
Method Article
Un saggio di reazione a catena della polimerasi digitale a goccia validabile per la rilevazione di vettori virali adeno-associati negli studi di bioshedding delle lacrime
In questo articolo
Riepilogo
Qui, presentiamo un protocollo per lo sviluppo e la convalida di buone pratiche di laboratorio nella rilevazione conforme di vettori virali adeno-associati in lacrime umane mediante reazione a catena della polimerasi digitale a supporto dello sviluppo clinico di vettori di terapia genica.
Abstract
L'uso di vettori virali per il trattamento di malattie genetiche è aumentato notevolmente negli ultimi anni, con oltre 2.000 studi registrati fino ad oggi. I vettori virali adeno-associati (AAV) hanno riscontrato particolare successo nel trattamento delle malattie correlate agli occhi, come esemplificato dall'approvazione di voretigene neparvovec-rzyl. Per portare nuove terapie sul mercato, le agenzie regolatorie in genere richiedono studi di bioshedding qualificati o convalidati per valutare il rilascio del vettore nell'ambiente. Tuttavia, nessuna linea guida ufficiale per lo sviluppo di saggi a base molecolare a supporto di tali studi di spargimento è stata rilasciata dalla Food and Drug Administration degli Stati Uniti, lasciando agli sviluppatori di determinare le migliori pratiche per se stessi. Lo scopo di questo protocollo è quello di presentare un protocollo validabile per la rilevazione di vettori AAV in lacrime umane mediante reazione a catena della polimerasi digitale a goccia (ddPCR) a supporto di studi clinici di bioshedding. Questo manoscritto discute gli attuali approcci industriali alla convalida dei saggi molecolari e dimostra che il metodo supera i criteri di accettazione del test target attualmente proposti nei white paper. Infine, vengono discussi i passaggi critici nell'esecuzione di qualsiasi test ddPCR, indipendentemente dall'applicazione.
Introduzione
Le definizioni di terapia genica variano, ma generalmente inducono un'alternanza permanente intenzionale e spesso prevista di una specifica sequenza di DNA del genoma cellulare per modificare o manipolare l'espressione di un gene o alterare le proprietà biologiche di una cellula vivente per uno scopo clinico 1,2. I vettori virali sono sempre più utilizzati come veicoli per la terapia genica a causa della loro efficienza di trasduzione, con un rapporto che suggerisce che oltre il 70% degli attuali studi clinici di terapia genica utilizza vettori virali3. L'interesse per i vettori virali per la terapia genica è in costante aumento. Il Quarter 4 2022 Quarterly Data Report on the Gene, Cell, and RNA therapy dell'American Society of Gene and Cell Therapy ha riportato che nel 2022 la pipeline di terapia genica, cellulare e RNA dalla preclinica alla pre-registrazione è cresciuta del 7%, portando il numero totale di terapie in fase di sviluppo a 3.726, di cui 2.053 (55%) erano terapie geniche4. La Food and Drug Administration degli Stati Uniti (USA FDA) ha attualmente approvato 27 terapie cellulari e geniche per uso clinico negli esseri umani, cinque delle quali utilizzano specificamente vettori virali5.
I virus adeno-associati (AAV) hanno guadagnato un interesse specifico come veicoli per la terapia genica. Una recente meta-analisi ha rivelato che ci sono stati circa 136 studi clinici che hanno studiato l'uso di AAV negli ultimi due decenni6. Inoltre, tre delle cinque terapie geniche approvate dalla FDA USA sono basate su AAV. Ciò è dovuto alla loro natura altamente modificabile, all'ampia gamma di ospiti che possono essere regolati in base all'uso di specifici vettori naturali o artificiali, alla bassa patogenicità e tossicità nell'uomo e generalmente alla bassa immunogenicità 7,8. Gli AAV sono stati anche utilizzati con successo per trattare le malattie oculari in un ambiente clinico approvato. Voretigene neparvovec-rzyl è una terapia a base di AAV2 approvata dalla FDA statunitense nel 2017 e dall'Agenzia europea per i medicinali (EMA) nel 2018 per il trattamento della distrofia retinica associata alla mutazione biallelica RPE65 9.
Con il crescente interesse per lo sviluppo di terapie basate su AAV arriva la necessità di linee guida normative sui saggi. Il rilevamento e la quantificazione accurati di qualsiasi vettore virale sono parte integrante delle fasi di scoperta, produzione e test preclinico/clinico dello sviluppo del prodotto. La FDA statunitense ha iniziato a pubblicare alcune linee guida per le terapie geniche, tra cui la chimica, la produzione e il controllo per le applicazioni sperimentali di nuovi farmaci per la terapia genica umana10, il follow-up a lungo termine dopo la somministrazione della terapia genica 11, i test retrovirus competenti per la replicazione 12 e le raccomandazioni per i vettori microbici utilizzati nelle terapie geniche 13. L'EMA ha anche pubblicato una serie di linee guida riguardanti lo sviluppo di prodotti di terapia genica che generalmente si allineano con le raccomandazioni della FDA, sebbene esistano alcune differenze14. È importante notare che, sebbene queste linee guida non stabiliscano responsabilità legalmente applicabili, tranne quando si fa riferimento a normative specifiche, forniscono chiarezza sull'attuale pensiero delle agenzie di regolamentazione sull'argomento e le loro aspettative per i test richiesti per i depositi di farmaci e l'approvazione normativa.
La FDA raccomanda specificamente che siano condotti studi per valutare la distribuzione, la persistenza e la clearance di un vettore dal sito di somministrazione per colpire i tessuti oculari e non oculari, i fluidi intraoculari e il sangue15. Questi assumono la forma di studi di biodistribuzione e spargimento. Gli studi di biodistribuzione valutano l'esposizione studiando come un prodotto si diffonde in tutto il corpo di un paziente dal sito di somministrazione. Lo spargimento valuta specificamente il rilascio del prodotto dal paziente nell'ambiente e aumenta la possibilità di trasmissione del vettore a individui non trattati16. La FDA formula raccomandazioni per la progettazione di studi di biodistribuzione e spargimento per quanto riguarda la frequenza della raccolta dei campioni, la durata della raccolta dei campioni, i tipi di campioni raccolti e le condizioni di conservazione.
Inoltre, la FDA raccomanda l'uso della reazione a catena della polimerasi quantitativa (qPCR o real-time PCR) per la rilevazione quantitativa dei genomi vettoriali grazie alla sua facilità di prestazioni, al formato ad alta produttività, ai rapidi tempi di consegna e alla sensibilità del test. Tuttavia, vi è una relativa mancanza di raccomandazioni per la progettazione e la valutazione delle prestazioni dei metodi molecolari rispetto a quelli esistenti per le molecole piccole e grandi. Molte delle linee guida per tali studi sono difficili da applicare ai metodi molecolari a causa della progettazione unica e complessa sia dei prodotti che dei saggi stessi, sollevando dubbi sull'adeguatezza delle piattaforme disponibili per le valutazioni raccomandate e dei metodi appropriati per la convalida dei saggi. Ad oggi, la FDA non ha richiesto la convalida formale dei test basati sulla PCR, sebbene l'EMA abbia imposto questo requisito17. Alla luce di questo vuoto, diversi gruppi e workshop hanno pubblicato white paper e raccomandazioni che i produttori e le organizzazioni di ricerca a contratto hanno cercato di seguire 18,19,20,21,22,23,24,25. La maggior parte di queste raccomandazioni sono scritte specificamente con saggi qPCR in mente, con suggerimenti o modifiche per piattaforme emergenti, come droplet digital PCR (ddPCR), inclusi solo se ritenuto rilevante. Le raccomandazioni più recenti si sono concentrate sulle considerazioni per i saggi ddPCR, ma si sono in gran parte concentrate sulle loro applicazioni alla quantificazione del genoma vettoriale in un ambiente di produzione piuttosto che nelle complesse matrici biologiche incontrate negli studi di bioshedding.
A seconda dell'applicazione clinica e degli obiettivi, la ddPCR può essere preferita alla qPCR a supporto degli studi di biodistribuzione e spargimento a causa della maggiore sensibilità e capacità di gestire l'interferenza della matrice rispetto alla qPCR. Inoltre, grazie alla suddivisione dei campioni in circa 20.000 goccioline, è possibile ottenere una quantificazione accurata del numero di copie senza l'uso di una curva standard utilizzando la statistica di Poisson, semplificando lo sviluppo e la convalida del metodo. L'obiettivo di questo protocollo è quello di descrivere un approccio standardizzato per lo sviluppo e la validazione di un metodo basato su ddPCR per la rilevazione di vettori AAV in lacrime raccolte dalla superficie oculare a supporto di studi clinici di bioshedding.
Protocollo
1. Preparazione di un frammento di DNA sintetico
- Progettare e ordinare un frammento di DNA sintetico contenente la regione di amplificazione target da utilizzare come controllo di qualità.
- Assicurarsi che la sequenza contenga l'intera sequenza di ampliconi dal primer in avanti al primer inverso del gene bersaglio di interesse, con un'estensione da quattro a sei coppie di basi di sequenza alle estremità 5' di ogni sequenza di legame del primer.
- Evitare omopolimeri di adenina e timina superiori a 12 paia di basi o coppie di basi guanina e citosina superiori a otto paia di basi, poiché gli omopolimeri possono interferire con la sintesi del frammento genico.
NOTA: Se gli ampliconi contengono tali sequenze, le sostituzioni di base possono essere effettuate purché vengano mantenuti i siti di ricottura per i primer e le sonde. - In alternativa, preparare un plasmide linearizzato contenente l'amplicone utilizzando strategie di clonazione tipiche.
- Centrifugare il tubo contenente il frammento di DNA sintetico in una microcentrifuga per ~ 10 s per garantire che il materiale venga raccolto sul fondo del tubo.
- Risospendere il frammento di DNA sintetico utilizzando tampone tris-EDTA (TE) a una concentrazione di 1,0 ×10 10 copie/μL o, se del caso, in base all'intervallo di analisi target.
- Vortice brevemente, quindi incubare a 50 °C per 20 ± 5 min. Raffreddare sul ghiaccio.
- Preparare aliquote multiple, idealmente monouso e conservare a -70-90 °C fino all'uso.
NOTA: I frammenti di DNA sintetico preparati in questo modo sono generalmente stabili per almeno 24 mesi dalla data di risospensione. - Se lo si desidera, determinare l'esatta concentrazione dello stock di DNA sintetico preparato prima dell'uso come controllo di qualità o stimare la concentrazione nominale in base alla risospensione utilizzata.
2. Preparazione di primer e sonda
- Progettare e ordinare primer e una sonda di idrolisi per indirizzare la regione di amplificazione desiderata utilizzando strategie di progettazione tipiche26,27.
- Utilizzare un colorante reporter fluorescente da 5' (ad esempio, FAM) e un quencher da 3 '(ad esempio, quencher scuro nero dell'Iowa) compatibile con il sistema ddPCR.
NOTA: Esistono numerosi pacchetti software per la progettazione di saggi PCR e possono essere utilizzati. Ad esempio, Primer-BLAST del National Center for Biotechnology Information28 è ampiamente utilizzato grazie alle robuste opzioni per la progettazione del saggio e alla facilità con cui la specificità può essere valutata bioinformaticamente per identificare possibili effetti fuori bersaglio. Va notato che la preparazione di primer e sonde può variare dai passaggi elencati qui a seconda del formato in cui vengono forniti.
- Utilizzare un colorante reporter fluorescente da 5' (ad esempio, FAM) e un quencher da 3 '(ad esempio, quencher scuro nero dell'Iowa) compatibile con il sistema ddPCR.
- Centrifugare i tubi contenenti il primer anteriore, il primer inverso e la sonda in una microcentrifuga per ~ 10 s al materiale pellet sul fondo del tubo.
- Risospendere i primer a 20 μM utilizzando il buffer TE. Vortice brevemente.
- Risospendere la sonda a 10 μM utilizzando il buffer TE. Vortice brevemente.
- Preparare aliquote multiple, idealmente monouso e conservare ad almeno -20 °C fino all'uso.
NOTA: I primer e le sonde preparati in questo modo sono generalmente stabili per almeno 24 mesi dalla data di risospensione.
3. Preparazione del tampone di diluizione del campione
- Tampone PCR di scongelamento e DNA di sperma di salmone tosato a temperatura ambiente. Vortice accuratamente per mescolare.
- Preparare un tampone di diluizione del campione, come da Tabella 1.
- Vortice a fondo. Conservare a 2-8 °C fino a 1 mese dopo la preparazione.
Tabella 1: Preparazione del tampone di diluizione del campione. Clicca qui per scaricare questa tabella.
4. Preparazione del master mix
- Scongelare la miscela master ddPCR per sonde, primer in avanti, primer inverso e sonda a temperatura ambiente e lasciare riscaldare per almeno 10 minuti dopo lo scongelamento prima dell'uso. Conservare a temperatura ambiente fino all'uso.
NOTA: Questi reagenti devono essere completamente portati a temperatura ambiente per garantire un'efficiente formazione di goccioline. Non tenere i reagenti sul ghiaccio durante la preparazione.- Vortex accuratamente e brevemente centrifugare in una mini centrifuga prima dell'uso.
NOTA: Gli enzimi di restrizione sono tipicamente forniti in glicerolo e devono essere rimossi dalla conservazione immediatamente prima dell'uso. Mescolare delicatamente. Non vortice.
- Vortex accuratamente e brevemente centrifugare in una mini centrifuga prima dell'uso.
- Preparare un master mix PCR per ogni target di amplificazione. Vedere la Tabella 2 per una composizione suggerita della miscela principale PCR e modificare le concentrazioni di primer e sonde come richiesto.
- Accuratamente vortice e centrifugare brevemente prima dell'aggiunta dell'enzima di restrizione. Aggiungere l'enzima di restrizione e invertire per mescolare.
NOTA: In questa fase, sono necessari 22 μL di reazione PCR per ottenere un volume finale di 40 μL di reazione PCR dopo la formazione di goccioline (costituito da 15 μL di master mix PCR, 5,0 μL di modello e 20 μL di olio per la generazione di goccioline).
- Accuratamente vortice e centrifugare brevemente prima dell'aggiunta dell'enzima di restrizione. Aggiungere l'enzima di restrizione e invertire per mescolare.
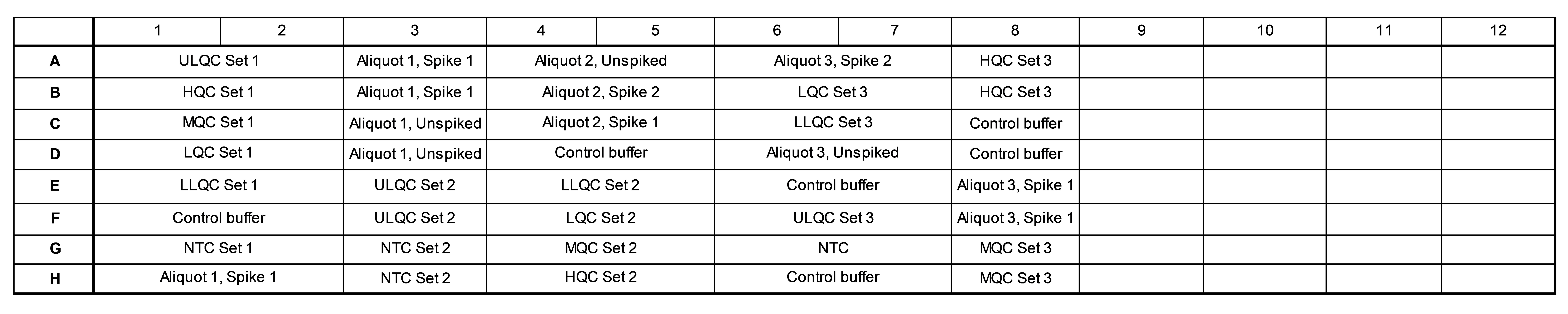
- Aggiungere 16,5 μL di miscela master a ciascun pozzetto secondo la mappa della piastra. Vedere la Figura 1 per una mappa delle piastre di esempio per l'accuratezza della convalida e l'esecuzione di precisione.
- Assicurarsi che una piastra contenga tre preparati indipendenti della serie di controllo qualità (QC), tre aliquote lacrimali endogene testate in modo indipendente, aumentate a un livello alto e basso e senza punte e tre controlli indipendenti senza modello (NTC).
- Variare la disposizione di questi pozzetti attraverso la piastra, dove il Set 1 viene caricato in ordine di concentrazione decrescente, il Set 2 viene caricato in ordine di concentrazione crescente e il Set 3 viene caricato in ordine casuale per valutare se ci sono effetti specifici della posizione della piastra.
- Disporre i campioni per riempire il maggior numero possibile di colonne e riempire i pozzetti inutilizzati all'interno di una colonna con buffer di controllo. Includere più lotti di controllo endogeni (ad esempio, più pozze di lacrime o lacrime raccolte da individui) nei pozzi rimanenti, se lo si desidera.
- Sigillare la piastra con pellicola adesiva trasparente. Tenere la piastra a temperatura ambiente durante la preparazione del modello. In alternativa, tenere la piastra per un massimo di 4 ore a 2-8 °C, ma riportarla a temperatura ambiente per almeno 10 minuti prima dell'aggiunta della dima.
Tabella 2: Esempio di preparazione della miscela master PCR. Clicca qui per scaricare questa tabella.

Figura 1: Esempio di mappa delle piastre per l'accuratezza della convalida e la precisione dell'esecuzione. Abbreviazioni: ULQC = controllo di qualità del limite superiore; HQC = controllo di alta qualità; MQC = controllo qualità medio; LQC = basso controllo di qualità; LLQC = controllo di qualità del limite inferiore; NTC = nessun controllo modello. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
5. Preparazione dei CQ
- Scongelare frammenti di DNA sintetico o plasmidi linearizzati a temperatura ambiente e lasciare riscaldare per almeno 10 minuti dopo lo scongelamento prima dell'uso. Portare i modelli a temperatura ambiente per garantire un'efficiente formazione di gocce.
- Conservare a temperatura ambiente fino all'uso. Vortex accuratamente e brevemente centrifugare in una mini centrifuga prima dell'uso.
- Preparare le diluizioni QC utilizzando il tampone di diluizione del campione come diluente. Un esempio delle concentrazioni raccomandate per preparare un'accuratezza di convalida e una corsa di precisione è presentato nella Tabella 3.
NOTA: dopo aver completato con successo le esecuzioni di accuratezza e precisione, su ogni piastra è necessario eseguire solo il controllo di alta qualità (HQC), il controllo di qualità media (MQC) e il controllo di bassa qualità (LQC). Per l'accuratezza e la precisione, sono incluse almeno tre diluizioni indipendenti dei QC per la valutazione dell'accuratezza e della precisione intra-test. Dopo l'accuratezza e la precisione, è necessario includere una sola serie di diluizioni. - Dopo la preparazione, conservare le diluizioni a temperatura ambiente fino ad aggiungerle alla piastra.
- Conservare le diluizioni su ghiaccio o a 2-8 °C se necessario. Prima dell'uso successivo, lasciare che le diluizioni si riscaldino a temperatura ambiente per almeno 10 minuti prima dell'uso. Scartare i QC alla fine della giornata.
Tabella 3: Esempio di preparazione del controllo di qualità (QC) utilizzando frammenti sintetici di DNA a doppio filamento. Abbreviazioni: ULQC = controllo di qualità del limite superiore; HQC = controllo di alta qualità; MQC = controllo qualità medio; LQC = basso controllo di qualità; LLQC = controllo di qualità del limite inferiore; NTC = nessun controllo modello. Clicca qui per scaricare questa tabella.
6. Preparazione dei campioni
- Scongelare i campioni di lacrime raccolti da uno studio clinico a temperatura ambiente fino allo scongelamento e lasciare riscaldare per almeno 10 minuti dopo lo scongelamento prima dell'uso.
- Conservare a temperatura ambiente fino all'uso. Vortex accuratamente e brevemente centrifugare in una microcentrifuga prima dell'uso.
- Diluire i campioni di lacrime 1:10 (o superiore) utilizzando il tampone di diluizione del campione come diluente in provette PCR da 0,2 ml o strisce PCR a 8 pozzetti. Sigillare i tubi.
NOTA: A seconda della concentrazione prevista del target nelle lacrime, potrebbe essere necessario diluire ulteriormente i campioni o testare più diluizioni di ciascun campione. - Riscaldare i campioni in un termociclatore a 95 °C per 10 minuti, quindi tenere a 4 °C per almeno 5 minuti per raffreddare. Utilizzare una velocità di rampa di 3 °C/s.
NOTA: I campioni possono rimanere nel termociclatore a 4 °C fino all'uso nello stesso giorno o possono essere congelati a -70-90 °C per una conservazione più lunga. Questo passaggio serve a denaturare il capside vettore, rilasciando il genoma. Poiché i frammenti di DNA sintetico QC o plasmidi linearizzati sono a doppio filamento, non dovrebbero subire questa fase di riscaldamento. - Riportare i campioni dopo il raffreddamento a temperatura ambiente (o se congelati, scongelarli a temperatura ambiente) e lasciare riscaldare per almeno 10 minuti.
NOTA: I campioni devono essere completamente portati a temperatura ambiente per garantire un'efficiente formazione di goccioline.
7. Aggiunta di modelli
- Recuperare la piastra ddPCR contenente la miscela master. Vortice ogni campione o provetta di diluizione QC accuratamente e brevemente centrifugare per raccogliere il materiale.
- Rimuovere la pellicola adesiva e aggiungere 5,5 μL di QC o campioni ai pozzetti appropriati della piastra a 96 pozzetti, come da mappa della piastra.
NOTA: fare riferimento al passaggio 4.2.1 per la spiegazione dei volumi richiesti - Aggiungere 5,5 μL di tampone di diluizione del campione ai pozzetti NTC.
- La generazione di goccioline richiede che tutti i pozzetti di una colonna abbiano un controllo di reazione o buffer. Se i pozzetti di una colonna non contengono reazioni del campione, diluire 2x controllo tampone ddPCR 1:2 usando acqua priva di nucleasi. Aggiungere 22 μL di 1x controllo buffer ddPCR a qualsiasi pozzetto vuoto di una colonna.
Nota : se non viene utilizzata un'intera colonna, non è necessario aggiungere il controllo buffer a questi pozzetti. - Aggiungere un sigillo di alluminio perforabile al piatto. Posizionare la piastra nel sigillante e sigillare per 5 secondi a 180 °C.
- In alternativa, sigillare la piastra secondo le raccomandazioni del produttore del sistema ddPCR.
- Vortice la piastra alla massima velocità per almeno 30 s (utilizzando l'impostazione del vortice continuo; non utilizzare il vortice a contatto) e centrifugare brevemente in un filatore a piastre.
NOTA: La miscelazione accurata e completa della piastra in questa fase è fondamentale per una corretta ripartizione della reazione PCR in goccioline. Assicurarsi che non ci siano bolle visibili nei pozzi. Se necessario, la piastra può essere tenuta a 2-8 °C prima della generazione di goccioline per un massimo di 4 ore. Se tenuta, lasciare che la piastra raggiunga la temperatura ambiente per un minimo di 10 minuti prima della generazione di goccioline.
8. Generazione automatica di goccioline, cicli termici e lettura delle gocce
- Generare goccioline nel generatore automatico di goccioline come segue.
- Sul touch screen, selezionare le colonne sulla mappa della lastra contenente i campioni. Il deck dello strumento si illuminerà per indicare quali materiali di consumo (cartucce DG32, punte, contenitore dei rifiuti, olio per la generazione di gocce) sono necessari. Le luci gialle indicano che è necessario aggiungere un materiale di consumo, mentre le luci verdi indicano che sono disponibili materiali di consumo sufficienti.
- Caricare il generatore di gocce da dietro a davanti.
- Per le sonde di idrolisi, assicurarsi che sia installato l'olio di generazione di goccioline per le sonde e che rimanga olio sufficiente per il numero di pozzi. Se vengono utilizzati prodotti chimici PCR alternativi, assicurarsi che sia installato un olio per la generazione di gocce compatibile.
- Posizionare un blocco freddo nel supporto della piastra a goccia. Assicurati che il blocco sia completamente di colore blu e che non sia visibile alcun rosa. Posizionare una nuova piastra ddPCR a 96 pozzetti nel blocco freddo.
- Posizionare la piastra PCR preparata nel supporto della piastra del campione. Chiudere il coperchio della macchina. Premere start per la generazione di goccioline.
- Dopo la formazione di goccioline, un totale di 40 μL per reazione viene trasferito automaticamente alla nuova piastra PCR.
- Entro 30 minuti dal completamento della generazione di goccioline, rimuovere la piastra contenente le goccioline dal blocco freddo. Lavora delicatamente poiché le goccioline sono più fragili in questa fase.
- Aggiungere un sigillo di alluminio perforabile al piatto. Posizionare la piastra nel sigillante e sigillare per 5 secondi a 180 °C.
- In alternativa, sigillare la piastra secondo le raccomandazioni del produttore del sistema ddPCR.
- Posizionare la piastra in un termociclatore compatibile. Inserire le condizioni di ciclismo (vedi tabella 4).
- Dopo la fine del ciclo termico, tenere la piastra nel termociclatore, trasferita a 2-8 °C, o leggerla immediatamente.
NOTA: Tenere la piastra per 12 ore a 4-12 °C può migliorare la conta delle goccioline, ma questo non è necessario. Goccioline sufficienti dovrebbero essere ottenute senza la presa. - Caricare la piastra nel lettore di gocce, assicurandosi che rimanga sufficiente olio per il lettore e che il contenitore dei rifiuti abbia spazio sufficiente. Leggi le goccioline. Eseguire la lettura delle goccioline entro 24 ore dall'inizio del ciclo termico.
Tabella 4: Condizioni tipiche del ciclo termico. Clicca qui per scaricare questa tabella.
9. Analisi dei dati
NOTA: Un minimo di 10.000 goccioline per pozzetto è necessario per il corretto calcolo della concentrazione utilizzando la statistica di Poisson. Non tentare l'analisi su pozzi con meno di 10.000 goccioline.
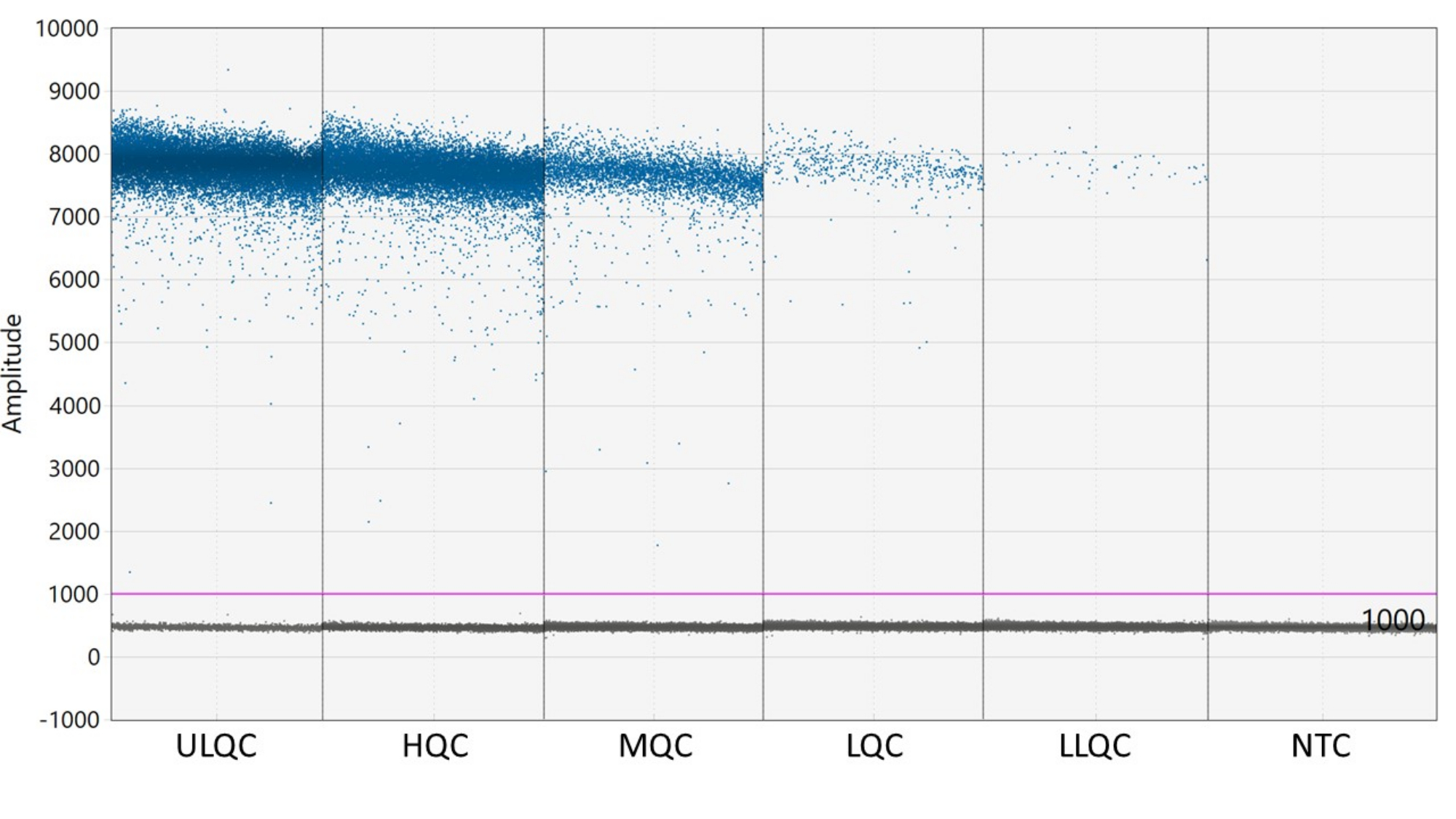
- È necessaria una soglia per definire le goccioline come positive o negative. Il software di analisi ddPCR applica automaticamente una soglia che può variare tra i pozzi. Tuttavia, impostare manualmente una soglia per tutti i pozzetti della piastra leggermente superiore all'intensità fluorescente dei pozzetti NTC per risultati più coerenti, accurati e precisi.
NOTA: Il corretto posizionamento della soglia può richiedere un'ottimizzazione a seconda della separazione delle goccioline positive e negative e della quantità di pioggia di goccioline presente (vedere la Figura 2). In questo esempio, il grafico dell'ampiezza delle gocce mostra pozzetti di esempio a ciascun livello QC e NTC. La linea viola indica una soglia di 1.000, posta leggermente al di sopra della popolazione di goccioline negative. - La modellazione statistica di Poisson richiede almeno tre goccioline positive per calcolare la concentrazione con un'affidabilità del 95%. Considera tutti i pozzetti contenenti zero, una o due goccioline positive come negativi e impostati su una concentrazione di zero27.
- Calcola il numero di copie in ciascun campione di strappo.
- La concentrazione, in copie/μL, è fornita nel rapporto sui dati. Utilizzare questo valore per determinare la concentrazione in copie/μL del campione originale (cioè nel campione lacrimale).
- Per calcolare la diluizione della reazione ddPCR, dividere il volume iniziale della reazione PCR prima della formazione di goccioline per il volume di modello aggiunto. Quando vengono utilizzati i volumi presentati in questo metodo, si ottiene un valore pari a 4.

- Determinare il fattore di diluizione seriale dal campione originale (punto 6.2).
- Per determinare le copie/μL nel campione, moltiplicare le copie/μL per la diluizione della reazione ddPCR, quindi per il fattore di diluizione seriale. Ad esempio, la concentrazione in copie/μL generata nel rapporto sui dati era 966; Sono stati aggiunti 5,5 μL di modello per 22 μL di reazione. È stata utilizzata una diluizione seriale 1:50.000 del campione.


- Se sono state testate più diluizioni dello stesso campione, analizzare tutte le diluizioni valide nell'intervallo e calcolare la media.
- Per ogni QC, calcolare le copie attese/μL di reazione PCR dividendo la concentrazione della diluizione QC data (in copie/μL) per il volume di reazione ddPCR (20 μL). Ciò consente il confronto diretto di questo valore nominale con il valore copie/μL fornito nel rapporto dati senza ulteriori calcoli.
NOTA: Questo approccio è stato utilizzato anche per l'analisi dei campioni di lacrime a spillo utilizzati nei risultati rappresentativi. - Determinare il valore medio, la deviazione standard, il coefficiente di variazione (%CV) e l'errore relativo percentuale rispetto alla concentrazione nominale (%RE) del campione o del valore QC utilizzando i pozzetti replicati (includere più diluizioni se applicabile).
- Per la valutazione della precisione tra pozzetti, determinarla per ciascuno dei duplicati del pozzo, se inclusi.
- Per la valutazione dell'accuratezza e della precisione intra-test, determinarla per ciascuna serie di diluizione o aliquota utilizzata all'interno di un lotto.
- Per la valutazione dell'accuratezza e della precisione tra test, determinarla utilizzando i mezzi intra-test di ciascuno dei lotti inclusi.

Figura 2: Esempio di impostazione della soglia. Abbreviazioni: ULQC = controllo di qualità del limite superiore; HQC = controllo di alta qualità; MQC = controllo qualità medio; LQC = basso controllo di qualità; LLQC = controllo di qualità del limite inferiore; NTC = nessun controllo modello. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
10. Criteri di accettazione del saggio
- Utilizzare le seguenti specifiche per i dati calcolati per ciascun lotto per determinare se il batch è accettabile. Se queste condizioni non sono soddisfatte, invalidare e ripetere il batch.
NOTA: Questi criteri sono stati determinati come consenso dai white paper pubblicati sulla convalida dei saggi basati sulla PCR 18,19,20,21,22,23,24,25. Può essere necessario modificare i criteri obiettivo in base all'applicazione clinica. - Nessun controllo modello (NTC)
- Assicurarsi che ogni pozzo NTC abbia almeno 10.000 goccioline.
- Assicurarsi che ogni pozzo NTC abbia meno di 3 goccioline positive.
- QC e gamma di test
- Assicurati che ogni pozzo QC abbia almeno 10.000 goccioline.
- Assicurarsi che la precisione dei pozzetti replicati di una concentrazione di controllo qualità sia ≤25,0% CV, tranne ai limiti superiore e inferiore della quantificazione, dove ≤30,0% è accettabile. Valutarlo in modo indipendente per ogni set di controllo qualità e livello di concentrazione.
- Assicurarsi che l'errore relativo della concentrazione retrocalcolata a ciascun livello medio di QC sia compreso tra ±25,0% RE della concentrazione nominale (copia / reazione PCR), tranne ai limiti superiore e inferiore della quantificazione, dove ±30,0% RE è accettabile. Valutarlo in modo indipendente per ogni set di controllo qualità e livello di concentrazione.
- Assicurati che almeno 2/3 dei campioni QC (ad esempio, quattro risultati su sei) e il 50% dei campioni QC a ciascun livello (basso, medio, alto) soddisfino queste linee guida.
- Campioni
- Assicurarsi che i pozzetti campione da analizzare abbiano almeno 10.000 goccioline.
- Assicurarsi che la precisione dei pozzetti replicati di una diluizione del campione da analizzare sia ≤25,0% CV.
- Assicurarsi che almeno una diluizione inclusa del campione dato rientri nell'intervallo di quantificazione definito del test, come definito sopra in base ai QC limite superiore e inferiore.
- Se tutte le diluizioni includevano risultati di resa superiori al limite superiore definito di quantificazione e se rimane un volume sufficiente del campione, ripetere il saggio utilizzando una diluizione più elevata del campione.
- Se tutte le diluizioni incluse danno un risultato inferiore al limite inferiore di quantificazione e se rimane un volume sufficiente del campione, ripetere il saggio utilizzando una diluizione inferiore del campione.
NOTA: I campioni contenenti più di tre goccioline positive, ma che hanno una concentrazione inferiore al limite inferiore di quantificazione, possono essere descritti come rilevabili, ma non quantificabili.
Risultati
A scopo dimostrativo, è stato sviluppato un test progettato per rilevare un vettore AAV2 che esprime una proteina fluorescente verde potenziata (eGFP) disponibile in commercio, con un frammento di DNA sintetico a doppio filamento contenente eGFP come controllo di qualità. Attualmente, c'è un dibattito in corso sul fatto che il vettore stesso o un frammento di DNA sintetico o plasmide linearizzato sia più appropriato per l'uso come QC. Generalmente, un frammento di DNA sintetico o un plasmide linearizzato possono essere utilizzati se l'equivalenza al vettore è dimostrata nello sviluppo del metodo (dati non mostrati). Primer e sonde sono stati progettati e ottimizzati per rilevare il transgene eGFP. Fare riferimento alla tabella supplementare S1 per le sequenze utilizzate in questo lavoro. La concentrazione dello stock di frammenti QC è stata determinata empiricamente utilizzando ddPCR. Tutti i test sono stati eseguiti utilizzando le concentrazioni e le condizioni PCR fornite come esempi nella sezione del protocollo.
Per i saggi qPCR, si raccomanda di valutare la linearità, la sensibilità, la gamma dinamica, l'accuratezza e la precisione della curva standard. Poiché la ddPCR non si basa su una curva standard per la quantificazione del target, queste raccomandazioni devono essere modificate. Invece, i QC costituiti da frammenti di DNA sintetico a doppio filamento diluiti a varie concentrazioni per coprire l'intervallo quantificabile atteso di una reazione ddPCR basata sulla modellazione statistica di Poisson sono stati utilizzati 29,30,31,32 per definire la gamma dinamica e la sensibilità e per valutare accuratezza e precisione. La scelta delle concentrazioni di QC si basava principalmente sul rapporto atteso tra goccioline positive e totali all'interno di un pozzo a una data concentrazione. Matematicamente, la ddPCR è teoricamente più accurata quando circa l'80% delle partizioni si amplifica positivamente. Poiché il rapporto tra goccioline positive e totali aumenta al di sopra di 0,8, l'accuratezza diminuisce a causa della saturazione delle partizioni, con la quantificazione non possibile una volta che il 100% delle goccioline è positivo. Nella fascia bassa, teoricamente, può essere rilevata e quantificata solo una goccia positiva, sebbene l'accuratezza sia inferiore e il test sia soggetto a falsi positivi di basso livello. In genere, almeno tre goccioline devono essere positive affinché un risultato venga calcolato con un'affidabilità del 95%, che è la soglia per calcolare una concentrazione che abbiamo usato qui.
È stata preparata una serie di cinque diverse concentrazioni di QC, con il numero di copie target / volumi di reazione PCR calcolati che dovrebbero produrre rapporti di goccioline positivi / totali che coprono l'intervallo quantificabile di ddPCR, come mostrato nella Tabella 5. Questi sono stati utilizzati per valutare l'accuratezza e la precisione del test. Nella valutazione qui, i limiti superiore e inferiore della quantificazione non sono stati spinti al massimo teorico possibile nella ddPCR. Una quantificazione accurata può essere possibile a livelli più alti e più bassi di quelli dimostrati qui. La gamma dovrebbe essere sviluppata in linea con le applicazioni a valle di questo metodo.
Un totale di tre serie di diluizione preparate in modo indipendente di questi QC sono state preparate in tampone di diluizione del campione per ciascun lotto per valutare l'accuratezza e la precisione intra-test. Sono stati inclusi pozzetti duplicati di ciascuna diluizione QC. Per simulare un protocollo di convalida effettivo, un totale di sei batch di accuratezza e precisione sono stati eseguiti da più analisti in più giorni. I risultati di questi sei lotti sono stati analizzati per definire l'accuratezza e la precisione intra-test e inter-saggio del metodo e per definire la gamma dinamica del test.
Le prestazioni intra-test sono state valutate per ciascun lotto a ciascun livello di controllo qualità. Ci aspettavamo che tutti i pozzi QC e NTC avrebbero avuto almeno 10.000 goccioline. Questo è stato raggiunto in 216 dei 216 pozzi testati in tutti e sei i lotti, con un conteggio medio di goccioline di 19.748 goccioline / pozzetto (Tabella 6). Successivamente, l'inter-pozzetto %CV di ciascun set di pozzi duplicati di ciascun QC era previsto essere ≤25,0%, ad eccezione del QC limite superiore e inferiore, dove era previsto il ≤30,0%. Questo è stato raggiunto in gruppi di 90 su 90 pozzi testati in tutti e sei i lotti per i QC, con una percentuale di CV media inter-pozzo del 3,9% in tutti i livelli di QC (Tabella 7). Tutti i QC hanno prodotto rapporti goccioline medi positivi / totali all'interno degli intervalli previsti sopra descritti (Tabella 6).
All'interno di ciascun lotto, la media intra-saggio e la deviazione standard sono state calcolate per ciascuno dei punti della serie di diluizione preparati in modo indipendente, e questi sono stati utilizzati per calcolare una media intra-saggio per ciascuna concentrazione in ciascun test. Questo è stato utilizzato per valutare l'accuratezza e la precisione del saggio (Tabella 8). La precisione si riferisce alla variabilità dei dati provenienti da repliche dello stesso campione omogeneo in condizioni normali di analisi e viene valutata calcolando il %CV delle aliquote multiple incluse. Ci aspettavamo che le tre aliquote testate all'interno di ciascun lotto avrebbero prodotto un intra-test %CV ≤25,0%, ad eccezione del limite superiore e inferiore QC in cui era previsto ≤30,0%. Questo è stato soddisfatto per tutti e cinque i livelli di controllo qualità in ciascuno dei 60 lotti (30 su 30 prestazioni totali). In generale, è stato possibile ottenere una maggiore precisione intra-test rispetto ai criteri target, con una percentuale media intra-test %CV del 7,7% in tutti i livelli di controllo qualità. L'accuratezza si riferisce alla vicinanza dell'accordo tra il valore determinato sperimentalmente e il valore nominale. Questo viene valutato calcolando l'errore relativo percentuale (%RE, o %Bias) tra le concentrazioni calcolate di ciascun QC e le loro concentrazioni nominali teoricamente attese. Ci si aspettava che la media intra-saggio delle tre aliquote sarebbe stata ±25,0% RE della concentrazione nominale, ad eccezione del QC limite superiore e inferiore in cui era previsto il ±30,0%. Questo è stato soddisfatto per tutti e cinque i livelli di controllo qualità in ciascuno dei 60 lotti (30 su 30 prestazioni totali). In generale, è stato possibile raggiungere una maggiore accuratezza intra-test rispetto al nostro obiettivo, con una percentuale media assoluta intra-test %RE del 4,2% in tutti i livelli di QC. In tutte le prestazioni del NTC (30 totali), non sono state rilevate goccioline positive.
L'accuratezza e la precisione tra i test sono state calcolate utilizzando anche la media intra-test di ciascun livello di controllo qualità all'interno di ciascun lotto. La precisione tra i test doveva essere di ≤25,0% CV, ad eccezione del QC limite superiore e inferiore in cui era previsto il ≤30,0%. Allo stesso modo, per l'accuratezza tra i test, era previsto un RE del ±25,0%, ad eccezione del QC del limite superiore e inferiore in cui era previsto il ±30,0%. È stata osservata un'accuratezza e una precisione inter-test significativamente maggiori rispetto a questi obiettivi (Tabella 9), con una precisione inter-test compresa tra il 4,0% e l'8,5% e un'accuratezza assoluta tra l'1,0% e il 3,2%. Collettivamente, questi risultati dimostrano che questo metodo può raggiungere un'accuratezza e una precisione sufficienti all'interno e all'interno del saggio entro gli attuali obiettivi del settore. Sulla base di questi risultati può essere definito un intervallo dinamico di questo test di 2.500-2,5 copie per μL di reazione PCR, con una sensibilità complessiva del saggio di 2,5 copie per μL di reazione PCR. Come accennato in precedenza, potrebbe essere possibile convalidare intervalli dinamici più ampi.
Successivamente, è stato necessario valutare l'accuratezza e la precisione del saggio all'interno della matrice target - in questo caso, lacrime. In genere, i test vengono convalidati prima dell'inizio degli studi clinici, il che significa che è improbabile che le lacrime raccolte da pazienti trattati con vettori siano disponibili a scopo di convalida. Questo può essere creato artificialmente facendo esplodere il vettore AAV target in lacrime raccolte da donatori volontari per creare QC a matrice. Le lacrime umane in pool sono state raccolte da una terza parte (BioIVT). Per la prova di principio, è stato utilizzato un vettore AAV2 che esprime eGFP acquisito da una fonte commerciale. La concentrazione dello stock vettoriale AAV2 è stata determinata empiricamente utilizzando ddPCR, senza l'uso di una fase di isolamento del DNA, come descritto in questo protocollo. In ogni run, l'AAV2 è stato indipendentemente inserito nelle tre aliquote lacrimali ad un livello elevato (previsto 1,41 x 103 copie/μL reazione PCR) e basso (28,2 copie/μL reazione PCR). Le aliquote senza spillo sono state incluse come controllo per dimostrare la specificità del metodo.
Le prestazioni intra-test sono state valutate per ciascun lotto a ciascun livello di picco. Ci si aspettava che tutti i campioni di lacrime avrebbero avuto almeno 10.000 goccioline. Questo è stato raggiunto in 108 dei 108 pozzi testati in tutti e sei i lotti, con un numero medio totale di goccioline di 20.208 goccioline / pozzetto (Tabella 10). Successivamente, il CV inter-pozzo di ciascun set di pozzi duplicati di ciascun QC dovrebbe essere del ≤25,0% per i livelli di picco alti e bassi. Questo è stato raggiunto in 36 dei 36 set di pozzi testati in tutti e sei i lotti per i QC, con una percentuale CV media inter-pozzo del 3,2% (Tabella 11).
All'interno di ciascun lotto, la media intra-test e la deviazione standard sono state calcolate per ciascuno dei picchi lacrimali preparati in modo indipendente, e questi sono stati utilizzati per calcolare una media intra-test per ciascuna concentrazione in ciascun test. Questo è stato utilizzato per valutare l'accuratezza e la precisione del saggio in matrice (Tabella 12). Ci aspettavamo che il test intra-test %CV fosse del ≤25,0% e livelli di picco alti e bassi. Questo è stato soddisfatto in sei lotti su sei per ogni livello. In generale, è stato possibile raggiungere una maggiore precisione intra-test nella matrice rispetto al target, con una media intra-test %CV del 3,7% al livello alto e del 12,2% al livello basso (complessivamente 8,0%). Ci si aspettava inoltre che l'intra-test %RE sarebbe stato del ±25,0% a entrambi i livelli di picco. Questo è stato soddisfatto in sei lotti su sei per ogni livello. Allo stesso modo, è stato generalmente riscontrato che è possibile raggiungere una maggiore accuratezza intra-test nella matrice rispetto all'obiettivo, con una %RE assoluta media intra-test dell'8,1% al livello basso e dell'11,3% al livello alto (complessivamente 9,7%). Per il controllo senza spike, nessun segnale eGFP era rilevabile in nessuna delle aliquote (Tabella 12), dimostrando la specificità del metodo nella matrice lacrimale umana.
L'accuratezza e la precisione tra i saggi nella matrice lacrimale sono state calcolate utilizzando la media intra-test di ciascun livello di picco all'interno di ciascun lotto. Ci si aspettava che la precisione inter-test sarebbe stata ≤25,0% CV, e per l'accuratezza inter-test, ci aspettavamo ±25,0% RE. È stata osservata un'accuratezza e una precisione inter-test significativamente maggiori rispetto a questi obiettivi (Tabella 13), con una precisione inter-test del 5,5% al livello alto e del 7,1% al livello basso, e con un'accuratezza assoluta inter-test dell'11,3% al livello alto e dell'8,1% al livello basso. Collettivamente, questi risultati dimostrano l'accuratezza, la precisione e la specificità del metodo nella matrice lacrimale.
Tabella 5: Controlli di qualità utilizzati per definire l'intervallo dinamico del saggio. Abbreviazioni: ULQC = controllo di qualità del limite superiore; HQC = controllo di alta qualità; MQC = controllo qualità medio; LQC = basso controllo di qualità; LLQC = controllo di qualità del limite inferiore; NTC = nessun controllo modello. Clicca qui per scaricare questa tabella.
Tabella 6: Conteggio totale delle goccioline e rapporti tra goccioline positive e totali del controllo di qualità del DNA sintetico a doppio filamento e NTC. Abbreviazioni: ULQC = controllo di qualità del limite superiore; HQC = controllo di alta qualità; MQC = controllo qualità medio; LQC = basso controllo di qualità; LLQC = controllo di qualità del limite inferiore; NTC = nessun controllo modello. Clicca qui per scaricare questa tabella.
Tabella 7: Statistiche inter-pozzetto QC (bersagli di copia/reazione PCR μL). Abbreviazioni: ULQC = controllo di qualità del limite superiore; HQC = controllo di alta qualità; MQC = controllo qualità medio; LQC = basso controllo di qualità; LLQC = controllo di qualità del limite inferiore; NTC = nessun controllo modello. Clicca qui per scaricare questa tabella.
Tabella 8: Accuratezza intra-saggio e precisione dei QC (copy targets/μL PCR reaction). Abbreviazioni: ULQC = controllo di qualità del limite superiore; HQC = controllo di alta qualità; MQC = controllo qualità medio; LQC = basso controllo di qualità; LLQC = controllo di qualità del limite inferiore; NTC = nessun controllo modello. Clicca qui per scaricare questa tabella.
Tabella 9: Accuratezza e precisione tra i saggi dei QC (bersagli di copia/reazione PCR μL). Abbreviazioni: ULQC = controllo di qualità del limite superiore; HQC = controllo di alta qualità; MQC = controllo qualità medio; LQC = basso controllo di qualità; LLQC = controllo di qualità del limite inferiore; NTC = nessun controllo modello. Clicca qui per scaricare questa tabella.
Tabella 10: Conteggio totale delle goccioline dei campioni lacrimali. Clicca qui per scaricare questa tabella.
Tabella 11: Statistiche inter-pozzetto del campione lacrimale (bersagli di copia/reazione PCR μL). Clicca qui per scaricare questa tabella.
Tabella 12: Accuratezza intra-saggio e precisione dei campioni lacrimali (bersagli di copia/reazione PCR μL). Clicca qui per scaricare questa tabella.
Tabella 13: Accuratezza e precisione tra i saggi dei campioni lacrimali (bersagli di copia/reazione PCR μL). Clicca qui per scaricare questa tabella.
Tabella supplementare S1: Sequenze di primer, sonde e controllo di qualità del DNA sintetico a doppio filamento utilizzati in questo studio. Clicca qui per scaricare questa tabella.
Discussione
Ci sono diversi passaggi del protocollo ddPCR che sono fondamentali per la corretta esecuzione del test. Il primo passo critico è la progettazione e l'ottimizzazione dei primer e della sonda. In generale, l'uso della chimica basata sulla sonda di idrolisi rispetto alla chimica a base di coloranti (ad esempio, SYBR Green) in un contesto preclinico o clinico è raccomandato a causa della loro specificità superiore. Inoltre, la scelta del target di amplificazione è critica. Tipicamente, il transgene di interesse del vettore è mirato. Tuttavia, in fasi precliniche precedenti o in vettori in cui potrebbe non essere possibile distinguere il transgene vettoriale rispetto al DNA genomico, può essere appropriato utilizzare bersagli vettoriali standardizzati. Ad esempio, si potrebbe prendere di mira la regione di ripetizione terminale invertita, il promotore, la coda poli-A o le giunzioni inter-segmento tra questi componenti vettoriali. La scelta del target varierà in base al design vettoriale. Le tradizionali strategie e software di progettazione di primer e sonda qPCR sono in genere appropriati per la ddPCR. I parametri di progettazione che dovrebbero produrre una temperatura di ricottura costante (ad esempio, 60 °C) devono essere selezionati per ridurre la quantità di ottimizzazione richiesta. È stato inoltre raccomandato di progettare, ordinare e valutare almeno tre diversi set per ciascun target. Si dovrebbe quindi selezionare il set che mostra la maggiore specificità (nessuna amplificazione nel pozzo di controllo negativo o in una matrice di DNA bersaglio correlato) e sensibilità (cioè limite di rilevazione)20.
Se è vantaggioso essere in grado di trasferire il test tra qPCR e ddPCR, si raccomanda di ottimizzare prima le condizioni del test utilizzando qPCR e di identificare le condizioni per il set selezionato che si traducono in efficienze di amplificazione del 90% -110% con un R2 ≥ 0,98. Tuttavia, la ddPCR come metodo endpoint è tipicamente meno sensibile della qPCR a causa delle variazioni nelle efficienze di amplificazione. Come minimo, si raccomanda di eseguire un gradiente di temperatura termica nella fase di ricottura/estensione per coprire temperature superiori e inferiori alle temperature di ricottura previste e di valutare la separazione dell'ampiezza della pioggia e della fluorescenza tra i cluster di goccioline negativi e positivi in funzione della temperatura. Se l'area di lavoro lo consente, si consiglia di disporre di singole workstation dedicate per la preparazione del master mix, l'aggiunta di modelli e l'amplificazione. Ove possibile, questi dovrebbero essere fisicamente separati da un flusso di lavoro unidirezionale con controlli tecnici integrati, come l'accesso controllato e le pressioni dell'aria differenziale, per ridurre il rischio di contaminazione incrociata e falsi positivi. Se ciò non è possibile, è necessario prestare estrema attenzione per prevenire la contaminazione incrociata.
Ci sono due passaggi in questo protocollo che possono sembrare insoliti per coloro che sono più abituati allo sviluppo del test qPCR. Il primo è l'inclusione di un enzima di restrizione nel master mix PCR. Durante l'amplificazione ddPCR, ogni goccia viene termociclizzata all'endpoint. In un test opportunamente ottimizzato, ciò si traduce in due popolazioni di goccioline, una serie che mostra un livello costantemente elevato di segnali fluorescenti - i positivi - e un altro che mostra costantemente un basso livello di segnale fluorescente - i negativi. Se si verifica un'interferenza PCR, può desincronizzare l'inizio dell'amplificazione PCR, con il risultato che la goccia non raggiunge un plateau di amplificazione e quindi endpoint fluorescenti incoerenti. In questo caso, le goccioline saranno distribuite tra negativi e positivi, risultando in un fenomeno chiamato pioggia ddPCR. Ciò può comportare una quantificazione imprecisa dell'obiettivo e soglie applicate in modo incoerente e soggettivo. La nostra raccomandazione di impostare la soglia leggermente al di sopra del segnale del NTC, che dovrebbe ridurre al minimo gli effetti della pioggia nella quantificazione finale poiché tutte le goccioline sono ancora considerate positive, anche se non completamente ciclizzate all'endpoint. Gli AAV hanno una struttura secondaria altamente complessa che, a seconda del target di amplificazione, può ridurre l'accessibilità ai primer e alle sonde, con conseguente interferenza PCR e quindi pioggia. L'inclusione dell'enzima di restrizione nella miscela principale fende questa struttura secondaria per aumentare l'accesso da parte dei primer e delle sonde, riducendo la pioggia, che può quindi migliorare l'accuratezza del test. Gli effetti dell'inclusione di un enzima di restrizione nella reazione ddPCR sono stati descritti in precedenza 25,32. Qualsiasi enzima di restrizione può essere utilizzato, purché sia confermato che non taglia all'interno della regione di amplificazione target. Non sono necessari passaggi di predigestione o composizioni tampone alternative.
Il secondo passo insolito è la preparazione del campione lacrimale contenente AAV. In questo protocollo, è stato utilizzato un rapporto 1:10 (o superiore) di lacrime e successivamente il campione è stato riscaldato. Tipicamente, quando le lacrime vengono raccolte attraverso un tubo capillare, che è un metodo di raccolta ampiamente utilizzato, in media circa 10,0 μL possono essere raccolti33. La diluizione aiuta a risolvere il volume limitato del campione e fornisce materiale sufficiente per i test dei pozzi duplicati. Mentre questo riduce il limite teorico di rilevazione, la robusta sensibilità della ddPCR dovrebbe comunque portare alla rilevazione di tutte le particelle vettoriali tranne un numero estremamente esiguo. Questo approccio crea inoltre un pozzo di "backup" se uno dovesse fallire inaspettatamente. In questo caso, o in caso di volume di campione insufficiente per far funzionare due pozzi, l'errore di Poisson potrebbe essere utilizzato per valutare la precisione. Inoltre, nei casi in cui la concentrazione è inferiore al limite di rilevamento, crea l'opportunità di unire i dati dei pozzi per determinare una concentrazione. È necessario liberare i vettori AAV dai capsidi virali per il rilevamento della ddPCR. Alcuni metodi per la quantificazione dell'AAV hanno incluso una fase di digestione della proteinasi K per rimuovere il capside virale34,35,36. Tutti i sierotipi AAV presenti in natura hanno temperature di fusione pari o inferiori a circa 90 °C, con la maggior parte che scende al di sotto di 80 °C; Pertanto, questa sembra essere un'inclusione non necessaria37. Il riscaldamento da solo sembra essere sufficiente per rilasciare DNA vettore.
Inoltre, la ddPCR è generalmente meno suscettibile agli inibitori della PCR che possono essere presenti in un campione che può influenzare un test qPCR. Se viene inclusa una specifica fase di isolamento del DNA, ciò richiederebbe anche una convalida specifica, che viene evitata in questo protocollo. I campioni vengono diluiti prima del riscaldamento a causa della cinetica di diffusione dei genomi vettori in un liquido. Durante il processo di riscaldamento e successivo raffreddamento, i filamenti di senso positivi e negativi del genoma del DNA a singolo filamento possono ricottura insieme per produrre un intermedio a doppio filamento se le concentrazioni sono sufficientemente elevate. La diluizione prima del riscaldamento riduce le concentrazioni e rende matematicamente improbabile che si formino abbastanza intermedi a doppio filamento da avere un effetto negativo sull'accuratezza della quantificazione. Va notato che i frammenti di DNA sintetico o i plasmidi linearizzati utilizzati come controlli di qualità non devono essere sottoposti a questa fase di riscaldamento. Poiché si tratta di due filamenti, il riscaldamento comporterebbe la conversione in intermedi a singolo filamento. A seguito della suddivisione indipendente di questi QC a singolo filamento in goccioline, ciò dovrebbe comportare un aumento doppio della concentrazione di QC rispetto alla concentrazione nominale. In alternativa, se i QC devono essere riscaldati per standardizzare il metodo, questo deve essere preso in considerazione nella ricostituzione e nell'assegnazione di una concentrazione nominale.
Infine, per quanto riguarda la preparazione del campione, molti protocolli raccomandano anche l'inclusione di una fase di trattamento della DNasi per rimuovere qualsiasi DNA vettoriale non capsidata. Questo passaggio è fondamentale nei casi in cui non si desidera quantificare il DNA libero associato alla preparazione del vettore (come durante la quantificazione ai fini del dosaggio). Tuttavia, nel contesto degli studi di biodistribuzione e bioshedding, in genere si desidera sapere dove ha viaggiato il DNA di un vettore, indipendentemente dal fatto che sia incapsulato o meno. Pertanto, si suggerisce di non eseguire in genere una fase di trattamento della DNasi durante tali studi. Se si ritiene necessario includere una fase DNase, questa fase deve essere prima delle diluizioni e del riscaldamento.
In questo articolo vengono presentati dati rappresentativi dell'approccio alla valutazione della gamma dinamica, della sensibilità, dell'accuratezza e della precisione del metodo nel contesto di una convalida conforme alle buone pratiche di laboratorio adatte allo scopo. L'attuale mancanza di linee guida su questo argomento lascia che i laboratori di convalida determinino i criteri di analisi target per se stessi, in linea con l'attuale pensiero del settore. Diversi gruppi hanno posto criteri target sia più alti che più bassi rispetto a quelli utilizzati in questo studio 19,20,21,22,23,24,25. I criteri di analisi target, fino a quando non saranno definiti in modo più rigido, devono essere selezionati prima della convalida in base alle applicazioni cliniche previste del metodo. A seconda delle decisioni a valle da prendere sulla base dei dati, potrebbero essere necessari livelli più elevati di accuratezza e precisione. Al contrario, un semplice risultato positivo contro negativo può essere sufficiente.
L'approccio ha anche affrontato raccomandazioni per la valutazione della specificità e di un effetto matrice. Un pool di lacrime raccolte da individui non trattati non è riuscito a produrre un risultato positivo in questo test, mentre il bersaglio poteva essere rilevato quando il vettore è stato inserito nelle lacrime ad una concentrazione alta e bassa entro i tassi di recupero raccomandati. Idealmente, anche la matrice contenente vettore endogeno (ad esempio, raccolta dopo il trattamento con vettore virale) dovrebbe essere inclusa in queste valutazioni. Tuttavia, è improbabile che tali campioni siano disponibili per l'uso in una convalida. Per aumentare la robustezza della convalida, è stato possibile valutare più pool di lacrime, o lacrime raccolte da una varietà di individui, per determinare se si verifica un effetto della matrice specifico del paziente. Infine, si raccomanda di valutare la stabilità. Nei flussi di lavoro in cui l'estrazione del DNA avviene al di fuori della matrice biologica, può essere necessario valutare la stabilità sia del campione che del DNA estratto. In questo flusso di lavoro, il campione viene testato direttamente nel test senza la necessità di estrarre il DNA. Pertanto, in considerazione della valutazione della stabilità per questo metodo, è necessario valutare la stabilità dei campioni lacrimali. In genere, si raccomandano valutazioni di stabilità da banco, frigorifero, congelamento / disgelo e a lungo termine. Questi non sono stati eseguiti come parte di questo studio, ma i metodi sviluppati qui possono essere utilizzati in questa valutazione, a seguito di manipolazioni ai campioni di input.
Nel complesso, questo metodo ha dimostrato di essere un test robusto, ripetibile e validabile per rilevare vettori basati su AAV in campioni lacrimali. Può servire come piattaforma da adattare a vettori specifici per supportare le sperimentazioni cliniche e fornisce una base per la convalida di un test coerente con le buone pratiche di laboratorio.
Divulgazioni
Tutti gli autori sono dipendenti di KCAS Bioanalytical and Biomarker Services, un'organizzazione di ricerca a contratto che fornisce servizi completi di sviluppo conformi alle buone pratiche di laboratorio / buone pratiche cliniche dal supporto alla scoperta precoce fino alla registrazione del prodotto, incluso il supporto di saggi basati su AAV, come descritto in questo protocollo. KCAS ha finanziato questo progetto e la pubblicazione di questo protocollo. Sebbene non esistano linee guida attuali della FDA per la progettazione e la convalida degli esperimenti presentati in questo documento, gli esperti e i leader di pensiero che sviluppano tali studi presso KCAS hanno adottato questo approccio come approccio di base. Ulteriori criteri e parametri sono discussi progetto per progetto e possono essere inclusi in base all'uso previsto.
Riconoscimenti
Vorremmo ringraziare Nick Russell e Brandon McKethan di Bio-Rad per le loro utili discussioni durante lo sviluppo di questo metodo.
Materiali
| Name | Company | Catalog Number | Comments |
| AAV-eGFP Vector | Charles River Laboratories | RS-AAV2-FL | Lot AAV2-0720-FL, used as a proof of principle vector |
| AutoDG Droplet Digital PCR system | Bio-Rad | QX200 | Alternative ddPCR system may be used following manufacturer’s protocol. |
| AutoDG Oil for Probes | Bio-Rad | 1864110 | Or use material compatible with ddPCR system. |
| ddPCR Buffer Control for Probes | Bio-Rad | 1863052 | Or use material compatible with ddPCR system and PCR chemistry. |
| ddPCR Droplet Reader Oil | Bio-Rad | 1863004 | Or use material compatible with ddPCR system. |
| ddPCR Piercable Foil Seals | Bio-Rad | 1814040 | Or use material compatible with ddPCR system. |
| ddPCR Plates 96-Well, Semi-Skirted | Bio-Rad | 12001925 | Or use material compatible with ddPCR system. |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023, 1863024, or 1863025 | Use master mix compatible with primers/probes and ddPCR system. |
| DG32 AutoDG Cartidges | Bio-Rad | 1864108 | Or use material compatible with ddPCR system. |
| Droplet Reader | Bio-Rad | QX200 | Alternative ddPCR system may be used following manufacturer’s protocol. |
| GeneAmp PCR Buffer | Applied Biosystems | N8080129 | N/A |
| Nuclease-Free Water | Ambion | AM9906 | N/A |
| PCR Plate Sealer | Bio-Rad | PX1 | Or use material compatible with ddPCR system. |
| Pipet Tips for AutoDG | Bio-Rad | 1864120 | Or use material compatible with ddPCR system. |
| Pluronic F-68 Non-ionic Surfactant | Gibco | 24040 | N/A |
| Primer and Hydrolysis Probes | Various | Various | Design based on target sequence using general approaches for primer/probe design. Select fluorphores and quenchers compatible with ddPCR system. |
| Restriction Enzyme | Various | Various | Varies with target amplification sequence. Use restriction enzyme that does not cut in the amplified sequence |
| Sheared salmon sperm DNA | ThermoFisher | AM9680 | N/A |
| Synthetic DNA gene fragment or linearized plasmid | Various | Various | Design a synthetic DNA fragment containing the target amplification region for use as a quality control |
| TE Buffer | Teknova | T0224 | Ensure prepared or purchases nuclease free. 10 mM Tris-HCl, 1.0 mM EDTA, pH=8.0 |
| Touch Thermal Cycler | Bio-Rad | C1000 | Or use material compatible with ddPCR system. |
Riferimenti
- Sherkow, J. S., Zettler, P. J., Greeley, H. T. Is it 'gene therapy. Journal of Law and the Biosciences. 5 (3), 786-793 (2018).
- Ginn, S. L., Amaya, A. K., Alexander, I. E., Edelstein, M., Abedi, M. R. Gene therapy clinical trials worldwide to 2017: an update. The Journal of Gene Medicine. 20 (5), 3015 (2018).
- Ghosh, S., Brown, A. M., Jenkins, C., Campbell, K. Viral vector systems for gene therapy: a comprehensive literature review of progress and biosafety challenges. Applied Biosafety. 25 (1), 7-18 (2020).
- Gene, Cell, & RNA therapy landscape, Q4 2022 quarterly data report. American Society for Gene & Cell Therapy Available from: https://asgct.org/global/documents/asgct_citeline-q4-2022-report_final.aspx (2022)
- Approved Cellular and Gene Therapy Products. United States Food and Drug Administration Available from: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (2022)
- Au, H. K. E., Isalan, M., Meilcarek, M. Gene therapy advances: a meta-analysis of AAV usage in clinical settings. Frontiers in Medicine. 8, 809118 (2022).
- Lundstrom, K. Viral vectors in gene therapy. Diseases. 6 (2), 42 (2018).
- Naso, M. F., Tomkowicz, B., Perry, W. L., Strohl, W. R. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 31, 317-334 (2017).
- Russell, S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomized, controlled, open-label, phase 3 trial. Lancet. 390 (10097), 849-860 (2017).
- Chemistry, manufacturing, and control (CMC) information for human gene therapy investigational new drugs (INDs). United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/chemistry-manufacturing-and-control-cmc-information-human-gene-therapy-investigationsal-new-drug (2020)
- Long term follow-up after administration of human gene therapy products. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/long-term-follow-after-adminstration-human-gene-therapy-products (2020)
- Testing of retroviral vector-based human gene therapy products for replication competent retrovirus during product manufacture and patient follow-up. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/testing-retroviral-vector-based-human-gene-therapy-products-replication-competent-retrovirus-during (2020)
- Recommendations for microbial vectors used in gene therapy. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/recommendations-microbial-vectors-used-gene-therapy (2016)
- Multidisciplinary: gene therapy. European Medicines Agency Available from: https://www.europa.eu/en/human-regulatory-development/scientific-guidelines/multidisciplinary/multidisciplinary-gene-therapy (2023)
- Human gene therapy for retinal disorders. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-retinal-disorders (2020)
- Design and analysis of shedding studies for virus or bacterial-based gene therapy and oncolytic products. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-and-analysis-shedding-studies-virus-or-bacteria-based-gene-therapy-and-oncolytic-products (2015)
- Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products. European Medicines Agency Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-clinical-aspects-gene-therapy-medicinal-products_en.pdf (2018)
- Kaur, S. white paper on recent issues in bioanalysis: mass spec of proteins, extracellular vesicles, CRISPR, chiral assays, oligos; nanomedicines bioanalysis; ICH M10 section 7.1; non-liquid & rare matrices; regulatory inputs (part 1A - recommendations on endogenous compounds, small molecules, complex methods, regulated mass spec of large molecules, small molecule, PoC & part 1B - regulatory agencies' inputs on bioanalysis, biomarkers, immunogenicity, gene & cell therapy and vaccine). Bioanalysis. 14 (9), 505-580 (2022).
- Hays, A., Islam, R., Matys, K., Williams, D. Best practices in qPCR and qPCR validation in regulated bio analytical laboratories. The AAPS Journal. 24 (2), 36 (2022).
- Ma, H., Bell, K. N., Loker, R. N. qPCR and qRT-PCR analysis: Regulatory points to consider when conducting biodistribution and vector shedding studies. Molecular Therapy. Methods & Clinical Development. 17 (20), 152-168 (2020).
- Wissel, M. Recommendations on qPCR/ddPCR assay validation by GCC. Bioanalysis. 14 (12), 853-863 (2022).
- Expectations for biodistribution (BD) assessments for gene therapy (GT) products. International Pharmaceutical Regulators Programme Available from: https://admin.iprp.global/sites/default/files/2018-09/IPRP_GTWG_ReflectionPaper_BD_Final_2018_0713.pdf (2018)
- Pinheiro, L., Emslie, K. R. Basic concepts and validation of digital PCR measurements. Methods in Molecular Biology. 1768, 11-24 (2018).
- Tzonev, S. Fundamentals of counting statistics in digital PCR: I measured two target copies-what does it mean. Methods in Molecular Biology. 1768, 25-43 (2018).
- Prantner, A., Marr, D. Genome concentration, characterization, and integrity analysis of recombinant adeno-associated viral vectors using droplet digital PCR. PLoS One. 18, 0280242 (2023).
- Primer-BLAST. National Library of Medicine, National Center for Biotechnology Information Available from: https://www.ncbi.nim.nih.gov/tools/primer-blast/ (2023)
- Koressaar, T., Remm, M. Enhancements and modifications of primer design program Primer 3. Bioinformatics. 23 (10), 1289-1291 (2007).
- Ye, J. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 13, 134 (2012).
- Droplet Digital PCR Application Guide. Bio-Rad Available from: https://www.bio-rad.com/webroot/we/pdf/lsr/literature/Bulletin_6407.pdf (2023)
- Qian, P. L., Sauzade, M., Brouzes, E. dPCR: A technology review. Sensors. 18 (4), 1271 (2018).
- Basu, A. Digital assays part I: portioning statistics and digital PCR. SLAS Technology. 22 (4), 369-386 (2017).
- Sanmiguel, J., Gao, G., Vandeberghe, L. H. Quantitative and digital droplet-based AAV genome titration. Methods in Molecular Biology. 1950, 51-83 (2019).
- Bachhuber, F., Huss, A., Senel, M., Tumani, H. Diagnostic biomarkers in tear fluid: from sampling to preanalytical processing. Scientific Reports. 11, 10064 (2021).
- Martinez-Fernandez de la Camara, C., McClements, M. E., MacLaren, R. E. Accurate quantification of AAV vector genomes by quantitative PCR. Genes. 12 (4), 601 (2021).
- Ai, J., Ibraheim, R., Tai, P. W. L., Gao, G. A scalable and accurate method for quantifying vector genomes of recombinant adeno-associated viruses in crude lysate. Human Gene TherapyMethods. 28 (3), 139-147 (2017).
- Dobnik, D., et al. Accurate quantification and characterization of adeno-associated viral vectors. Frontiers in Microbiology. 10, 1570 (2019).
- Bennett, A., et al. Thermal stability as a determinant of AAV serotype identity. Molecular Therapy Methods and Clinical Development. 6, 171-182 (2017).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati