
Method Article
Visualisierung einzelsträngiger DNA-Herde in der G1-Phase des Zellzyklus
In diesem Artikel
Zusammenfassung
Das folgende Protokoll zeigt den Nachweis einzelsträngiger DNA-Foci in der G1-Phase des Zellzyklus unter Verwendung von Zellzyklussynchronisation, gefolgt von RPA2-Immunfluoreszenzfärbung.
Zusammenfassung
Die DNA verfügt über spezielle zelluläre Reparaturwege, die in der Lage sind, mit Läsionen fertig zu werden, die sowohl aus endogenen als auch aus exogenen Quellen stammen können. Die DNA-Reparatur erfordert die Zusammenarbeit zwischen zahlreichen Proteinen, die für eine breite Palette von Aufgaben verantwortlich sind, von der Erkennung und Signalisierung des Vorhandenseins einer DNA-Läsion bis hin zur physischen Reparatur. Während dieses Prozesses entstehen oft Spuren von einzelsträngiger DNA (ssDNA), die schließlich von DNA-Polymerasen gefüllt werden. Die Art dieser ssDNA-Spuren (sowohl in Bezug auf die Länge als auch auf die Anzahl) sowie die Polymerase, die rekrutiert wird, um diese Lücken zu füllen, sind spezifisch für den Reparaturweg. Die Visualisierung dieser ssDNA-Spuren kann uns helfen, die komplizierte Dynamik der DNA-Reparaturmechanismen zu verstehen.
Dieses Protokoll bietet eine detaillierte Methode für die Präparation von G1-synchronisierten Zellen zur Messung der Bildung von ssDNA-Foci bei genotoxischem Stress. Mit Hilfe eines einfach anzuwendenden Immunfluoreszenzansatzes visualisieren wir ssDNA durch Färbung auf RPA2, eine Komponente des heterotrimeren Replikationsprotein-A-Komplexes (RPA). RPA2 bindet und stabilisiert ssDNA-Zwischenprodukte, die bei genotoxischem Stress oder Replikation entstehen, um die DNA-Reparatur und die Aktivierung von DNA-Schadens-Checkpoints zu kontrollieren. Die 5-Ethynyl-2'-Desoxyuridin (EdU)-Färbung wird verwendet, um die DNA-Replikation sichtbar zu machen, um S-Phase-Zellen auszuschließen. Dieses Protokoll bietet einen alternativen Ansatz zu den herkömmlichen, nicht-denaturierenden 5-Brom-2'-Desoxyuridin (BrdU)-basierten Assays und eignet sich besser für den Nachweis von ssDNA-Herden außerhalb der S-Phase.
Einleitung
Um Leben zu erhalten, untersuchen und reparieren Zellen ständig DNA, um ihre genomische Integrität zu erhalten. Zellen können verschiedene Arten von DNA-Schäden ansammeln, die sowohl auf endogene (z. B. Oxidation, Alkylierung, Desaminierung, Replikationsfehler) als auch auf exogene (z. B. UV, ionisierende Bestrahlung) Quellen von DNA-Stressoren zurückzuführen sind. Wenn diese Läsionen nicht repariert werden, führt dies entweder zu Apoptose, Zellzyklusstillstand oder Seneszenz und kann zu Krankheiten führen1. DNA-Läsionen können durch einen der folgenden Haupt-DNA-Reparaturwege behandelt werden: DR (Direct Reversal Repair), die hauptsächlich alkylierte Basen repariert2; BER (Base Excision Repair), das auf nicht sperrige DNA-Basenfehler und einzelsträngige DNA-Brüche (SSBs) abzielt3; NER (Nukleotid-Exzisionsreparatur) zur Korrektur sperriger, Helix-verzerrender DNA-Läsionen4; MMR (Mismatch Repair), die hauptsächlich auf DNA-Fehlanpassungen, Insertions-/Deletionsschleifen (IDLs) und bestimmte Basenschäden abzielt5; NHEJ (nicht-homologe Endverbindung) und HRR (homologe Rekombinationsreparatur), die beide an doppelsträngigen DNA-Brüchen (DSBs) aktiv sind6; und TLS (Transläsionssynthese), bei der es sich um einen Bypass-Mechanismus für DNA-Läsionen handelt7. Obwohl diese Wege unterschiedliche Substratspezifitäten aufweisen, gibt es bestimmte Überschneidungen zwischen ihnen, um Redundanz für eine effiziente Reparatur zu gewährleisten. Das Verständnis der Wirkung der verschiedenen DNA-Reparaturwege in verschiedenen Phasen des Zellzyklus ist von entscheidender Bedeutung, da diese DNA-Reparaturfaktoren als wesentliche Ziele für therapeutische Ansätze zur Behandlung von Krebs, Alterung und neurologischen Erkrankungen dienen könnten 8,9.
Einzelsträngige DNA (ssDNA) wird während des gesamten Zellzyklus durch die Reparatur von DNA-Läsionen erzeugt, die sowohl durch endogene als auch durch exogene DNA-schädigende Substanzen erzeugt werden. Bei genotoxischem Stress wird ssDNA reichlich in den S- und G2-Phasen erzeugt, in denen HRR und MMR ihre höchste Aktivität aufweisen und wenn die Replikationsmaschinerie abwürgt oder kollabiert, wenn DNA-Läsionen auftreten 6,10,11. Andere DNA-Reparaturwege (z. B. NHEJ/Microhomology-Mediated End Joining (MMEJ)/Single-Strand Annealing [SSA]) erzeugen während der DSB-Reparatur ebenfalls ssDNA12. Diese ssDNA-Spuren entstehen in der Regel durch DNA-Resektion, die durch Exonukleasen wie EXO1, DNA2 und CtIP während HR und MMR, Endonukleasen wie XPF und XPG während NER oder durch die kombinierte Wirkung von POLB und FEN1 während BER 4,13,14,15,16,17,18,19 durchgeführt wird . Aufgrund der Arbeit der Replikationsmaschinerie werden ssDNA-Spuren auch erzeugt, wenn die DNA-Helikasen DNA vor den PCNA-gebundenen replikativen Polymerasen20 abwickeln. Im Gegensatz dazu verringern in der G1-Phase das Fehlen von HRR und DNA-Replikation sowie die begrenzte Aktivität von MMR das Ausmaß der erzeugten ssDNA-Spuren und sind daher schwieriger zu detektieren 10,11,21.
Zelluläre ssDNA-Spuren sind hochempfindliche Strukturen, die geschützt werden müssen, um die Bildung von DSBs zu vermeiden. Dies wird erreicht, indem die ssDNA-Spuren mit RPA beschichtet werden. RPA ist ein reichlich vorhandener heterotrimerer Proteinkomplex, der aus mehreren Untereinheiten (RPA1, RPA2 und RPA3, auch als RPA70, RPA32 bzw. RPA14 bezeichnet) besteht, die während des gesamten Zellzyklus ubiquitär exprimiert werden22. Jede RPA-Untereinheit enthält eine DNA-Bindungsdomäne (DBD), die mit 4-6 Nukleotiden interagieren kann, und die kombinierten Untereinheiten bilden einen stabilen Trimerisierungskern. Insgesamt bindet RPA an ca. 20-30 Nukleotide mit sub-nanomolarer Affinität23,24.
Konventionelle Methoden verwenden die Immunfluoreszenzmikroskopie (IF), um ssDNA-Foci sichtbar zu machen, indem 5-Brom-2'-Desoxyuridin (BrdU), das mit BrdU-Antikörpern in die genomische DNA eingebaut wird, markiertwerden 25. Dieser Ansatz beruht auf der Tatsache, dass BrdU-Antikörper BrdU nur in exponierter ssDNA25 nachweisen können. Obwohl dieser Ansatz einfach ist, weist er auch gewisse Einschränkungen auf. Zum Beispiel werden die Zellen vor Beginn des Experiments vorbehandelt, um BrdU einzubauen, was zeitaufwändig ist und nachgeschaltete Effektoren stören kann. Daher ist die BrdU-basierte ssDNA-Detektion auf replizierende Zellen beschränkt und kann nicht für ruhende Zellen verwendet werden. Dies schließt die Anwendung dieser Methode zur Untersuchung der DNA-Reparatur in nicht-replizierenden Zellen aus, obwohl sie für verschiedene Krankheiten wie Krebs und Neurodegeneration von Bedeutung ist 5,26. Da die Strukturen von BrdU und EdU sehr ähnlich sind, zeigen die meisten BrdU-Antikörper eine Kreuzreaktivität gegenüber EdU, was bei der Durchführung von Experimenten mit doppelter Markierung berücksichtigt werden muss27. Die RPA-Färbung wurde bisher verwendet, um ssDNA-Foci hauptsächlich in S-Phase-Zellen zu zeigen. Einige Arbeiten haben es jedoch auch außerhalb der S-Phase erfolgreich eingesetzt 28,29,30,31,32,33,34,35. Das folgende Protokoll nutzt die Eigenschaften von RPA effizient aus und ermöglicht die Visualisierung von ssDNA-Herden nach DNA-Schäden in der G1-Phase des Zellzyklus (obwohl es in allen Zellzyklusphasen verwendet werden kann).
Protokoll
1. Erhaltung von hTERT-immortalisierten retinalen Pigmentepithelzellen (RPE1)
- Bewahren Sie RPE1-Zelllinien in Dulbeccos Modified Eagle Medium (DMEM) auf, ergänzt mit 10 % hitzeinaktiviertem fötalem Kälberserum (Hi-FBS) und 100 μg/ml Penicillin-Streptomycin (im Folgenden als Kultivierungsmedium bezeichnet) in einem befeuchteten Inkubator mit 5 % CO2 bei 37 °C. Für die routinemäßige Kultivierung züchten Sie RPE1-Zellen in einer 15-cm-Gewebekultur-behandelten Schale und teilen Sie sie, wenn Sie eine Konfluenz von 80-90 % erreichen (~16-18 × 106 Zellen pro 15-cm-Schale).
- Bei der Spaltung das Medium entfernen und die Zellen mit 10 ml 1x phosphatgepufferter Kochsalzlösung (PBS) spülen.
- Fügen Sie 3 ml 0,05 % Trypsin-EDTA hinzu, um die gesamte Oberfläche der Schale zu bedecken. Halten Sie die Zellen mit dem Trypsin bei 37 °C, bis sie sich ablösen.
- Nach der Trypsinisierung werden die Zellen mit Nährmedium resuspendiert und bei 150 × g für 5 min bei Raumtemperatur (RT, 22-25 °C) heruntergeschleudert. Entfernen Sie den Überstand und resuspendieren Sie die Zellen vorsichtig in 10 ml Nährmedium.
- 1,6-1,8 × 106 Zellen in eine neue 15-cm-Schale (~1 ml der Zellsuspension) aussäen.
HINWEIS: Alle Gewebekulturarbeiten sollten unter BSL-2-Sicherheitsstufen durchgeführt werden. Die Inkubationszeit für die Trypsinisierung hängt von der Zellkonfluenz ab. Normalerweise dauert der Prozess 2-3 Minuten für eine 90% konfluierende Platte. Die Zellen sollten regelmäßig mit kommerziell erhältlichen Kits auf Mykoplasmenkontamination untersucht werden (siehe Beispiele in der Materialtabelle).
2. siRNA-Knock-down des interessierenden Gens (GOI)
- 1,0 × 106 RPE1-Zellen am Tag vor der Transfektion in eine 10 cm große, mit Gewebekultur behandelte Platte mit 10 ml Nährmedium aussäen.
- Am Tag der Transfektion komplexieren Sie die siRNA. Für eine 10-cm-Platte wird eine Endkonzentration von 20 nM siRPA2 und 12 μl lipidbasiertem Transfektionsreagenz in 500 μl niedrigem Serumtransfektionsmedium verwendet. Mischen Sie alle Komponenten vorsichtig durch Schnippen des Röhrchens und inkubieren Sie 5 Minuten lang bei RT (22-25 °C).
- Geben Sie das komplexierte siRNA-Gemisch tropfenweise zu den Zellen und inkubieren Sie die Zellen 48 h lang mit der siRNA.
3. Synchronisation von RPE1-Zellen in die G0-Phase
- Trypsinisieren Sie die RPE1-Zellen aus Schritt 2.3 wie in Abschnitt 1 beschrieben (~2 × 106 Zellen).
- Die Zellsuspension wird in 15-ml-Zentrifugenröhrchen überführt und bei 150 × g, RT (22-25 °C) 5 min lang zentrifugiert.
- Entfernen Sie den Überstand und resuspendieren Sie die Zellen in 12 ml PBS. Die Zellen werden bei 150 × g bei RT (22-25 °C) für 5 min zentrifugiert. Wiederholen Sie die Entfernung des Überstandes und die Zentrifugation zweimal.
- Resuspendieren Sie die Zellen in 10 ml serumfreiem DMEM, ergänzt mit 100 μg/ml Penicillin-Streptomycin, 1 mM Natriumpyruvat und 15 mM HEPES, und legen Sie sie auf eine 10 cm große Gewebekulturschale.
HINWEIS: Wenn die Zellen dazu neigen, zu verklumpen, resuspendieren Sie sie in nur 1 ml serumfreiem DMEM und pipettieren Sie sie 5x mit einer P1000-Spitze, um die Klumpen zu lösen, bevor Sie die Suspension bis zu einem Endvolumen von 10 ml verdünnen. - Nach 24 Stunden Serumhunger wird die zweite Runde des Silencings mit dem gleichen Verfahren wie in Abschnitt 2 beschrieben eingeführt, indem die komplexierte siRNA zu den serumhungernden Zellen hinzugefügt wird.
- Halten Sie die RPE1-Zellen 72 Stunden lang in serumfreiem DMEM, bevor Sie mit der G1-Freisetzung fortfahren.
4. Deckglasbeschichtung und Freisetzung der Zellen in die G1-Phase
- Sterilisieren Sie die Pinzette mit 70 % Ethanol und legen Sie ein einzelnes Deckglas (12 mm Durchmesser und #1,5 Dicke [0,17 mm]) in eine Vertiefung einer 24-Well-Platte.
- Die Vitronektin-Beschichtungsmatrix wird mit PBS verdünnt, um eine Endkonzentration von 10 μg/ml zu erhalten. Geben Sie 500 μl der Vitronektinlösung in jede Vertiefung, die die Deckgläser enthält, und inkubieren Sie 1 h lang bei RT.
- Entfernen Sie die Beschichtungslösung und waschen Sie die Deckgläser mit 1 ml PBS.
- Lösen Sie die serumarmen RPE1-Zellen von der 10 cm großen, mit Gewebekultur behandelten Platte mit 1 ml 0,05%igem Trypsin nach einer PBS-Wäsche für 1 Minute bei 37 °C.
HINWEIS: Zellen lösen sich nach Serummangel viel schneller ab. Seien Sie vorsichtig, wenn Sie die Zellen mit PBS waschen und verwenden Sie kurze Trypsinisierungszeiten. - Um das Trypsin zu inaktivieren, resuspendieren Sie die RPE1-Zellen in insgesamt 6 ml Nährmedium. Entfernen Sie das inaktivierte Trypsin, indem Sie die Zellen mit 150 × g bei RT (22-25 °C) für 5 Minuten herunterschleudern.
- Resuspendieren Sie die Zellen in 1 ml Nährmedium und messen Sie die Zellzahl.
- Säen Sie 4 × 104 RPE1-Zellen in insgesamt 500 μl Nährmedium auf das beschichtete Deckglas.
HINWEIS: Stellen Sie sicher, dass die Zellviabilität über 90 % liegt, bevor Sie mit den nachgelagerten Schritten fortfahren. Die Zellviabilität konnte durch Trypanblau-Färbung während des Zellzählschritts schnell beurteilt werden. - Nach 6 h des Einbringens der Zellen in das Kulturmedium befinden sich die G0-freigesetzten Zellen in der frühen G1-Phase. Führen Sie Experimente in G1 in diesem 6-12-Stunden-Fenster durch, bevor die Zellen in die S-Phase eintreten.
- Vor dem Einbringen von DNA-Schäden werden die Zellen mit 10 μM 5-Ethinyl-2'-desoxyuridin (EdU) für 30 min bei 37 °C, verdünnt in Nährmedium, gepulst.
- Entfernen Sie das EdU-haltige Medium und jagen Sie die Zellen mit 10 μM Thymidin für 10 min bei 37 °C, um zu verhindern, dass während der Induktion von DNA-Schäden ein verbleibender EdU-Einbau erfolgt.
- Entfernen Sie das Medium mit Thymidin und behandeln Sie die Zellen 1 h lang mit 250 μMH2O2, verdünnt in Nährmedium.
5. Immunfluoreszenzfärbung von ssDNA
- Waschen Sie die Zellen einmal mit 1 ml RT (22-25 °C) PBS, um das Medium und die Serumkomponenten zu entfernen.
HINWEIS: Gehen Sie beim Waschen der Zellen vorsichtig vor, um ein Ablösen und Austrocknen zu vermeiden. Verarbeiten Sie nicht viele Vertiefungen gleichzeitig. - Vorextraktion: Inkubieren Sie die gewaschenen Zellen in 1 ml CSK-Extraktionspuffer (Tabelle 1) für 5 Minuten bei RT (22-25 °C).
HINWEIS: Bei der CSK-Vorextraktion werden alle nicht chromatingebundenen Proteine, einschließlich löslichem RPA2, entfernt.
VORSICHT: Triton X-100 ist beim Verschlucken schädlich und kann Hautreizungen und Augenschäden verursachen. - Entfernen Sie den CSK-Puffer aus den Zellen und fixieren Sie sie direkt, indem Sie 0,5 ml 3,6%ige Paraformaldehydlösung (in PBS) mit 0,05% Triton X-100 für 10 min bei RT (22-25 °C) hinzufügen.
ACHTUNG: Es ist wichtig, 3,6 % PFA aus 32 % PFA-Brühe frisch zuzubereiten. Paraformaldehyd kann schwere Augenschäden, Hautreizungen und Atemwegsreizungen verursachen. - Waschen Sie die Zellen einmal mit 1 ml PBS mit 0,05 % Triton X-100, um das PFA zu entfernen.
- Weitere Permeabilisierung der Zellen mit 1 ml PBS mit 0,5 % Triton X-100 für 15 Minuten bei RT (22-25 °C).
- EdU-Click-IT-Reaktion zur Visualisierung replizierender Zellen (S-Phase)
- Entfernen Sie die Permeabilisierungslösung und waschen Sie die Zellen 2x mit 1 ml Blockierungspuffer (Tabelle 1).
VORSICHT: Rinderserumalbumin (BSA) kann Atemwegsreizungen verursachen. - Fügen Sie 1 ml Blockierpuffer hinzu (Tabelle 1) und rocken Sie die Deckglasplatte vorsichtig 10 Minuten lang bei RT (22-25 °C).
- Entfernen Sie den Blockierungspuffer und fügen Sie 500 μl Click-Reaktionscocktail hinzu, der Picolylazid 647 enthält (Tabelle 1). Inkubieren Sie die Deckgläser für 30 min bei RT (22-25 °C) mit leichtem Schaukeln und führen Sie nachgeschaltete Inkubationen im Dunkeln durch.
HINWEIS: Verwenden Sie bei der Verwendung von BrdU-Antikörpern die doppelte Menge (1 ml) und Zeit (60 min) für die Click-Reaktion, wie vom Hersteller empfohlen, um sicherzustellen, dass die Reaktion gesättigt ist und das eingebaute EdU markiert ist. Dies schränkt die Kreuzreaktivität der BrdU-Antikörperein 27.
- Entfernen Sie die Permeabilisierungslösung und waschen Sie die Zellen 2x mit 1 ml Blockierungspuffer (Tabelle 1).
- Entfernen Sie die Click-Reaktionsmischung und waschen Sie die Zellen 2x mit PBS mit 0,05% Triton X-100 für 10 min bei RT (22-25 °C) (Abbildung 1 und Abbildung 2).
- 1 ml Blockierungspuffer zugeben und bei RT (22-25 °C) 30 Minuten lang inkubieren. Alternativ können Sie die Zellen über Nacht im Blockierpuffer bei 4 °C aufbewahren.
- Primärer Antikörper (Anti-RPA2-Ratte, 1:1.000-Verdünnung) für 2 h bei RT (22-25 °C) in 250-500 μl Blockierungspuffer mit leichtem Schaukeln applizieren.
- Waschen Sie die Zellen 2x mit PBS mit 0,05% Triton X-100, um den größten Teil der Antikörperlösung schnell zu entfernen.
- Waschen Sie die Zellen 3 x 10 Minuten lang mit Blockierungspuffer bei RT (22-25 °C).
- Sekundärer Antikörper (Anti-Ratten-Alexa-488, 1:1.000-Verdünnung) in 250-500 μl Blockierungspuffer bei RT (22-25 °C) für 2 h mit leichtem Schaukeln applizieren.
- Waschen Sie die Zellen 2x mit Blockierungspuffer, um den größten Teil des sekundären Antikörpers schnell zu entfernen. Waschen Sie die Zellen 3 x 10 Minuten lang mit PBS mit 0,05 % Triton X-100 bei RT (22-25 °C).
- Um die Zellkerne zu verfärben, waschen Sie die Zellen einmal mit PBS mit 0,05 % Triton X-100 und 1 μg/ml 4',6-Diamidino-2-phenylindol (DAPI) für 10 Minuten bei RT (22-25 °C). Waschen Sie die Zellen einmal mit PBS für 5 Minuten bei RT (22-25 °C).
- Montieren Sie das Deckglas mit 10 μl Eindeckmedium/Deckglas auf Objektträgern. Tauchen Sie die Deckgläser vor dem Montieren in destilliertes Wasser, um Salzkristalle zu entfernen. Fotografieren Sie die Objektträger am nächsten Tag und lagern Sie sie wochenlang bei 4 °C (Abbildung 3).
6. Bildaufnahme und Quantifizierung
- Verwenden Sie zum Aufnehmen von Bildern ein beliebiges verfügbares Epifluoreszenzmikroskop, das mit Routinefiltersätzen ausgestattet ist, um DAPI-, FITC- und Cy5-Kanäle mit mindestens 60- bis 63-facher Vergrößerung, hoher numerischer Apertur und Ölobjektiven abzubilden, um Kernherde zu visualisieren.
HINWEIS: Die optimale DAPI-Anregung beträgt ~359 nm; Die Anregung von Alexa 488 beträgt ~488 nm; während die Anregung von Alexa 647 ~647 nm beträgt. - Öffnen Sie für die Bildanalyse die Bilddateien in Fiji/ImageJ.
- Herstellung von Nuklearmasken mit der DAPI-Färbung (Abbildung 4A-F und ergänzendes Video S1).
- Öffnen Sie das DAPI-Image.
- Wählen Sie Prozess | Erhöhen Sie den Kontrast und stellen Sie gesättigte Pixel auf 0,35 ein.
- Klicken Sie auf Prozess | Binär | In Maske konvertieren. Wählen Sie Binär | Löcher füllen und klicken Sie auf Analysieren | Analysieren Sie Partikel. Legen Sie die Größe auf 10-Unendlich fest.
- Klicken Sie im ROI-Manager auf Alle anzeigen.
- Auffinden von RPA2-Herden im Zellkern (Abbildung 4G,H und ergänzendes Video S1)
- Öffnen Sie das RPA2-Image.
- Wählen Sie Prozess | Finde Maxima. Legen Sie die Prominenz auf einen Wert fest, der die RPA2-Brennpunkte (zwischen 500 und 750) hervorhebt und vom Hintergrund trennt.
- Klicken Sie abschließend auf die Schaltfläche Messen im ROI-Manager.
- Berechnen Sie die Gesamtzahl der nukleären ssDNA-Foci, indem Sie den Wert in der RawinDen-Spalte durch 255 dividieren (den maximalen Wert der Pixelintensität in jedem Foci).
- Führen Sie statistische Analysen mit dem bevorzugten Statistik-Software-Tool durch.
HINWEIS: Schließen Sie alle EdU-positiven Zellen und falsch segmentierten DAPI-Masken von der Analyse aus.
- Herstellung von Nuklearmasken mit der DAPI-Färbung (Abbildung 4A-F und ergänzendes Video S1).
Ergebnisse
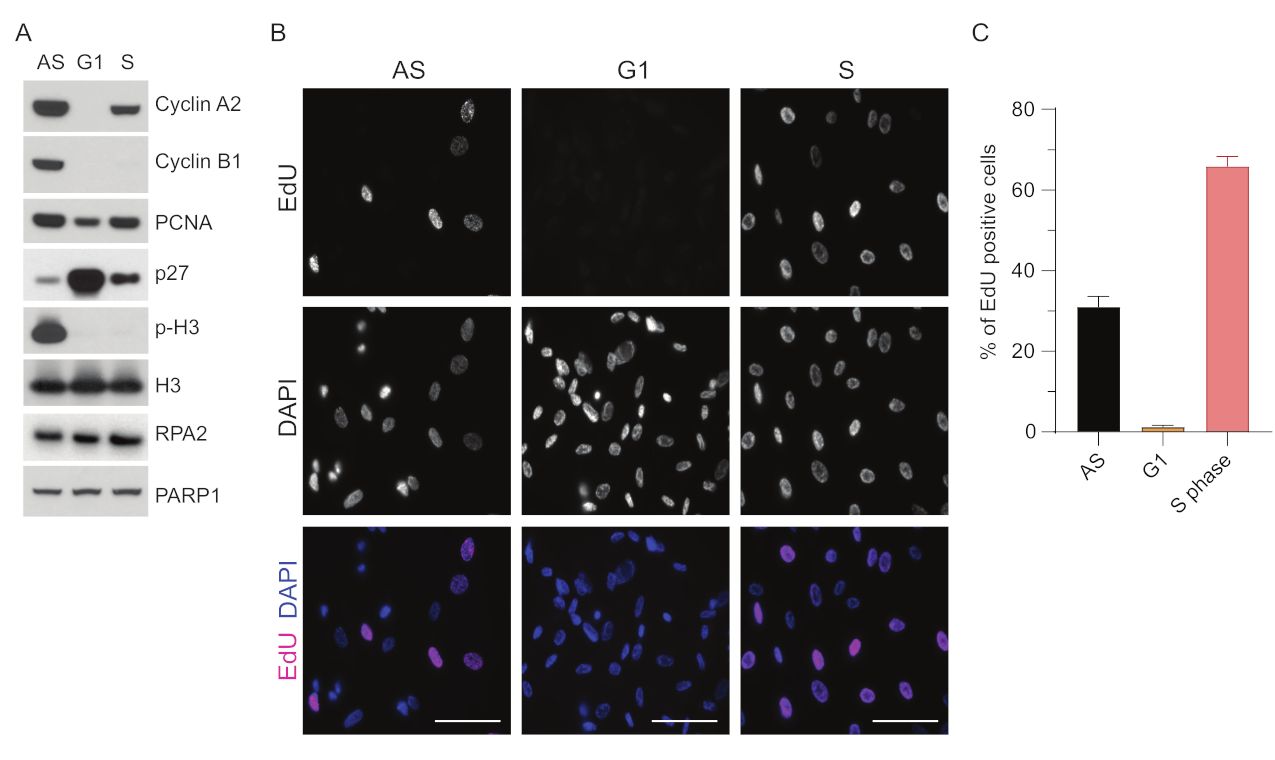
Um die Einschränkungen beim Nachweis von ssDNA in G1 zu überwinden, haben wir RPA2 verwendet, das sowohl die Spezifität als auch die Intensität des Nachweises von ssDNA-Foci verbessert35. Um eine präzise Zellsynchronisation zu erreichen, haben wir RPE1-Zellen verwendet, die effizient serumgehungert und in die G0-Phase synchronisiert werden können. Sie können dann durch die Zugabe von Serum nach Serumentzug dazu gebracht werden, wieder in den Zellzyklus einzutreten. Um die Synchronisationseffizienz zu bestätigen, markierten wir die Zellen mit EdU und ihren DNA-Gehalt mit Propidium-Iodid. Des Weiteren wurden qualitative und quantitative Ergebnisse mittels Durchflusszytometrie erhoben (Ergänzende Abbildung S1A). Die Punktdiagramme zeigen, dass sich nach 72 h Serumhunger ~98% der Zellen in der G0-Phase befinden. Nach der Zugabe von serumhaltigen Medien für 6 h treten die Zellen wieder in den Zellzyklus ein (wie durch den Anstieg der p27-Spiegel in Abbildung 1A zu sehen ist), wobei ~97 % der Zellen in G1 vorhanden sind, während sie nur <1 % Zellen in der S-Phase und <2 % Zellen in der G2-Phase aufweisen (ergänzende Abbildung S1A). Nach 20-28 Stunden Zugabe von Serum zu den Zellen durchlaufen sie allmählich die S-Phase, wie die Durchflusszytometrie-Diagramme zeigen (Ergänzende Abbildung S1A). Dieses Zellsynchronisationsprotokoll ergibt eine ~97% reine G1-Population (6 Stunden nach Serumzugabe nach 72 Stunden Serumhunger). Um die Synchronisationseffizienz weiter zu validieren, verglichen wir die Expression von Zellzyklusmarkern nach Serumfreisetzung mit Western Blot (Abbildung 1A und ergänzende Abbildung S1B) und führten parallel dazu einen EdU-Inkorporationsassay durch, um die DNA-Replikation zu visualisieren. Die EdU-Färbung unterstreicht auch die Synchronisationseffizienz und die fehlende DNA-Replikation in der G1-Phase (Abbildung 1B,C).
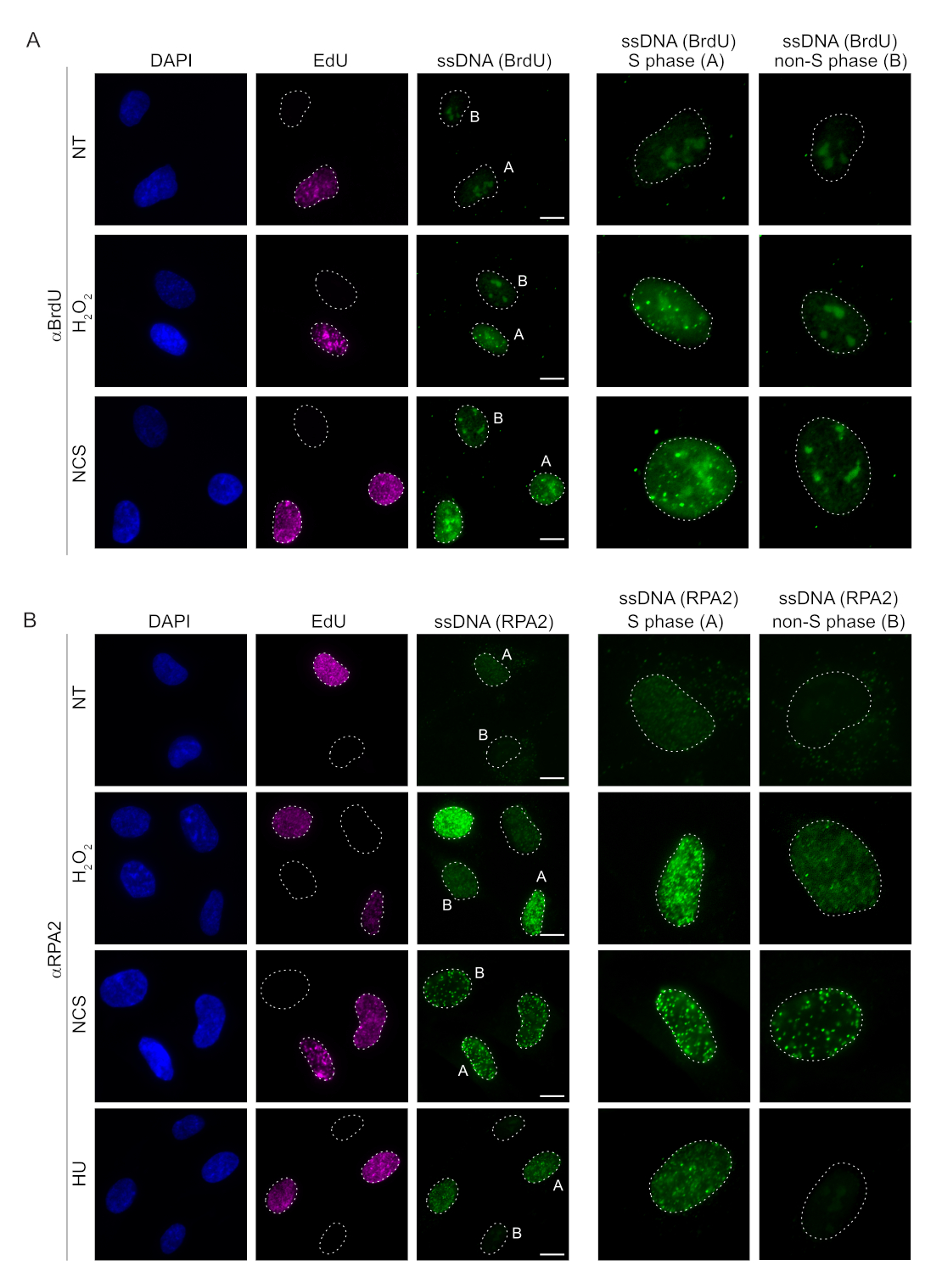
Herkömmliche Methoden zum Nachweis von ssDNA in Säugetierzellen beruhen auf dem Nachweis von BrdU in ssDNA. Abbildung 2A zeigt, dass nach der Behandlung mitH2O2und Neocarzinostatin (NCS) die BrdU-Foci nur in S-Phase-Zellen nachweisbar waren, während in Nicht-S-Phase-Zellen keine ssDNA-Foci nachweisbar waren. Die BrdU-Antikörper-Färbung zeigte auch eine auffällige nukleoläre Hintergrundfärbung, die in allen Zellkernen nachgewiesen werden konnte, unabhängig vom Zellzyklusstadium oder den angewendeten Behandlungen. Mit dem hier beschriebenen EdU-Click-Protokoll konnten wir keine kolokalisierenden EdU- und BrdU-Herde nachweisen, was in den unbehandelten Proben in Abbildung 2A deutlich wird. Um ein BrdU-Signal, das durch Kreuzreaktivität entstanden ist, vollständig auszuschließen, haben wir auf eine EdU-Markierung verzichtet und stattdessen Cyclin A2 als S-G2-Marker verwendet. Die Cyclin-A2-Färbung erlaubte jedoch keine CSK-Vorextraktion, und unter dieser Bedingung sahen wir auch nach genotoxischem Stress keine BrdU-Herde (Ergänzende Abbildung S2A). Dies unterstreicht die Tatsache, dass die CSK-Vorextraktion für die Anti-BrdU-basierte ssDNA-Färbung notwendig ist. Als Kontrolle testeten wir die BrdU-Antikörperfärbung unter denaturierenden Bedingungen. Dadurch wird die DNA geöffnet, um das eingebaute BrdU freizulegen, was zeigt, dass BrdU gleichmäßig eingebaut wurde (Ergänzende Abbildung S2B).
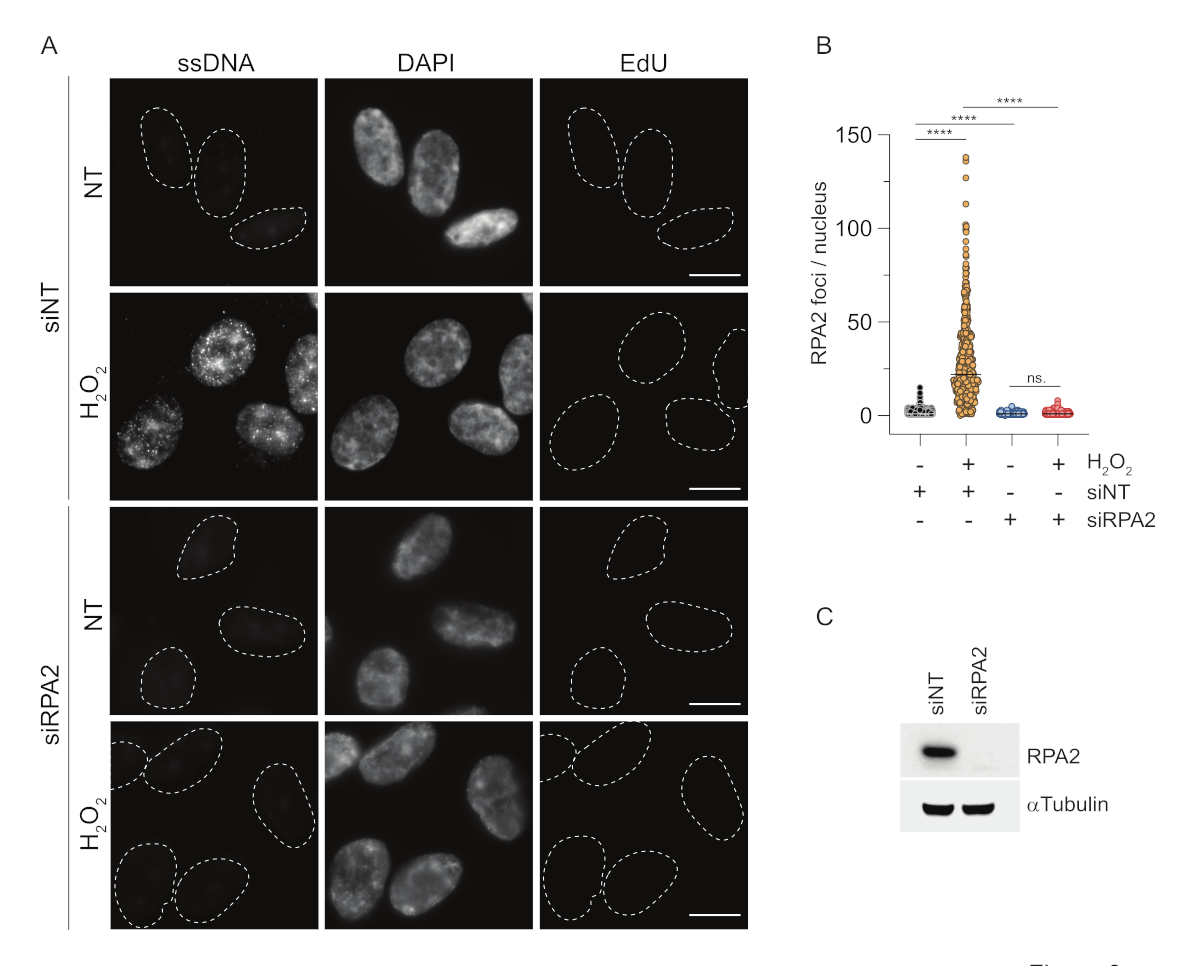
Im Gegensatz dazu zeigt die RPA2-Färbung NCS- undH2O2-abhängige Foci-Bildung nicht nur in der S-Phase, sondern auch in anderen Zellzyklusphasen (Abbildung 2B). Als Kontrolle behandelten wir die Zellen auch mit HU, was nur zu einer ssDNA-Akkumulation in Zellen führt, die sich replizieren. Wie erwartet, konnten wir nur in EdU-positiven Zellen einen Signalanstieg nach HU-Behandlung mit dem RPA2-Antikörper feststellen, was die Spezifität dieses Ansatzes unterstreicht. Der RPA2-Antikörper kann auch die natürlich vorkommende ssDNA-Bildung während der Replikation in Abwesenheit von exogenem genotoxischem Stress nachweisen (Abbildung 2B). Die hohe Sensitivität des RPA2-Antikörpers veranlasste uns, ihn in der G1-Phase einzusetzen, in der die konventionelle BrdU-Färbung kein Signal für genotoxischen Stress nachweisen konnte (Ergänzende Abbildung S2C). Abbildung 3A zeigt, dass die Bildung von ssDNA-Foci nachH2O2-Behandlungbei Verwendung eines Anti-RPA2-Antikörpers auch in G1 nachweisbar war. Es gab einen signifikanten Anstieg der Anzahl der RPA2-Herde in diesen Kernen nachH2O2-Behandlung (Abbildung 3B). Diese Foci waren spezifisch für RPA2, da die Stummschaltung von RPA2 das IF-Signal abschaffte (Abbildung 3A,B). Abbildung 3C und ergänzende Abbildung S1C zeigen die Effizienz der RPA2-Stummschaltung in diesen Zellen. Im Vergleich zu herkömmlichen Methoden ist der RPA2-basierte Nachweis von ssDNA hochsensitiv und kann daher auf G1-Phasenzellen ausgeweitet werden.

Abbildung 1: Synchronisationseffizienz von RPE1-Zellen nach Serumhunger . (A) Immunoblots zeigen angezeigte Proteinspiegel in asynchronen, G1- und S-Phasen-synchronisierten RPE1-Zellen. (B) Repräsentative Bilder zeigen asynchrone, G1- und S-Phasen-synchronisierte RPE1-Zellen, die vor der Fixierung 30 Minuten lang bei 10 μM EdU exponiert und durch Click-IT-Reaktion visualisiert wurden. DAPI wurde verwendet, um die Kern-DNA zu entfärben. Maßstabsbalken = 50 μm. (C) Die Grafik zeigt den prozentualen Anteil der EdU-positiven Zellen an der gesamten Zellpopulation, die mit DAPI bewertet wurde. Der Fehlerbalken stellt den Standardfehler des Mittelwerts dar, und die analysierte Anzahl der Kerne war die folgende: AS n = 219, G1 n = 630, S n = 437. Abkürzungen: RPE1 = hTERT-immortalisierte retinale Pigmentepithelzellen; AS = asynchron; EdU = 5-Ethinyl-2'-desoxyuridin; DAPI = 4',6-Diamidino-2-phenylindol. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: ssDNA-Nachweis mit BrdU-Antikörper oder RPA2-Antikörper bei DNA-Schädigung. (A) Repräsentative Bilder veranschaulichen ssDNA-Foci mit αBrdU (grün), S-Phasenzellen werden durch EdU hervorgehoben (violett) und DAPI wurde verwendet, um nukleäre DNA gegenfärbend zu färben (blau). RPE1-Zellen wurden vor einer weiteren Behandlung 48 Stunden lang in 10 μM BrdU aufbewahrt. Nach 48 h wurden die Zellen mit 10 μM EdU für 30 min gepulst, gefolgt von einer Behandlung mitH2O2(250 μM) für 1 h oder Neocarzinostatin (0,5 μg/ml) für 4 h. Die Zellen wurden nach der CSK-Vorextraktion fixiert. Eine weiß gestrichelte Linie markiert den Rand jedes Kerns. Maßstabsbalken = 5 μm. Die Tafeln auf der rechten Seite zeigen vergrößerte Bilder der angedeuteten S-Phasen- oder Nicht-S-Phasenkerne. (B) Repräsentative Bilder veranschaulichen ssDNA-Herde mit αRPA2-Antikörpern (grün). S-Phase-Zellen werden durch EdU (violett) hervorgehoben, und DAPI wurde verwendet, um die Kern-DNA (blau) zu entfärben. RPE1-Zellen wurden 30 Minuten lang mit 10 μM EdU gepulst, gefolgt von entweder 1 hH2O2(250 μM), 4 h Hydroxyharnstoff (2 mM) oder 4 h NCS (0,5 μg/ml). Die Zellen wurden nach der CSK-Vorextraktion fixiert. Eine weiß gestrichelte Linie markiert den Rand jedes Kerns. Maßstabsleiste = 10 μm. Die Tafeln auf der rechten Seite zeigen vergrößerte Bilder der angedeuteten S-Phasen- oder Nicht-S-Phasenkerne. Abkürzungen: ssDNA = einzelsträngige DNA; BrdU = 5-Brom-2'-desoxyuridin; DAPI = 4',6-Diamidino-2-phenylindol; RPE1 = hTERT-immortalisierte retinale Pigmentepithelzellen; EdU = 5-Ethinyl-2'-desoxyuridin; NCS = Neocarzinostatin; HU = Hydroxyharnstoff. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Nachweis von ssDNA-Herden in der G1-Phase mittels RPA2-Antikörper. (A) RPE1-Zellen wurden entweder mit siRNAs, die auf RPA2 abzielen, oder mit einer nicht-zielgerichteten siRNA-Kontrolle transfiziert und anschließend in G1 synchronisiert und 30 min lang mit 10 μM EdU gepulst, bevor sie mit H2O2 (250μM) für 1 h behandelt wurden, wo dies angezeigt war. DAPI wurde verwendet, um die Kern-DNA zu entfärben. Die Zellen wurden nach der CSK-Vorextraktion fixiert. Eine weiß gestrichelte Linie markiert den Rand jedes Kerns. Maßstabsbalken = 5 μm. (B) Die Messungen für die Anzahl der RPA2-Herde/-Kerne wurden in zwei unabhängigen Experimenten durchgeführt. Bei der Analyse wurden nur EdU-negative Zellen berücksichtigt. Linien stellen den Mittelwert in den Diagrammen dar. Für die statistische Analyse wurde ein nicht-parametrischer ANOVA-Test (Kruskal-Wallis) durchgeführt. Die Sterne zeigen P < 0,0001 an. Die Anzahl der untersuchten Kerne war wie folgt: siNT noH2O2n= 513, siNT H2 O2 n = 603, siRPA2 no H2O2 n = 266, siRPA2 H2O2 n = 536. (C) Die Effizienz des siRNA-Knockdowns zeigt sich im Immunoblotting. Abkürzungen: siNT = non-targeting siRNA control; BrdU = 5-Brom-2'-desoxyuridin; DAPI = 4',6-Diamidino-2-phenylindol; RPE1 = hTERT-immortalisierte retinale Pigmentepithelzellen; EdU = 5-Ethinyl-2'-desoxyuridin. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Quantifizierung von ssDNA-Herden anhand von Fidschi. Detaillierte Schritte in Fidschi, die zeigen, wie die Anzahl der RPA2-Foci im Zellkern zu beurteilen ist. (A-E) Die Erstellung einer nuklearen Maske unter Verwendung des DAPI-Kanals. (F-H) Schwellenwerte zur Identifizierung einzelner nukleärer ssDNA-Foci aus dem Hintergrundsignal. Abkürzungen: ssDNA = einzelsträngige DNA; DAPI = 4',6-Diamidino-2-phenylindol. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
| Zytoskelett-Puffer (CSK) | |
| ROHRE pH 7,0 | 10 mM |
| NaCl | 100 mM |
| EDTA pH 8 | 1 mM |
| MgCl2 | 3 mM |
| D-Saccharose | 300 mM |
| Triton X-100 | 0.20% |
| Phosphatase-Hemmer-Cocktail | 1 Tablette pro 10 ml |
| Protease-Inhibitor-Cocktail | 1 Tablette pro 10 ml |
| verdünnt inddH2O | n/a |
| Wasch-Puffer | |
| Triton X-100 | 0.05% |
| verdünnt in PBS | n/a |
| Permeabilisierungspuffer | |
| Triton X-100 | 0.50% |
| verdünnt in PBS | n/a |
| Fixierungslösung | |
| Paraformaldehyd | 3.60% |
| Triton X-100 | 0.05% |
| verdünnt in PBS | n/a |
| Blockieren des Puffers | |
| Rinderserumalbumin (BSA) | 5% |
| Triton X-100 | 0.10% |
| verdünnt in PBS | n/a |
| Click-iT Plus Reaktions-Cocktail | |
| 1x Click-iT Reaktionspuffer | 435 ml |
| Alexa Fluor PCA-Lösung | 5 ml |
| CuSO 4-Kupfer-Schutzmittel-Vormischung | 10 ml |
| 1x Click-iT Puffer-Additiv | 50 ml |
| Gesamtvolumen | 500 ml |
Tabelle 1: Zusammensetzung der in diesem Protokoll verwendeten Puffer.
Ergänzende Abbildung S1. (A) RPE1-Zellen wurden mit der G0-Phase synchronisiert, indem sie 72 Stunden lang ausgehungert und anschließend durch erneute Einführung von Serum in verschiedene Zellzyklusphasen freigesetzt wurden. Punktdiagramme zeigen Zellen in G0/G1-, S- oder G2/M-Phasen, wobei Stunden die Zeit nach der erneuten Zugabe von Serum nach Serumhunger angeben. Das Diagramm auf der rechten Seite zeigt den Prozentsatz der G0/G1-, S- und G2/M-Zellen in jeder Bedingung. Die FACS-Analyse wurde mit einem handelsüblichen Zellproliferationskit unter Verwendung von EdU und Propidiumiodid gemäß den Empfehlungen des Herstellers durchgeführt. (B) Unbeschnittene Western-Blot-Scans für Abbildung 1. Die Zahlen zeigen Molekulargewichtsmarker in kDa. PARP1 wurde als Beladungskontrolle verwendet und auf das Gel geladen, das auch gegen CCNA2, p27 (für PCNA weiter abgestreift) und pH3 (S10) (weiter abgestreift für H3) entwickelt wurde, indem die Membran zerschnitten wurde. CCNB1 und RPA2 wurden auf ein separates Gel geladen, wobei die gleiche Menge an Proteinlysat verwendet wurde, um die Vergleichbarkeit zu gewährleisten. (C) Unbeschnittene Western-Blot-Scans für Abbildung 3. Die Zahlen zeigen Molekulargewichtsmarker in kDa. Abkürzung: EdU = 5-Ethinyl-2'-desoxyuridin. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung S2: (A) Repräsentative Bilder veranschaulichen ssDNA-Herde unter Verwendung von BrdU-Antikörpern (grün); Zellen der S-Phase werden durch Cyclin A2 (rot) hervorgehoben; und DAPI wurde verwendet, um Kern-DNA (blau) zu konterkarieren. RPE1-Zellen wurden vor der weiteren Behandlung 48 Stunden lang in 10 μM BrdU aufbewahrt. Nach 48 h wurden die Zellen vor der Fixierung für 1 h mitH2O2(250 μM) oder Neocarzinostatin (0,5 μg/ml) für 4 h behandelt. Eine weiß gestrichelte Linie markiert den Rand jedes Kerns. Skalenbalken = 5 μm. (B) BrdU-Färbung von RPE1-Zellen mit und ohne denaturierende Bedingung. Asynchrone RPE1-Zellen wurden mit 10 μM BrdU für 48 h vorbehandelt. Maßstabsbalken = 10 μm. (C) Die Messungen für die Anzahl der BrdU-Herde/-Kerne wurden aus zwei unabhängigen Experimenten in G1-synchronisierten RPE1-Zellen durchgeführt. Bei der Analyse wurden nur EdU-negative Zellen berücksichtigt. Linien stellen den Mittelwert in den Diagrammen dar. Für die statistische Analyse wurde ein nicht-parametrischer ANOVA-Test (Kruskal-Wallis) durchgeführt. Das "ns" steht für einen nicht signifikanten Unterschied. Die Anzahl der untersuchten Kerne war wie folgt: NT n = 52, NCS n = 105, H2O2 n = 82. Abkürzungen: siNT = non-targeting siRNA control; BrdU = 5-Brom-2'-desoxyuridin; DAPI = 4',6-Diamidino-2-phenylindol; RPE1 = hTERT-immortalisierte retinale Pigmentepithelzellen; NCS = Neocarzinostatin. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzendes Video S1: Bildschirmaufzeichnung der RPA2-Fokusanalyse auf den Fidschi-Inseln. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Die Aufrechterhaltung einer gesunden, mykoplasmenfreien Zellkultur ist für alle oben beschriebenen Experimente von entscheidender Bedeutung. RPE1-Zellen haben unter normalen Kultivierungsmedien eine starke Bindung an mit Gewebekulturen behandelte Kunststoffe; Ihre Bindungseigenschaften nehmen jedoch deutlich ab, wenn sie unter serumfreien Bedingungen aufbewahrt werden. Um hochauflösende Bilder von ssDNA-Foci unter einem Mikroskop aufzunehmen, müssen die Zellen auf ein 0,17 mm dickes Deckglas aufgebracht werden, das nicht hydrophil genug ist, um eine ordnungsgemäße Anheftung von RPE1-Zellen zu unterstützen. Ohne richtig abgeflachte und gleichmäßig verteilte Zellen ist es sehr schwierig, einzelne ssDNA-Herde sichtbar zu machen. Daher ist es wichtig, das richtige Beschichtungsmaterial (z. B. Vitronektin) zu wählen und den Zellen ausreichend Zeit (6-12 h) zu lassen, um sich auszubreiten und anzuheften, nachdem sie in die G1-Phase entlassen wurden.
Ein schwieriger Teil des Protokolls besteht darin, homogene G1-synchronisierte RPE1-Zellen zu erhalten. Dies erfordert zwei kritische Schritte. Zunächst müssen die Zellen für eine effiziente Serumhungerung trypsinisiert, gründlich mit PBS gewaschen und mit serumfreien Medien direkt auf neue Gewebekulturschalen ausgesät werden. Das direkte Waschen der Zellen in Gewebekulturschalen, um das Serum zu entfernen, führt nicht zu einer effizienten G0-Synchronisation. Zweitens müssen die Zellen bei der Freisetzung von Zellen in die G1-Phase erneut trypsinisiert und auf frische Gewebekulturplatten ausgesät werden. In ähnlicher Weise führt der bloße Wechsel des Mediums und die Zugabe von serumhaltigem Kultivierungsmedium zu den Zellen nicht zu einem synchronen G1-Eintrag. Darüber hinaus muss für einen ordnungsgemäßen G1-Eintritt die Aussaatdichte der Zellen auf den beschichteten Deckgläsern bestimmte Konfluenzniveaus aufweisen. Während eine perfekte Zellsynchronisation im Allgemeinen nicht erreichbar ist, ergibt dieses hier beschriebene Synchronisationsprotokoll eine ~97% reine G1-Population. Die empfohlene Aussaatdichte für RPE1 auf einem Deckglas mit einem Durchmesser von 12 mm beträgt ~4 × 104 , um ein homogenes Sichtfeld für die Bildgebung mit ca. 70 % Konfluenz zu erhalten. Eine höhere Seeding-Dichte führt dazu, dass sich die Zellen nach der CSK-Extraktion ablösen und "ablösen", was zu einem höheren Hintergrundsignal während der Bildaufnahme führt.
Um ein Hintergrundsignal zu reduzieren und ein günstiges Signal-Rausch-Verhältnis zu erreichen, ist eine gründliche Reinigung nach der Inkubation von Primär- und Sekundärantikörpern unerlässlich. Da zahlreiche Waschschritte durchgeführt werden müssen, ist es auch wichtig, dass die Vertiefung bei jedem Waschschritt nicht austrocknet. Wir minimieren dieses Artefakt, indem wir in allen Wasch- und Inkubationsschritten mindestens 0,05 % Triton X-100 auftragen. Sobald die Vertiefungen ausgetrocknet waren, zeigten die Zellen ein verändertes Signal-Rausch-Verhältnis; Dies führt unter dem Mikroskop zu einem mosaikartigen Muster und könnte die Auswertung stören. Die Z-Stapel-Bilderfassung in Kombination mit der Dekonvolution kann dazu beitragen, Brennpunkte in verschiedenen Fokusebenen zu erfassen, um die Analyse zu verbessern.
Herkömmliche Methoden beruhen auf dem Nachweis von inkorporiertem BrdU unter nicht-denaturierenden Bedingungen. Diese Methoden sind jedoch auf die Vorbehandlung der Zellen mit hohen Dosierungen von BrdU für mindestens 1-2 Tage (oder Zeit, die einem vollständigen Zellzyklus in der verwendeten Zelllinie entspricht) angewiesen, um einen gleichmäßigen genomischen Einbau zu gewährleisten. Unerwünschterweise kann ein umfangreicher BrdU-Einbau zu Zellzyklusinterferenzenführen 36. Um diese Einschränkungen zu umgehen, nutzt diese Methode endogenes RPA2, um ssDNA-Foci zu detektieren. Dieser Ansatz erfordert keine replikationsgetriebene BrdU-Inkorporation, sondern kann auch in post-mitotischen Zellen eingesetzt werden. Da eine umfangreiche BrdU-Einarbeitung nicht erforderlich ist, spart dies Zeit und reduziert die experimentelle Komplexität. Durch die Verwendung von RPA2-Färbung zur Visualisierung von ssDNA können wir 2′-Desoxy-5-ethynyluridin (EdU) und Click-Chemie verwenden, um die DNA-Replikation zu markieren und gleichzeitig eine mögliche Kreuzreaktivität der BrdU-Antikörper gegen EdU 27,37,38 zu vermeiden. Es muss besonders darauf geachtet werden, dass das eingebaute EdU während der Klickreaktion richtig maskiert wird, damit die BrdU-Antikörper nicht mit EdU27,39 kreuzreagieren.
Schließlich ist ein wichtiger Vorteil der Verwendung von RPA2 anstelle von BrdU einfach ein besseres Signal-Rausch-Verhältnis im Vergleich zur BrdU-Färbung außerhalb der S-Phase. Wir fanden heraus, dass die nicht-denaturierende BrdU-Färbung und ihre Fähigkeit, ssDNA sichtbar zu machen, auch in replizierenden Zellen auf die S-Phase beschränkt ist (Abbildung 2). Der BrdU-Antikörper bindet nur an den ausreichend exponierten BrdU in ssDNA-Abschnitten. Die Bindung von Reparaturproteinen, einschließlich RPA2, an die ssDNA-Abschnitte kann die ausreichende Exposition von BrdU in ssDNA unterdrücken oder erschweren. Wir fanden auch heraus, dass die CSK-Vorextraktion für die ssDNA-Visualisierung mit BrdU-Antikörpern notwendig ist. Dies ist möglich, weil die ssDNA-Spuren für den Antikörper nicht zugänglich sind, ohne leicht gebundene Proteinbestandteile aus ihnen zu entfernen.
Nichtsdestotrotz gibt es einige Einschränkungen, die mit diesem Protokoll verbunden sind. Eine Einschränkung bei der Verwendung von RPA2 für den ssDNA-Nachweis ist die Notwendigkeit, den CSK-Vorextraktionsschritt zu optimieren. Ungebundenes, überschüssiges RPA2 muss vor der Fixierung der Zellen von der DNA abgewaschen werden. Zum einen führt die Unterextraktion zu einem hohen Hintergrund aufgrund der RPA2-Proteinfraktion, die nicht an ssDNA gebunden ist. Auf der anderen Seite führt eine Überextraktion zu Signalverlusten. Für den BrdU-Nachweis ist dies keine Variable, da BrdU stabil in die DNA eingebaut ist und nicht durch Vorextraktion weggespült werden kann. Daher müssen die Zeit der CSK-Vorextraktion, die Menge an Triton X-100 im Puffer, das Volumen und die Temperatur, bei der die Vorextraktion durchgeführt wird, sorgfältig berücksichtigt werden. Die CSK-Vorextraktion schränkt auch die Verwendung der Zellkerngröße ein, um G0/G1-Zellen von S/G2-Zellen zu unterscheiden.
Darüber hinaus können wir nicht ausschließen, dass ein Teil des Signals, das von RPA2 kommt, von der Bindung an andere Chromatin-bindende Proteininteraktoren herrührt. Man muss auch die Speziesspezifität des RPA2-Antikörpers berücksichtigen. Der in diesem Protokoll verwendete Antikörper kann RPA2 von Menschen, Mäusen, Ratten, Hamstern und Affen erkennen. Eine weitere Einschränkung dieses Ansatzes besteht darin, dass nicht alle Zelllinien für die G0-Synchronisation mit Serum ausgehungert werden können. Die meisten Krebszelllinien können Zellzyklus-Checkpoints umgehen und sich sogar in serumarmen Medien vermehren. Obwohl Serummangel vorteilhaft ist, da er keine DNA-Schäden verursacht, muss man ihre Zellsynchronisationseffizienz sorgfältig überwachen, um sicherzustellen, dass eine ordnungsgemäße Anreicherung der Zellzyklusphase erreicht wird. Für Zellen, die nicht auf Serumentzug ansprechen, müssen andere Zellsynchronisationsmethoden in Betracht gezogen werden (z. B. mitotisches Abschütteln, CDK1-Hemmung für den G2-Arrest oder nicht-invasive Techniken wie zentrifugale Abschlämmung). Ein weiteres mögliches Verfahren ist die Verwendung von High-Content-Imaging zur Messung des EdU- und Kern-DNA-Gehalts für die Erstellung von Zellzyklusprofilen von asynchronen Zellen31. Man muss die Auswirkungen der Verwendung alternativer Synchronisierungsmethoden berücksichtigen, um Interferenzen mit der nachgelagerten Analyse zu vermeiden. Zum Beispiel führt die Verwendung von doppeltem Thymidinblock oder Aphidicolin, das häufig in der Literatur verwendet wird, zu Replikationsstress und DNA-Schäden40.
Die Erforschung von DNA-Reparaturmechanismen ist nach wie vor ein Diskussionsschwerpunkt in den Bereichen Krebs und Zellbiologie. Das hier vorgestellte Protokoll bietet einen wertvollen Ansatz für die Präparation von Zellen, der die Visualisierung und quantitative Analyse von ssDNA unter Exposition gegenüber DNA-schädigenden Substanzen ermöglicht. Dieses Protokoll unterstreicht insbesondere die Verwendung des ssDNA-bindenden Proteins, RPA2, und demonstriert seine hohe Spezifität, um kleine Mengen von ssDNA-Foci sichtbar zu machen und gleichzeitig unerwünschte Kreuzreaktivität in allen Zellzyklusphasen zu vermeiden. Der Einsatz von RPA2 bietet zahlreiche Vorteile, insbesondere für Forscher, die Zellen in der G1-Phase des Zellzyklus analysieren wollen. Dieses Protokoll berücksichtigt mehrere Einschränkungen und berücksichtigt Bedenken im Zusammenhang mit Signalstörungen, unerwünschtem Hintergrundrauschen und Kreuzreaktivität bei der Verwendung von RPA2- oder BrdU-Färbung zum Nachweis von ssDNA.
Offenlegungen
Die Autoren haben keine konkurrierenden Interessen zu deklarieren.
Danksagungen
Die Autoren danken Michele Pagano für seine Unterstützung und seine hilfreichen Einblicke, Ashley Chui und Sharon Kaisari für die kritische Lektüre des Manuskripts sowie Jeffrey Estrada und Vilma Diaz für ihre kontinuierliche Unterstützung. Diese Arbeit wurde durch einen Diversity-Zusatz zum National Institutes of Health Grant GM136250 unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Alpha-tubulin antibody | Sigma-Aldrich | T6074 | primary antibody (1:5,000) |
| Axio Observer Inverted Microscope | Zeiss | na | microscope |
| Bis-Tris Plus Mini Protein Gels, 4-12% | Invitrogen | NW04127BOX | Western Blot |
| Bovine Serum Albumin | Jackson ImmunoResearch | 001-000-162 | blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Sigma-Aldrich | B5002-100MG | nucleotide analogue |
| BrdU antibody BU1/75 | Abcam | ab6326 | primary antibody (1:500) |
| CellAdhere Dilution Buffer | Stemcell Technologies | 07183 | coating reagent |
| Click-iT Plus EdU Flow Cytometry Assay Kits | Invitrogen | C10632 | flow cytomery |
| Click-iT Plus EdU Cell Proliferation Kit for Imaging, Alexa Fluor 647 dye | Thermo Fisher Scientific | C10640 | click-reaction kit |
| cOmplete ULTRA Protease inhibitor tablets | Sigma-Aldrich | 5892791001 | reagent |
| Countess 3 Automated cell counter | Thermo Scientific | AMQAX2000 | cell counter |
| Coverslip | neuVitro | GG12PRE | tissue culture |
| Cyclin A2 antibody | Santa Cruz Biotechnology | sc-271682 | primary antibody (1:1,000) for IF and WB |
| Cyclin B1 antibody | Santa Cruz Biotechnology | sc-245 | primary antibody (1:5,000) |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D2650-100ML | vehicle control |
| DMEM, high glucose, with HEPES | Gibco | 12430051 | cell culture medium for RPE cells |
| DPBS, no calcium, no magnesium | Gibco | 14190144 | the PBS used throughout the protocol |
| D-Sucrose | Thermo Fisher Scientific | bp220-1 | reagent |
| Eclipse Ti2 Series Epifluorescent Microscope | Nikon | na | microscope |
| EdU (5-Ethynyl-2'-deoxyuridine) | Thermo Fisher Scientific | C10637 | nucleotide analogue |
| Falcon 24-well plate | Corning | 351147 | tissue culture |
| Falcon Cell Culture Dishes 100 mm | Corning | 353003 | tissue culture |
| Fetal Bovine Serum, heat inactivated | Gibco | 16140071 | media supplement |
| Fiji (ImageJ) | NIH | version 1.54f | software and algorithms |
| FxCycle PI/RNase Staining Solution | Invitrogen | F10797 | PI staining |
| Goat anti-mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 555 | Thermo Fisher Scientific | A21422 | secondary antibody (1:1,000) |
| Goat anti-rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A48262 | secondary antibody (1:1,000) |
| Histone H3 antibody | Abcam | ab1791 | primary antibody (1:10,000) |
| hTERT RPE1 | ATCC | CRL-3216 | cell line |
| Hydrochloric acid | Sigma-Aldrich | H1758-100ML | reagent |
| Hydrogen peroxide 30% soultion | Sigma-Aldrich | H1009-100ML | reagent |
| Hydroxyurea,98% powder | Sigma-Aldrich | H8627-5G | reagent |
| Invitrogen Ultra Pure 0.5 M EDTA pH 8.0 | Thermo Fisher Scientific | 15-575-020 | reagent |
| Lipfectamine RNAiMAX Transfection Reagent | Invitrogen | 13778150 | transfection reagent |
| Magnesium chloride solution 1 M | Sigma-Aldrich | M1028-100ML | reagent |
| MycoFluor | Thermo Fisher | M7006 | Mycoplasma Detection Kit |
| Neocarzinostatin from Streptomyces carzinostaticus | Sigma-Aldrich | N9162-100UG | reagent |
| NuPage MES SDS Running Buffer (20x) | Invitrogen | NP0002 | Western Blot |
| onTARGETplus Human RPA2 siRNA | Dharmacon | L-017058-01-0005 | siRNA |
| p27 antibody | BD Biosciences | 610241 | primary antibody (1:1,000) |
| Paraformaldehyde aqueous solution (32%) | Electron Microscopy Sciences | 50-980-494 | fixative |
| PARP1 antibody | Cell Signaling Technology | 9542S | primary antibody (1:1,000) |
| PCNA antibody | Cell Signaling Technology | 13110S | primary antibody (1:2,000) |
| Penicillin-Streptomycin | Gibco | 15140163 | media supplement |
| pH3 antibody | Cell Signaling Technology | 3377S | primary antibody (1:2,000) |
| PhosSTOP phosphatase inhibitor tablets | Sigma-Aldrich | 4906837001 | reagent |
| PIPES Buffer 0.5 M solution, pH 7.0 | Bioworld | 41620034-1 | reagent |
| Precision Plus Protein Kaleidoscope Prestained Protein Standards | Bio-Rad | 1610395 | Western Blot |
| Prism | GraphPad | version 10 | statistical analysis and graph |
| ProLong Diamond Antifade Mountant | Thermo Scientific | P36961 | mounting media |
| Reduced serum media (Opti-MEM) | Gibco | 31985070 | used for transfection |
| Rpa32/rpa2 antibody (mouse) | EMD Millipore | NA19L | primary antibody (1:1,000) for WB |
| Rpa32/rpa2 antibody (rat) | Cell Signaling Technology | 2208S | primary antibody (1:1,000) for IF |
| Sodium Chloride solution (5 M) | Sigma-Aldrich | S5150 | reagent |
| Sodium Pyruvate (100 mM) | Gibco | 11360070 | media supplement |
| Sodium tetraborate decahydrate | Sigma-Aldrich | B3535-500G | reagent |
| Thermo Scientific Pierce DAPI Nuclear Counterstain | Thermo Scientific | 62248 | nucleic acid stain |
| Thymidine,powder | Sigma-Aldrich | T1985-1G | reagent |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | detergent |
| Trypsin-EDTA (0.5%), no phenol red | Gibco | 1540054 | cell dissociation agent |
| Vitronectin XF | Stemcell Technologies | 07180 | coating reagent |
| ZE5 Cell Analyzer | Bio-Rad | na | flow cytomery |
Referenzen
- Hakem, R. DNA-damage repair; the good, the bad, and ugly. EMBO J. 27 (4), 589-605 (2008).
- Gutierrez, R., O'Connor, T. R. DNA direct reversal repair and alkylating agent drug resistance. Cancer Drug Resist. 4 (2), 414-423 (2021).
- Krokan, H. E., Bjoras, M. Base excision repair. Cold Spring Harb Perspect Biol. 5 (4), a012583 (2013).
- Marteijn, J. A., Lans, H., Vermeulen, W., Hoeijmakers, J. H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 15 (7), 465-481 (2014).
- Li, G. M. Mechanisms and functions of DNA mismatch repair. Cell Res. 18 (1), 85-98 (2008).
- Hustedt, N., Durocher, D. The control of DNA repair by the cell cycle. Nat Cell Biol. 19 (1), 1-9 (2016).
- Yang, W., Gao, Y. Translesion and repair DNA polymerases: diverse structure and mechanism. Annu Rev Biochem. 87, 239-261 (2018).
- Bhat, D. S., et al. Therapeutic disruption of RAD52-ssDNA complexation via novel drug-like inhibitors. NAR Cancer. 5 (2), zcad018 (2023).
- Gupta, P., Saha, B., Chattopadhyay, S., Patro, B. S. Pharmacological targeting of differential DNA repair, radio-sensitizes WRN-deficient cancer cells in vitro and in vivo. Biochem Pharmacol. 186, 114450 (2021).
- Pena-Diaz, J., et al. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol Cell. 47 (5), 669-680 (2012).
- Schroering, A. G., Edelbrock, M. A., Richards, T. J., Williams, K. J. The cell cycle and DNA mismatch repair. Exp Cell Res. 313 (2), 292-304 (2007).
- Scully, R., Panday, A., Elango, R., Willis, N. A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 20 (11), 698-714 (2019).
- Escribano-Diaz, C., et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 49 (5), 872-883 (2013).
- Genschel, J., Modrich, P. Mechanism of 5'-directed excision in human mismatch repair. Mol Cell. 12 (5), 1077-1086 (2003).
- Hu, J., et al. Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J Biol Chem. 288 (29), 20918-20926 (2013).
- Huertas, P., Jackson, S. P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 284 (14), 9558-9565 (2009).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in DNA replication with putative roles in cancer. Int J Mol Sci. 20 (1), 74 (2018).
- Symington, L. S. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol. 6 (8), a016436 (2014).
- Liu, Y., et al. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem. 280 (5), 3665-3674 (2005).
- Wold, M. S., Kelly, T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc Natl Acad Sci U S A. 85 (8), 2523-2527 (1988).
- Wienholz, F., Vermeulen, W., Marteijn, J. A. Amplification of unscheduled DNA synthesis signal enables fluorescence-based single cell quantification of transcription-coupled nucleotide excision repair. Nucleic Acids Res. 45 (9), e68 (2017).
- Wold, M. S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 66, 61-92 (1997).
- Chen, R., Wold, M. S. Replication protein A: single-stranded DNA's first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays. 36 (12), 1156-1161 (2014).
- Kang, Y., et al. Alteration of replication protein A binding mode on single-stranded DNA by NSMF potentiates RPA phosphorylation by ATR kinase. Nucleic Acids Res. 51 (15), 7936-7950 (2023).
- Kilgas, S., Kiltie, A. E., Ramadan, K. Immunofluorescence microscopy-based detection of ssDNA foci by BrdU in mammalian cells. STAR Protoc. 2 (4), 100978 (2021).
- Madabhushi, R., Pan, L., Tsai, L. H. DNA damage and its links to neurodegeneration. Neuron. 83 (2), 266-282 (2014).
- Liboska, R., Ligasova, A., Strunin, D., Rosenberg, I., Koberna, K. Most anti-BrdU antibodies react with 2'-deoxy-5-ethynyluridine -- the method for the effective suppression of this cross-reactivity. PLoS One. 7 (12), e51679 (2012).
- Biehs, R., et al. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol Cell. 65 (4), 671-684.e5 (2017).
- Cruz-Garcia, A., Lopez-Saavedra, A., Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 9 (2), 451-459 (2014).
- Ercilla, A., et al. Physiological tolerance to ssDNA enables strand uncoupling during DNA replication. Cell Rep. 30 (7), 2416-2429.e7 (2020).
- Lezaja, A., et al. RPA shields inherited DNA lesions for post-mitotic DNA synthesis. Nat Commun. 12 (1), 3827 (2021).
- Mukherjee, B., Tomimatsu, N., Burma, S. Immunofluorescence-based methods to monitor DNA end resection. Methods Mol Biol. 1292, 67-75 (2015).
- Ochs, F., et al. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol. 23 (8), 714-721 (2016).
- Raderschall, E., Golub, E. I., Haaf, T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad Sci U S A. 96 (5), 1921-1926 (1999).
- Forment, J. V., Walker, R. V., Jackson, S. P. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytometry A. 81 (10), 922-928 (2012).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Sci Rep. 6, 19567 (2016).
- Aten, J. A., Bakker, P. J., Stap, J., Boschman, G. A., Veenhof, C. H. DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem J. 24 (5), 251-259 (1992).
- Podgorny, O., Peunova, N., Park, J. H., Enikolopov, G. Triple S-phase labeling of dividing stem cells. Stem Cell Reports. 10 (2), 615-626 (2018).
- Cappella, P., Gasparri, F., Pulici, M., Moll, J. Cell proliferation method: click chemistry based on BrdU coupling for multiplex antibody staining. Curr Protoc Cytom. Chapter 7, (2008).
- Ligasova, A., Koberna, K. Strengths and weaknesses of cell synchronization protocols based on inhibition of DNA synthesis. Int J Mol Sci. 22 (19), 10759 (2021).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten