
Method Article
Visualizando focos de DNA de fita simples na fase G1 do ciclo celular
Neste Artigo
Resumo
O protocolo a seguir apresenta a detecção de focos de DNA de fita simples na fase G1 do ciclo celular utilizando sincronização do ciclo celular seguida de coloração por imunofluorescência RPA2.
Resumo
O DNA possui vias de reparo celular dedicadas, capazes de lidar com lesões que podem advir de fontes endógenas e/ou exógenas. O reparo do DNA requer a colaboração entre várias proteínas, responsáveis por cobrir uma ampla gama de tarefas, desde reconhecer e sinalizar a presença de uma lesão de DNA até repará-la fisicamente. Durante esse processo, traços de DNA de fita simples (ssDNA) são frequentemente criados, que são eventualmente preenchidos por DNA polimerases. A natureza dessas trilhas de ssDNA (em termos de comprimento e número), juntamente com a polimerase recrutada para preencher essas lacunas, são específicas da via de reparo. A visualização desses rastros de ssDNA pode nos ajudar a entender a complicada dinâmica dos mecanismos de reparo do DNA.
Este protocolo fornece um método detalhado para a preparação de células sincronizadas G1 para medir a formação de focos de ssDNA após estresse genotóxico. Usando uma abordagem de imunofluorescência fácil de utilizar, visualizamos o ssDNA pela coloração para RPA2, um componente do complexo de proteína A de replicação heterotrimérica (RPA). RPA2 se liga e estabiliza intermediários de ssDNA que surgem após estresse genotóxico ou replicação para controlar o reparo de DNA e ativação de pontos de verificação de danos ao DNA. A coloração 5-etinil-2'-desoxiuridina (EdU) é usada para visualizar a replicação do DNA para excluir quaisquer células da fase S. Este protocolo fornece uma abordagem alternativa para os ensaios convencionais baseados em 5-bromo-2'-desoxiuridina (BrdU) não desnaturantes e é mais adequado para a detecção de focos de ssDNA fora da fase S.
Introdução
Para sustentar a vida, as células constantemente pesquisam e reparam o DNA para manter sua integridade genômica. As células podem acumular vários tipos de danos ao DNA devido a fontes endógenas (por exemplo, oxidação, alquilação, desaminação, erros de replicação) e exógenas (por exemplo, UV, irradiação ionizante) de estressores de DNA. A falha no reparo dessas lesões resulta em apoptose, parada do ciclo celular ou senescência, podendo levar adoenças1. As lesões de DNA poderiam ser abordadas por qualquer uma das seguintes vias principais de reparo do DNA: DR (direct reversal repair), que repara principalmente bases alquiladas2; BER (reparo por excisão de bases), que tem como alvo erros de bases de DNA não volumosas e quebras de DNA de fita simples (SSBs)3; NER (nucleotide excision repair) corrigindo lesões volumosas de DNA que distorcem a hélice4; MMR (reparo de incompatibilidade) visando principalmente incompatibilidades de DNA, laços de inserção/exclusão (IDLs) e certos danos de base5; NHEJ (non-homologous end joining) e HRR (homologous recombination repair) que são ambos ativos em quebras de DNA de fita dupla (DSBs)6; e TLS (síntese translesional), que é um mecanismo de bypass de lesão de DNA7. Embora essas vias tenham especificidades distintas de substrato, há certas sobreposições entre elas para garantir redundância para um reparo eficiente. A compreensão da ação das diferentes vias de reparo do DNA nas diversas fases do ciclo celular é crucial, pois esses fatores de reparo do DNA podem servir como alvos essenciais para abordagens terapêuticas no tratamento de câncer, envelhecimento e distúrbiosneurológicos8,9.
O DNA de fita simples (ssDNA) é gerado ao longo do ciclo celular devido ao reparo de lesões de DNA geradas por agentes endógenos e exógenos prejudiciais ao DNA. Diante do estresse genotóxico, o ssDNA é gerado abundantemente nas fases S e G2, onde a HRR e a RMM têm sua maior atividade e quando a maquinaria de replicação trava ou entra em colapso ao encontrar lesões no DNA6,10,11. Outras vias de reparo de DNA (por exemplo, NHEJ/junção final mediada por microhomologia (MMEJ)/recozimento de fita simples [SSA]) também geram ssDNA durante o reparo do DSB12. Essas trilhas de ssDNA geralmente surgem da ressecção do DNA, realizada por exonucleases como EXO1, DNA2 e CtIP durante FC e RMM, endonucleases como XPF e XPG durante NER, ou pela ação combinada de POLB e FEN1 durante BER 4,13,14,15,16,17,18,19 . Devido ao trabalho da maquinaria de replicação, trilhas de ssDNA também são geradas quando as helicases de DNA desenrolam o DNA na frente das polimerases replicativas ligadas ao PCNA20. Em contraste, na fase G1, a falta de FCR e replicação do DNA e a atividade limitada da RMM reduzem a extensão das trilhas de ssDNA geradas e, portanto, são mais difíceis de detectar 10,11,21.
Os rastros celulares de ssDNA são estruturas altamente sensíveis que devem ser protegidas para evitar a formação de DSBs. Isto é conseguido revestindo as faixas de ssDNA com RPA. RPA é um abundante complexo proteico heterotrimérico composto por múltiplas subunidades (RPA1, RPA2 e RPA3, também referidas como RPA70, RPA32 e RPA14, respectivamente), que são ubíquas expressas ao longo do ciclo celular22. Cada subunidade RPA contém um domínio de ligação ao DNA (DBD), capaz de interagir com 4-6 nucleotídeos, e as subunidades combinadas formam um núcleo estável de trimerização. Em conjunto, o RPA liga-se a aproximadamente 20-30 nucleotídeos com afinidade subnanomolar23,24.
Os métodos convencionais utilizam microscopia de imunofluorescência (IF) para visualizar focos de ssDNA marcando 5-bromo-2'-desoxiuridina (BrdU) incorporada ao DNA genômico usando anticorpos BrdU25. Essa abordagem baseia-se no fato de que os anticorpos BrdU só podem detectar BrdU no ssDNA25 exposto. Embora essa abordagem seja direta, ela também apresenta certas limitações. Por exemplo, as células são pré-tratadas para incorporar BrdU antes do início do experimento, o que é demorado e pode interferir com os efetores a jusante. Portanto, a detecção de ssDNA baseada em BrdU é limitada a células replicantes e não pode ser usada para células quiescentes. Isso exclui a aplicação desse método para estudar o reparo do DNA em células não replicantes, apesar de sua importância em várias doenças, como câncer e neurodegeneração5,26. Além disso, como as estruturas de BrdU e EdU são muito semelhantes, a maioria dos anticorpos BrdU exibe reatividade cruzada em relação à EdU, o que deve ser considerado quando se pretende experimentar a dupla marcação27. A coloração de RPA foi previamente utilizada para mostrar focos de ssDNA principalmente em células da fase S; entretanto, alguns trabalhos também a utilizaram com sucesso fora da fase S 28,29,30,31,32,33,34,35. O protocolo a seguir utiliza eficientemente as propriedades do RPA, permitindo a visualização de focos de ssDNA após danos ao DNA na fase G1 do ciclo celular (embora possa ser usado em todas as fases do ciclo celular).
Protocolo
1. Manutenção das células epiteliais pigmentares da retina imortalizadas por hTERT-(EPR1)
- Manter linhagens celulares de RPE1 em meio Dulbecco's Modified Eagle Medium (DMEM) suplementado com 10% de soro fetal bovino inativado pelo calor (Hi-FBS) e 100 μg/mL de Penicilina-Estreptomicina (doravante denominada meio de cultura) em estufa umidificada com 5% de CO2 a 37 °C. Para o cultivo de rotina, cultivar células RPE1 em uma placa tratada com cultura de tecido de 15 cm e dividir quando atingir 80-90% de confluência (~16-18 × 106 células por placa de 15 cm).
- Ao dividir, remova o meio e lave as células com 10 mL de solução salina tamponada com fosfato (PBS) 1x.
- Adicionar 3 mL de tripsina-EDTA a 0,05% para cobrir toda a superfície do prato. Manter as células a 37 °C com a tripsina até que se desprendam.
- Após tripsinização, ressuspender as células com meio de cultura e girá-las para baixo a 150 × g por 5 min à temperatura ambiente (TR, 22-25 °C). Retire o sobrenadante e ressuspenda suavemente as células em 10 mL de meio de cultura.
- Semear 1,6-1,8 × 106 células em um novo prato de 15 cm (~1 mL da suspensão celular).
NOTA: Todo o trabalho de cultura de tecidos deve ser feito sob os níveis de segurança BSL-2. O tempo de incubação para tripsinização depende da confluência celular. Normalmente, o processo leva 2-3 min para ser concluído para uma placa confluente de 90%. As células devem ser rastreadas para contaminação por Mycoplasma regularmente com kits disponíveis comercialmente (ver exemplos na Tabela de Materiais).
2. derrubada de siRNA do gene de interesse (GOI)
- Sementes 1,0 × 106 células RPE1 em placa tratada com cultura de tecidos de 10 cm com 10 mL de meio de cultura no dia anterior à transfecção.
- No dia da transfecção, complexo o siRNA. Para uma placa de 10 cm, utilizar uma concentração final de 20 nM de siRPA2 e 12 μL de reagente de transfecção à base de lipídios em 500 μL de meio de transfecção de soro baixo. Misture suavemente todos os componentes agitando o tubo e incube em RT (22-25 °C) por 5 min.
- Adicione a mistura complexada de siRNA às células gota a gota e incube as células com o siRNA por 48 h.
3. Sincronização das células RPE1 na fase G0
- Tripsinize as células RPE1 do passo 2.3 conforme descrito na secção 1 (~2 × 106 células).
- Transfira a suspensão celular para tubos de centrífuga de 15 mL e centrifuga-a a 150 × g, RT (22-25 °C) por 5 min.
- Remover o sobrenadante e ressuspender as células em 12 mL de PBS. Centrifugar as células a 150 × g em TR (22-25 °C) durante 5 min. Repetir a remoção do sobrenadante e a centrifugação duas vezes.
- Ressuspender as células em 10 mL de DMEM sem soro suplementado com 100 μg/mL de Penicilina-Estreptomicina, 1 mM de Piruvato de Sódio, 15 mM de HEPES e colocá-las em uma placa de cultura de tecidos de 10 cm.
NOTA: Se as células tenderem a se aglomerar, ressuspendê-las em apenas 1 mL de DMEM livre de soro e pipetá-las para cima e para baixo 5x usando uma ponta P1000 para desalojar os aglomerados antes de diluir a suspensão até um volume final de 10 mL. - Após 24 h de inanição de soro, introduzir a segunda rodada de silenciamento usando o mesmo procedimento descrito na seção 2, adicionando o siRNA complexado às células famintas de soro.
- Manter as células RPE1 em DMEM sem soro por 72 h antes de proceder à liberação de G1.
4. Revestimento de deslizamento de cobertura e liberação de células na fase G1
- Esterilizar pinças com etanol 70% e colocar uma única lamínula de vidro (12 mm de diâmetro e espessura #1,5 [0,17 mm]) em um poço de uma placa de 24 poços.
- Diluir a matriz de revestimento de vitronectina com PBS para obter uma concentração final de 10 μg/mL. Adicionar 500 μL da solução de vitronectina em cada poço contendo as lamínulas e incubar durante 1 h em RT.
- Retire a solução de revestimento e lave as lamínulas com 1 mL de PBS.
- Separar as células RPE1 sem soro da placa tratada cultivada com tecido de 10 cm usando 1 mL de tripsina a 0,05% após uma lavagem com PBS por 1 min a 37 °C.
NOTA: As células se desprendem muito mais rápido após a inanição do soro. Tenha cuidado ao lavar as células com PBS e use tempos de tripsinização curtos. - Para inativar a tripsina, ressuspender as células RPE1 em um total de 6 mL de meio de cultura. Remova a tripsina inativada girando as células usando 150 × g em TR (22-25 °C) por 5 min.
- Ressuspender as células em 1 mL de meio de cultura e medir o número de células.
- Sementes 4 × 104 células RPE1 sobre a lamínula revestida num total de 500 μL de meio de cultura.
NOTA: Certifique-se de que a viabilidade da célula está acima de 90% antes de prosseguir para as etapas a jusante. A viabilidade celular pode ser rapidamente avaliada pela coloração com azul de tripano durante a etapa de contagem celular. - Após 6 h de plaqueamento das células no meio de cultura, as células liberadas de G0 estarão na fase inicial de G1. Realizar experimentos no G1 nesta janela de 6-12 h antes que as células comecem a entrar na fase S.
- Antes de introduzir danos no DNA, pulsar as células com 10 μM de 5-etinil-2'-desoxiuridina (EdU) por 30 min a 37 °C, diluídas em meio de cultura.
- Remover o meio que contém EdU e perseguir as células com 10 μM de timidina durante 10 min a 37 °C para evitar a incorporação de EdU remanescente durante a indução de danos no DNA.
- Retirar o meio com timidina e tratar as células com 250 μM H2O2 por 1 h, diluído em meio de cultura.
5. Coloração por imunofluorescência do ssDNA
- Lavar as células uma vez com 1 mL de RT (22-25 °C) PBS para remover os componentes do meio e do soro.
NOTA: Seja gentil ao lavar as células para evitar descolamento e secagem. Não processe muitos poços ao mesmo tempo. - Pré-extração: Incubar as células lavadas em 1 mL de tampão de extração CSK (Tabela 1) por 5 min em TR (22-25 °C).
NOTA: A pré-extração de CSK remove todas as proteínas não ligadas à cromatina, incluindo RPA2 solúvel.
CUIDADO: Triton X-100 é prejudicial se ingerido e pode causar irritação da pele e danos nos olhos. - Remova o tampão CSK das células e fixe-as diretamente adicionando 0,5 mL de solução de paraformaldeído a 3,6% (em PBS) contendo Triton X-100 a 0,05% por 10 min em TR (22-25 °C).
CUIDADO: É importante preparar 3,6% de PFA a partir de 32% de PFA fresco. O paraformaldeído pode causar danos oculares graves, irritação da pele e irritação respiratória. - Lavar as células uma vez com 1 mL de PBS contendo Triton X-100 a 0,05% para remover o PFA.
- Permeabilizar ainda mais as células usando 1 mL de PBS contendo Triton X-100 a 0,5% por 15 min em TR (22-25 °C).
- Reação de EdU click-IT para visualizar células replicantes (fase S)
- Retirar a solução de permeabilização e lavar as células 2x com 1 mL de tampão bloqueador (Tabela 1).
CUIDADO: A albumina de soro bovino (BSA) pode causar irritação respiratória. - Adicionar 1 mL de tampão de bloqueio (Tabela 1) e balançar suavemente a placa contendo lamínula por 10 min no TR (22-25 °C).
- Remover o tampão de bloqueio e adicionar 500 μL de cocktail de reacção de clique contendo picolyl azide 647 (Tabela 1). Incubar as lamínulas por 30 min em RT (22-25 °C) usando balanço suave e realizar incubações a jusante no escuro.
NOTA: Ao usar anticorpos BrdU, use o dobro da quantidade (1 mL) e do tempo (60 min) para a reação de clique, conforme recomendado pelo fabricante, para garantir que a reação esteja saturada e a EdU incorporada seja rotulada. Isso limita a reatividade cruzada dos anticorpos BrdU27.
- Retirar a solução de permeabilização e lavar as células 2x com 1 mL de tampão bloqueador (Tabela 1).
- Remova a mistura de reação de clique e lave as células 2x com PBS com Triton X-100 a 0,05% por 10 min em TR (22-25 °C) (Figura 1 e Figura 2).
- Adicionar 1 mL de tampão de bloqueio e incubar em TR (22-25 °C) por 30 min. Alternativamente, mantenha as células em tampão de bloqueio a 4 °C durante a noite.
- Aplicar anticorpo primário (rato anti-RPA2, diluição de 1:1.000) durante 2 h em TR (22-25 °C) em 250-500 μL de tampão de bloqueio com balanço suave.
- Lave as células 2x com PBS contendo 0,05% Triton X-100 para remover rapidamente a maior parte da solução de anticorpos.
- Continue lavando as células por 3 x 10 min com tampão de bloqueio em TR (22-25 °C).
- Aplicar anticorpo secundário (anti-rato Alexa-488, diluição 1:1.000) em 250-500 μL de tampão de bloqueio em RT (22-25 °C) por 2 h com balanço suave.
- Lave as células com tampão de bloqueio 2x para remover rapidamente a maior parte do anticorpo secundário. Continuar lavando as células por 3 x 10 min com PBS contendo 0,05% de Triton X-100 em TR (22-25 °C).
- Para contracoloração dos núcleos, lavar as células uma vez com PBS contendo Triton X-100 a 0,05% e 4',6-diamidino-2-fenilindol (DAPI) 1 μg/mL por 10 min a 22-25 °C. Lavar as células uma vez com PBS durante 5 minutos em RT (22-25 °C).
- Monte o vidro da tampa nas lâminas do microscópio usando 10 μL de meio/lamínula de montagem. Mergulhe as tampas em água destilada antes de montar para se livrar de quaisquer cristais de sal. Fotografe as lâminas no dia seguinte e armazene-as a 4 °C por semanas (Figura 3).
6. Aquisição e quantificação das imagens
- Para capturar imagens, use qualquer microscópio epifluorescente disponível equipado com conjuntos de filtros de rotina para obter imagens de canais DAPI, FITC e Cy5 com pelo menos 60-63x de aumento, alta abertura numérica e objetivas de óleo para visualizar focos nucleares.
NOTA: A excitação DAPI ideal é de ~359 nm; Alexa 488 excitação é ~ 488 nm; enquanto a excitação do Alexa 647 é de ~647 nm. - Para análise de imagem, abra arquivos de imagem em Fiji/ImageJ.
- Confecção de máscaras nucleares utilizando a coloração DAPI (Figura 4A-F e Vídeo Suplementar S1).
- Abra a imagem DAPI.
- Selecionar Processo | Melhore o contraste e defina o pixel saturado como 0,35.
- Clique em Processo | Binário | Converter em Máscara. Escolha Binário | Preencha Buracos e clique em Analisar | Analise partículas. Defina o tamanho como 10-Infinito.
- No gerenciador de ROI, clique em Mostrar tudo.
- Localizando focos de RPA2 no núcleo (Figura 4G,H e Vídeo Suplementar S1)
- Abra a imagem RPA2.
- Escolha o Processo | Encontre o Maxima. Defina a proeminência para um valor que destaque os focos RPA2 (entre 500 e 750), separando-o do plano de fundo.
- Por fim, clique no botão Medir no ROI Manager.
- Calcule o número total de focos nucleares de ssDNA dividindo o valor na coluna RawinDen por 255 (o valor máximo de intensidade de pixel em cada foco).
- Realizar análise estatística usando a ferramenta de software estatístico preferida.
NOTA: Exclua todas as células EdU-positivas e máscaras DAPI segmentadas incorretamente da análise.
- Confecção de máscaras nucleares utilizando a coloração DAPI (Figura 4A-F e Vídeo Suplementar S1).
Resultados
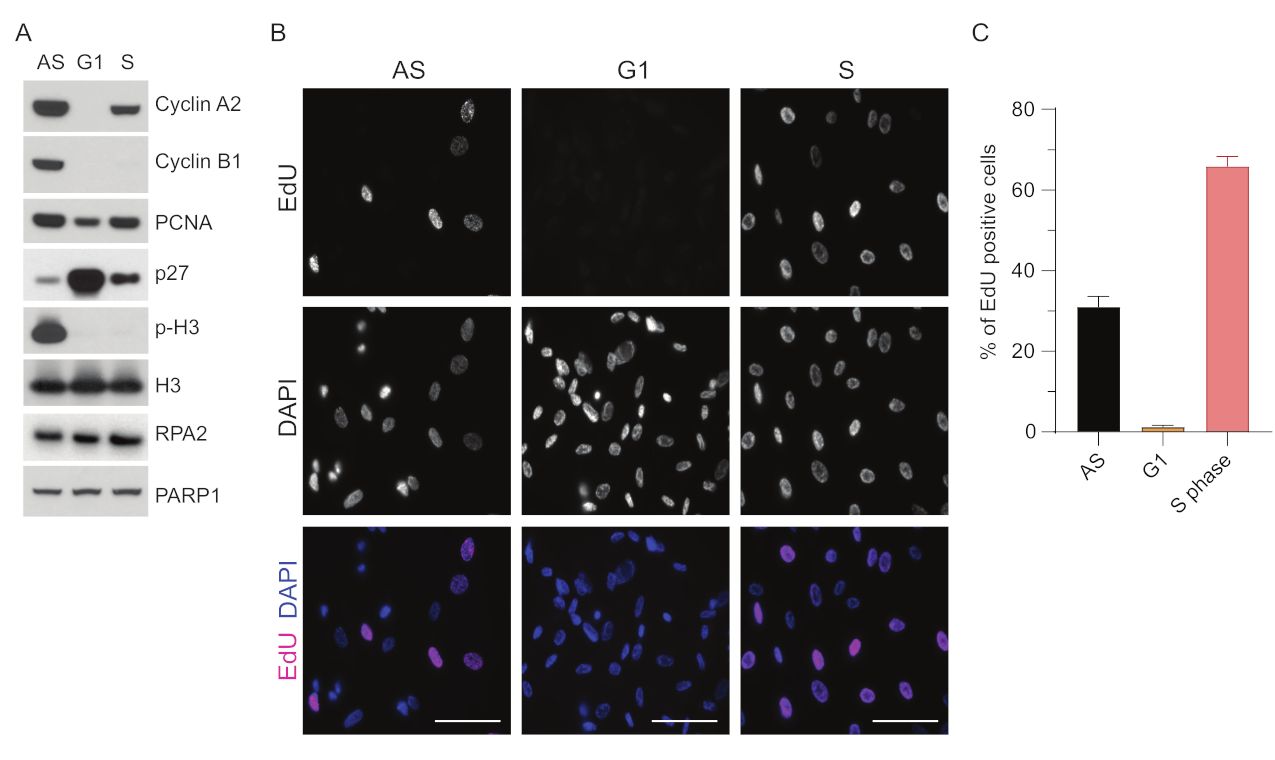
Para superar as limitações da detecção de ssDNA no G1, utilizamos RPA2, que aumenta tanto a especificidade quanto a intensidade da detecção de focos de ssDNA35. Para obter uma sincronização celular precisa, usamos células RPE1 que podem ser eficientemente carentes de soro e sincronizadas na fase G0. Eles podem então ser induzidos a reentrar no ciclo celular pela adição de soro após a privação do soro. Para confirmar a eficiência da sincronização, marcamos as células com EdU e seu conteúdo de DNA com iodeto de propídio. Posteriormente, foram coletados resultados qualitativos e quantitativos por citometria de fluxo (Figura Suplementar S1A). Os dot-plots mostram que após 72 h de inanição sérica, ~98% das células estão na fase G0. Após a adição do meio contendo soro por 6 h, as células reentram no ciclo celular (como visto pelo aumento dos níveis de p27 na Figura 1A), tendo ~97% de células no G1, enquanto tendo apenas <1% de células na fase S <2% células na fase G2 (Figura Suplementar S1A). Após 20-28 h de adição de soro às células, elas passam gradualmente pela fase S, como mostram os gráficos de citometria de fluxo (Figura Suplementar S1A). Este protocolo de sincronização celular fornece uma população de G1 pura de ~97% (6 h após a adição de soro após 72 h de inanição de soro). Para validar ainda mais a eficiência da sincronização, comparamos a expressão de marcadores do ciclo celular após a liberação do soro usando western blotting (Figura 1A e Figura Suplementar S1B) e, em paralelo, realizamos um ensaio de incorporação de EdU para visualizar a replicação do DNA. A coloração de EdU também destaca a eficiência da sincronização e a ausência de replicação do DNA na fase G1 (Figura 1B,C).
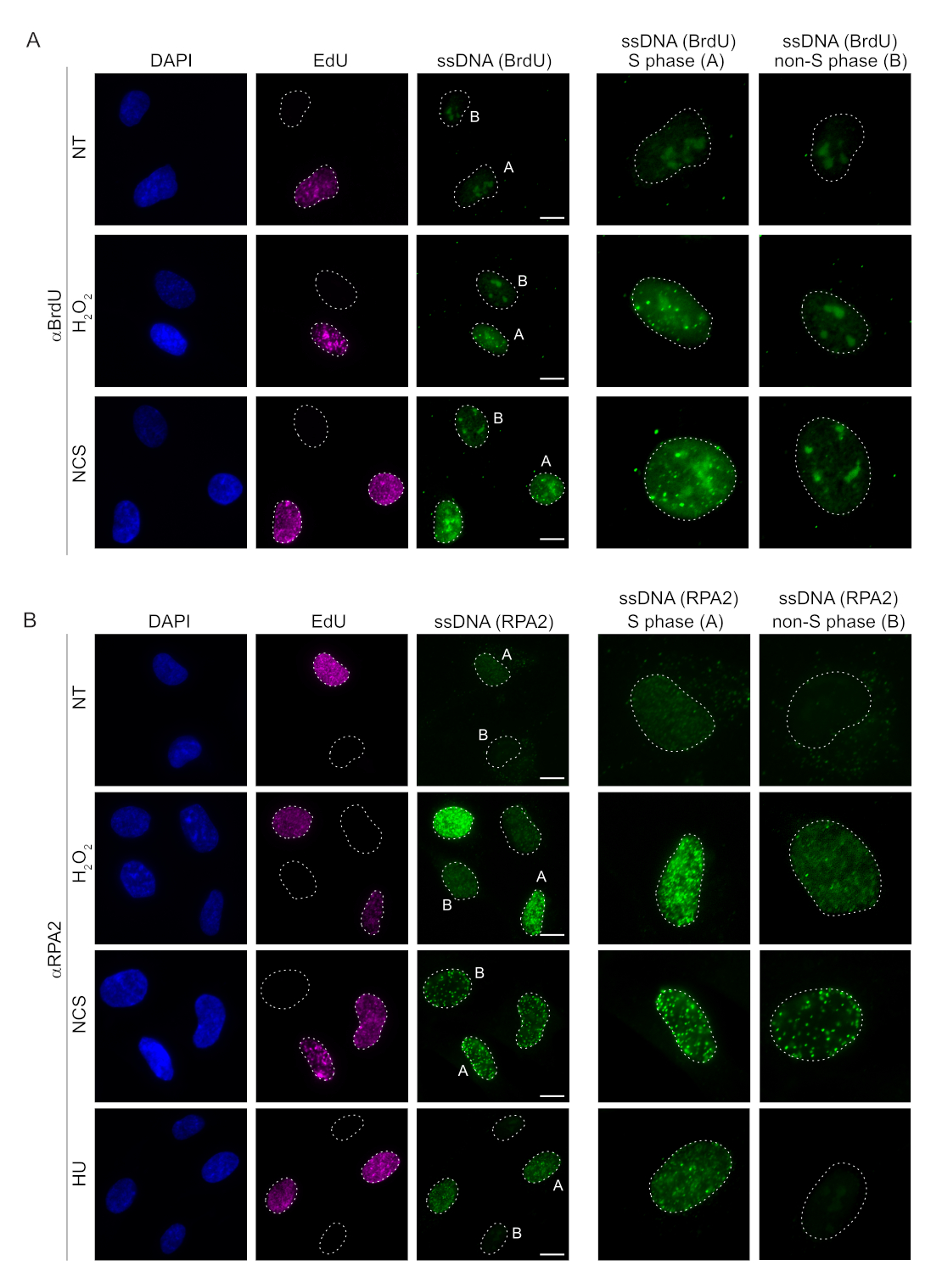
Métodos convencionais para detectar ssDNA em células de mamíferos baseiam-se na detecção de BrdU em ssDNA. A Figura 2A demonstra que, no tratamento com H2O2 e neocarzinostatina (NCS), os focos de BrdU foram detectáveis apenas em células de fase S, enquanto nenhum foco de ssDNA foi detectável em células de fase não-S. A coloração de anticorpos BrdU também mostrou uma coloração de fundo nucleolar perceptível que pode ser detectada em todos os núcleos, independentemente do estágio do ciclo celular ou dos tratamentos aplicados. Usando o protocolo de clique de EdU descrito aqui, não foi possível detectar focos de EdU e BrdU co-localizados, o que é evidente nas amostras não tratadas da Figura 2A. Para descartar completamente qualquer sinal de BrdU que surgisse de reatividade cruzada, evitamos a marcação de EdU e usamos a ciclina A2 como marcador S-G2. Entretanto, a coloração com ciclina A2 não permitiu a pré-extração da CSK e, nessa condição, não foram observados focos de BrdU, mesmo após estresse genotóxico (Figura Suplementar S2A). Isso destaca o fato de que a pré-extração de CSK é necessária para a coloração de ssDNA anti-BrdU. Como controle, testamos a coloração de anticorpos BrdU sob condições de desnaturação. Isso abre o DNA para expor o BrdU incorporado, o que revela que o BrdU foi uniformemente incorporado (Figura Suplementar S2B).
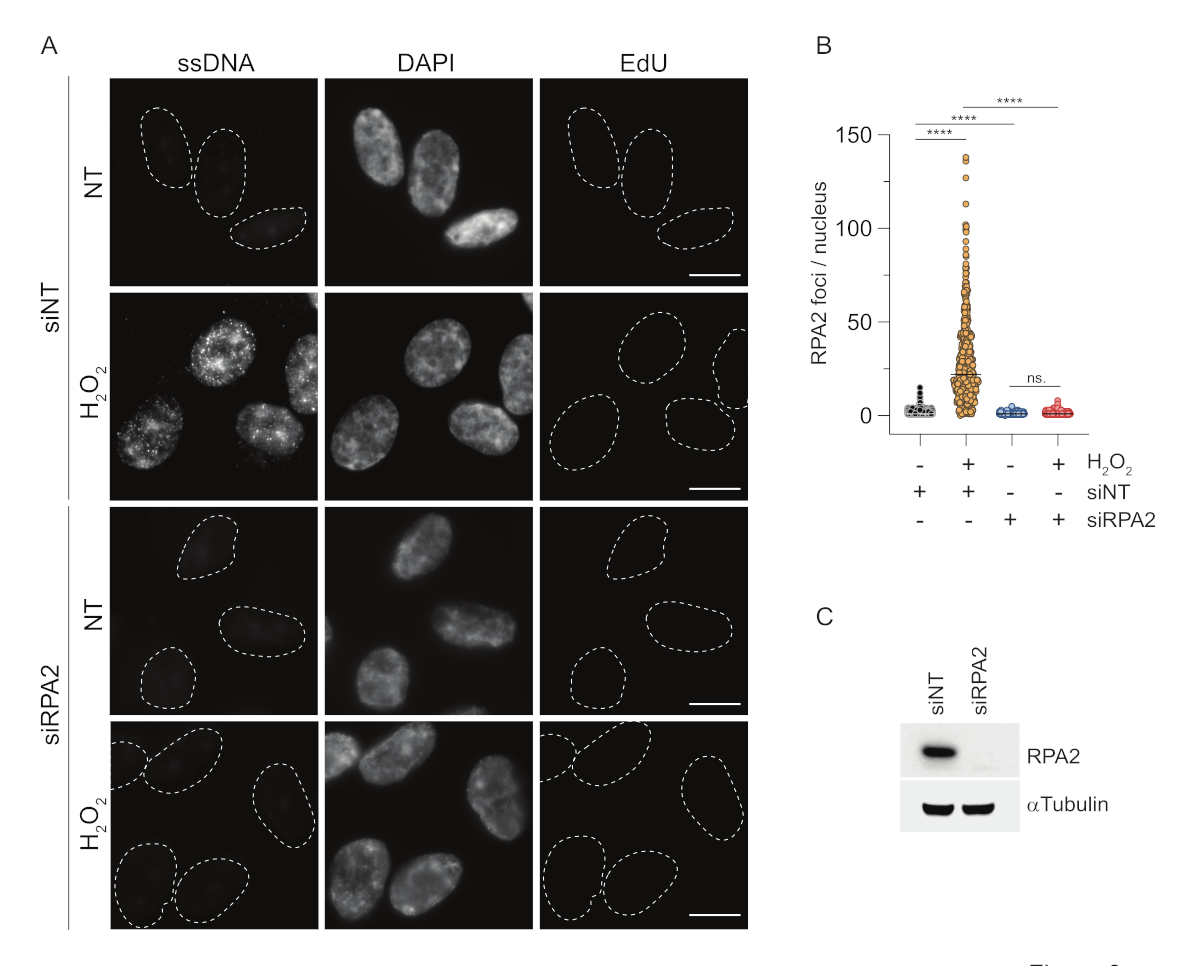
Em contraste, a coloração RPA2 mostra a formação de focos dependentes de NCS e H2O2 não apenas na fase S, mas também em outras fases do ciclo celular (Figura 2B). Como controle, também tratamos as células com HU, que só causa acúmulo de ssDNA em células em replicação. Como esperado, detectamos apenas um aumento de sinal no tratamento da HU com o anticorpo RPA2 em células EdU-positivas, ressaltando a especificidade dessa abordagem. O anticorpo RPA2 também pode detectar a formação natural de ssDNA durante a replicação na ausência de estresse genotóxico exógeno (Figura 2B). A natureza altamente sensível do anticorpo RPA2 levou-nos a tentar utilizá-lo na fase G1, onde a coloração convencional de BrdU não detectou nenhum sinal sobre estresse genotóxico (Figura Suplementar S2C). A Figura 3A mostra que a formação de focos de ssDNA após o tratamento com H2O2 foi detectável com o uso de anticorpo anti-RPA2, mesmo no G1. Houve um aumento significativo no número de focos de RPA2 nesses núcleos com o tratamento com H2O2 (Figura 3B). Esses focos foram específicos para RPA2, pois o silenciamento de RPA2 aboliu o sinal de FI (Figura 3A,B). A Figura 3C e a Figura Suplementar S1C mostram a eficiência do silenciamento de RPA2 nessas células. Em comparação com os métodos convencionais, a detecção de ssDNA baseada em RPA2 é altamente sensível, e sua aplicação pode, portanto, ser estendida para células de fase G1.

Figura 1: Eficiência de sincronização das células RPE1 após a inanição do soro. (A) Os immunoblots mostram os níveis de proteína indicados em células RPE1 sincronizadas em fase G1 assíncrona, G1 e S. (B) Imagens representativas mostram células RPE1 sincronizadas com fase G1 e S assíncronas que foram expostas a 10 μM de EdU por 30 min antes da fixação e visualizadas pela reação Click-IT. DAPI foi usado para contra-corar o DNA nuclear. Barras de escala = 50 μm. (C) O gráfico mostra a porcentagem de células positivas para EdU sobre a população celular total avaliada pelo DAPI. A barra de erro representa o erro padrão da média, e os números de núcleos analisados foram os seguintes: AS n = 219, G1 n = 630, S n = 437. Abreviações: RPE1 = células epiteliais pigmentares da retina imortalizadas por hTERT; EA = assíncrona; EdU = 5-etinil-2'-desoxiuridina; DAPI = 4',6-diamidino-2-fenilindol. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Detecção de ssDNA com anticorpo BrdU ou anticorpo RPA2 após dano ao DNA. (A) Imagens representativas ilustram focos de ssDNA usando αBrdU (verde), células da fase S são destacadas por EdU (roxo) e DAPI foi usado para contracoloração de DNA nuclear (azul). As células RPE1 foram mantidas em BrdU 10 μM por 48 h antes de qualquer tratamento adicional. Após 48 h, as células foram pulsadas com 10 μM de EdU por 30 min, seguido de tratamento de H2O2 (250 μM) por 1 h ou Neocarzinostatin (0,5 μg/mL) por 4 h. As células foram fixadas após pré-extração de CSK. Uma linha tracejada branca denota a borda de cada núcleo. Barra de escala = 5 μm. Os painéis à direita são imagens ampliadas dos núcleos de fase S ou não-S indicados. (B) Imagens representativas ilustram focos de ssDNA usando o anticorpo αRPA2 (verde). As células da fase S são destacadas por EdU (roxo), e DAPI foi usado para contra-coloração de DNA nuclear (azul). As células RPE1 foram pulsadas com 10 μM de EdU por 30 min, seguidas por 1 h H2O2 (250 μM), 4 h de Hidroxiureia (2 mM) ou 4 h de NCS (0,5 μg/mL). As células foram fixadas após pré-extração de CSK. Uma linha tracejada branca denota a borda de cada núcleo. Barra de escala = 10 μm. Os painéis à direita são imagens ampliadas dos núcleos de fase S ou não-S indicados. Abreviações: ssDNA = DNA de fita simples; BrdU = 5-bromo-2'-desoxiuridina; DAPI = 4',6-diamidino-2-fenilindol; RPE1 = células epiteliais pigmentares da retina imortalizadas por hTERT; EdU = 5-etinil-2'-desoxiuridina; NCS = Neocarzinostatina; UH = hidroxiureia. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Detecção de focos de ssDNA na fase G1 utilizando o anticorpo RPA2. (A) As células RPE1 foram transfectadas com siRNAs direcionados para RPA2 ou um controle de siRNA não direcionado e, posteriormente, sincronizadas em G1 e marcadas com pulso com 10 μM EdU por 30 min antes de tratá-las com H2O2 (250 μM) por 1 h quando indicado. DAPI foi usado para contra-corar o DNA nuclear. As células foram fixadas após pré-extração de CSK. Uma linha tracejada branca denota a borda de cada núcleo. Barra de escala = 5 μm. (B) As medidas do número de focos/núcleo de RPA2 foram realizadas a partir de dois experimentos independentes. Apenas células EdU-negativas foram consideradas durante a análise. As linhas representam o valor médio nos gráficos. Para análise estatística foi realizado o teste ANOVA não paramétrico (Kruskal-Wallis). As estrelas indicam P < 0,0001. Os núcleos analisados foram: siNT no H2O2 n = 513, siNT H2O2 n = 603, siRPA2 no H2O2 n = 266, siRPA2 H2O2 n = 536. (C) A eficiência do knockdown de siRNA é demonstrada no immunoblotting. Abreviações: siNT = controle de siRNA não direcionado; BrdU = 5-bromo-2'-desoxiuridina; DAPI = 4',6-diamidino-2-fenilindol; RPE1 = células epiteliais pigmentares da retina imortalizadas por hTERT; EdU = 5-etinil-2'-desoxiuridina. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Quantificação dos focos de ssDNA usando Fiji. Etapas detalhadas em Fiji mostrando como avaliar os números de focos RPA2 no núcleo. (A-E) A criação de uma máscara nuclear usando o canal DAPI. (F-H) Limiares para identificar focos individuais de ssDNA nuclear a partir do sinal de fundo. Abreviações: ssDNA = DNA de fita simples; DAPI = 4',6-diamidino-2-fenilindol. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Tampão citoesquelético (CSK) | |
| TUBOS pH 7,0 | 10 mM |
| NaCl | 100 mM |
| EDTA pH 8 | 1 mM |
| MgCl2 | 3 mM |
| D-sacarose | 300 mM |
| Tritão X-100 | 0.20% |
| Coquetel inibidor de fosfatase | 1 comprimido por 10 ml |
| Coquetel inibidor de protease | 1 comprimido por 10 ml |
| diluído em ddH2O | n/d |
| Tampão de lavagem | |
| Tritão X-100 | 0.05% |
| diluído em PBS | n/d |
| Tampão de permeabilização | |
| Tritão X-100 | 0.50% |
| diluído em PBS | n/d |
| Solução de fixação | |
| Paraformaldeído | 3.60% |
| Tritão X-100 | 0.05% |
| diluído em PBS | n/d |
| Buffer de bloqueio | |
| Albumina de soro bovino (BSA) | 5% |
| Tritão X-100 | 0.10% |
| diluído em PBS | n/d |
| Coquetel de reação Click-iT Plus | |
| 1x Buffer de reação Click-iT | 435 mL |
| Solução Alexa Fluor PCA | 5 mL |
| Pré-mistura protetora CuSO4-cobre | 10 mL |
| 1x Click-iT aditivo de buffer | 50 mL |
| Volume total | 500 mL |
Tabela 1: Composição dos buffers utilizados neste protocolo.
Figura suplementar S1. (A) As células RPE1 foram sincronizadas à fase G0 usando inanição de soro por 72 h e posteriormente liberadas em diferentes fases do ciclo celular pela reintrodução do soro. Os gráficos de pontos mostram células nas fases G0/G1, S ou G2/M, onde horas indicam o tempo após a readição de soro após a inanição do soro. O gráfico à direita mostra a porcentagem de células G0/G1, S e G2/M em cada condição. A análise da FACS foi realizada utilizando um kit de proliferação celular comercialmente disponível usando EdU e iodeto de propídio de acordo com as recomendações do fabricante. (B) Western blot não cortado para a Figura 1. Os números mostram marcadores de peso molecular em kDa. PARP1 foi usado como controle de carga e carregado no gel que também foi desenvolvido contra CCNA2, p27 (mais despojado para PCNA) e pH3 (S10) (posteriormente descascado para H3) cortando a membrana. CCNB1 e RPA2 foram carregados em um gel separado, usando a mesma quantidade de lisado proteico para garantir a comparabilidade. (C) Western blot não cortado para a Figura 3. Os números mostram marcadores de peso molecular em kDa. Abreviação: EdU = 5-etinil-2'-desoxiuridina. Clique aqui para baixar este arquivo.
Figura Suplementar S2: (A) Imagens representativas ilustram focos de ssDNA usando o anticorpo BrdU (verde); As células da fase S são destacadas pela ciclina A2 (vermelho); e DAPI foi usado para contra-corar o DNA nuclear (azul). As células RPE1 foram mantidas em BrdU 10 μM por 48 h antes do tratamento adicional. Após 48 h, as células foram tratadas com H2O2 (250 μM) por 1 h ou Neocarzinostatin (0,5 μg/mL) por 4 h antes da fixação. Uma linha tracejada branca denota a borda de cada núcleo. Barra de escala = 5 μm. (B) Coloração BrdU de células RPE1 com e sem condição de desnaturação. Células RPE1 assíncronas foram pré-tratadas com 10 μM BrdU por 48 h. Barra de escala = 10 μm. (C) As medidas para o número de focos/núcleo de BrdU foram realizadas a partir de dois experimentos independentes em células RPE1 sincronizadas G1. Apenas células EdU-negativas foram consideradas durante a análise. As linhas representam o valor médio nos gráficos. Para análise estatística foi realizado o teste ANOVA não paramétrico (Kruskal-Wallis). O 'ns' indica diferença não significativa. O número de núcleos analisados foi o seguinte: NT n = 52, NCS n = 105, H2O2 n = 82. Abreviações: siNT = controle de siRNA não direcionado; BrdU = 5-bromo-2'-desoxiuridina; DAPI = 4',6-diamidino-2-fenilindol; RPE1 = células epiteliais pigmentares da retina imortalizadas por hTERT; NCS = Neocarzinostatina. Clique aqui para baixar este arquivo.
Vídeo Suplementar S1: Gravação de tela da análise de focos RPA2 baseada em Fiji. Clique aqui para baixar este arquivo.
Discussão
A manutenção de uma cultura de células saudável e livre de micoplasma é fundamental para todos os experimentos descritos acima. As células RPE1 têm uma forte ligação a produtos plásticos tratados com cultura de tecidos em meios de cultura normais; no entanto, suas características de ligação diminuem significativamente quando mantidas em condições livres de soro. Além disso, para capturar imagens de alta resolução de focos de ssDNA sob um microscópio, as células precisam ser plaqueadas em um vidro de cobertura de 0,17 mm de espessura, que não é hidrofílico o suficiente para suportar a fixação adequada de células RPE1. Sem células devidamente achatadas e uniformemente distribuídas, é muito desafiador visualizar focos individuais de ssDNA. Portanto, é fundamental escolher o material de revestimento adequado (por exemplo, vitronectina) e deixar tempo adequado (6-12 h) para que as células se espalhem e se fixem após liberá-las na fase G1.
Uma parte desafiadora do protocolo é obter células G1 RPE1 homogêneas sincronizadas. Isso requer duas etapas críticas. Primeiro, para uma inanição de soro eficiente, as células precisam ser tripsinizadas, lavadas cuidadosamente com PBS e semeadas diretamente em novas placas de cultura de tecidos usando meios livres de soro. A lavagem direta das células em placas de cultura de tecidos para remoção do soro não produzirá sincronização eficiente de G0. Em segundo lugar, ao liberar células para a fase G1, as células devem ser tripsinizadas novamente e semeadas em placas de cultura de tecidos frescos. Da mesma forma, apenas mudar o meio e adicionar meio de cultura contendo soro às células não resultará em uma entrada G1 síncrona. Além disso, para a entrada adequada do G1, a densidade de semeadura das células nos vidros de cobertura revestidos deve estar em certos níveis de confluência. Embora a sincronização celular perfeita seja geralmente inatingível, este protocolo de sincronização descrito aqui dá uma população de G1 ~97% pura. A densidade de semeadura recomendada para RPE1 em uma lamínula de 12 mm de diâmetro é de ~4 × 104 para adquirir um campo de visão homogêneo para aquisição de imagens, com aproximadamente 70% de confluência. A maior densidade de semeadura faz com que as células se desprendam e "descasquem" após a extração de CSK e resultará em um sinal de fundo mais alto durante a aquisição da imagem.
Para reduzir qualquer sinal de fundo e alcançar uma relação sinal-ruído favorável, a lavagem completa após a incubação de anticorpos primários e secundários é essencial. Como várias etapas de lavagem devem ser aplicadas, também é essencial evitar que o poço seque durante cada etapa de lavagem. Minimizamos este artefato aplicando um mínimo de 0,05% Triton X-100 em todas as etapas de lavagem e incubação. Uma vez que os poços secaram, as células apresentaram uma relação sinal-ruído alterada; Isso leva a um padrão semelhante a um mosaico sob o microscópio e pode interferir na avaliação. A aquisição de imagens Z-stack combinada com deconvolução pode auxiliar na captura de focos em diferentes planos focais para melhorar a análise.
Os métodos convencionais baseiam-se na detecção de BrdU incorporado em condições não desnaturantes. Esses métodos, no entanto, dependem do pré-tratamento das células com altas doses de BrdU por pelo menos 1-2 dias (ou tempo equivalente a um ciclo celular completo na linhagem celular utilizada) para garantir uma incorporação genômica uniforme. Indesejavelmente, a incorporação extensiva de BrdU pode causar interferência no ciclo celular36. Para resolver essas limitações, este método utiliza RPA2 endógeno para detectar focos de ssDNA. Esta abordagem não requer a incorporação de BrdU orientada pela replicação, ela também pode ser usada em células pós-mitóticas. Como a incorporação extensiva de BrdU não é necessária, isso economiza tempo e reduz a complexidade experimental. Usando a coloração RPA2 para visualizar o ssDNA, podemos usar 2′-deoxi-5-etiniluridina (EdU) e click-chemistry para marcar a replicação do DNA, evitando possível reatividade cruzada dos anticorpos BrdU contra EdU 27,37,38. Cuidados especiais devem ser tomados para mascarar adequadamente a EdU incorporada durante a clique-reação para que os anticorpos BrdU não reajam de forma cruzada com a EdU27,39.
Finalmente, um benefício importante da utilização de RPA2 em vez de BrdU é simplesmente ter uma relação sinal-ruído superior quando comparado à coloração BrdU fora da fase S. Observamos que a coloração BrdU não desnaturante e sua capacidade de visualizar o ssDNA é restrita à fase S mesmo em células replicantes (Figura 2). O anticorpo BrdU liga-se apenas ao BrdU suficientemente exposto em trechos de ssDNA. A ligação de proteínas de reparo, incluindo RPA2, aos trechos de ssDNA pode suprimir ou dificultar a exposição suficiente de BrdU no ssDNA. Verificamos também que a pré-extração de CSK é necessária para a visualização do ssDNA usando o anticorpo BrdU. Isso é possível porque as trilhas de ssDNA não são acessíveis para o anticorpo sem remover componentes proteicos levemente ligados a eles.
No entanto, existem algumas limitações associadas a esse protocolo. Uma limitação do uso de RPA2 para detecção de ssDNA é a necessidade de otimizar a etapa de pré-extração de CSK. O excesso de RPA2 não ligado deve ser lavado do DNA antes de fixar as células. Por um lado, a subextração leva a um fundo elevado devido à fração da proteína RPA2 que não está ligada ao ssDNA. Por outro lado, a extração excessiva levará à perda de sinal. Para a detecção de BrdU, esta não é uma variável, uma vez que o BrdU é incorporado de forma estável ao DNA e não pode ser lavado por pré-extração. Portanto, o tempo da pré-extração do CSK, a quantidade de Triton X-100 no tampão, o volume e a temperatura em que a pré-extração é realizada devem ser cuidadosamente considerados. A pré-extração de CSK também limita o uso do tamanho do núcleo para discriminar células G0/G1 de células S/G2.
Além disso, não podemos excluir a possibilidade de que parte do sinal que vem de RPA2 se origine de estar ligado a outros interatores de proteínas ligadoras de cromatina. Deve-se considerar também a especificidade por espécie do anticorpo RPA2. O anticorpo usado neste protocolo pode reconhecer RPA2 humano, rato, rato, hamster e macaco. Outra limitação dessa abordagem é que nem todas as linhagens celulares podem ser carentes de soro para sincronização G0. A maioria das linhagens de células cancerosas pode contornar os pontos de verificação do ciclo celular e proliferar mesmo em meios privados de soro. Embora a inanição de soro seja benéfica, uma vez que não causa danos ao DNA, deve-se monitorar cuidadosamente sua eficiência de sincronização celular para garantir que o enriquecimento adequado da fase do ciclo celular seja alcançado. Para células que não respondem à privação sérica, outros métodos de sincronização celular devem ser considerados (por exemplo, shake off mitótico, inibição da CDK1 para parada de G2 ou técnicas não invasivas, como elutriação centrífuga). Outro método possível é o uso de imagens de alto conteúdo para medir o conteúdo de EdU e DNA nuclear para o perfil do ciclo celular de células assíncronas31. Deve-se considerar as implicações da utilização de métodos alternativos de sincronização para evitar interferência na análise a jusante. Por exemplo, o uso de bloqueio duplo de timidina ou afrodicolina, frequentemente utilizado na literatura, resultará em estresse de replicação e danos ao DNA40.
A investigação dos mecanismos de reparo do DNA continua sendo um ponto focal de discussão nos campos do câncer e da biologia celular. O protocolo aqui apresentado oferece uma abordagem valiosa para a preparação de células, permitindo a visualização e análise quantitativa do ssDNA após exposição a agentes prejudiciais ao DNA. Notavelmente, este protocolo destaca a utilização da proteína ligadora de ssDNA, RPA2, demonstrando sua alta especificidade para visualizar pequenas quantidades de focos de ssDNA, evitando reatividade cruzada indesejada em todas as fases do ciclo celular. O uso do RPA2 confere inúmeras vantagens, principalmente para os pesquisadores que pretendem analisar células na fase G1 do ciclo celular. Este protocolo considera várias limitações e aborda preocupações relacionadas à interferência de sinal, ruído de fundo indesejado e reatividade cruzada ao usar a coloração RPA2 ou BrdU para detectar ssDNA.
Divulgações
Os autores não têm interesses concorrentes a declarar.
Agradecimentos
Os autores agradecem a Michele Pagano por seu apoio e insights úteis, Ashley Chui e Sharon Kaisari pela leitura crítica do manuscrito, e Jeffrey Estrada e Vilma Diaz por seu apoio contínuo. Este trabalho foi apoiado por um suplemento de diversidade para o National Institutes of Health grant GM136250.
Materiais
| Name | Company | Catalog Number | Comments |
| Alpha-tubulin antibody | Sigma-Aldrich | T6074 | primary antibody (1:5,000) |
| Axio Observer Inverted Microscope | Zeiss | na | microscope |
| Bis-Tris Plus Mini Protein Gels, 4-12% | Invitrogen | NW04127BOX | Western Blot |
| Bovine Serum Albumin | Jackson ImmunoResearch | 001-000-162 | blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Sigma-Aldrich | B5002-100MG | nucleotide analogue |
| BrdU antibody BU1/75 | Abcam | ab6326 | primary antibody (1:500) |
| CellAdhere Dilution Buffer | Stemcell Technologies | 07183 | coating reagent |
| Click-iT Plus EdU Flow Cytometry Assay Kits | Invitrogen | C10632 | flow cytomery |
| Click-iT Plus EdU Cell Proliferation Kit for Imaging, Alexa Fluor 647 dye | Thermo Fisher Scientific | C10640 | click-reaction kit |
| cOmplete ULTRA Protease inhibitor tablets | Sigma-Aldrich | 5892791001 | reagent |
| Countess 3 Automated cell counter | Thermo Scientific | AMQAX2000 | cell counter |
| Coverslip | neuVitro | GG12PRE | tissue culture |
| Cyclin A2 antibody | Santa Cruz Biotechnology | sc-271682 | primary antibody (1:1,000) for IF and WB |
| Cyclin B1 antibody | Santa Cruz Biotechnology | sc-245 | primary antibody (1:5,000) |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D2650-100ML | vehicle control |
| DMEM, high glucose, with HEPES | Gibco | 12430051 | cell culture medium for RPE cells |
| DPBS, no calcium, no magnesium | Gibco | 14190144 | the PBS used throughout the protocol |
| D-Sucrose | Thermo Fisher Scientific | bp220-1 | reagent |
| Eclipse Ti2 Series Epifluorescent Microscope | Nikon | na | microscope |
| EdU (5-Ethynyl-2'-deoxyuridine) | Thermo Fisher Scientific | C10637 | nucleotide analogue |
| Falcon 24-well plate | Corning | 351147 | tissue culture |
| Falcon Cell Culture Dishes 100 mm | Corning | 353003 | tissue culture |
| Fetal Bovine Serum, heat inactivated | Gibco | 16140071 | media supplement |
| Fiji (ImageJ) | NIH | version 1.54f | software and algorithms |
| FxCycle PI/RNase Staining Solution | Invitrogen | F10797 | PI staining |
| Goat anti-mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 555 | Thermo Fisher Scientific | A21422 | secondary antibody (1:1,000) |
| Goat anti-rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A48262 | secondary antibody (1:1,000) |
| Histone H3 antibody | Abcam | ab1791 | primary antibody (1:10,000) |
| hTERT RPE1 | ATCC | CRL-3216 | cell line |
| Hydrochloric acid | Sigma-Aldrich | H1758-100ML | reagent |
| Hydrogen peroxide 30% soultion | Sigma-Aldrich | H1009-100ML | reagent |
| Hydroxyurea,98% powder | Sigma-Aldrich | H8627-5G | reagent |
| Invitrogen Ultra Pure 0.5 M EDTA pH 8.0 | Thermo Fisher Scientific | 15-575-020 | reagent |
| Lipfectamine RNAiMAX Transfection Reagent | Invitrogen | 13778150 | transfection reagent |
| Magnesium chloride solution 1 M | Sigma-Aldrich | M1028-100ML | reagent |
| MycoFluor | Thermo Fisher | M7006 | Mycoplasma Detection Kit |
| Neocarzinostatin from Streptomyces carzinostaticus | Sigma-Aldrich | N9162-100UG | reagent |
| NuPage MES SDS Running Buffer (20x) | Invitrogen | NP0002 | Western Blot |
| onTARGETplus Human RPA2 siRNA | Dharmacon | L-017058-01-0005 | siRNA |
| p27 antibody | BD Biosciences | 610241 | primary antibody (1:1,000) |
| Paraformaldehyde aqueous solution (32%) | Electron Microscopy Sciences | 50-980-494 | fixative |
| PARP1 antibody | Cell Signaling Technology | 9542S | primary antibody (1:1,000) |
| PCNA antibody | Cell Signaling Technology | 13110S | primary antibody (1:2,000) |
| Penicillin-Streptomycin | Gibco | 15140163 | media supplement |
| pH3 antibody | Cell Signaling Technology | 3377S | primary antibody (1:2,000) |
| PhosSTOP phosphatase inhibitor tablets | Sigma-Aldrich | 4906837001 | reagent |
| PIPES Buffer 0.5 M solution, pH 7.0 | Bioworld | 41620034-1 | reagent |
| Precision Plus Protein Kaleidoscope Prestained Protein Standards | Bio-Rad | 1610395 | Western Blot |
| Prism | GraphPad | version 10 | statistical analysis and graph |
| ProLong Diamond Antifade Mountant | Thermo Scientific | P36961 | mounting media |
| Reduced serum media (Opti-MEM) | Gibco | 31985070 | used for transfection |
| Rpa32/rpa2 antibody (mouse) | EMD Millipore | NA19L | primary antibody (1:1,000) for WB |
| Rpa32/rpa2 antibody (rat) | Cell Signaling Technology | 2208S | primary antibody (1:1,000) for IF |
| Sodium Chloride solution (5 M) | Sigma-Aldrich | S5150 | reagent |
| Sodium Pyruvate (100 mM) | Gibco | 11360070 | media supplement |
| Sodium tetraborate decahydrate | Sigma-Aldrich | B3535-500G | reagent |
| Thermo Scientific Pierce DAPI Nuclear Counterstain | Thermo Scientific | 62248 | nucleic acid stain |
| Thymidine,powder | Sigma-Aldrich | T1985-1G | reagent |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | detergent |
| Trypsin-EDTA (0.5%), no phenol red | Gibco | 1540054 | cell dissociation agent |
| Vitronectin XF | Stemcell Technologies | 07180 | coating reagent |
| ZE5 Cell Analyzer | Bio-Rad | na | flow cytomery |
Referências
- Hakem, R. DNA-damage repair; the good, the bad, and ugly. EMBO J. 27 (4), 589-605 (2008).
- Gutierrez, R., O'Connor, T. R. DNA direct reversal repair and alkylating agent drug resistance. Cancer Drug Resist. 4 (2), 414-423 (2021).
- Krokan, H. E., Bjoras, M. Base excision repair. Cold Spring Harb Perspect Biol. 5 (4), a012583 (2013).
- Marteijn, J. A., Lans, H., Vermeulen, W., Hoeijmakers, J. H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 15 (7), 465-481 (2014).
- Li, G. M. Mechanisms and functions of DNA mismatch repair. Cell Res. 18 (1), 85-98 (2008).
- Hustedt, N., Durocher, D. The control of DNA repair by the cell cycle. Nat Cell Biol. 19 (1), 1-9 (2016).
- Yang, W., Gao, Y. Translesion and repair DNA polymerases: diverse structure and mechanism. Annu Rev Biochem. 87, 239-261 (2018).
- Bhat, D. S., et al. Therapeutic disruption of RAD52-ssDNA complexation via novel drug-like inhibitors. NAR Cancer. 5 (2), zcad018 (2023).
- Gupta, P., Saha, B., Chattopadhyay, S., Patro, B. S. Pharmacological targeting of differential DNA repair, radio-sensitizes WRN-deficient cancer cells in vitro and in vivo. Biochem Pharmacol. 186, 114450 (2021).
- Pena-Diaz, J., et al. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol Cell. 47 (5), 669-680 (2012).
- Schroering, A. G., Edelbrock, M. A., Richards, T. J., Williams, K. J. The cell cycle and DNA mismatch repair. Exp Cell Res. 313 (2), 292-304 (2007).
- Scully, R., Panday, A., Elango, R., Willis, N. A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 20 (11), 698-714 (2019).
- Escribano-Diaz, C., et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 49 (5), 872-883 (2013).
- Genschel, J., Modrich, P. Mechanism of 5'-directed excision in human mismatch repair. Mol Cell. 12 (5), 1077-1086 (2003).
- Hu, J., et al. Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J Biol Chem. 288 (29), 20918-20926 (2013).
- Huertas, P., Jackson, S. P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 284 (14), 9558-9565 (2009).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in DNA replication with putative roles in cancer. Int J Mol Sci. 20 (1), 74 (2018).
- Symington, L. S. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol. 6 (8), a016436 (2014).
- Liu, Y., et al. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem. 280 (5), 3665-3674 (2005).
- Wold, M. S., Kelly, T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc Natl Acad Sci U S A. 85 (8), 2523-2527 (1988).
- Wienholz, F., Vermeulen, W., Marteijn, J. A. Amplification of unscheduled DNA synthesis signal enables fluorescence-based single cell quantification of transcription-coupled nucleotide excision repair. Nucleic Acids Res. 45 (9), e68 (2017).
- Wold, M. S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 66, 61-92 (1997).
- Chen, R., Wold, M. S. Replication protein A: single-stranded DNA's first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays. 36 (12), 1156-1161 (2014).
- Kang, Y., et al. Alteration of replication protein A binding mode on single-stranded DNA by NSMF potentiates RPA phosphorylation by ATR kinase. Nucleic Acids Res. 51 (15), 7936-7950 (2023).
- Kilgas, S., Kiltie, A. E., Ramadan, K. Immunofluorescence microscopy-based detection of ssDNA foci by BrdU in mammalian cells. STAR Protoc. 2 (4), 100978 (2021).
- Madabhushi, R., Pan, L., Tsai, L. H. DNA damage and its links to neurodegeneration. Neuron. 83 (2), 266-282 (2014).
- Liboska, R., Ligasova, A., Strunin, D., Rosenberg, I., Koberna, K. Most anti-BrdU antibodies react with 2'-deoxy-5-ethynyluridine -- the method for the effective suppression of this cross-reactivity. PLoS One. 7 (12), e51679 (2012).
- Biehs, R., et al. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol Cell. 65 (4), 671-684.e5 (2017).
- Cruz-Garcia, A., Lopez-Saavedra, A., Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 9 (2), 451-459 (2014).
- Ercilla, A., et al. Physiological tolerance to ssDNA enables strand uncoupling during DNA replication. Cell Rep. 30 (7), 2416-2429.e7 (2020).
- Lezaja, A., et al. RPA shields inherited DNA lesions for post-mitotic DNA synthesis. Nat Commun. 12 (1), 3827 (2021).
- Mukherjee, B., Tomimatsu, N., Burma, S. Immunofluorescence-based methods to monitor DNA end resection. Methods Mol Biol. 1292, 67-75 (2015).
- Ochs, F., et al. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol. 23 (8), 714-721 (2016).
- Raderschall, E., Golub, E. I., Haaf, T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad Sci U S A. 96 (5), 1921-1926 (1999).
- Forment, J. V., Walker, R. V., Jackson, S. P. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytometry A. 81 (10), 922-928 (2012).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Sci Rep. 6, 19567 (2016).
- Aten, J. A., Bakker, P. J., Stap, J., Boschman, G. A., Veenhof, C. H. DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem J. 24 (5), 251-259 (1992).
- Podgorny, O., Peunova, N., Park, J. H., Enikolopov, G. Triple S-phase labeling of dividing stem cells. Stem Cell Reports. 10 (2), 615-626 (2018).
- Cappella, P., Gasparri, F., Pulici, M., Moll, J. Cell proliferation method: click chemistry based on BrdU coupling for multiplex antibody staining. Curr Protoc Cytom. Chapter 7, (2008).
- Ligasova, A., Koberna, K. Strengths and weaknesses of cell synchronization protocols based on inhibition of DNA synthesis. Int J Mol Sci. 22 (19), 10759 (2021).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados