens 10 Minuten lang erwärmen.
Anmerkungen: Die Proben müssen vollständig auf Raumtemperatur gebracht werden, um eine effiziente Tröpfchenbildung zu gewährleisten.
7. Hinzufügen von Vorlagen
- Holen Sie sich die ddPCR-Platte mit dem Mastermix. Jede Probe oder jedes QC-Verdünnungsröhrchen gründlich vortexen und kurz zentrifugieren, um das Material zu gewinnen.
- Entfernen Sie die Klebefolie und geben Sie 5,5 μl QCs oder Proben gemäß der Plattenkarte in die entsprechenden Vertiefungen der 96-Well-Platte.
HINWEIS: In Schritt 4.2.1 finden Sie Erläuterungen zu den erforderlichen Volumes - Geben Sie 5,5 μl Probenverdünnungspuffer in die NTC-Vertiefungen.
- Die Tröpfchenerzeugung erfordert, dass alle Wells einer Säule über eine Reaktions- oder Puffersteuerung verfügen. Wenn Vertiefungen einer Säule keine Probenreaktionen enthalten, verdünnen Sie 2x ddPCR-Pufferkontrolle 1:2 mit nukleasefreiem Wasser. Fügen Sie 22 μl 1x ddPCR-Pufferkontrolle zu allen leeren Vertiefungen einer Säule hinzu.
HINWEIS: Wenn nicht eine ganze Spalte verwendet wird, ist es nicht erforderlich, diesen Wells eine Puffersteuerung hinzuzufügen. - Fügen Sie der Platte eine durchstechbare Foliendichtung hinzu. Legen Sie die Platte in die Plattenversiegelung und versiegeln Sie sie 5 s lang bei 180 °C.
- Alternativ können Sie die Platte gemäß den Empfehlungen des Herstellers des ddPCR-Systems versiegeln.
- Die Platte mindestens 30 s lang mit maximaler Geschwindigkeit vortexen (bei kontinuierlicher Vortexing-Einstellung; kein Touch-Vortexing verwenden) und kurz in einem Plattenschleuder zentrifugieren.
HINWEIS: Das gründliche und vollständige Mischen der Platte in diesem Schritt ist entscheidend für die ordnungsgemäße Aufteilung der PCR-Reaktion in Tröpfchen. Stellen Sie sicher, dass in den Vertiefungen keine Blasen sichtbar sind. Bei Bedarf kann die Platte vor der Tröpfchenbildung für maximal 4 h bei 2-8 °C gehalten werden. Wenn die Platte gehalten wird, lassen Sie sie mindestens 10 Minuten lang Raumtemperatur erreichen, bevor sich Tröpfchen bilden.
8. Automatisierte Tröpfchenerzeugung, Temperaturwechsel und Tröpfchenablesung
- Erzeugen Sie Tröpfchen im automatischen Tröpfchengenerator wie folgt.
- Wählen Sie auf dem Touchscreen die Spalten auf der Plattenkarte aus, die die Proben enthalten. Das Deck des Instruments leuchtet auf, um anzuzeigen, welche Verbrauchsmaterialien (DG32-Kartuschen, Spitzen, Abfallbehälter, Tröpfchenerzeugungsöl) erforderlich sind. Gelbe Lichter zeigen an, dass ein Verbrauchsmaterial hinzugefügt werden muss, während grüne Lichter anzeigen, dass genügend Verbrauchsmaterial verfügbar ist.
- Laden Sie den Tröpfchengenerator von hinten nach vorne.
- Stellen Sie bei Hydrolysesonden sicher, dass das Tröpfchenerzeugungsöl für Sonden installiert ist und genügend Öl für die Anzahl der Vertiefungen vorhanden ist. Wenn alternative PCR-Chemikalien verwendet werden, stellen Sie sicher, dass ein kompatibles Tröpfchenerzeugungsöl installiert ist.
- Legen Sie einen kalten Block in den Tröpfchenplattenhalter. Stellen Sie sicher, dass der Block vollständig blau gefärbt ist und kein Rosa sichtbar ist. Legen Sie eine neue 96-Well-ddPCR-Platte in den kalten Block.
- Legen Sie die vorbereitete PCR-Platte in den Probenplattenhalter. Schließen Sie den Maschinendeckel. Drücken Sie Start für die Tröpfchenerzeugung.
- Nach der Tröpfchenbildung werden insgesamt 40 μL pro Reaktion automatisch auf die neue PCR-Platte übertragen.
- Entfernen Sie innerhalb von 30 Minuten nach Abschluss der Tröpfchenerzeugung die Platte mit den Tröpfchen aus dem Kühlblock. Arbeiten Sie vorsichtig, da die Tröpfchen in diesem Stadium am zerbrechlichsten sind.
- Fügen Sie der Platte eine durchstechbare Foliendichtung hinzu. Legen Sie die Platte in die Plattenversiegelung und versiegeln Sie sie 5 s lang bei 180 °C.
- Alternativ können Sie die Platte gemäß den Empfehlungen des Herstellers des ddPCR-Systems versiegeln.
- Legen Sie die Platte in einen kompatiblen Thermocycler. Geben Sie die Fahrbedingungen ein (siehe Tabelle 4).
- Halten Sie die Platte nach dem Ende des Temperaturwechsels in den Thermocycler, der auf 2-8 °C umgefüllt wurde, oder lesen Sie sie sofort ab.
Anmerkungen: Wenn Sie die Platte 12 Stunden lang bei 4-12 °C halten, kann dies die Tröpfchenzahl verbessern, dies ist jedoch nicht erforderlich. Es sollten genügend Tröpfchen ohne Halt erhalten werden. - Legen Sie die Platte in das Tröpfchenlesegerät ein und stellen Sie sicher, dass genügend Leseöl verbleibt und der Abfallbehälter ausreichend Platz hat. Lesen Sie die Tröpfchen. Führen Sie die Tröpfchenmessung innerhalb von 24 Stunden nach Beginn des Temperaturwechsels durch.
Tabelle 4: Typische Temperaturwechselbedingungen. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
9. Datenanalyse
HINWEIS: Mindestens 10.000 Tröpfchen pro Bohrung sind für die ordnungsgemäße Berechnung der Konzentration anhand der Poisson-Statistik erforderlich. Versuchen Sie nicht, Bohrungen mit weniger als 10.000 Tröpfchen durchzuführen.
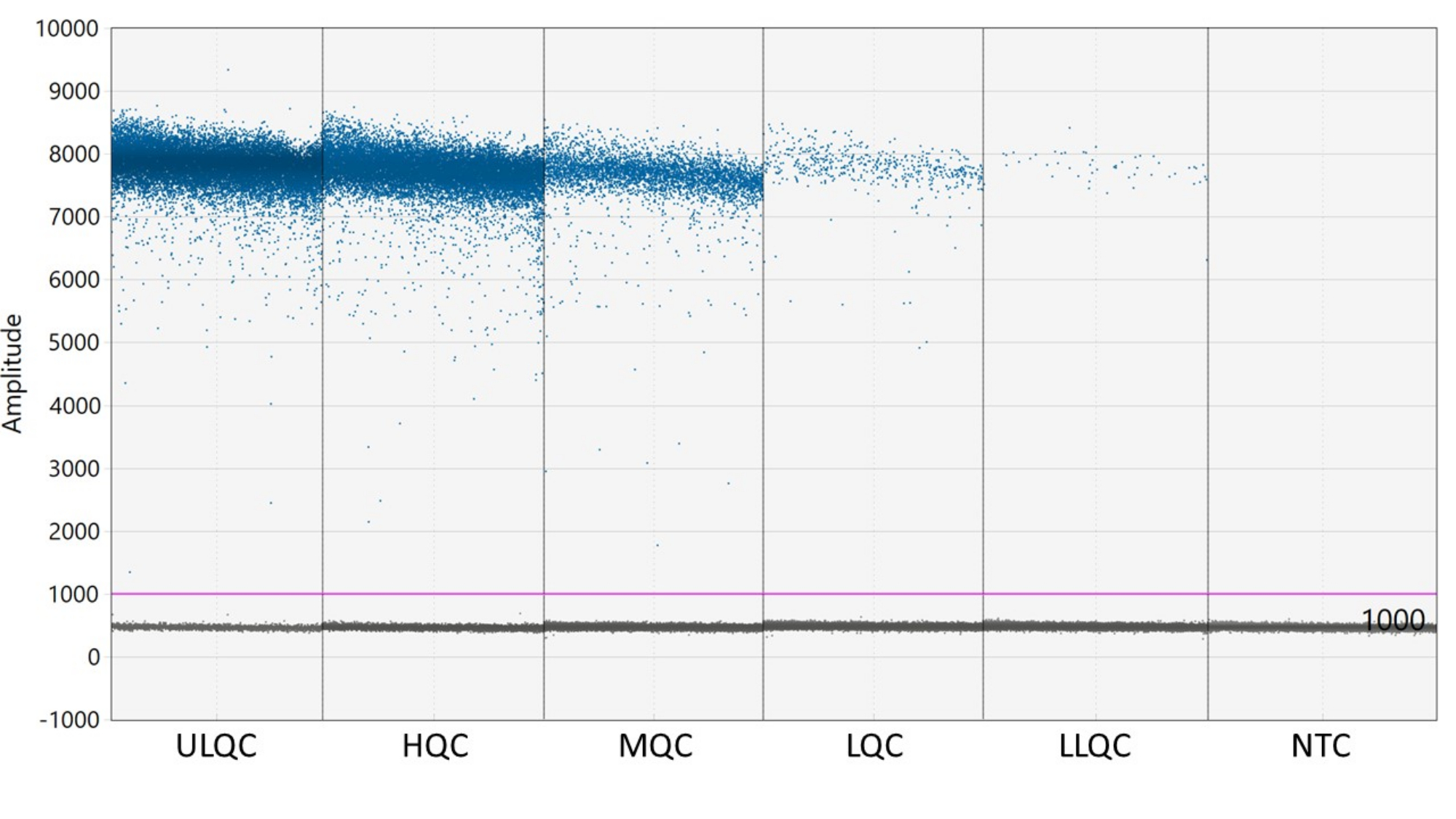
- Ein Schwellenwert ist erforderlich, um die Tröpfchen als positiv oder negativ zu definieren. Die ddPCR-Analysesoftware wendet automatisch einen Schwellenwert an, der je nach Bohrloch variieren kann. Legen Sie jedoch manuell einen Schwellenwert für alle Vertiefungen der Platte fest, der leicht über der Fluoreszenzintensität der NTC-Vertiefungen liegt, um konsistentere, genauere und präzisere Ergebnisse zu erzielen.
HINWEIS: Die richtige Platzierung des Schwellenwerts kann je nach Trennung der positiven und negativen Tröpfchen und der vorhandenen Menge an Tröpfchenregen optimiert werden müssen (siehe Abbildung 2). In diesem Beispiel zeigt das Tröpfchenamplitudendiagramm Beispielwells auf jeder QC-Ebene und im NTC. Die violette Linie zeigt einen Schwellenwert von 1.000 an, der leicht über der negativen Tröpfchenpopulation liegt. - Für die statistische Poisson-Modellierung sind mindestens drei positive Tröpfchen erforderlich, um die Konzentration mit einer Sicherheit von 95 % zu berechnen. Betrachten Sie alle Vertiefungen, die null, ein oder zwei positive Tröpfchen enthalten, als negativ und auf eine Konzentration von Null27 eingestellt.
- Berechnen Sie die Kopienzahl in jeder Tränenprobe zurück.
- Die Konzentration in Kopien/μl ist im Datenbericht angegeben. Verwenden Sie diesen Wert, um die Konzentration in Kopien/μL der Originalprobe (d. h. in der Tränenprobe) zu bestimmen.
- Um die ddPCR-Reaktionsverdünnung zu berechnen, teilen Sie das anfängliche PCR-Reaktionsvolumen vor der Tröpfchenbildung durch das Volumen der zugegebenen Vorlage. Wenn die in dieser Methode dargestellten Volumina verwendet werden, ergibt sich ein Wert von 4.

- Bestimmen Sie den seriellen Verdünnungsfaktor aus der Originalprobe (Schritt 6.2).
- Um die Kopien/μL in der Probe zu bestimmen, multiplizieren Sie die Kopien/μL mit der ddPCR-Reaktionsverdünnung und dann mit dem seriellen Verdünnungsfaktor. Beispielsweise betrug die im Datenbericht erzeugte Konzentration in Kopien/μl 966; Pro 22 μl Reaktion wurden 5,5 μl Template zugegeben. Es wurde eine serielle Verdünnung der Probe im Verhältnis 1:50.000 verwendet.


- Wenn mehrere Verdünnungen derselben Probe getestet wurden, analysieren Sie alle gültigen Verdünnungen innerhalb des Bereichs und berechnen Sie den Mittelwert.
- Berechnen Sie für jede QC die erwarteten Kopien/μL PCR-Reaktion, indem Sie die Konzentration der angegebenen QC-Verdünnung (in Kopien/μL) durch das ddPCR-Reaktionsvolumen (20 μL) dividieren. Dies ermöglicht einen direkten Vergleich dieses Sollwertes mit dem im Datenbericht angegebenen Kopien/μL-Wert ohne weitere Berechnungen.
HINWEIS: Dieser Ansatz wurde auch für die Analyse der Tränenproben verwendet, die in den repräsentativen Ergebnissen verwendet wurden. - Bestimmen Sie den Mittelwert, die Standardabweichung, den Variationskoeffizienten (%CV) und den prozentualen relativen Fehler zur Nennkonzentration (%RE) der Probe oder den QC-Wert unter Verwendung der Wiederholungsvertiefungen (ggf. einschließlich mehrerer Verdünnungen).
- Für die Beurteilung der Präzision zwischen den Bohrlöchern ist diese für jedes der Bohrlochduplikate zu bestimmen, falls vorhanden.
- Um die Genauigkeit und Präzision innerhalb des Assays zu beurteilen, bestimmen Sie diese für jede Verdünnungsserie oder jedes Aliquot, das innerhalb einer Charge verwendet wird.
- Für die Beurteilung der Genauigkeit und Präzision zwischen den Assays ist diese mit den Intra-Assay-Mittelwerten jeder der eingeschlossenen Chargen zu bestimmen.

Abbildung 2: Beispiel für die Einstellung des Schwellenwerts. Abkürzungen: ULQC = obere Grenze der Qualitätskontrolle; HQC = hohe Qualitätskontrolle; MQC = mittlere Qualitätskontrolle; LQC = niedrige Qualitätskontrolle; LLQC = untere Grenze der Qualitätskontrolle; NTC = keine Vorlagensteuerung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
10. Akzeptanzkriterien für Assays
- Verwenden Sie die folgenden Spezifikationen für die berechneten Daten für jede Charge, um zu bestimmen, ob die Charge akzeptabel ist. Wenn diese Bedingungen nicht erfüllt sind, machen Sie den Batch ungültig, und wiederholen Sie ihn.
HINWEIS: Diese Kriterien wurden als Konsens aus veröffentlichten Whitepapern zur PCR-basierten Assay-Validierung 18,19,20,21,22,23,24,25 festgelegt. Es kann erforderlich sein, die Zielkriterien entsprechend der klinischen Anwendung zu modifizieren. - Keine Vorlagensteuerung (NTC)
- Stellen Sie sicher, dass jede NTC-Bohrung mindestens 10.000 Tröpfchen enthält.
- Stellen Sie sicher, dass jede NTC-Vertiefung weniger als 3 positive Tröpfchen enthält.
- QCs und Assay-Bereich
- Stellen Sie sicher, dass jede QC-Bohrung mindestens 10.000 Tröpfchen enthält.
- Stellen Sie sicher, dass die Genauigkeit der Wiederholungsbohrungen einer QC-Konzentration ≤25,0 % CV beträgt, außer an der oberen und unteren Grenze der Quantifizierung, wo ≤30,0 % akzeptabel sind. Beurteilen Sie dies unabhängig für jedes QC-Set und jede Konzentrationsstufe.
- Stellen Sie sicher, dass der relative Fehler der rückberechneten Konzentration bei jedem mittleren QK-Wert innerhalb von ±25,0 % RE der Nennkonzentration (Kopien/PCR-Reaktion) liegt, außer an den oberen und unteren Grenzen der Quantifizierung, wo ±30,0 % RE akzeptabel sind. Beurteilen Sie dies unabhängig für jedes QC-Set und jede Konzentrationsstufe.
- Stellen Sie sicher, dass mindestens 2/3 der QC-Proben (z. B. vier von sechs Ergebnissen) und 50 % der QC-Proben auf jeder Stufe (niedrig, mittel, hoch) diesen Richtlinien entsprechen.
- Proben
- Stellen Sie sicher, dass die zu analysierenden Probenvertiefungen mindestens 10.000 Tröpfchen enthalten.
- Stellen Sie sicher, dass die Genauigkeit der Wiederholungsvertiefungen einer zu analysierenden Probenverdünnung ≤25,0 % CV beträgt.
- Stellen Sie sicher, dass mindestens eine eingeschlossene Verdünnung der gegebenen Probe innerhalb des definierten Bestimmungsbereichs des Assays liegt, wie oben definiert, basierend auf den oberen und unteren Grenz-QCs.
- Wenn alle enthaltenen Verdünnungen Ergebnisse ergeben, die größer sind als die definierte obere Bestimmungsgrenze, und wenn ein ausreichendes Probenvolumen verbleibt, wiederholen Sie den Assay mit einer höheren Verdünnung der Probe.
- Wenn alle enthaltenen Verdünnungen zu einem Ergebnis führen, das unter der unteren Bestimmungsgrenze liegt, und wenn ein ausreichendes Probenvolumen verbleibt, wiederholen Sie den Assay mit einer geringeren Verdünnung der Probe.
HINWEIS: Proben, die mehr als drei positive Tröpfchen enthalten, aber eine Konzentration unterhalb der unteren Bestimmungsgrenze aufweisen, können als nachweisbar, aber nicht quantifizierbar bezeichnet werden.
Ergebnisse
Zu Demonstrationszwecken wurde ein Assay entwickelt, der einen kommerziell erhältlichen, enhanced green fluorescent protein (eGFP)-exprimierenden AAV2-Vektor mit einem synthetischen doppelsträngigen DNA-Fragment mit eGFP als Qualitätskontrolle nachweisen kann. Derzeit gibt es eine anhaltende Debatte darüber, ob der Vektor selbst oder ein synthetisches DNA-Fragment oder ein linearisiertes Plasmid für die Verwendung als QC am besten geeignet ist. Im Allgemeinen kann ein synthetisches DNA-Fragment oder ein linearisiertes Plasmid verwendet werden, wenn die Äquivalenz zum Vektor in der Methodenentwicklung nachgewiesen wird (Daten nicht gezeigt). Primer und Sonden wurden für den Nachweis des eGFP-Transgens entwickelt und optimiert. In der Ergänzungstabelle S1 finden Sie die in dieser Arbeit verwendeten Sequenzen. Die Konzentration des QC-Fragmentbestandes wurde mittels ddPCR empirisch bestimmt. Alle Assays wurden unter Verwendung der Konzentrationen und PCR-Bedingungen durchgeführt, die im Protokollabschnitt beispielhaft aufgeführt sind.
Für qPCR-Assays wird empfohlen, die Linearität, Sensitivität, den Dynamikbereich, die Genauigkeit und die Präzision der Standardkurve zu bewerten. Da die ddPCR nicht auf eine Standardkurve zur Zielquantifizierung angewiesen ist, müssen diese Empfehlungen modifiziert werden. Stattdessen wurden QCs, bestehend aus synthetischen doppelsträngigen DNA-Fragmenten, die auf verschiedene Konzentrationen verdünnt wurden, um den erwarteten quantifizierbaren Bereich einer ddPCR-Reaktion auf der Grundlage der statistischen Poisson-Modellierungabzudecken, 29,30,31,32 verwendet, um den dynamischen Bereich und die Sensitivität zu definieren und die Genauigkeit und Präzision zu bewerten. Die Auswahl der QC-Konzentrationen basierte in erster Linie auf dem erwarteten Verhältnis von positiven zu Gesamttröpfchen innerhalb eines Bohrlochs bei einer bestimmten Konzentration. Mathematisch gesehen ist die ddPCR theoretisch am genauesten, wenn etwa 80% der Partitionen positiv amplifizieren. Wenn das Verhältnis von positiven zu Gesamttröpfchen über 0,8 steigt, nimmt die Genauigkeit aufgrund der Sättigung der Trennwände ab, wobei eine Quantifizierung nicht mehr möglich ist, sobald 100 % der Tröpfchen positiv sind. Am unteren Ende kann theoretisch nur ein positives Tröpfchen nachgewiesen und quantifiziert werden, obwohl die Genauigkeit schlechter ist und der Assay falsch positiven Ergebnissen auf niedrigem Niveau unterliegt. In der Regel müssen mindestens drei Tröpfchen positiv sein, damit ein Ergebnis mit einer Konfidenz von 95 % berechnet werden kann, was der Schwellenwert für die Berechnung einer hier verwendeten Konzentration ist.
Es wurde eine Reihe von fünf verschiedenen QC-Konzentrationen erstellt, wobei die berechneten Zielkopienzahl/μL PCR-Reaktionsvolumina voraussichtlich ein positives zu einem Gesamttröpfchenverhältnis ergeben, das den quantifizierbaren Bereich der ddPCR abdeckt, wie in Tabelle 5 dargestellt. Diese wurden verwendet, um die Genauigkeit und Präzision des Assays zu bewerten. In der Bewertung wurden hier die oberen und unteren Grenzen der Bestimmungsfähigkeit nicht auf das theoretisch mögliche Maximum in der ddPCR verschoben. Eine genaue Quantifizierung kann auf höheren und niedrigeren Niveaus als hier gezeigt möglich sein. Das Sortiment sollte in Übereinstimmung mit den nachgelagerten Anwendungen dieser Methode entwickelt werden.
Insgesamt drei unabhängig voneinander hergestellte Verdünnungsserien dieser QCs wurden für jede Charge in Probenverdünnungspuffer hergestellt, um die Genauigkeit und Präzision innerhalb des Assays zu bewerten. Doppelte Vertiefungen jeder QC-Verdünnung wurden eingeschlossen. Um ein tatsächliches Validierungsprotokoll zu simulieren, wurden insgesamt sechs Genauigkeits- und Präzisionschargen von mehreren Analysten über mehrere Tage hinweg durchgeführt. Die Ergebnisse dieser sechs Chargen wurden analysiert, um die Genauigkeit und Präzision der Methode innerhalb und zwischen den Assays zu definieren und den dynamischen Bereich des Assays zu definieren.
Die Intra-Assay-Leistung wurde für jede Charge auf jeder QC-Ebene bewertet. Wir gingen davon aus, dass alle QC- und NTC-Bohrungen mindestens 10.000 Tröpfchen aufweisen würden. Dies wurde in 216 von 216 untersuchten Bohrlöchern in allen sechs Chargen mit einer durchschnittlichen Tröpfchenzahl von 19.748 Tröpfchen pro Bohrloch erreicht (Tabelle 6). Als nächstes wurde erwartet, dass der %CV zwischen den Bohrlöchern jedes Satzes von doppelten Bohrlöchern jeder Qualitätskontrolle ≤25,0 % betragen würde, mit Ausnahme der oberen und unteren Qualitätskontrolle, bei der ≤30,0 % erwartet wurden. Dies wurde in Sätzen von 90 von 90 Bohrlöchern erreicht, die in allen sechs Chargen für die Qualitätskontrollen getestet wurden, mit einem durchschnittlichen %CV zwischen den Bohrlöchern von 3,9 % über alle Qualitätskontrollstufen hinweg (Tabelle 7). Alle QCs ergaben ein mittleres positives zu einem Gesamttröpfchenverhältnis innerhalb der oben beschriebenen erwarteten Bereiche (Tabelle 6).
Innerhalb jeder Charge wurden der Intra-Assay-Mittelwert und die Standardabweichung für jeden der unabhängig voneinander erstellten Verdünnungsreihenpunkte berechnet, und diese wurden verwendet, um einen Intra-Assay-Mittelwert für jede Konzentration in jedem Assay zu berechnen. Dies wurde verwendet, um die Genauigkeit und Präzision des Assays zu beurteilen (Tabelle 8). Die Präzision bezieht sich auf die Variabilität der Daten von Wiederholungen derselben homogenen Probe unter normalen Assay-Bedingungen und wird durch die Berechnung des %CV der mehrfach eingeschlossenen Aliquots bewertet. Wir erwarteten, dass die drei Aliquote, die in jeder Charge getestet wurden, einen Intra-Assay-%CV von ≤25,0 % ergeben würden, mit Ausnahme der oberen und unteren Grenze, bei der ≤30,0 % erwartet wurden. Dies wurde für alle fünf Qualitätsstufen in jeder der 60 Chargen (30 von 30 Gesamtleistungen) erreicht. Im Allgemeinen konnte eine höhere Intra-Assay-Präzision als die Zielkriterien erreicht werden, mit einem mittleren Intra-Assay-CV von 7,7 % über alle QC-Stufen hinweg. Die Genauigkeit bezieht sich auf die Übereinstimmung zwischen dem experimentell ermittelten Wert und dem Sollwert. Dies wird durch die Berechnung des prozentualen relativen Fehlers (%RE oder %Bias) zwischen den berechneten Konzentrationen jeder QC und ihren theoretisch erwarteten nominalen Konzentrationen bewertet. Es wurde erwartet, dass der Intra-Assay-Mittelwert der drei Aliquote ±25,0 % RE der nominalen Konzentration betragen würde, mit Ausnahme der oberen und unteren Grenze, bei der ±30,0 % erwartet wurden. Dies wurde für alle fünf Qualitätsstufen in jeder der 60 Chargen (30 von 30 Gesamtleistungen) erreicht. Im Allgemeinen konnte eine höhere Genauigkeit innerhalb des Assays als unser Ziel erreicht werden, mit einem mittleren absoluten Intra-Assay-%RE von 4,2 % über alle QC-Stufen hinweg. Bei allen Durchführungen des NTC (insgesamt 30) waren keine positiven Tröpfchen nachweisbar.
Die Genauigkeit und Präzision zwischen den Proben wurde auch anhand des Intra-Assay-Mittelwerts jeder QC-Stufe innerhalb jeder Charge berechnet. Es wurde erwartet, dass die Präzision zwischen den Proben ≤25,0 % CV betragen würde, mit Ausnahme der oberen und unteren Grenze, bei der ≤30,0 % erwartet wurden. Ebenso wurde für die Genauigkeit zwischen den Assays ±25,0 % RE erwartet, mit Ausnahme der oberen und unteren Grenze, bei der ±30,0 % erwartet wurden. Es wurde eine deutlich höhere Genauigkeit und Präzision als bei diesen Zielen beobachtet (Tabelle 9), mit einer Inter-Assay-Genauigkeit von 4,0 % bis 8,5 % und einer absoluten Inter-Assay-Genauigkeit von 1,0 % bis 3,2 %. Zusammengenommen zeigen diese Ergebnisse, dass diese Methode eine ausreichende Intra- und Interassay-Genauigkeit und -Präzision erreichen kann, die weit innerhalb der aktuellen Industrieziele liegt. Auf der Grundlage dieser Ergebnisse kann ein dynamischer Bereich dieses Assays von 2.500 bis 2,5 Kopien pro μl PCR-Reaktion definiert werden, mit einer Gesamtsensitivität von 2,5 Kopien pro μl PCR-Reaktion. Wie bereits erwähnt, ist es möglicherweise möglich, größere Dynamikbereiche zu validieren.
Als nächstes war es notwendig, die Genauigkeit und Präzision des Assays innerhalb der Zielmatrix - in diesem Fall Risse - zu bewerten. In der Regel werden Assays vor Beginn klinischer Studien validiert, was bedeutet, dass Tränen von vektorbehandelten Patienten wahrscheinlich nicht für Validierungszwecke zur Verfügung stehen. Dies kann künstlich erzeugt werden, indem der Ziel-AAV-Vektor in Tränen gestochen wird, die von freiwilligen Spendern gesammelt wurden, um matrixgespickte QCs zu erzeugen. Gepoolte menschliche Tränen wurden von einem Dritten (BioIVT) gesammelt. Als Prinzipiennachweis wurde ein eGFP-exprimierender AAV2-Vektor aus einer kommerziellen Quelle verwendet. Die Konzentration des AAV2-Vektorstocks wurde empirisch mittels ddPCR bestimmt, ohne die Verwendung eines DNA-Isolationsschritts, wie in diesem Protokoll beschrieben. In jedem Durchlauf wurde das AAV2 unabhängig voneinander in die drei Tränenaliquote auf hohem (erwartete 1,41 x 103 Kopien/μl PCR-Reaktion) und niedrigem (28,2 Kopien/μl PCR-Reaktion) Niveau gestochen. Ungespießte Aliquots wurden als Kontrolle einbezogen, um die Spezifität der Methode zu demonstrieren.
Die Intra-Assay-Leistung wurde für jede Charge auf jeder Spike-Ebene bewertet. Es wurde erwartet, dass alle Tränenproben mindestens 10.000 Tröpfchen aufweisen würden. Dies wurde in 108 von 108 untersuchten Bohrlöchern in allen sechs Chargen mit einer durchschnittlichen Gesamttröpfchenzahl von 20.208 Tröpfchen pro Bohrloch erreicht (Tabelle 10). Als nächstes wurde erwartet, dass der %CV zwischen den Bohrlöchern jedes Satzes von doppelten Bohrlöchern jeder Qualitätskontrolle ≤25,0 % für die hohen und niedrigen Spike-Gehalte betragen würde. Dies wurde in 36 von 36 Sätzen von Bohrlöchern erreicht, die in allen sechs Chargen für die QCs getestet wurden, mit einem durchschnittlichen %CV zwischen den Bohrlöchern von 3,2 % (Tabelle 11).
Innerhalb jeder Charge wurden der Intra-Assay-Mittelwert und die Standardabweichung für jeden der unabhängig voneinander hergestellten Tränenspitzen berechnet, und diese wurden verwendet, um einen Intra-Assay-Mittelwert für jede Konzentration in jedem Assay zu berechnen. Dies wurde verwendet, um die Genauigkeit und Präzision des Assays in der Matrix zu bewerten (Tabelle 12). Wir erwarteten einen Intra-Assay-Wert von %CV von ≤25,0 % sowie hohe und niedrige Spike-Werte. Dies wurde in sechs von sechs Chargen für jede Stufe erfüllt. Im Allgemeinen konnte eine höhere Intra-Assay-Präzision in der Matrix als das Ziel erreicht werden, mit einem mittleren Intra-Assay-%CV von 3,7 % auf hohem Niveau und 12,2 % auf niedrigem Niveau (insgesamt 8,0 %). Es wurde auch erwartet, dass der Intra-Assay-%RE bei beiden Spike-Stufen ±25,0 % betragen würde. Dies wurde in sechs von sechs Chargen für jede Stufe erfüllt. Ebenso wurde allgemein festgestellt, dass eine höhere Intra-Assay-Genauigkeit in der Matrix als das Ziel erreicht werden konnte, mit einem mittleren Intra-Assay-absoluten %RE von 8,1 % auf niedrigem Niveau und 11,3 % auf hohem Niveau (insgesamt 9,7 %). Bei der Kontrolle ohne Spikes war in keinem der Aliquots ein eGFP-Signal nachweisbar (Tabelle 12), was die Spezifität der Methode in der humanen Tränenmatrix demonstriert.
Die Genauigkeit und Präzision der Tear-Matrix zwischen den Proben wurde ebenfalls anhand des Intra-Assay-Mittelwerts jedes Spike-Levels innerhalb jeder Charge berechnet. Es wurde erwartet, dass die Genauigkeit zwischen den Proben ≤25,0 % CV betragen würde, und für die Genauigkeit zwischen den Proben erwarteten wir ±25,0 % RE. Es wurde eine deutlich höhere Genauigkeit und Präzision zwischen den Proben beobachtet (Tabelle 13), mit einer Präzision von 5,5 % auf hohem und 7,1 % auf niedrigem Niveau sowie mit einer absoluten Genauigkeit von 11,3 % auf hohem Niveau und 8,1 % auf niedrigem Niveau. Zusammengenommen demonstrieren diese Ergebnisse die Genauigkeit, Präzision und Spezifität der Methode in der Tränenmatrix.
Tabelle 5: Qualitätskontrollen, die zur Definition des Dynamikbereichs des Assays verwendet werden. Abkürzungen: ULQC = obere Grenze der Qualitätskontrolle; HQC = hohe Qualitätskontrolle; MQC = mittlere Qualitätskontrolle; LQC = niedrige Qualitätskontrolle; LLQC = untere Grenze der Qualitätskontrolle; NTC = keine Vorlagensteuerung. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 6: Gesamtzahl der Tröpfchen und Verhältnis von positiven zu Gesamttröpfchen der synthetischen doppelsträngigen DNA-Qualitätskontrolle und NTC. Abkürzungen: ULQC = obere Grenze der Qualitätskontrolle; HQC = hohe Qualitätskontrolle; MQC = mittlere Qualitätskontrolle; LQC = niedrige Qualitätskontrolle; LLQC = untere Grenze der Qualitätskontrolle; NTC = keine Vorlagensteuerung. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 7: QC-Interwell-Statistik (Copy Targets/μL PCR-Reaktion). Abkürzungen: ULQC = obere Grenze der Qualitätskontrolle; HQC = hohe Qualitätskontrolle; MQC = mittlere Qualitätskontrolle; LQC = niedrige Qualitätskontrolle; LLQC = untere Grenze der Qualitätskontrolle; NTC = keine Vorlagensteuerung. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 8: Intra-Assay-Genauigkeit und Präzision von QCs (Copy Targets/μL PCR-Reaktion). Abkürzungen: ULQC = obere Grenze der Qualitätskontrolle; HQC = hohe Qualitätskontrolle; MQC = mittlere Qualitätskontrolle; LQC = niedrige Qualitätskontrolle; LLQC = untere Grenze der Qualitätskontrolle; NTC = keine Vorlagensteuerung. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 9: Interassay-Genauigkeit und Präzision von QCs (Copy Targets/μL PCR-Reaktion). Abkürzungen: ULQC = obere Grenze der Qualitätskontrolle; HQC = hohe Qualitätskontrolle; MQC = mittlere Qualitätskontrolle; LQC = niedrige Qualitätskontrolle; LLQC = untere Grenze der Qualitätskontrolle; NTC = keine Vorlagensteuerung. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 10: Gesamtzahl der Tröpfchenzahlen von Tränenproben. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 11: Statistik der Tränenproben zwischen den Bohrlöchern (Kopienziele/μL-PCR-Reaktion). Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 12: Intra-Assay-Genauigkeit und Präzision von Tränenproben (Copy-Targets/μL-PCR-Reaktion). Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 13: Interassay-Genauigkeit und Präzision von Tränenproben (Copy-Targets/μL-PCR-Reaktion). Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Ergänzende Tabelle S1: Sequenzen von Primern, Sonden und synthetischer doppelsträngiger DNA-Qualitätskontrolle, die in dieser Studie verwendet wurden. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Diskussion
Es gibt mehrere Schritte des ddPCR-Protokolls, die für die ordnungsgemäße Durchführung des Assays entscheidend sind. Der erste kritische Schritt ist das Design und die Optimierung der Primer und der Sonde. Im Allgemeinen wird die Verwendung von Hydrolysesonden-basierter Chemie gegenüber farbstoffbasierter Chemie (z. B. SYBR Green) in einem präklinischen oder klinischen Umfeld aufgrund ihrer überlegenen Spezifität empfohlen. Darüber hinaus ist die Wahl des Amplifikationsziels von entscheidender Bedeutung. Typischerweise wird das Transgen des interessierenden Vektors anvisiert. In früheren präklinischen Stadien oder bei Vektoren, bei denen es möglicherweise nicht möglich ist, Vektortransgen von genomischer DNA zu unterscheiden, kann es jedoch sinnvoll sein, standardisierte Vektorziele zu verwenden. Zum Beispiel könnte man auf die invertierte terminale Wiederholungsregion, den Promotor, den Poly-A-Schwanz oder die Verbindungen zwischen den Segmenten zwischen diesen Vektorkomponenten abzielen. Die Wahl des Ziels hängt vom Vektordesign ab. Herkömmliche qPCR-Primer- und Sondendesignstrategien und -software sind in der Regel für die ddPCR geeignet. Konstruktionsparameter, von denen erwartet wird, dass sie eine konstante Glühtemperatur (z. B. 60 °C) ergeben, sollten ausgewählt werden, um den erforderlichen Optimierungsaufwand zu reduzieren. Es wurde auch empfohlen, mindestens drei verschiedene Sets für jedes Ziel zu entwerfen, zu bestellen und zu bewerten. Man sollte dann das Set auswählen, das die größte Spezifität (keine Amplifikation in der Negativkontrollvertiefung oder in einer Matrix verwandter Ziel-DNA) und Sensitivität (d. h. Nachweisgrenze) aufweist20.
Wenn es vorteilhaft ist, den Assay zwischen qPCR und ddPCR wechseln zu können, wird empfohlen, zuerst die Assay-Bedingungen mit qPCR zu optimieren und Bedingungen für das ausgewählte Set zu identifizieren, die zu Amplifikationseffizienzen von 90%-110% mit einemR2-≥ 0,98 führen. Die ddPCR als Endpunktmethode ist jedoch aufgrund von Varianzen in der Amplifikationseffizienz in der Regel weniger empfindlich als die qPCR. Es wird empfohlen, mindestens einen thermischen Temperaturgradienten im Glüh-/Verlängerungsschritt zu fahren, um Temperaturen über und unter den erwarteten Glühtemperaturen abzudecken und den Regen- und Fluoreszenzamplitudenabstand zwischen den negativen und positiven Tröpfchenclustern in Abhängigkeit von der Temperatur zu bewerten. Wenn der Arbeitsbereich es zulässt, empfiehlt es sich, individuelle dedizierte Arbeitsplätze für die Mastermix-Vorbereitung, das Hinzufügen von Vorlagen und die Verstärkung zu haben. Wenn möglich, sollten diese durch einen unidirektionalen Arbeitsablauf mit integrierten technischen Kontrollen, wie z. B. kontrolliertem Zugang und unterschiedlichen Luftdrücken, physisch getrennt werden, um das Risiko von Kreuzkontaminationen und Fehlalarmen zu verringern. Wenn dies nicht möglich ist, muss äußerste Vorsicht walten lassen, um eine Kreuzkontamination zu vermeiden.
Es gibt zwei Schritte in diesem Protokoll, die für diejenigen, die eher an die Entwicklung von qPCR-Assays gewöhnt sind, ungewöhnlich erscheinen mögen. Die erste ist die Aufnahme eines Restriktionsenzyms in den PCR-Mastermix. Während der ddPCR-Amplifikation wird jedes Tröpfchen bis zum Endpunkt thermozyklisiert. In einem richtig optimierten Assay führt dies zu zwei Populationen von Tröpfchen, von denen eine Gruppe ein konstant hohes Maß an Fluoreszenzsignalen aufweist - die positiven Signale - und ein anderes konsistent ein niedriges Fluoreszenzsignal aufweist - die negativen Signale. Wenn eine PCR-Interferenz auftritt, kann dies die Initiierung der PCR-Amplifikation desynchronisieren, was dazu führt, dass das Tröpfchen kein Amplifikationsplateau erreicht und somit inkonsistente Fluoreszenzendpunkte aufweist. In diesem Fall werden die Tröpfchen zwischen den Negativen und den Positiven verteilt, was zu einem Phänomen führt, das als ddPCR-Regen bezeichnet wird. Dies kann zu einer ungenauen Quantifizierung des Ziels und zu inkonsistenten und subjektiv angewandten Schwellenwerten führen. Unsere Empfehlung, den Schwellenwert leicht über dem Signal des NTC zu setzen, was die Auswirkungen von Regen in der endgültigen Quantifizierung minimieren sollte, da alle Tröpfchen immer noch als positiv angesehen werden, auch wenn sie nicht vollständig bis zum Endpunkt zyklisiert sind. AAVs haben eine hochkomplexe Sekundärstruktur, die je nach Amplifikationsziel die Zugänglichkeit zu den Primern und Sonden einschränken kann, was zu PCR-Interferenzen und damit zu Regen führen kann. Die Aufnahme des Restriktionsenzyms in die Mastermischung spaltet diese Sekundärstruktur, um den Zugang für die Primer und Sonden zu verbessern und den Regen zu reduzieren, was dadurch die Genauigkeit des Assays verbessern kann. Die Effekte des Einschlusses eines Restriktionsenzyms in die ddPCR-Reaktion wurden bereits beschrieben25,32. Jedes Restriktionsenzym kann verwendet werden, solange bestätigt wird, dass es nicht innerhalb des Zielamplifikationsbereichs schneidet. Es sind keine Voraufschlussschritte oder alternative Pufferzusammensetzungen erforderlich.
Der zweite ungewöhnliche Schritt ist die Aufbereitung der AAV-haltigen Tränenprobe. In diesem Protokoll wurde ein Tränenverhältnis von 1:10 (oder mehr) verwendet und anschließend die Probe erhitzt. Wenn Tränen über ein Kapillarröhrchen gesammelt werden, was eine weit verbreitete Entnahmemethode ist, können in der Regel durchschnittlich etwa 10,0 μl gesammelt werden33. Die Verdünnung trägt dazu bei, das begrenzte Probenvolumen zu beheben und genügend Material für die Untersuchung von Doppelbohrungen bereitzustellen. Dies verringert zwar die theoretische Nachweisgrenze, aber die robuste Sensitivität der ddPCR sollte immer noch dazu führen, dass alle bis auf eine extrem wenige Anzahl von Vektorpartikeln nachgewiesen werden können. Dieser Ansatz schafft zusätzlich einen "Backup"-Brunnen, falls einer unerwartet ausfallen sollte. In diesem Fall oder bei unzureichendem Probenvolumen für den Betrieb von zwei Bohrlöchern könnte der Poisson-Fehler zur Beurteilung der Präzision verwendet werden. Darüber hinaus bietet es in Fällen, in denen die Konzentration unterhalb der Nachweisgrenze liegt, die Möglichkeit, Bohrlochdaten zusammenzuführen, um eine Konzentration zu bestimmen. Für den ddPCR-Nachweis ist es notwendig, die AAV-Vektoren von den viralen Kapsiden zu befreien. Einige Methoden zur Quantifizierung von AAV beinhalten einen Proteinase-K-Verdauungsschritt, um das virale Kapsid34,35,36 zu entfernen. Alle natürlich vorkommenden AAV-Serotypen haben Schmelztemperaturen bei oder unter etwa 90 °C, wobei die meisten unter 80 °C fallen; Daher scheint dies eine unnötige Aufnahme zu sein37. Erhitzen allein scheint auszureichen, um Vektor-DNA freizusetzen.
Darüber hinaus ist die ddPCR im Allgemeinen weniger anfällig für PCR-Inhibitoren, die in einer Probe vorhanden sein können und einen qPCR-Assay beeinflussen können. Wenn ein bestimmter DNA-Isolationsschritt enthalten ist, würde dies ebenfalls eine spezifische Validierung erfordern, die in diesem Protokoll vermieden wird. Die Proben werden vor dem Erhitzen aufgrund der Kinetik der Diffusion der Vektorgenome in einer Flüssigkeit verdünnt. Während des Erhitzungs- und anschließenden Abkühlprozesses können die positiven und negativen Sense-Stränge des einzelsträngigen DNA-Genoms zusammenglühen, um ein doppelsträngiges Zwischenprodukt zu erzeugen, wenn die Konzentrationen ausreichend hoch sind. Die Verdünnung vor dem Erhitzen verringert die Konzentrationen und macht es mathematisch unwahrscheinlich, dass sich genügend doppelsträngige Zwischenprodukte bilden, um die Genauigkeit der Quantifizierung nachteilig zu beeinflussen. Es ist zu beachten, dass die synthetischen DNA-Fragmente oder linearisierten Plasmide, die als Qualitätskontrollen verwendet werden, diesem Erhitzungsschritt nicht unterzogen werden dürfen. Da diese doppelsträngig sind, würde eine Erwärmung zu einer Umwandlung in einzelsträngige Zwischenprodukte führen. Nach der unabhängigen Aufteilung dieser einzelsträngigen QCs in Tröpfchen wäre zu erwarten, dass dies zu einer Verdoppelung der QC-Konzentration im Vergleich zur Nennkonzentration führen würde. Wenn QCs erhitzt werden sollen, um die Methode zu standardisieren, muss dies alternativ bei der Rekonstitution und Zuweisung einer Nennkonzentration berücksichtigt werden.
In Bezug auf die Probenvorbereitung empfehlen viele Protokolle auch die Aufnahme eines DNase-Behandlungsschritts, um nicht verkapselte Vektor-DNA zu entfernen. Dieser Schritt ist kritisch in Fällen, in denen es nicht erwünscht ist, freie DNA zu quantifizieren, die mit der Vektorpräparation assoziiert ist (z. B. während der Quantifizierung zu Dosierungszwecken). Im Zusammenhang mit Biodistributions- und Bioshedding-Studien möchte man jedoch in der Regel wissen, wohin eine Vektor-DNA gereist ist, unabhängig davon, ob sie eingekapselt ist oder nicht. Daher wird empfohlen, während solcher Studien in der Regel keinen DNase-Behandlungsschritt durchzuführen. Wenn festgestellt wird, dass ein DNase-Schritt erforderlich ist, sollte dieser Schritt vor der Verdünnung und Erhitzung erfolgen.
In diesem Beitrag werden Daten vorgestellt, die repräsentativ für den Ansatz zur Bewertung des Dynamikbereichs, der Sensitivität, der Genauigkeit und der Präzision der Methode im Kontext einer zweckmäßigen, der guten Laborpraxis entsprechenden Validierung sind. Der derzeitige Mangel an Leitlinien zu diesem Thema überlässt es den validierenden Laboratorien, die Ziel-Assay-Kriterien für sich selbst festzulegen, im Einklang mit dem aktuellen Denken der Branche. Verschiedene Gruppen haben sowohl höhere als auch niedrigere Zielkriterien als in dieser Studie verwendet 19,20,21,22,23,24,25. Die Ziel-Assay-Kriterien sollten, bis sie strenger definiert sind, vor der Validierung auf der Grundlage der beabsichtigten klinischen Anwendungen der Methode ausgewählt werden. Abhängig von den nachgelagerten Entscheidungen, die auf der Grundlage der Daten zu treffen sind, kann ein höheres Maß an Genauigkeit und Präzision erforderlich sein. Umgekehrt kann ein einfaches positives versus negatives Ergebnis ausreichend sein.
Der Ansatz befasste sich auch mit Empfehlungen zur Bewertung der Spezifität und eines Matrixeffekts. Eine Lache von Tränen, die von unbehandelten Personen gesammelt wurden, führte in diesem Assay nicht zu einem positiven Ergebnis, während das Ziel nachgewiesen werden konnte, wenn der Vektor in einer hohen und niedrigen Konzentration innerhalb der empfohlenen Wiederfindungsraten in die Tränen gestochen wurde. Idealerweise wird auch eine Matrix, die einen endogenen Vektor enthält (z. B. nach einer Behandlung mit einem viralen Vektor) in diese Bewertungen einbezogen. Es ist jedoch unwahrscheinlich, dass solche Proben für die Verwendung in einer Validierung zur Verfügung stehen. Um die Robustheit der Validierung zu erhöhen, könnten mehrere Tränenpools oder Tränen, die von einer Vielzahl von Individuen gesammelt wurden, ausgewertet werden, um festzustellen, ob ein patientenspezifischer Matrixeffekt auftritt. Schließlich empfiehlt es sich, die Stabilität zu bewerten. Bei Arbeitsabläufen, bei denen die DNA-Extraktion aus der biologischen Matrix erfolgt, kann es erforderlich sein, die Stabilität sowohl der Probe als auch der extrahierten DNA zu bewerten. Bei diesem Workflow wird die Probe direkt im Assay getestet, ohne dass eine DNA-Extraktion erforderlich ist. Daher muss bei der Bewertung der Stabilität für diese Methode die Stabilität der Tränenproben bewertet werden. In der Regel werden Tisch-, Kühlschrank-, Gefrier-/Auftau- und Langzeitstabilitätsbewertungen empfohlen. Diese wurden nicht im Rahmen dieser Studie durchgeführt, aber die hier entwickelten Methoden können nach Manipulationen an den Eingangsproben in dieser Bewertung verwendet werden.
Insgesamt hat sich diese Methode als robuster, wiederholbarer und validierbarer Assay zum Nachweis von AAV-basierten Vektoren in Tränenproben erwiesen. Es kann als Plattform dienen, die an bestimmte Vektoren angepasst werden kann, um klinische Studien zu unterstützen, und bietet eine Grundlage für die Validierung eines Assays im Einklang mit der guten Laborpraxis.
Offenlegungen
Alle Autoren sind Mitarbeiter von KCAS Bioanalytical and Biomarker Services, einem Auftragsforschungsinstitut, das umfassende Entwicklungsdienstleistungen anbietet, die den Good Laboratory Practices/Good Clinical Practices entsprechen, von der Unterstützung bei der frühen Entdeckung bis zur Produktregistrierung, einschließlich der Unterstützung von AAV-basierten Assays, wie in diesem Protokoll beschrieben. KCAS finanzierte dieses Projekt und die Veröffentlichung dieses Protokolls. Obwohl es keine aktuellen FDA-Richtlinien für das Design und die Validierung der in diesem Artikel vorgestellten Experimente gibt, haben die Experten und Vordenker, die solche Studien am KCAS entwickeln, diesen Ansatz als Basisansatz übernommen. Zusätzliche Kriterien und Parameter werden projektbezogen diskutiert und können je nach Verwendungszweck einbezogen werden.
Danksagungen
Wir bedanken uns bei Nick Russell und Brandon McKethan von Bio-Rad für die hilfreichen Gespräche während der Entwicklung dieser Methode.
Materialien
| Name | Company | Catalog Number | Comments |
| AAV-eGFP Vector | Charles River Laboratories | RS-AAV2-FL | Lot AAV2-0720-FL, used as a proof of principle vector |
| AutoDG Droplet Digital PCR system | Bio-Rad | QX200 | Alternative ddPCR system may be used following manufacturer’s protocol. |
| AutoDG Oil for Probes | Bio-Rad | 1864110 | Or use material compatible with ddPCR system. |
| ddPCR Buffer Control for Probes | Bio-Rad | 1863052 | Or use material compatible with ddPCR system and PCR chemistry. |
| ddPCR Droplet Reader Oil | Bio-Rad | 1863004 | Or use material compatible with ddPCR system. |
| ddPCR Piercable Foil Seals | Bio-Rad | 1814040 | Or use material compatible with ddPCR system. |
| ddPCR Plates 96-Well, Semi-Skirted | Bio-Rad | 12001925 | Or use material compatible with ddPCR system. |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023, 1863024, or 1863025 | Use master mix compatible with primers/probes and ddPCR system. |
| DG32 AutoDG Cartidges | Bio-Rad | 1864108 | Or use material compatible with ddPCR system. |
| Droplet Reader | Bio-Rad | QX200 | Alternative ddPCR system may be used following manufacturer’s protocol. |
| GeneAmp PCR Buffer | Applied Biosystems | N8080129 | N/A |
| Nuclease-Free Water | Ambion | AM9906 | N/A |
| PCR Plate Sealer | Bio-Rad | PX1 | Or use material compatible with ddPCR system. |
| Pipet Tips for AutoDG | Bio-Rad | 1864120 | Or use material compatible with ddPCR system. |
| Pluronic F-68 Non-ionic Surfactant | Gibco | 24040 | N/A |
| Primer and Hydrolysis Probes | Various | Various | Design based on target sequence using general approaches for primer/probe design. Select fluorphores and quenchers compatible with ddPCR system. |
| Restriction Enzyme | Various | Various | Varies with target amplification sequence. Use restriction enzyme that does not cut in the amplified sequence |
| Sheared salmon sperm DNA | ThermoFisher | AM9680 | N/A |
| Synthetic DNA gene fragment or linearized plasmid | Various | Various | Design a synthetic DNA fragment containing the target amplification region for use as a quality control |
| TE Buffer | Teknova | T0224 | Ensure prepared or purchases nuclease free. 10 mM Tris-HCl, 1.0 mM EDTA, pH=8.0 |
| Touch Thermal Cycler | Bio-Rad | C1000 | Or use material compatible with ddPCR system. |
Referenzen
- Sherkow, J. S., Zettler, P. J., Greeley, H. T. Is it 'gene therapy. Journal of Law and the Biosciences. 5 (3), 786-793 (2018).
- Ginn, S. L., Amaya, A. K., Alexander, I. E., Edelstein, M., Abedi, M. R. Gene therapy clinical trials worldwide to 2017: an update. The Journal of Gene Medicine. 20 (5), 3015 (2018).
- Ghosh, S., Brown, A. M., Jenkins, C., Campbell, K. Viral vector systems for gene therapy: a comprehensive literature review of progress and biosafety challenges. Applied Biosafety. 25 (1), 7-18 (2020).
- Gene, Cell, & RNA therapy landscape, Q4 2022 quarterly data report. American Society for Gene & Cell Therapy Available from: https://asgct.org/global/documents/asgct_citeline-q4-2022-report_final.aspx (2022)
- Approved Cellular and Gene Therapy Products. United States Food and Drug Administration Available from: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (2022)
- Au, H. K. E., Isalan, M., Meilcarek, M. Gene therapy advances: a meta-analysis of AAV usage in clinical settings. Frontiers in Medicine. 8, 809118 (2022).
- Lundstrom, K. Viral vectors in gene therapy. Diseases. 6 (2), 42 (2018).
- Naso, M. F., Tomkowicz, B., Perry, W. L., Strohl, W. R. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 31, 317-334 (2017).
- Russell, S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomized, controlled, open-label, phase 3 trial. Lancet. 390 (10097), 849-860 (2017).
- Chemistry, manufacturing, and control (CMC) information for human gene therapy investigational new drugs (INDs). United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/chemistry-manufacturing-and-control-cmc-information-human-gene-therapy-investigationsal-new-drug (2020)
- Long term follow-up after administration of human gene therapy products. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/long-term-follow-after-adminstration-human-gene-therapy-products (2020)
- Testing of retroviral vector-based human gene therapy products for replication competent retrovirus during product manufacture and patient follow-up. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/testing-retroviral-vector-based-human-gene-therapy-products-replication-competent-retrovirus-during (2020)
- Recommendations for microbial vectors used in gene therapy. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/recommendations-microbial-vectors-used-gene-therapy (2016)
- Multidisciplinary: gene therapy. European Medicines Agency Available from: https://www.europa.eu/en/human-regulatory-development/scientific-guidelines/multidisciplinary/multidisciplinary-gene-therapy (2023)
- Human gene therapy for retinal disorders. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-retinal-disorders (2020)
- Design and analysis of shedding studies for virus or bacterial-based gene therapy and oncolytic products. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-and-analysis-shedding-studies-virus-or-bacteria-based-gene-therapy-and-oncolytic-products (2015)
- Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products. European Medicines Agency Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-clinical-aspects-gene-therapy-medicinal-products_en.pdf (2018)
- Kaur, S. white paper on recent issues in bioanalysis: mass spec of proteins, extracellular vesicles, CRISPR, chiral assays, oligos; nanomedicines bioanalysis; ICH M10 section 7.1; non-liquid & rare matrices; regulatory inputs (part 1A - recommendations on endogenous compounds, small molecules, complex methods, regulated mass spec of large molecules, small molecule, PoC & part 1B - regulatory agencies' inputs on bioanalysis, biomarkers, immunogenicity, gene & cell therapy and vaccine). Bioanalysis. 14 (9), 505-580 (2022).
- Hays, A., Islam, R., Matys, K., Williams, D. Best practices in qPCR and qPCR validation in regulated bio analytical laboratories. The AAPS Journal. 24 (2), 36 (2022).
- Ma, H., Bell, K. N., Loker, R. N. qPCR and qRT-PCR analysis: Regulatory points to consider when conducting biodistribution and vector shedding studies. Molecular Therapy. Methods & Clinical Development. 17 (20), 152-168 (2020).
- Wissel, M. Recommendations on qPCR/ddPCR assay validation by GCC. Bioanalysis. 14 (12), 853-863 (2022).
- Expectations for biodistribution (BD) assessments for gene therapy (GT) products. International Pharmaceutical Regulators Programme Available from: https://admin.iprp.global/sites/default/files/2018-09/IPRP_GTWG_ReflectionPaper_BD_Final_2018_0713.pdf (2018)
- Pinheiro, L., Emslie, K. R. Basic concepts and validation of digital PCR measurements. Methods in Molecular Biology. 1768, 11-24 (2018).
- Tzonev, S. Fundamentals of counting statistics in digital PCR: I measured two target copies-what does it mean. Methods in Molecular Biology. 1768, 25-43 (2018).
- Prantner, A., Marr, D. Genome concentration, characterization, and integrity analysis of recombinant adeno-associated viral vectors using droplet digital PCR. PLoS One. 18, 0280242 (2023).
- Primer-BLAST. National Library of Medicine, National Center for Biotechnology Information Available from: https://www.ncbi.nim.nih.gov/tools/primer-blast/ (2023)
- Koressaar, T., Remm, M. Enhancements and modifications of primer design program Primer 3. Bioinformatics. 23 (10), 1289-1291 (2007).
- Ye, J. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 13, 134 (2012).
- Droplet Digital PCR Application Guide. Bio-Rad Available from: https://www.bio-rad.com/webroot/we/pdf/lsr/literature/Bulletin_6407.pdf (2023)
- Qian, P. L., Sauzade, M., Brouzes, E. dPCR: A technology review. Sensors. 18 (4), 1271 (2018).
- Basu, A. Digital assays part I: portioning statistics and digital PCR. SLAS Technology. 22 (4), 369-386 (2017).
- Sanmiguel, J., Gao, G., Vandeberghe, L. H. Quantitative and digital droplet-based AAV genome titration. Methods in Molecular Biology. 1950, 51-83 (2019).
- Bachhuber, F., Huss, A., Senel, M., Tumani, H. Diagnostic biomarkers in tear fluid: from sampling to preanalytical processing. Scientific Reports. 11, 10064 (2021).
- Martinez-Fernandez de la Camara, C., McClements, M. E., MacLaren, R. E. Accurate quantification of AAV vector genomes by quantitative PCR. Genes. 12 (4), 601 (2021).
- Ai, J., Ibraheim, R., Tai, P. W. L., Gao, G. A scalable and accurate method for quantifying vector genomes of recombinant adeno-associated viruses in crude lysate. Human Gene TherapyMethods. 28 (3), 139-147 (2017).
- Dobnik, D., et al. Accurate quantification and characterization of adeno-associated viral vectors. Frontiers in Microbiology. 10, 1570 (2019).
- Bennett, A., et al. Thermal stability as a determinant of AAV serotype identity. Molecular Therapy Methods and Clinical Development. 6, 171-182 (2017).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten