
Method Article
Visualización de focos de ADN monocatenario en la fase G1 del ciclo celular
En este artículo
Resumen
El siguiente protocolo presenta la detección de focos de ADN monocatenario en la fase G1 del ciclo celular utilizando la sincronización del ciclo celular seguida de la tinción inmunofluorescente de RPA2.
Resumen
El ADN tiene vías de reparación celular específicas capaces de hacer frente a las lesiones que podrían surgir tanto de fuentes endógenas como exógenas. La reparación del ADN requiere la colaboración entre numerosas proteínas, responsables de cubrir una amplia gama de tareas, desde el reconocimiento y la señalización de la presencia de una lesión en el ADN hasta su reparación física. Durante este proceso, a menudo se crean rastros de ADN monocatenario (ssDNA), que finalmente son llenados por ADN polimerasas. La naturaleza de estas huellas de ssDNA (en términos de longitud y número), junto con la polimerasa reclutada para llenar estos vacíos, son específicas de la vía de reparación. La visualización de estas huellas de ssDNA puede ayudarnos a comprender la complicada dinámica de los mecanismos de reparación del ADN.
Este protocolo proporciona un método detallado para la preparación de células sincronizadas G1 para medir la formación de focos de ADNss en caso de estrés genotóxico. Utilizando un enfoque de inmunofluorescencia fácil de utilizar, visualizamos el ssDNA mediante la tinción de RPA2, un componente del complejo de replicación heterotrimérica de la proteína A (RPA). RPA2 se une y estabiliza los intermedios de ssDNA que surgen tras el estrés genotóxico o la replicación para controlar la reparación del ADN y la activación del punto de control del daño del ADN. La tinción con 5-etinil-2'-desoxiuridina (EdU) se utiliza para visualizar la replicación del ADN y excluir cualquier célula de fase S. Este protocolo proporciona un enfoque alternativo a los ensayos convencionales basados en 5-bromo-2'-desoxiuridina (BrdU) no desnaturalizantes y es más adecuado para la detección de focos de ADNss fuera de la fase S.
Introducción
Para mantener la vida, las células estudian y reparan constantemente el ADN para mantener su integridad genómica. Las células pueden acumular varios tipos de daño en el ADN debido a fuentes endógenas (p. ej., oxidación, alquilación, desaminación, errores de replicación) y exógenas (p. ej., UV, irradiación ionizante) de factores estresantes del ADN. La falta de reparación de estas lesiones da lugar a la apoptosis, la detención del ciclo celular o la senescencia y puede dar lugar a enfermedades1. Las lesiones del ADN pueden abordarse mediante cualquiera de las siguientes vías principales de reparación del ADN: DR (reparación de reversión directa), que repara principalmente las bases alquiladas2; BER (reparación por escisión de bases), que se dirige a los errores de bases de ADN no voluminosos y a las roturas de ADN monocatenario (SSB)3; NER (reparación por escisión de nucleótidos) que corrige lesiones voluminosas de ADN que distorsionan la hélice4; MMR (reparación de errores de emparejamiento) que se dirige principalmente a las discordancias de ADN, los bucles de inserción/deleción (IDL) y ciertos daños en la base5; NHEJ (unión de extremos no homólogos) y HRR (reparación de recombinación homóloga) que son activos en las roturas de ADN bicatenario (DSB)6; y TLS (síntesis translesional), que es un mecanismo de derivación de la lesión del ADN7. Aunque estas vías tienen distintas especificidades de sustrato, existen ciertas superposiciones entre ellas para garantizar la redundancia para una reparación eficiente. Comprender la acción de las diferentes vías de reparación del ADN en varias fases del ciclo celular es crucial, ya que estos factores de reparación del ADN podrían servir como objetivos esenciales para los enfoques terapéuticos para tratar el cáncer, el envejecimiento y los trastornos neurológicos 8,9.
El ADN monocatenario (ssDNA) se genera a lo largo del ciclo celular debido a la reparación de las lesiones del ADN generadas por agentes que dañan el ADN tanto endógenos como exógenos. Ante el estrés genotóxico, el ssDNA se genera abundantemente en las fases S y G2 donde HRR y MMR tienen su mayor actividad y cuando la maquinaria de replicación se detiene o colapsa al encontrarse con lesiones de ADN 6,10,11. Otras vías de reparación del ADN (p. ej., NHEJ/unión de extremos mediada por microhomología (MMEJ)/recocido monocatenario [SSA]) también generan ssDNA durante la reparación de DSB12. Estas trazas de ADNss suelen surgir de la resección del ADN, llevada a cabo por exonucleasas como EXO1, ADN2 y CtIP durante la FC y la MMR, endonucleasas como XPF y XPG durante la NER, o a través de la acción combinada de POLB y FEN1 durante la BER 4,13,14,15,16,17,18,19 . Debido al trabajo de la maquinaria de replicación, también se generan rastros de ssDNA cuando las helicasas de ADN desenrollan el ADN frente a las polimerasas replicativas unidas a PCNA20. Por el contrario, en la fase G1, la falta de HRR y replicación del ADN y la actividad limitada de la MMR reducen la extensión de las huellas de ADNs generadas y, por lo tanto, son más difíciles de detectar 10,11,21.
Las trazas celulares de ssDNA son estructuras altamente sensibles que deben protegerse para evitar la formación de DSB. Esto se logra recubriendo las pistas de ssDNA con RPA. RPA es un abundante complejo proteico heterotriménico compuesto por múltiples subunidades (RPA1, RPA2 y RPA3, también denominadas RPA70, RPA32 y RPA14, respectivamente), que se expresan de forma ubicua a lo largo del ciclo celular22. Cada subunidad de RPA contiene un dominio de unión al ADN (DBD), capaz de interactuar con 4-6 nucleótidos, y las subunidades combinadas forman un núcleo de trimerización estable. En total, el RPA se une a aproximadamente 20-30 nucleótidos con afinidad subnanomolar23,24.
Los métodos convencionales utilizan microscopía de inmunofluorescencia (IF) para visualizar focos de ssDNA mediante el marcaje de 5-bromo-2'-desoxiuridina (BrdU) incorporado en el ADN genómico utilizando anticuerpos BrdU25. Este enfoque se basa en el hecho de que los anticuerpos BrdU solo pueden detectar BrdU en el ssDNA25 expuesto. Aunque este enfoque es sencillo, también muestra ciertas limitaciones. Por ejemplo, las células se tratan previamente para incorporar BrdU antes del inicio del experimento, lo que lleva mucho tiempo y puede interferir con los efectores posteriores. Por lo tanto, la detección de ssDNA basada en BrdU se limita a las células replicantes y no se puede utilizar para las células inactivas. Esto excluye la aplicación de este método para estudiar la reparación del ADN en células no replicantes, a pesar de su importancia en varias enfermedades como el cáncer y la neurodegeneración 5,26. Además, debido a que las estructuras de BrdU y EdU son muy similares, la mayoría de los anticuerpos BrdU muestran reactividad cruzada hacia EdU, lo que debe tenerse en cuenta cuando se apunta a experimentos de marcaje dual27. La tinción de RPA se ha utilizado previamente para mostrar focos de ssDNA principalmente en células de fase S; sin embargo, algunos trabajos también lo han utilizado con éxito fuera de la fase S 28,29,30,31,32,33,34,35. El siguiente protocolo utiliza eficientemente las propiedades de RPA, permitiendo la visualización de focos de ssDNA después del daño al ADN en la fase G1 del ciclo celular (aunque se puede usar en todas las fases del ciclo celular).
Protocolo
1. Mantenimiento de las células epiteliales pigmentarias de la retina inmortalizadas por hTERT (RPE1)
- Mantener las líneas celulares RPE1 en el Medio Eagle Modificado (DMEM) de Dulbecco suplementado con un 10% de suero fetal bovino inactivado por calor (Hi-FBS) y 100 μg/mL de penicilina-estreptomicina (denominado medio de cultivo a partir de ahora) en una incubadora humidificada con 5% de CO2 a 37 °C. Para el cultivo rutinario, cultive células RPE1 en una placa tratada con cultivo de tejidos de 15 cm y divida cuando alcance una confluencia del 80-90% (~16-18 × 106 células por placa de 15 cm).
- Al dividir, retire el medio y enjuague las células con 10 ml de solución salina tamponada con fosfato (PBS) 1x.
- Añadir 3 mL de tripsina-EDTA al 0,05% para cubrir toda la superficie del plato. Mantener las células a 37 °C con la tripsina hasta que se desprendan.
- Después de la tripsinización, resuspender las células con medio de cultivo y centrifugarlas a 150 × g durante 5 min a temperatura ambiente (RT, 22-25 °C). Retirar el sobrenadante y resuspender suavemente las células en 10 mL de medio de cultivo.
- Siembre 1,6-1,8 × 106 células en un nuevo plato de 15 cm (~1 ml de la suspensión celular).
NOTA: Todo el trabajo de cultivo de tejidos debe realizarse bajo los niveles de seguridad BSL-2. El tiempo de incubación de la tripsinización depende de la confluencia celular. Por lo general, el proceso tarda entre 2 y 3 minutos en completarse para obtener una placa confluente del 90%. Las células deben ser examinadas para detectar la contaminación por Mycoplasma de forma regular con kits disponibles comercialmente (ver ejemplos en la Tabla de Materiales).
2. Eliminación del gen de interés (GOI) por siRNA
- Siembre 1,0 × 106 células RPE1 en una placa tratada con cultivo de tejidos de 10 cm con 10 mL de medio de cultivo el día anterior a la transfección.
- El día de la transfección, acompleja el siRNA. Para una placa de 10 cm, utilizar una concentración final de 20 nM de siRPA2 y 12 μL de reactivo de transfección a base de lípidos en 500 μL de medio de transfección con bajo contenido sérico. Mezcle suavemente todos los componentes moviendo el tubo e incube a RT (22-25 °C) durante 5 min.
- Agregue la mezcla de siRNA complejado a las células gota a gota e incube las células con el siRNA durante 48 h.
3. Sincronización de células RPE1 en fase G0
- Tripsinar las células RPE1 del paso 2.3 como se describe en la sección 1 (~2 × 106 células).
- Transfiera la suspensión celular a tubos de centrífuga de 15 ml y centrifugue a 150 × g, RT (22-25 °C) durante 5 min.
- Retirar el sobrenadante y resuspender las células en 12 mL de PBS. Centrifugar las células a 150 × g a RT (22-25 °C) durante 5 min. Repita la eliminación del sobrenadante y la centrifugación dos veces.
- Resuspender las células en 10 mL de DMEM sin suero suplementado con 100 μg/mL de penicilina-estreptomicina, 1 mM de piruvato de sodio, 15 mM de HEPES y colocarlas en una placa de cultivo de tejidos de 10 cm.
NOTA: Si las células tienden a aglutinarse, vuelva a suspenderlas en solo 1 ml de DMEM sin suero y pipetee hacia arriba y hacia abajo 5 veces con una punta P1000 para desalojar los grumos antes de diluir la suspensión hasta un volumen final de 10 ml. - Después de 24 h de inanición sérica, introducir la segunda ronda de silenciamiento utilizando el mismo procedimiento descrito en la sección 2 añadiendo el siRNA complejado a las células hambrientas de suero.
- Mantenga las células RPE1 en DMEM sin suero durante 72 h antes de proceder a la liberación de G1.
4. Recubrimiento de cubreobjetos y células de liberación en fase G1
- Esterilice las pinzas con etanol al 70% y coloque un solo cubreobjetos de vidrio (12 mm de diámetro y #1,5 de grosor [0,17 mm]) en un pocillo de una placa de 24 pocillos.
- Diluir la matriz de recubrimiento de vitronectina con PBS para obtener una concentración final de 10 μg/mL. Añadir 500 μL de la solución de vitronectina en cada pocillo que contenga los cubreobjetos e incubar durante 1 h a RT.
- Retire la solución de recubrimiento y lave los cubreobjetos con 1 ml de PBS.
- Separe las células RPE1 carentes de suero de la placa tratada con cultivo de tejido de 10 cm con 1 ml de tripsina al 0,05% después de un lavado con PBS durante 1 min a 37 °C.
NOTA: Las células se desprenden mucho más rápido después de la inanición del suero. Tenga cuidado al lavar las células con PBS y utilice tiempos de tripsinización cortos. - Para inactivar la tripsina, resuspender las células RPE1 en un total de 6 mL de medio de cultivo. Eliminar la tripsina inactivada girando las células con 150 × g a RT (22-25 °C) durante 5 min.
- Resuspenda las células en 1 ml de medio de cultivo y mida el número de células.
- Siembre 4 × 104 células RPE1 en el cubreobjetos recubierto en un total de 500 μL de medio de cultivo.
NOTA: Asegúrese de que la viabilidad de la celda esté por encima del 90% antes de continuar con los pasos posteriores. La viabilidad celular podría evaluarse rápidamente mediante la tinción con azul de tripano durante la etapa de recuento celular. - Después de 6 h de sembrar las células en el medio de cultivo, las células liberadas por G0 estarán en la fase G1 temprana. Realice experimentos en G1 en esta ventana de 6 a 12 horas antes de que las células comiencen a entrar en la fase S.
- Antes de introducir el daño en el ADN, pulsar las células con 10 μM de 5-etinil-2'-desoxiuridina (EdU) durante 30 min a 37 °C, diluidas en medio de cultivo.
- Retirar el medio que contiene EdU y perseguir las células con timidina 10 μM durante 10 min a 37 °C para evitar la incorporación de EdU restante durante la inducción del daño en el ADN.
- Retirar el medio con timidina y tratar las células con 250 μMH2O2 durante 1 h, diluido en medio de cultivo.
5. Tinción por inmunofluorescencia del ssDNA
- Lave las células una vez con 1 ml de RT (22-25 °C) PBS para eliminar el medio y los componentes del suero.
NOTA: Sea cuidadoso al lavar las celdas para evitar que se desprendan y se sequen. No procese muchos pozos al mismo tiempo. - Pre-extracción: Incubar las células lavadas en 1 mL de tampón de extracción CSK (Tabla 1) durante 5 min a RT (22-25 °C).
NOTA: La preextracción de CSK elimina todas las proteínas no unidas a la cromatina, incluida la RPA2 soluble.
PRECAUCIÓN: Triton X-100 es dañino si se ingiere y puede causar irritación de la piel y daño ocular. - Retire el tampón CSK de las celdas y fíjelas directamente añadiendo 0,5 ml de solución de paraformaldehído al 3,6% (en PBS) que contenga Triton X-100 al 0,05% durante 10 min a RT (22-25 °C).
PRECAUCIÓN: Es importante preparar PFA al 3,6 % a partir de un stock de PFA al 32 % recién hecho. El paraformaldehído puede causar daños oculares graves, irritación de la piel e irritación respiratoria. - Lave las células una vez con 1 ml de PBS que contenga 0.05% de Triton X-100 para eliminar el PFA.
- Permeabilizar aún más las células utilizando 1 ml de PBS que contenga Triton X-100 al 0,5% durante 15 min a RT (22-25 °C).
- Reacción click-IT de EdU para visualizar células replicantes (fase S)
- Retire la solución de permeabilización y lave las células 2 veces con 1 ml de tampón de bloqueo (Tabla 1).
PRECAUCIÓN: La albúmina sérica bovina (BSA) puede causar irritación respiratoria. - Añadir 1 ml de tampón de bloqueo (Tabla 1) y mecer suavemente la placa que contiene cubreobjetos durante 10 min a RT (22-25 °C).
- Retirar el tampón de bloqueo y añadir 500 μL de cóctel de reacción de clic que contenga picolilazida 647 (Tabla 1). Incubar los cubreobjetos durante 30 min a RT (22-25 °C) con un balanceo suave y realizar incubaciones posteriores en la oscuridad.
NOTA: Cuando use anticuerpos BrdU, use el doble de la cantidad (1 ml) y el tiempo (60 min) para la reacción de clic según lo recomendado por el fabricante para asegurarse de que la reacción esté saturada y que el EdU incorporado esté etiquetado. Esto limita la reactividad cruzada de los anticuerpos BrdU27.
- Retire la solución de permeabilización y lave las células 2 veces con 1 ml de tampón de bloqueo (Tabla 1).
- Retire la mezcla de reacción de clic y lave las celdas 2 veces con PBS con Triton X-100 al 0,05% durante 10 min a RT (22-25 °C) (Figura 1 y Figura 2).
- Añadir 1 mL de tampón de bloqueo e incubar a RT (22-25 °C) durante 30 min. Alternativamente, mantenga las celdas en tampón de bloqueo a 4 °C durante la noche.
- Aplicar anticuerpo primario (rata anti-RPA2, dilución 1:1.000) durante 2 h a RT (22-25 °C) en 250-500 μL de tampón de bloqueo con balanceo suave.
- Lave las células 2 veces con PBS que contenga Triton X-100 al 0,05 % para eliminar rápidamente la mayor parte de la solución de anticuerpos.
- Continuar lavando las celdas durante 3 x 10 min con tampón de bloqueo a RT (22-25 °C).
- Aplicar anticuerpo secundario (Alexa-488 anti-rata, dilución 1:1.000) en 250-500 μL de tampón de bloqueo a RT (22-25 °C) durante 2 h con balanceo suave.
- Lave las células con tampón de bloqueo 2 veces para eliminar rápidamente la mayor parte del anticuerpo secundario. Continúe lavando las celdas durante 3 x 10 min con PBS que contiene 0,05% de Triton X-100 a RT (22-25 °C).
- Para contratinción de los núcleos, lavar las células una vez con PBS que contenga 0,05% de Triton X-100 y 1 μg/ml de 4',6-diamidino-2-fenilindol (DAPI) durante 10 min a RT (22-25 °C). Lave las celdas una vez con PBS durante 5 min a RT (22-25 °C).
- Monte el cubreobjetos en portaobjetos de microscopio utilizando 10 μL de medio de montaje/cubreobjetos. Sumerja los cubreobjetos en agua destilada antes de montarlos para deshacerse de los cristales de sal. Tome imágenes de los portaobjetos al día siguiente y guárdelos a 4 °C durante semanas (Figura 3).
6. Adquisición y cuantificación de imágenes
- Para capturar imágenes, utilice cualquier microscopio epifluorescente disponible equipado con conjuntos de filtros de rutina para obtener imágenes de los canales DAPI, FITC y Cy5 con un aumento de al menos 60-63x, una apertura numérica alta y objetivos de aceite para visualizar focos nucleares.
NOTA: La excitación DAPI óptima es de ~359 nm; La excitación de Alexa 488 es de ~488 nm; mientras que la excitación de Alexa 647 es de ~647 nm. - Para el análisis de imágenes, abra archivos de imagen en Fiji/ImageJ.
- Hacer máscaras nucleares usando la tinción DAPI (Figura 4A-F y Video Suplementario S1).
- Abra la imagen DAPI.
- Seleccionar proceso | Mejore el contraste y establezca el píxel saturado en 0.35.
- Haga clic en Procesar | Binario | Convertir en máscara. Elija Binario | Rellena agujeros y haz clic en Analizar | Analizar partículas. Establece el tamaño en 10-Infinito.
- En el administrador de ROI, haga clic en Mostrar todo.
- Búsqueda de focos de RPA2 en el núcleo (Figura 4G,H y Video Suplementario S1)
- Abra la imagen RPA2.
- Elija Proceso | Encuentra a Maxima. Establezca la prominencia en un valor que resalte los focos de RPA2 (entre 500 y 750), separándolo del fondo.
- Finalmente, haga clic en el botón Medir en el Administrador de ROI.
- Calcule el número total de focos nucleares de ssDNA dividiendo el valor de la columna RawinDen por 255 (el valor máximo de la intensidad de píxeles en cada foco).
- Realice análisis estadísticos utilizando la herramienta de software estadístico preferida.
NOTA: Excluya del análisis todas las células positivas para EdU y las máscaras DAPI segmentadas incorrectamente.
- Hacer máscaras nucleares usando la tinción DAPI (Figura 4A-F y Video Suplementario S1).
Resultados
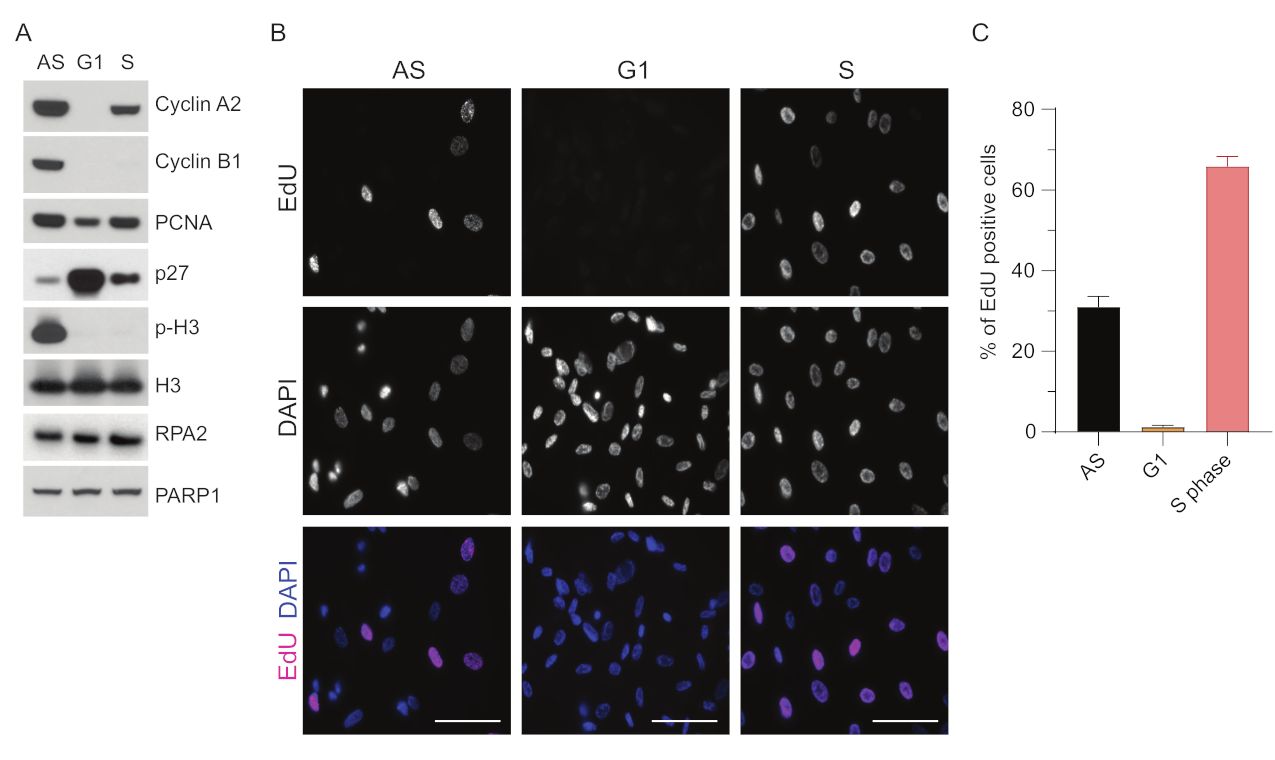
Para superar las limitaciones de la detección de ssDNA en G1, utilizamos RPA2, que mejora tanto la especificidad como la intensidad de la detección de focos de ssDNA35. Para lograr una sincronización celular precisa, utilizamos células RPE1 que pueden ser eficientemente privadas de suero y sincronizadas en la fase G0. A continuación, se les puede inducir a volver a entrar en el ciclo celular mediante la adición de suero después de la privación sérica. Para confirmar la eficiencia de la sincronización, marcamos las células con EdU y su contenido de ADN con yoduro de propidio. Además, recopilamos resultados cualitativos y cuantitativos mediante citometría de flujo (Figura suplementaria S1A). Los diagramas de puntos muestran que después de 72 h de inanición sérica, ~98% de las células están en fase G0. Después de la adición de medios que contienen suero durante 6 h, las células vuelven a entrar en el ciclo celular (como se ve por el aumento en los niveles de p27 en la Figura 1A), teniendo ~ 97% de células en G1, mientras que solo tienen <1% de células en la fase S, <2% de células en la fase G2 (Figura Suplementaria S1A). Después de 20-28 h de adición de suero a las células, pasan gradualmente a la fase S, como se muestra en los gráficos de citometría de flujo (Figura suplementaria S1A). Este protocolo de sincronización celular proporciona una población G1 con una pureza de ~97% (6 h después de la adición de suero después de 72 h de inanición sérica). Para validar aún más la eficiencia de la sincronización, comparamos la expresión de los marcadores del ciclo celular después de la liberación sérica mediante Western blot (Figura 1A y Figura Suplementaria S1B) y, en paralelo, realizamos un ensayo de incorporación de EdU para visualizar la replicación del ADN. La tinción de EdU también destaca la eficiencia de sincronización y la falta de replicación del ADN en la fase G1 (Figura 1B, C).
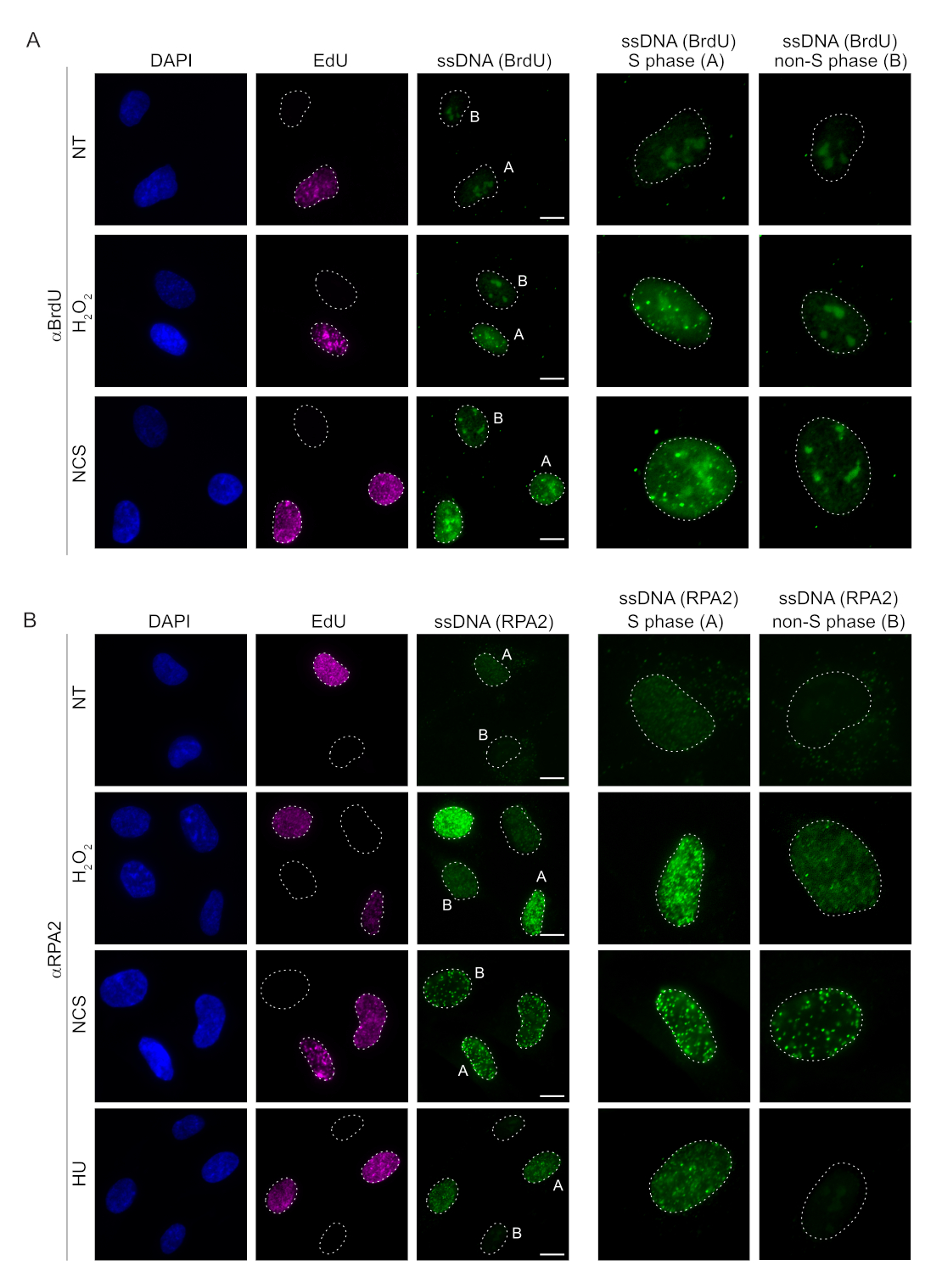
Los métodos convencionales para detectar el ssDNA en células de mamíferos se basan en la detección de BrdU en el ssDNA. La Figura 2A demuestra que tras el tratamiento conH2O2y neocarzinostatina (NCS), los focos de BrdU fueron detectables solo en las células de la fase S, mientras que no se detectaron focos de ssDNA en las células que no estaban en la fase S. La tinción de anticuerpos BrdU también mostró una notable tinción de fondo nucleolar que pudo detectarse en todos los núcleos, independientemente de la etapa del ciclo celular o de los tratamientos aplicados. Utilizando el protocolo de clic de EdU descrito aquí, no pudimos detectar la colocalización de los focos de EdU y BrdU, lo que es evidente en las muestras no tratadas de la Figura 2A. Para descartar por completo cualquier señal de BrdU que surgiera de la reactividad cruzada, evitamos el marcaje de EdU y en su lugar utilizamos ciclina A2 como marcador S-G2. Sin embargo, la tinción con ciclina A2 no permitió la preextracción de CSK y, en esta condición, no vimos ningún foco de BrdU, incluso después del estrés genotóxico (Figura suplementaria S2A). Esto pone de relieve el hecho de que la preextracción de CSK es necesaria para la tinción de ssDNA basada en anti-BrdU. Como control, probamos la tinción de anticuerpos BrdU en condiciones de desnaturalización. Esto abre el ADN para exponer el BrdU incorporado, lo que revela que el BrdU se incorporó uniformemente (Figura Suplementaria S2B).
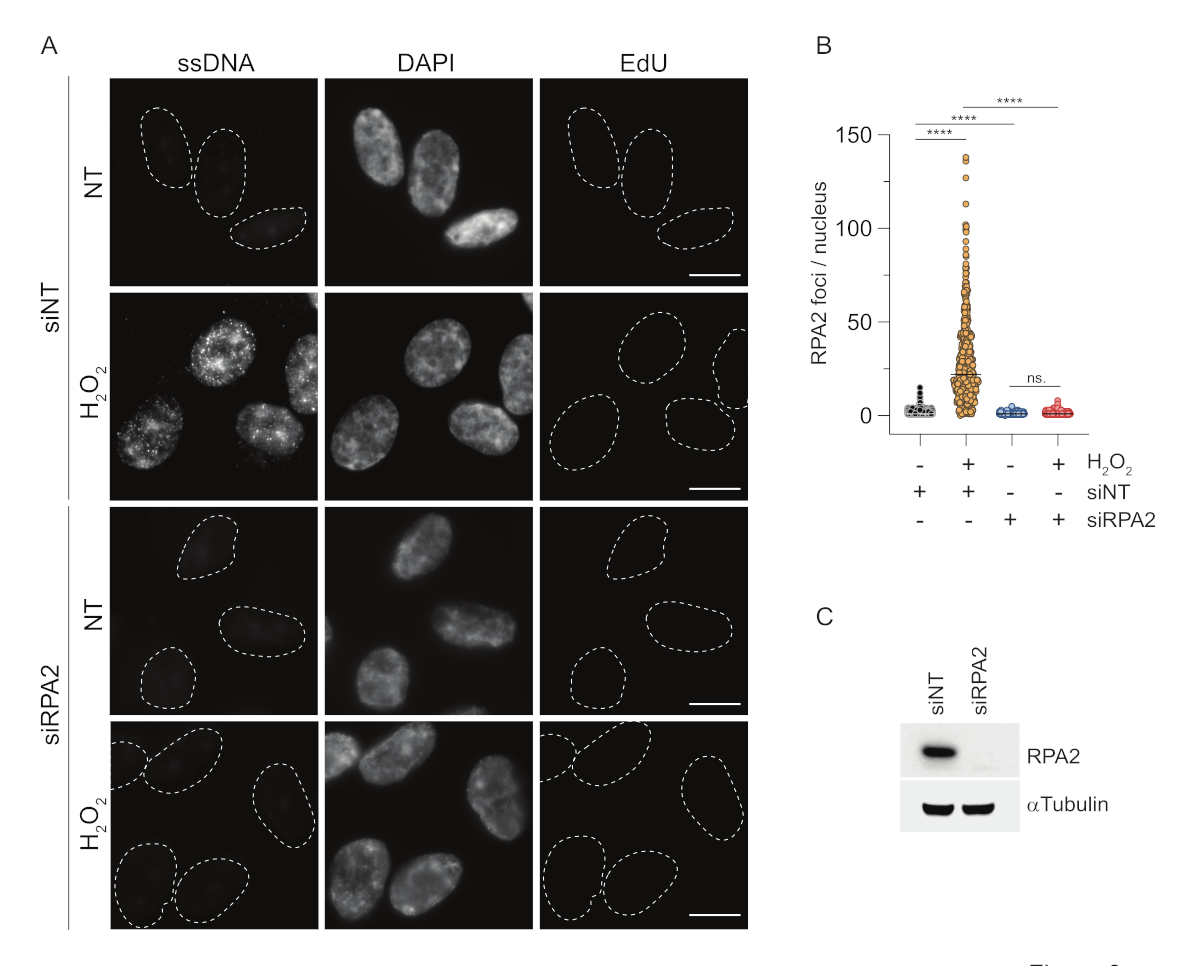
Por el contrario, la tinción de RPA2 muestra la formación de focos dependientes de NCS yH2O2 no solo en la fase S sino también en otras fases del ciclo celular (Figura 2B). Como control, también tratamos las células con HU, que solo causa la acumulación de ssDNA en las células en proceso de replicación. Como era de esperar, solo detectamos un aumento de la señal tras el tratamiento con el anticuerpo RPA2 en células EdU positivas, lo que pone de manifiesto la especificidad de este abordaje. El anticuerpo RPA2 también puede detectar la formación natural de ssDNA durante la replicación en ausencia de estrés genotóxico exógeno (Figura 2B). La naturaleza altamente sensible del anticuerpo RPA2 nos llevó a intentar utilizarlo en la fase G1, donde la tinción convencional de BrdU no detectó ninguna señal de estrés genotóxico (Figura suplementaria S2C). La Figura 3A muestra que la formación de focos de ADNss tras el tratamiento conH2O2 fue detectable cuando se utilizó un anticuerpo anti-RPA2, incluso en G1. Hubo un aumento significativo en el número de focos de RPA2 en estos núcleos tras el tratamiento conH2O2(Figura 3B). Estos focos eran específicos de RPA2, ya que el silenciamiento de RPA2 abolió la señal de FI (Figura 3A, B). La Figura 3C y la Figura Suplementaria S1C muestran la eficiencia del silenciamiento de RPA2 en estas células. En comparación con los métodos convencionales, la detección de ssDNA basada en RPA2 es altamente sensible y, por lo tanto, su aplicación puede extenderse a las células de fase G1.

Figura 1: Eficiencia de sincronización de las células RPE1 después de la inanición sérica. (A) Los inmunoblots muestran los niveles de proteínas indicados en las células RPE1 sincronizadas en fase asíncrona, G1 y S. (B) Las imágenes representativas muestran células RPE1 asíncronas, sincronizadas con las fases G1 y S que se expusieron a 10 μM de EdU durante 30 minutos antes de la fijación y se visualizaron mediante la reacción Click-IT. El DAPI se utilizó para contrarrestar la tinción del ADN nuclear. Barras de escala = 50 μm. (C) El gráfico muestra el porcentaje de células positivas para EdU sobre la población total de células evaluada por DAPI. La barra de error representa el error estándar de la media, y los números de núcleos analizados fueron los siguientes: AS n = 219, G1 n = 630, S n = 437. Abreviaturas: RPE1 = células epiteliales pigmentarias de la retina inmortalizadas por hTERT; AS = asíncrono; EdU = 5-etinil-2'-desoxiuridina; DAPI = 4',6-diamidino-2-fenilindol. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Detección de ssDNA con anticuerpo BrdU o anticuerpo RPA2 tras daño en el ADN. (A) Las imágenes representativas ilustran los focos de ADNss utilizando αBrdU (verde), las células de fase S se resaltan con EdU (púrpura) y DAPI se utilizó para contrarrestar la tinción del ADN nuclear (azul). Las células RPE1 se mantuvieron en 10 μM de BrdU durante 48 h antes de cualquier tratamiento adicional. Después de 48 h, las células se pulsaron con 10 μM de EdU durante 30 min seguido de tratamiento deH2O2 (250 μM) durante 1 h o Neocarzinostatina (0,5 μg/mL) durante 4 h. Las células se fijaron después de la preextracción de CSK. Una línea discontinua blanca denota el borde de cada núcleo. Barra de escala = 5 μm. Los paneles de la derecha son imágenes ampliadas de los núcleos indicados de fase S o no fase S. (B) Las imágenes representativas ilustran los focos de ADNss utilizando el anticuerpo αRPA2 (verde). Las células de la fase S se resaltan con EdU (púrpura), y DAPI se utilizó para contrarrestar la tinción del ADN nuclear (azul). Las células RPE1 se pulsaron con 10 μM de EdU durante 30 min, seguidas de 1 h H2O2 (250 μM), 4 h de hidroxiurea (2 mM) o 4 h de NCS (0,5 μg/mL). Las células se fijaron después de la preextracción de CSK. Una línea discontinua blanca denota el borde de cada núcleo. Barra de escala = 10 μm. Los paneles de la derecha son imágenes ampliadas de los núcleos indicados de fase S o no fase S. Abreviaturas: ssDNA = ADN monocatenario; BrdU = 5-bromo-2'-desoxiuridina; DAPI = 4',6-diamidino-2-fenilindol; RPE1 = células epiteliales pigmentarias de la retina inmortalizadas por hTERT; EdU = 5-etinil-2'-desoxiuridina; NCS = Neocarzinostatina; HU = hidroxiurea. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Detección de focos de ADNss en fase G1 utilizando el anticuerpo RPA2. (A) Las células RPE1 se transfectaron con siRNAs dirigidos a RPA2 o un control de siRNA no dirigido, y posteriormente se sincronizaron en G1 y se marcaron con pulsos con 10 μM de EdU durante 30 min antes de tratarlas conH2O2(250 μM) durante 1 h donde estaba indicado. El DAPI se utilizó para contrarrestar la tinción del ADN nuclear. Las células se fijaron después de la preextracción de CSK. Una línea discontinua blanca denota el borde de cada núcleo. Barra de escala = 5 μm. (B) Las mediciones del número de focos/núcleo de RPA2 se llevaron a cabo a partir de dos experimentos independientes. Durante el análisis solo se consideraron las células EdU negativas. Las líneas representan el valor medio de las gráficas. Para el análisis estadístico se realizó la prueba de ANOVA no paramétrica (Kruskal-Wallis). Las estrellas indican P < 0,0001. El número de núcleos analizados fue el siguiente: siNT no H2O2 n = 513, siNT H2O2 n = 603, siRPA2 no H2O2 n = 266, siRPA2 H2O2 n = 536. (C) La eficiencia de la eliminación del siRNA se muestra en el inmunotransferencia. Abreviaturas: siNT = control de siRNA no dirigido; BrdU = 5-bromo-2'-desoxiuridina; DAPI = 4',6-diamidino-2-fenilindol; RPE1 = células epiteliales pigmentarias de la retina inmortalizadas por hTERT; EdU = 5-etinil-2'-desoxiuridina. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Cuantificación de focos de ADNss utilizando Fiji. Pasos detallados en Fiji que muestran cómo evaluar el número de focos de RPA2 en el núcleo. (A-E) La creación de una máscara nuclear utilizando el canal DAPI. (F-H) Umbral para identificar focos individuales de ADNss nuclear a partir de la señal de fondo. Abreviaturas: ssDNA = ADN monocatenario; DAPI = 4',6-diamidino-2-fenilindol. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Tampón citoesquelético (CSK) | |
| TUBERÍAS pH 7.0 | 10 mM |
| NaCl | 100 mM |
| EDTA pH 8 | 1 mM |
| MgCl2 | 3 mM |
| D-sacarosa | 300 mM |
| Tritón X-100 | 0.20% |
| Cóctel de inhibidores de la fosfatasa | 1 comprimido por cada 10 ml |
| Cóctel de inhibidores de la proteasa | 1 comprimido por cada 10 ml |
| diluido en ddH2O | N/A |
| Tampón de lavado | |
| Tritón X-100 | 0.05% |
| diluido en PBS | N/A |
| Tampón de permeabilización | |
| Tritón X-100 | 0.50% |
| diluido en PBS | N/A |
| Solución de fijación | |
| Paraformaldehido | 3.60% |
| Tritón X-100 | 0.05% |
| diluido en PBS | N/A |
| Búfer de bloqueo | |
| Albúmina sérica bovina (BSA) | 5% |
| Tritón X-100 | 0.10% |
| diluido en PBS | N/A |
| Cóctel de reacción Click-iT Plus | |
| 1x tampón de reacción Click-iT | 435 mL |
| Solución Alexa Fluor PCA | 5 mL |
| Premezcla protectora de CuSO4-cobre | 10 mL |
| 1x Aditivo de búfer Click-iT | 50 mL |
| Volumen total | 500 ml |
Tabla 1: Composición de los tampones utilizados en este protocolo.
Figura complementaria S1. (A) Las células RPE1 se sincronizaron con la fase G0 mediante inanición sérica durante 72 h y posteriormente se liberaron en diferentes fases del ciclo celular mediante la reintroducción del suero. Los diagramas de puntos muestran las células en las fases G0/G1, S o G2/M, donde las horas indican el tiempo después de la readición de suero después de la inanición sérica. El gráfico de la derecha muestra el porcentaje de celdas G0/G1, S y G2/M en cada condición. El análisis de FACS se llevó a cabo utilizando un kit de proliferación celular disponible comercialmente con EdU y yoduro de propidio de acuerdo con las recomendaciones del fabricante. (B) Exploraciones de Western blot sin recortar para la Figura 1. Los números muestran marcadores de peso molecular en kDa. PARP1 se utilizó como control de carga y se cargó en el gel que también se desarrolló contra CCNA2, p27 (más despojado para PCNA) y pH3 (S10) (más despojado para H3) cortando la membrana. CCNB1 y RPA2 se cargaron en un gel separado, utilizando la misma cantidad de lisado de proteínas para garantizar la comparabilidad. (C) Exploraciones de Western blot sin recortar para la Figura 3. Los números muestran marcadores de peso molecular en kDa. Abreviatura: EdU = 5-etinil-2'-desoxiuridina. Haga clic aquí para descargar este archivo.
Figura suplementaria S2: (A) Imágenes representativas ilustran focos de ADNss utilizando anticuerpos BrdU (verde); Las células de la fase S están resaltadas por la ciclina A2 (rojo); y DAPI se utilizó para contrarrestar la tinción del ADN nuclear (azul). Las células RPE1 se mantuvieron en 10 μM de BrdU durante 48 h antes del tratamiento adicional. Después de 48 h, las células fueron tratadas conH2O2 (250 μM) durante 1 h o Neocarzinostatina (0,5 μg/mL) durante 4 h antes de la fijación. Una línea discontinua blanca denota el borde de cada núcleo. Barra de escala = 5 μm. (B) Tinción BrdU de células RPE1 con y sin condición desnaturalizante. Las células RPE1 asíncronas se trataron previamente con 10 μM de BrdU durante 48 h. Barra de escala = 10 μm. (C) Las mediciones del número de focos/núcleo de BrdU se llevaron a cabo a partir de dos experimentos independientes en células RPE1 sincronizadas G1. Durante el análisis solo se consideraron las células EdU negativas. Las líneas representan el valor medio de las gráficas. Para el análisis estadístico se realizó la prueba de ANOVA no paramétrica (Kruskal-Wallis). La 'ns' indica una diferencia no significativa. El número de núcleos analizados fue el siguiente: NT n = 52, NCS n = 105, H2O2 n = 82. Abreviaturas: siNT = control de siRNA no dirigido; BrdU = 5-bromo-2'-desoxiuridina; DAPI = 4',6-diamidino-2-fenilindol; RPE1 = células epiteliales pigmentarias de la retina inmortalizadas por hTERT; NCS = Neocarzinostatina. Haga clic aquí para descargar este archivo.
Video Suplementario S1: Grabación de pantalla del análisis de focos de RPA2 basado en Fiji. Haga clic aquí para descargar este archivo.
Discusión
Mantener un cultivo celular sano y libre de micoplasma es fundamental para todos los experimentos descritos anteriormente. Las células RPE1 tienen una fuerte adhesión a los materiales plásticos tratados con cultivo de tejidos en medios de cultivo normales; sin embargo, sus características de unión disminuyen significativamente cuando se mantienen en condiciones libres de suero. Además, para capturar imágenes de alta resolución de focos de ADNss bajo un microscopio, las células deben colocarse en un cubreobjetos de 0,17 mm de grosor, que no es lo suficientemente hidrófilo como para soportar la unión adecuada de las células RPE1. Sin células correctamente aplanadas y distribuidas uniformemente, es muy difícil visualizar los focos individuales de ADNs. Por lo tanto, es fundamental elegir el material de recubrimiento adecuado (por ejemplo, vitronectina) y dejar el tiempo adecuado (6-12 h) para que las células se extiendan y se adhieran después de liberarlas en la fase G1.
Una parte desafiante del protocolo es obtener células RPE1 sincronizadas con G1 homogéneas. Esto requiere dos pasos críticos. En primer lugar, para una inanición sérica eficaz, las células deben tripsinizarse, lavarse a fondo con PBS y sembrarse directamente en nuevas placas de cultivo de tejidos utilizando medios sin suero. Lavar las células directamente en placas de cultivo de tejidos para eliminar el suero no producirá una sincronización eficiente de G0. En segundo lugar, cuando se liberan células en la fase G1, las células deben tripsinizarse nuevamente y sembrarse en placas de cultivo de tejidos frescos. Del mismo modo, el simple hecho de cambiar el medio y añadir un medio de cultivo que contenga suero a las células no dará lugar a una entrada G1 sincrónica. Además, para una entrada adecuada de G1, la densidad de siembra de las células en los cubreobjetos recubiertos debe estar en ciertos niveles de confluencia. Si bien la sincronización celular perfecta es generalmente inalcanzable, este protocolo de sincronización descrito aquí da una población G1 pura de ~ 97%. La densidad de siembra recomendada para RPE1 en un cubreobjetos de 12 mm de diámetro es de ~4 × 104 para adquirir un campo de visión homogéneo para la obtención de imágenes, con aproximadamente un 70% de confluencia. Una mayor densidad de siembra hace que las células se desprendan y se "despeguen" después de la extracción de CSK y dará como resultado una señal de fondo más alta durante la adquisición de imágenes.
Para reducir cualquier señal de fondo y lograr una relación señal-ruido favorable, es esencial lavar a fondo después de la incubación de anticuerpos primarios y secundarios. Dado que se deben aplicar numerosos pasos de lavado, también es esencial evitar que el pozo se seque durante cada paso de lavado. Minimizamos este artefacto aplicando un mínimo de 0,05% de Triton X-100 en todos los pasos de lavado e incubación. Una vez que los pozos se secaron, las celdas mostraron una relación señal-ruido alterada; Esto conduce a un patrón similar a un mosaico bajo el microscopio y podría interferir con la evaluación. La adquisición de imágenes de pila Z combinada con la deconvolución puede ayudar a capturar focos en diferentes planos focales para mejorar el análisis.
Los métodos convencionales se basan en la detección de BrdU incorporado en condiciones no desnaturalizantes. Estos métodos, sin embargo, dependen del pretratamiento de las células con altas dosis de BrdU durante al menos 1-2 días (o tiempo equivalente a un ciclo celular completo en la línea celular utilizada) para garantizar una incorporación genómica uniforme. Indeseablemente, la incorporación extensiva de BrdU puede causar interferencia en el ciclo celular36. Para abordar estas limitaciones, este método utiliza RPA2 endógeno para detectar focos de ssDNA. Este enfoque no requiere la incorporación de BrdU impulsada por la replicación, también se puede utilizar en células postmitóticas. Dado que no es necesaria una incorporación extensa de BrdU, esto ahorra tiempo y reduce la complejidad experimental. Mediante el uso de la tinción de RPA2 para visualizar el ssDNA, podemos utilizar la 2′-desoxi-5-etiniluridina (EdU) y la química del clic para marcar la replicación del ADN evitando la posible reactividad cruzada de los anticuerpos BrdU contra el EdU 27,37,38. Se debe tener especial cuidado de enmascarar adecuadamente el EdU incorporado durante la reacción de clic para que los anticuerpos BrdU no reaccionen de forma cruzada con EdU27,39.
Por último, una ventaja importante de utilizar RPA2 en lugar de BrdU es simplemente tener una relación señal-ruido superior en comparación con la tinción de BrdU fuera de la fase S. Descubrimos que la tinción de BrdU no desnaturalizante y su capacidad para visualizar el ssDNA está restringida a la fase S incluso en células replicantes (Figura 2). El anticuerpo BrdU se une solo al BrdU suficientemente expuesto en tramos de ADNss. La unión de las proteínas de reparación, incluida la RPA2, a los tramos de ssDNA puede suprimir o dificultar la exposición suficiente de BrdU en el ssDNA. También encontramos que la preextracción de CSK es necesaria para la visualización del ssDNA utilizando el anticuerpo BrdU. Esto es posible porque las trazas de ssDNA no son accesibles para el anticuerpo sin eliminar de ellas los componentes proteicos ligeramente unidos.
No obstante, existen algunas limitaciones asociadas a este protocolo. Una limitación del uso de RPA2 para la detección de ssDNA es la necesidad de optimizar el paso previo a la extracción de CSK. Sin unir, el exceso de RPA2 debe eliminarse del ADN antes de fijar las células. Por un lado, la subextracción conduce a un alto fondo debido a la fracción de proteína RPA2 que no está unida al ssDNA. Por otro lado, la sobreextracción provocará la pérdida de señal. Para la detección de BrdU, esto no es una variable, ya que BrdU se incorpora de manera estable al ADN y no se puede lavar mediante la extracción previa. Por lo tanto, se debe considerar cuidadosamente el tiempo de la preextracción de CSK, la cantidad de Triton X-100 en el tampón, el volumen y la temperatura a la que se realiza la preextracción. La preextracción de CSK también limita el uso del tamaño del núcleo para discriminar las células G0/G1 de las células S/G2.
Además, no podemos excluir la posibilidad de que parte de la señal que proviene de RPA2 se origine al unirse a otros interactores de proteínas de unión a la cromatina. También hay que tener en cuenta la especificidad de especie del anticuerpo RPA2. El anticuerpo utilizado en este protocolo puede reconocer RPA2 en humanos, ratones, ratas, hámsteres y monos. Otra limitación de este enfoque es que no todas las líneas celulares pueden carecer de suero para la sincronización de G0. La mayoría de las líneas celulares cancerosas pueden eludir los puntos de control del ciclo celular y proliferar incluso en medios privados de suero. Aunque la inanición del suero es beneficiosa, ya que no causa daño en el ADN, se debe monitorear cuidadosamente la eficiencia de la sincronización celular para asegurarse de que se logre el enriquecimiento adecuado de la fase del ciclo celular. Para las células que no responden a la privación sérica, se deben considerar otros métodos de sincronización celular (p. ej., sacudida mitótica, inhibición de CDK1 para la detención de G2 o técnicas no invasivas como la elutriación centrífuga). Otro método posible es el uso de imágenes de alto contenido para medir el contenido de EdU y ADN nuclear para el perfil del ciclo celular de células asíncronas31. Hay que tener en cuenta las implicaciones de utilizar métodos de sincronización alternativos para evitar interferencias con el análisis posterior. Por ejemplo, el uso de doble bloqueo de timidina o afidicolina, a menudo utilizado en la literatura, dará lugar a estrés de replicación y daño en el ADN40.
La investigación de los mecanismos de reparación del ADN sigue siendo un punto focal de discusión en los campos del cáncer y la biología celular. El protocolo presentado aquí ofrece un enfoque valioso para la preparación de células, lo que permite la visualización y el análisis cuantitativo del ssDNA tras la exposición a agentes que dañan el ADN. En particular, este protocolo destaca la utilización de la proteína de unión al ADNss, RPA2, demostrando su alta especificidad para visualizar pequeñas cantidades de focos de ADNss, evitando al mismo tiempo la reactividad cruzada no deseada en todas las fases del ciclo celular. El uso de RPA2 confiere numerosas ventajas, especialmente para los investigadores que pretenden analizar células en la fase G1 del ciclo celular. Este protocolo tiene en cuenta varias limitaciones y aborda las preocupaciones relacionadas con la interferencia de la señal, el ruido de fondo no deseado y la reactividad cruzada cuando se utiliza la tinción de RPA2 o BrdU para detectar ssDNA.
Divulgaciones
Los autores no tienen intereses contrapuestos que declarar.
Agradecimientos
Los autores agradecen a Michele Pagano por su apoyo y sus útiles ideas, a Ashley Chui y Sharon Kaisari por la lectura crítica del manuscrito, y a Jeffrey Estrada y Vilma Díaz por su continuo apoyo. Este trabajo fue apoyado por un suplemento de diversidad de la GM136250 de subvenciones de los Institutos Nacionales de Salud.
Materiales
| Name | Company | Catalog Number | Comments |
| Alpha-tubulin antibody | Sigma-Aldrich | T6074 | primary antibody (1:5,000) |
| Axio Observer Inverted Microscope | Zeiss | na | microscope |
| Bis-Tris Plus Mini Protein Gels, 4-12% | Invitrogen | NW04127BOX | Western Blot |
| Bovine Serum Albumin | Jackson ImmunoResearch | 001-000-162 | blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Sigma-Aldrich | B5002-100MG | nucleotide analogue |
| BrdU antibody BU1/75 | Abcam | ab6326 | primary antibody (1:500) |
| CellAdhere Dilution Buffer | Stemcell Technologies | 07183 | coating reagent |
| Click-iT Plus EdU Flow Cytometry Assay Kits | Invitrogen | C10632 | flow cytomery |
| Click-iT Plus EdU Cell Proliferation Kit for Imaging, Alexa Fluor 647 dye | Thermo Fisher Scientific | C10640 | click-reaction kit |
| cOmplete ULTRA Protease inhibitor tablets | Sigma-Aldrich | 5892791001 | reagent |
| Countess 3 Automated cell counter | Thermo Scientific | AMQAX2000 | cell counter |
| Coverslip | neuVitro | GG12PRE | tissue culture |
| Cyclin A2 antibody | Santa Cruz Biotechnology | sc-271682 | primary antibody (1:1,000) for IF and WB |
| Cyclin B1 antibody | Santa Cruz Biotechnology | sc-245 | primary antibody (1:5,000) |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D2650-100ML | vehicle control |
| DMEM, high glucose, with HEPES | Gibco | 12430051 | cell culture medium for RPE cells |
| DPBS, no calcium, no magnesium | Gibco | 14190144 | the PBS used throughout the protocol |
| D-Sucrose | Thermo Fisher Scientific | bp220-1 | reagent |
| Eclipse Ti2 Series Epifluorescent Microscope | Nikon | na | microscope |
| EdU (5-Ethynyl-2'-deoxyuridine) | Thermo Fisher Scientific | C10637 | nucleotide analogue |
| Falcon 24-well plate | Corning | 351147 | tissue culture |
| Falcon Cell Culture Dishes 100 mm | Corning | 353003 | tissue culture |
| Fetal Bovine Serum, heat inactivated | Gibco | 16140071 | media supplement |
| Fiji (ImageJ) | NIH | version 1.54f | software and algorithms |
| FxCycle PI/RNase Staining Solution | Invitrogen | F10797 | PI staining |
| Goat anti-mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 555 | Thermo Fisher Scientific | A21422 | secondary antibody (1:1,000) |
| Goat anti-rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A48262 | secondary antibody (1:1,000) |
| Histone H3 antibody | Abcam | ab1791 | primary antibody (1:10,000) |
| hTERT RPE1 | ATCC | CRL-3216 | cell line |
| Hydrochloric acid | Sigma-Aldrich | H1758-100ML | reagent |
| Hydrogen peroxide 30% soultion | Sigma-Aldrich | H1009-100ML | reagent |
| Hydroxyurea,98% powder | Sigma-Aldrich | H8627-5G | reagent |
| Invitrogen Ultra Pure 0.5 M EDTA pH 8.0 | Thermo Fisher Scientific | 15-575-020 | reagent |
| Lipfectamine RNAiMAX Transfection Reagent | Invitrogen | 13778150 | transfection reagent |
| Magnesium chloride solution 1 M | Sigma-Aldrich | M1028-100ML | reagent |
| MycoFluor | Thermo Fisher | M7006 | Mycoplasma Detection Kit |
| Neocarzinostatin from Streptomyces carzinostaticus | Sigma-Aldrich | N9162-100UG | reagent |
| NuPage MES SDS Running Buffer (20x) | Invitrogen | NP0002 | Western Blot |
| onTARGETplus Human RPA2 siRNA | Dharmacon | L-017058-01-0005 | siRNA |
| p27 antibody | BD Biosciences | 610241 | primary antibody (1:1,000) |
| Paraformaldehyde aqueous solution (32%) | Electron Microscopy Sciences | 50-980-494 | fixative |
| PARP1 antibody | Cell Signaling Technology | 9542S | primary antibody (1:1,000) |
| PCNA antibody | Cell Signaling Technology | 13110S | primary antibody (1:2,000) |
| Penicillin-Streptomycin | Gibco | 15140163 | media supplement |
| pH3 antibody | Cell Signaling Technology | 3377S | primary antibody (1:2,000) |
| PhosSTOP phosphatase inhibitor tablets | Sigma-Aldrich | 4906837001 | reagent |
| PIPES Buffer 0.5 M solution, pH 7.0 | Bioworld | 41620034-1 | reagent |
| Precision Plus Protein Kaleidoscope Prestained Protein Standards | Bio-Rad | 1610395 | Western Blot |
| Prism | GraphPad | version 10 | statistical analysis and graph |
| ProLong Diamond Antifade Mountant | Thermo Scientific | P36961 | mounting media |
| Reduced serum media (Opti-MEM) | Gibco | 31985070 | used for transfection |
| Rpa32/rpa2 antibody (mouse) | EMD Millipore | NA19L | primary antibody (1:1,000) for WB |
| Rpa32/rpa2 antibody (rat) | Cell Signaling Technology | 2208S | primary antibody (1:1,000) for IF |
| Sodium Chloride solution (5 M) | Sigma-Aldrich | S5150 | reagent |
| Sodium Pyruvate (100 mM) | Gibco | 11360070 | media supplement |
| Sodium tetraborate decahydrate | Sigma-Aldrich | B3535-500G | reagent |
| Thermo Scientific Pierce DAPI Nuclear Counterstain | Thermo Scientific | 62248 | nucleic acid stain |
| Thymidine,powder | Sigma-Aldrich | T1985-1G | reagent |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | detergent |
| Trypsin-EDTA (0.5%), no phenol red | Gibco | 1540054 | cell dissociation agent |
| Vitronectin XF | Stemcell Technologies | 07180 | coating reagent |
| ZE5 Cell Analyzer | Bio-Rad | na | flow cytomery |
Referencias
- Hakem, R. DNA-damage repair; the good, the bad, and ugly. EMBO J. 27 (4), 589-605 (2008).
- Gutierrez, R., O'Connor, T. R. DNA direct reversal repair and alkylating agent drug resistance. Cancer Drug Resist. 4 (2), 414-423 (2021).
- Krokan, H. E., Bjoras, M. Base excision repair. Cold Spring Harb Perspect Biol. 5 (4), a012583 (2013).
- Marteijn, J. A., Lans, H., Vermeulen, W., Hoeijmakers, J. H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 15 (7), 465-481 (2014).
- Li, G. M. Mechanisms and functions of DNA mismatch repair. Cell Res. 18 (1), 85-98 (2008).
- Hustedt, N., Durocher, D. The control of DNA repair by the cell cycle. Nat Cell Biol. 19 (1), 1-9 (2016).
- Yang, W., Gao, Y. Translesion and repair DNA polymerases: diverse structure and mechanism. Annu Rev Biochem. 87, 239-261 (2018).
- Bhat, D. S., et al. Therapeutic disruption of RAD52-ssDNA complexation via novel drug-like inhibitors. NAR Cancer. 5 (2), zcad018 (2023).
- Gupta, P., Saha, B., Chattopadhyay, S., Patro, B. S. Pharmacological targeting of differential DNA repair, radio-sensitizes WRN-deficient cancer cells in vitro and in vivo. Biochem Pharmacol. 186, 114450 (2021).
- Pena-Diaz, J., et al. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol Cell. 47 (5), 669-680 (2012).
- Schroering, A. G., Edelbrock, M. A., Richards, T. J., Williams, K. J. The cell cycle and DNA mismatch repair. Exp Cell Res. 313 (2), 292-304 (2007).
- Scully, R., Panday, A., Elango, R., Willis, N. A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 20 (11), 698-714 (2019).
- Escribano-Diaz, C., et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 49 (5), 872-883 (2013).
- Genschel, J., Modrich, P. Mechanism of 5'-directed excision in human mismatch repair. Mol Cell. 12 (5), 1077-1086 (2003).
- Hu, J., et al. Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J Biol Chem. 288 (29), 20918-20926 (2013).
- Huertas, P., Jackson, S. P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 284 (14), 9558-9565 (2009).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in DNA replication with putative roles in cancer. Int J Mol Sci. 20 (1), 74 (2018).
- Symington, L. S. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol. 6 (8), a016436 (2014).
- Liu, Y., et al. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem. 280 (5), 3665-3674 (2005).
- Wold, M. S., Kelly, T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc Natl Acad Sci U S A. 85 (8), 2523-2527 (1988).
- Wienholz, F., Vermeulen, W., Marteijn, J. A. Amplification of unscheduled DNA synthesis signal enables fluorescence-based single cell quantification of transcription-coupled nucleotide excision repair. Nucleic Acids Res. 45 (9), e68 (2017).
- Wold, M. S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 66, 61-92 (1997).
- Chen, R., Wold, M. S. Replication protein A: single-stranded DNA's first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays. 36 (12), 1156-1161 (2014).
- Kang, Y., et al. Alteration of replication protein A binding mode on single-stranded DNA by NSMF potentiates RPA phosphorylation by ATR kinase. Nucleic Acids Res. 51 (15), 7936-7950 (2023).
- Kilgas, S., Kiltie, A. E., Ramadan, K. Immunofluorescence microscopy-based detection of ssDNA foci by BrdU in mammalian cells. STAR Protoc. 2 (4), 100978 (2021).
- Madabhushi, R., Pan, L., Tsai, L. H. DNA damage and its links to neurodegeneration. Neuron. 83 (2), 266-282 (2014).
- Liboska, R., Ligasova, A., Strunin, D., Rosenberg, I., Koberna, K. Most anti-BrdU antibodies react with 2'-deoxy-5-ethynyluridine -- the method for the effective suppression of this cross-reactivity. PLoS One. 7 (12), e51679 (2012).
- Biehs, R., et al. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol Cell. 65 (4), 671-684.e5 (2017).
- Cruz-Garcia, A., Lopez-Saavedra, A., Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 9 (2), 451-459 (2014).
- Ercilla, A., et al. Physiological tolerance to ssDNA enables strand uncoupling during DNA replication. Cell Rep. 30 (7), 2416-2429.e7 (2020).
- Lezaja, A., et al. RPA shields inherited DNA lesions for post-mitotic DNA synthesis. Nat Commun. 12 (1), 3827 (2021).
- Mukherjee, B., Tomimatsu, N., Burma, S. Immunofluorescence-based methods to monitor DNA end resection. Methods Mol Biol. 1292, 67-75 (2015).
- Ochs, F., et al. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol. 23 (8), 714-721 (2016).
- Raderschall, E., Golub, E. I., Haaf, T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad Sci U S A. 96 (5), 1921-1926 (1999).
- Forment, J. V., Walker, R. V., Jackson, S. P. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytometry A. 81 (10), 922-928 (2012).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Sci Rep. 6, 19567 (2016).
- Aten, J. A., Bakker, P. J., Stap, J., Boschman, G. A., Veenhof, C. H. DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem J. 24 (5), 251-259 (1992).
- Podgorny, O., Peunova, N., Park, J. H., Enikolopov, G. Triple S-phase labeling of dividing stem cells. Stem Cell Reports. 10 (2), 615-626 (2018).
- Cappella, P., Gasparri, F., Pulici, M., Moll, J. Cell proliferation method: click chemistry based on BrdU coupling for multiplex antibody staining. Curr Protoc Cytom. Chapter 7, (2008).
- Ligasova, A., Koberna, K. Strengths and weaknesses of cell synchronization protocols based on inhibition of DNA synthesis. Int J Mol Sci. 22 (19), 10759 (2021).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados