
Method Article
Visualisation des foyers d’ADN monocaténaire dans la phase G1 du cycle cellulaire
Dans cet article
Résumé
Le protocole suivant présente la détection de foyers d’ADN simple brin dans la phase G1 du cycle cellulaire en utilisant la synchronisation du cycle cellulaire suivie d’une coloration immunofluorescente RPA2.
Résumé
L’ADN possède des voies de réparation cellulaire dédiées capables de faire face aux lésions qui pourraient provenir de sources endogènes et/ou exogènes. La réparation de l’ADN nécessite la collaboration de nombreuses protéines, responsables de la couverture d’un large éventail de tâches allant de la reconnaissance et de la signalisation de la présence d’une lésion de l’ADN à sa réparation physique. Au cours de ce processus, des traces d’ADN simple brin (ADNss) sont souvent créées, qui sont finalement remplies par des ADN polymérases. La nature de ces traces d’ADNss (en termes de longueur et de nombre), ainsi que la polymérase recrutée pour combler ces lacunes, sont spécifiques à la voie de réparation. La visualisation de ces traces d’ADNss peut nous aider à comprendre la dynamique complexe des mécanismes de réparation de l’ADN.
Ce protocole fournit une méthode détaillée pour la préparation de cellules synchronisées G1 afin de mesurer la formation de foyers d’ADNss lors d’un stress génotoxique. À l’aide d’une approche d’immunofluorescence facile à utiliser, nous visualisons l’ADNss en colorant la RPA2, un composant du complexe de protéine de réplication hétérotrimérique A (RPA). La RPA2 se lie aux intermédiaires de l’ADNss qui surviennent lors d’un stress génotoxique ou d’une réplication et stabilise les éléments intermédiaires pour contrôler la réparation de l’ADN et l’activation des points de contrôle des dommages à l’ADN. La coloration à la 5-éthynyl-2'-désoxyuridine (EdU) est utilisée pour visualiser la réplication de l’ADN afin d’exclure toute cellule de la phase S. Ce protocole offre une approche alternative aux tests conventionnels à base de 5-bromo-2'-désoxyuridine (BrdU) non dénaturants et est mieux adapté à la détection des foyers d’ADNss en dehors de la phase S.
Introduction
Pour maintenir la vie, les cellules surveillent et réparent constamment l’ADN afin de maintenir leur intégrité génomique. Les cellules peuvent accumuler divers types de dommages à l’ADN en raison de sources endogènes (p. ex., oxydation, alkylation, désamination, erreurs de réplication) et exogènes (p. ex., UV, irradiation ionisante) de facteurs de stress de l’ADN. L’incapacité à réparer ces lésions entraîne soit l’apoptose, soit l’arrêt du cycle cellulaire, soit la sénescence et peut entraîner des maladies1. Les lésions de l’ADN peuvent être traitées par l’une des principales voies de réparation de l’ADN suivantes : DR (réparation directe par inversion), qui répare principalement les bases alkylées2 ; BER (réparation par excision de base), qui cible les erreurs de bases d’ADN non volumineuses et les cassures d’ADN simple brin (SSB)3 ; NER (réparation par excision de nucléotides) corrigeant les lésions volumineuses de l’ADN qui déforment l’hélice4 ; MMR (réparation des mésappariements) ciblant principalement les incompatibilités d’ADN, les boucles d’insertion/délétion (IDL) et certains dommages de base5 ; NHEJ (jointure d’extrémité non homologue) et HRR (réparation par recombinaison homologue) qui sont tous deux actifs au niveau des cassures d’ADN double brin (DSB)6 ; et TLS (synthèse de translésion), qui est un mécanisme de dérivation des lésions de l’ADN7. Bien que ces voies aient des spécificités de substrat distinctes, il existe certains chevauchements entre elles afin d’assurer la redondance pour une réparation efficace. Il est crucial de comprendre l’action des différentes voies de réparation de l’ADN dans les différentes phases du cycle cellulaire, car ces facteurs de réparation de l’ADN pourraient servir de cibles essentielles pour les approches thérapeutiques visant à traiter le cancer, le vieillissement et les troubles neurologiques.
L’ADN simple brin (ADNss) est généré tout au long du cycle cellulaire en raison de la réparation des lésions de l’ADN générées par des agents endommageant l’ADN endogène et exogène. Lors d’un stress génotoxique, l’ADNss est généré en abondance dans les phases S et G2 où HRR et MMR ont leur activité la plus élevée et lorsque la machinerie de réplication s’arrête ou s’effondre lorsqu’elle rencontre des lésions de l’ADN 6,10,11. D’autres voies de réparation de l’ADN (p. ex., NHEJ/jonction d’extrémités médiée par microhomologie (MMEJ)/recuit simple brin [SSA]) génèrent également de l’ADNss au cours de la réparation de la DSB12. Ces traces d’ADNss proviennent généralement de la résection de l’ADN, réalisée par des exonucléases telles que EXO1, DNA2 et CtIP pendant HR et MMR, des endonucléases telles que XPF et XPG pendant NER, ou par l’action combinée de POLB et FEN1 pendant BER 4,13,14,15,16,17,18,19 . En raison du travail de la machinerie de réplication, des traces d’ADNss sont également générées lorsque les hélicases d’ADN déroulent l’ADN devant les polymérases réplicatives liées au PCNA20. En revanche, dans la phase G1, l’absence de HRR et de réplication de l’ADN et l’activité limitée du MMR réduisent l’étendue des traces d’ADNss générées et sont donc plus difficiles à détecter 10,11,21.
Les traces d’ADNss cellulaires sont des structures très sensibles qui doivent être protégées pour éviter la formation de DSB. Ceci est réalisé en enrobant les pistes d’ADNss avec de la RPA. La RPA est un complexe protéique hétérotrimérique abondant composé de plusieurs sous-unités (RPA1, RPA2 et RPA3, également appelées RPA70, RPA32 et RPA14, respectivement), qui sont exprimées de manière ubiquitaire tout au long du cycle cellulaire22. Chaque sous-unité RPA contient un domaine de liaison à l’ADN (DBD), capable d’interagir avec 4 à 6 nucléotides, et les sous-unités combinées forment un noyau de trimérisation stable. Au total, la RPA se lie à environ 20 à 30 nucléotides avec une affinité sub-nanomolairede 23,24.
Les méthodes conventionnelles utilisent la microscopie à immunofluorescence (IF) pour visualiser les foyers d’ADNss en marquant la 5-bromo-2'-désoxyuridine (BrdU) incorporée dans l’ADN génomique à l’aide d’anticorps BrdU25. Cette approche repose sur le fait que les anticorps BrdU ne peuvent détecter BrdU que dans l’ADNss25 exposé. Bien que cette approche soit simple, elle présente également certaines limites. Par exemple, les cellules sont prétraitées pour incorporer du BrdU avant le début de l’expérience, ce qui prend du temps et peut interférer avec les effecteurs en aval. Par conséquent, la détection de l’ADNss basée sur BrdU est limitée aux cellules réplicatives et ne peut pas être utilisée pour les cellules quiescentes. Cela exclut l’application de cette méthode à l’étude de la réparation de l’ADN dans les cellules non réplicatives malgré son importance dans plusieurs maladies telles que le cancer et la neurodégénérescence 5,26. De plus, étant donné que les structures de BrdU et d’EdU sont très similaires, la plupart des anticorps BrdU présentent une réactivité croisée vis-à-vis de l’EdU, ce qui doit être pris en compte lors d’expériences de double marquage27. La coloration RPA a déjà été utilisée pour montrer des foyers d’ADNss principalement dans les cellules de la phase S ; cependant, certains articles l’ont également utilisé avec succès en dehors de la phase S 28,29,30,31,32,33,34,35. Le protocole suivant utilise efficacement les propriétés de la RPA, permettant la visualisation des foyers d’ADNss suite à des dommages à l’ADN dans la phase G1 du cycle cellulaire (bien qu’il puisse être utilisé dans toutes les phases du cycle cellulaire).
Protocole
1. Maintien des cellules épithéliales pigmentaires rétiniennes immortalisées par hTERT (RPE1)
- Maintenir les lignées cellulaires RPE1 dans le milieu Eagle modifié (DMEM) de Dulbecco, complétées par 10 % de sérum de veau fœtal inactivé par la chaleur (Hi-FBS) et 100 μg/mL de pénicilline-streptomycine (désormais appelée milieu de culture) dans un incubateur humidifié contenant 5 % de CO2 à 37 °C. Pour la culture de routine, cultivez les cellules RPE1 dans une boîte de 15 cm traitée par culture tissulaire et divisez-les lorsque vous atteignez une confluence de 80 à 90 % (~16-18 × 10 cellulesde 6 cellules par boîte de 15 cm).
- Lors de la fendillement, retirez le milieu et rincez les cellules avec 10 mL de solution saline tamponnée au phosphate (PBS) 1x.
- Ajouter 3 mL de trypsine-EDTA à 0,05 % pour couvrir toute la surface du plat. Maintenez les cellules à 37 °C avec la trypsine jusqu’à ce qu’elles se détachent.
- Après la trypsinisation, remettre les cellules en suspension avec un milieu de culture et les faire tourner à 150 × g pendant 5 min à température ambiante (RT, 22-25 °C). Retirer le surnageant et remettre délicatement les cellules en suspension dans 10 mL de milieu de culture.
- Semez 1,6 à 1,8 × 106 cellules dans une nouvelle boîte de 15 cm (~1 mL de suspension cellulaire).
REMARQUE : Tous les travaux de culture tissulaire doivent être effectués selon les niveaux de sécurité BSL-2. Le temps d’incubation de la trypsinisation dépend de la confluence cellulaire. Habituellement, le processus prend 2 à 3 minutes pour une plaque confluente à 90 %. Les cellules doivent faire l’objet d’un dépistage régulier de la contamination par les mycoplasmes à l’aide de trousses disponibles dans le commerce (voir les exemples dans le tableau des matériaux).
2. Inactivation par l’ARNi du gène d’intérêt (GOI)
- Semer 1,0 × 106 cellules RPE1 dans une plaque de 10 cm traitée pour la culture tissulaire avec 10 mL de milieu de culture la veille de la transfection.
- Le jour de la transfection, complexer le siRNA. Pour une plaque de 10 cm, utiliser une concentration finale de 20 nM siRPA2 et 12 μL de réactif de transfection à base de lipides dans 500 μL de milieu de transfection à faible teneur en sérum. Mélangez délicatement tous les composants en effleurant le tube et incubez à RT (22-25 °C) pendant 5 min.
- Ajouter le mélange d’ARNsi complexé aux cellules goutte à goutte et incuber les cellules avec l’ARNi pendant 48 h.
3. Synchronisation des cellules RPE1 en phase G0
- Trypsiniser les cellules RPE1 à partir de l’étape 2.3 comme indiqué dans la section 1 (~2 × 106 cellules).
- Transvaser la suspension cellulaire dans des tubes à centrifuger de 15 mL et la centrifuger à 150 × g, RT (22-25 °C) pendant 5 min.
- Retirer le surnageant et remettre les cellules en suspension dans 12 mL de PBS. Centrifuger les cellules à 150 × g à RT (22-25 °C) pendant 5 min. Répétez l’élimination du surnageant et la centrifugation deux fois.
- Remettre les cellules en suspension dans 10 mL de DMEM sans sérum complété par 100 μg/mL de pénicilline-streptomycine, 1 mM de pyruvate de sodium, 15 mM d’HEPES, et les déposer sur une boîte de culture tissulaire de 10 cm.
REMARQUE : Si les cellules ont tendance à s’agglutiner, remettez-les en suspension dans seulement 1 mL de DMEM sans sérum et pipetez-les de haut en bas 5 fois à l’aide d’un embout P1000 pour déloger les grumeaux avant de diluer la suspension jusqu’à un volume final de 10 mL. - Après 24 h de privation de sérum, introduire le deuxième cycle de silençage en utilisant la même procédure que celle décrite dans la section 2 en ajoutant l’ARNi complexé aux cellules affamées de sérum.
- Conservez les cellules RPE1 dans un DMEM sans sérum pendant 72 h avant de procéder à la libération de G1.
4. Revêtement de lamelles et libération des cellules en phase G1
- Stérilisez la pince à épiler avec de l’éthanol à 70 % et placez une seule lamelle de verre (12 mm de diamètre et #1,5 d’épaisseur [0,17 mm]) dans un puits d’une plaque de 24 puits.
- Diluer la matrice d’enrobage de vitronectine avec du PBS pour obtenir une concentration finale de 10 μg/mL. Ajouter 500 μL de la solution de vitronectine dans chaque puits contenant les lamelles et incuber pendant 1 h à RT.
- Retirez la solution d’enrobage et lavez les lamelles avec 1 mL de PBS.
- Détacher les cellules RPE1 affamées de sérum de la plaque traitée de culture tissulaire de 10 cm en utilisant 1 mL de trypsine à 0,05 % après un lavage PBS pendant 1 min à 37 °C.
REMARQUE : Les cellules se détachent beaucoup plus rapidement après la famine sérique. Soyez prudent lorsque vous lavez les cellules avec du PBS et utilisez des temps de trypsinisation courts. - Pour inactiver la trypsine, remettre en suspension les cellules RPE1 dans un total de 6 mL de milieu de culture. Retirez la trypsine inactivée en faisant tourner les cellules à l’aide de 150 × g à RT (22-25 °C) pendant 5 min.
- Remettre les cellules en suspension dans 1 mL de milieu de culture et mesurer le nombre de cellules.
- Semer 4 × 104 cellules RPE1 sur la lamelle enrobée dans un total de 500 μL de milieu de culture.
REMARQUE : Assurez-vous que la viabilité de la cellule est supérieure à 90% avant de passer aux étapes en aval. La viabilité cellulaire a pu être rapidement évaluée par coloration au bleu de trypan pendant l’étape de comptage cellulaire. - Après 6 h de placage des cellules dans le milieu de culture, les cellules libérées par G0 seront en phase G1 précoce. Effectuez des expériences en G1 dans cette fenêtre de 6 à 12 h avant que les cellules ne commencent à entrer dans la phase S.
- Avant d’introduire des dommages à l’ADN, pulser les cellules avec 10 μM de 5-éthynyl-2'-désoxyuridine (EdU) pendant 30 min à 37 °C, dilué dans un milieu de culture.
- Retirez le milieu contenant de l’EdU et poursuivez les cellules avec 10 μM de thymidine pendant 10 min à 37 °C pour empêcher l’incorporation restante d’EdU pendant l’induction des dommages à l’ADN.
- Retirer le milieu avec de la thymidine et traiter les cellules avec 250 μM H2O2 pendant 1 h, dilué dans un milieu de culture.
5. Marquage par immunofluorescence de l’ADNss
- Laver les cellules une fois avec 1 mL de RT (22-25 °C) PBS pour éliminer les composants du milieu et du sérum.
REMARQUE : Soyez doux lorsque vous lavez les cellules pour éviter qu’elles ne se détachent et ne sèchent. Ne traitez pas plusieurs puits en même temps. - Pré-extraction : Incuber les cellules lavées dans 1 mL de tampon d’extraction CSK (tableau 1) pendant 5 min à RT (22-25 °C).
REMARQUE : La pré-extraction CSK élimine toutes les protéines non liées à la chromatine, y compris la RPA2 soluble.
ATTENTION : Triton X-100 est nocif en cas d’ingestion et peut provoquer une irritation de la peau et des lésions oculaires. - Retirez le tampon CSK des cellules et fixez-les directement en ajoutant 0,5 mL de solution de paraformaldéhyde à 3,6 % (dans du PBS) contenant 0,05 % de Triton X-100 pendant 10 min à RT (22-25 °C).
ATTENTION : Il est important de préparer 3,6 % de PFA à partir de 32 % de PFA frais. Le paraformaldéhyde peut causer de graves lésions oculaires, une irritation de la peau et une irritation des voies respiratoires. - Lavez les cellules une fois avec 1 mL de PBS contenant 0,05 % de Triton X-100 pour éliminer le PFA.
- Perméabiliser davantage les cellules à l’aide de 1 mL de PBS contenant 0,5 % de Triton X-100 pendant 15 min à RT (22-25 °C).
- Réaction EdU click-IT pour visualiser les cellules qui se répliquent (phase S)
- Retirez la solution de perméabilisation et lavez les cellules 2 fois avec 1 mL de tampon bloquant (tableau 1).
ATTENTION : L’albumine sérique bovine (BSA) peut provoquer une irritation des voies respiratoires. - Ajouter 1 mL de tampon bloquant (tableau 1) et agiter doucement la plaque contenant la lamelle pendant 10 min à RT (22-25 °C).
- Retirer le tampon bloquant et ajouter 500 μL de cocktail de réaction par clic contenant de l’azoture de picolyle 647 (tableau 1). Incuber les lamelles pendant 30 min à RT (22-25 °C) en les berçant doucement et effectuer des incubations en aval dans l’obscurité.
REMARQUE : Lors de l’utilisation d’anticorps BrdU, utilisez le double de la quantité (1 ml) et de la durée (60 min) de la réaction de clic recommandée par le fabricant pour vous assurer que la réaction est saturée et que l’EdU incorporé est étiqueté. Cela limite la réactivité croisée des anticorps BrdU27.
- Retirez la solution de perméabilisation et lavez les cellules 2 fois avec 1 mL de tampon bloquant (tableau 1).
- Retirez le mélange de réaction d’encliquetage et lavez les cellules 2 fois avec du PBS avec 0,05 % de Triton X-100 pendant 10 min à RT (22-25 °C) (Figure 1 et Figure 2).
- Ajouter 1 mL de tampon bloquant et incuber à RT (22-25 °C) pendant 30 min. Vous pouvez également conserver les cellules dans un tampon bloquant à 4 °C pendant la nuit.
- Appliquer l’anticorps primaire (rat anti-RPA2, dilution de 1 :1 000) pendant 2 h à RT (22-25 °C) dans 250-500 μL de tampon bloquant avec un léger balancement.
- Lavez les cellules 2 fois avec du PBS contenant 0,05 % de Triton X-100 pour éliminer rapidement la majeure partie de la solution d’anticorps.
- Continuez à laver les cellules pendant 3 x 10 min avec un tampon de blocage à RT (22-25 °C).
- Appliquer l’anticorps secondaire (anti-rat Alexa-488, dilution 1 :1 000) dans 250-500 μL de tampon bloquant à RT (22-25 °C) pendant 2 h avec un léger balancement.
- Lavez les cellules avec un tampon bloquant 2 fois pour éliminer rapidement la majeure partie de l’anticorps secondaire. Continuez à laver les cellules pendant 3 x 10 min avec du PBS contenant 0,05 % de Triton X-100 à RT (22-25 °C).
- Pour contre-colorer les noyaux, laver les cellules une fois avec du PBS contenant 0,05 % de Triton X-100 et 1 μg/mL de 4',6-diamidino-2-phénylindole (DAPI) pendant 10 min à RT (22-25 °C). Laver les cellules une fois avec du PBS pendant 5 min à RT (22-25 °C).
- Montez la vitre de protection sur les lames de microscope à l’aide de 10 μL de support de montage/lamelle. Trempez les lamelles dans de l’eau distillée avant de les monter pour vous débarrasser des cristaux de sel. Visualisez les lames le lendemain et conservez-les à 4 °C pendant des semaines (figure 3).
6. Acquisition et quantification d’images
- Pour capturer des images, utilisez n’importe quel microscope à épifluorescence disponible équipé de jeux de filtres de routine pour imager les canaux DAPI, FITC et Cy5 avec un grossissement d’au moins 60 à 63x, une ouverture numérique élevée et des objectifs pétroliers afin de visualiser les foyers nucléaires.
REMARQUE : L’excitation DAPI optimale est de ~359 nm ; L’excitation d’Alexa 488 est de ~488 nm ; tandis que l’excitation d’Alexa 647 est de ~647 nm. - Pour l’analyse d’image, ouvrez les fichiers image dans Fiji/ImageJ.
- Fabriquer des masques nucléaires à l’aide de la coloration DAPI (figure 4A-F et vidéo supplémentaire S1).
- Ouvrez l’image DAPI.
- Sélectionnez Traiter | Améliorez le contraste et définissez le pixel saturé sur 0,35.
- Cliquez sur Traiter | Binaire | Convertir en masque. Choisissez Binaire | Remplissez les trous et cliquez sur Analyser | Analysez les particules. Définissez la taille sur 10-Infinity.
- Dans le gestionnaire de retour sur investissement, cliquez sur Afficher tout.
- Recherche de foyers RPA2 dans le noyau (Figure 4G, H et vidéo supplémentaire S1)
- Ouvrez l’image RPA2.
- Choisissez Processus | Trouvez Maxima. Définissez la proéminence sur une valeur qui met en évidence les foyers RPA2 (entre 500 et 750), en les séparant de l’arrière-plan.
- Enfin, cliquez sur le bouton Mesurer dans le Gestionnaire de retour sur investissement.
- Calculez le nombre total de foyers d’ADNss nucléaire en divisant la valeur de la colonne RawinDen par 255 (la valeur maximale de l’intensité des pixels dans chaque foyer).
- Effectuez des analyses statistiques à l’aide de l’outil logiciel statistique de votre choix.
REMARQUE : Exclure de l’analyse toutes les cellules EdU positives et les masques DAPI mal segmentés.
- Fabriquer des masques nucléaires à l’aide de la coloration DAPI (figure 4A-F et vidéo supplémentaire S1).
Résultats
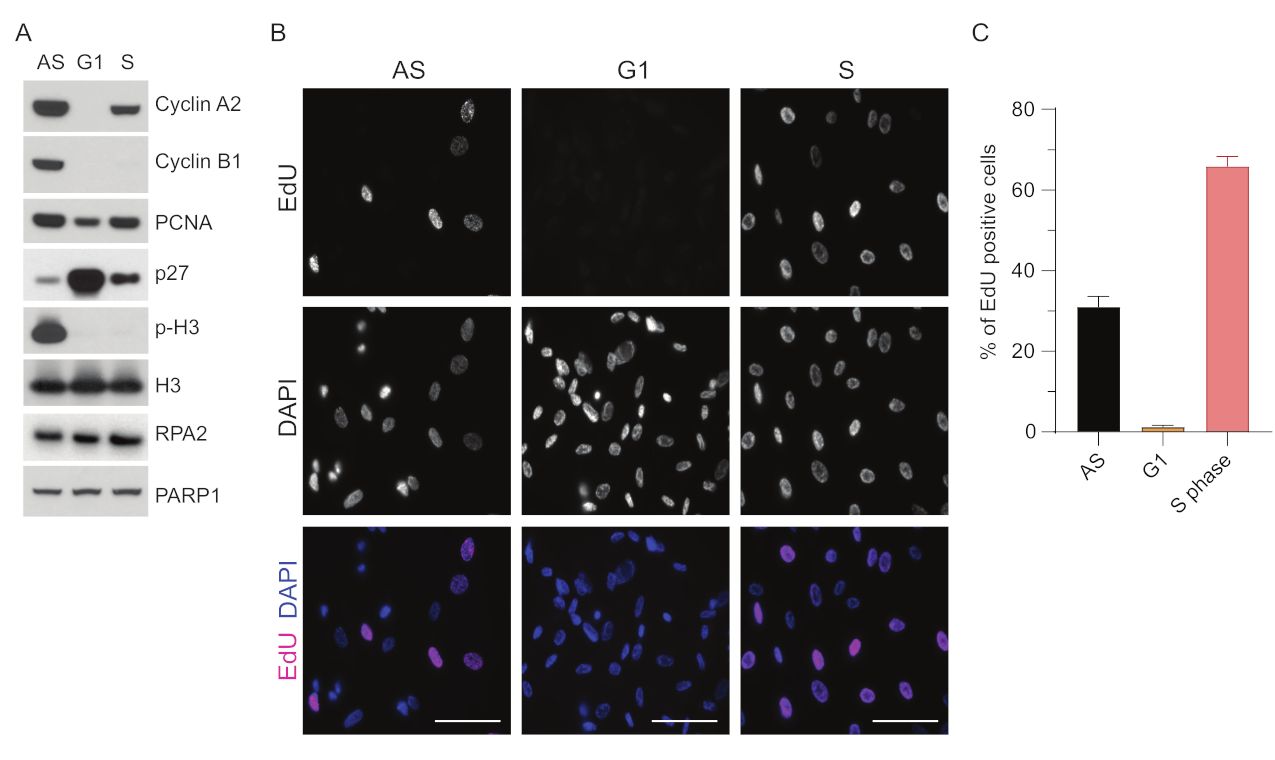
Pour surmonter les limites de la détection de l’ADNss dans G1, nous avons utilisé RPA2, qui améliore à la fois la spécificité et l’intensité de la détection des foyers d’ADNss35. Pour obtenir une synchronisation cellulaire précise, nous avons utilisé des cellules RPE1 qui peuvent être efficacement affamées de sérum et synchronisées en phase G0. Ils peuvent ensuite être amenés à réintégrer le cycle cellulaire par l’ajout de sérum après privation de sérum. Pour confirmer l’efficacité de la synchronisation, nous avons marqué les cellules avec de l’EdU et leur contenu en ADN avec de l’iodure de propidium. Nous avons ensuite recueilli des résultats qualitatifs et quantitatifs par cytométrie en flux (figure supplémentaire S1A). Les diagrammes à points montrent qu’après 72 h de privation de sérum, ~98% des cellules sont en phase G0. Après l’ajout de milieux contenant du sérum pendant 6 h, les cellules réintègrent le cycle cellulaire (comme le montre l’augmentation des niveaux de p27 dans la figure 1A), ayant ~ 97 % de cellules en G1, alors qu’elles n’ont que <1 % de cellules en phase S, <2 % de cellules en phase G2 (figure supplémentaire S1A). Après 20 à 28 h d’ajout de sérum aux cellules, celles-ci passent progressivement en phase S, comme le montrent les diagrammes de cytométrie en flux (Figure supplémentaire S1A). Ce protocole de synchronisation cellulaire donne une population G1 pure à ~97 % (6 h après l’ajout de sérum après 72 h de privation de sérum). Pour valider davantage l’efficacité de la synchronisation, nous avons comparé l’expression des marqueurs du cycle cellulaire après la libération du sérum à l’aide du western blot (Figure 1A et Figure supplémentaire S1B) et, en parallèle, nous avons effectué un test d’incorporation d’EdU pour visualiser la réplication de l’ADN. La coloration EdU met également en évidence l’efficacité de la synchronisation et l’absence de réplication de l’ADN en phase G1 (Figure 1B,C).
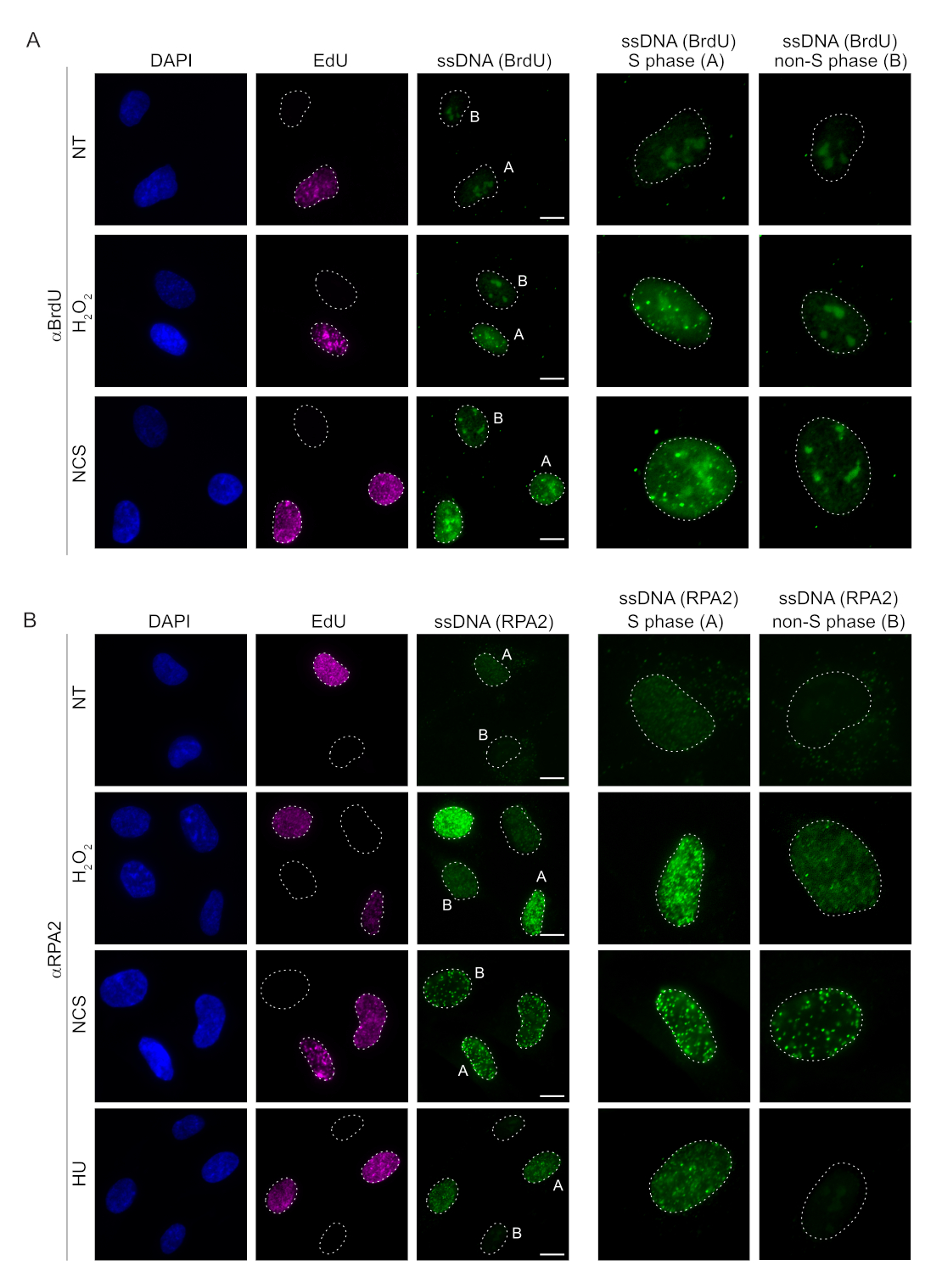
Les méthodes conventionnelles de détection de l’ADNss dans les cellules de mammifères reposent sur la détection de BrdU dans l’ADNss. La figure 2A montre qu’après le traitement par H2O2 et la néocarzinosatine (NCS), les foyers BrdU n’étaient détectables que dans les cellules en phase S, tandis qu’aucun foyer d’ADNss n’était détectable dans les cellules en phase non S. La coloration de l’anticorps BrdU a également montré une coloration nucléolaire de fond notable qui a pu être détectée dans tous les noyaux, indépendamment de l’étape du cycle cellulaire ou des traitements appliqués. En utilisant le protocole de clic EdU décrit ici, nous n’avons pas pu détecter de foyers EdU et BrdU co-localisés, ce qui est évident dans les échantillons non traités de la figure 2A. Pour exclure complètement tout signal BrdU résultant d’une réactivité croisée, nous avons évité le marquage EdU et avons plutôt utilisé la cycline A2 comme marqueur S-G2. Cependant, la coloration de la cycline A2 n’a pas permis la pré-extraction de CSK, et dans ces conditions, nous n’avons pas vu de foyers de BrdU, même après un stress génotoxique (Figure supplémentaire S2A). Cela met en évidence le fait que la pré-extraction CSK est nécessaire pour la coloration de l’ADNss à base d’anti-BrdU. À titre de contrôle, nous avons testé la coloration des anticorps BrdU dans des conditions de dénaturation. Cela ouvre l’ADN pour exposer le BrdU incorporé, ce qui révèle que le BrdU a été uniformément incorporé (Figure supplémentaire S2B).
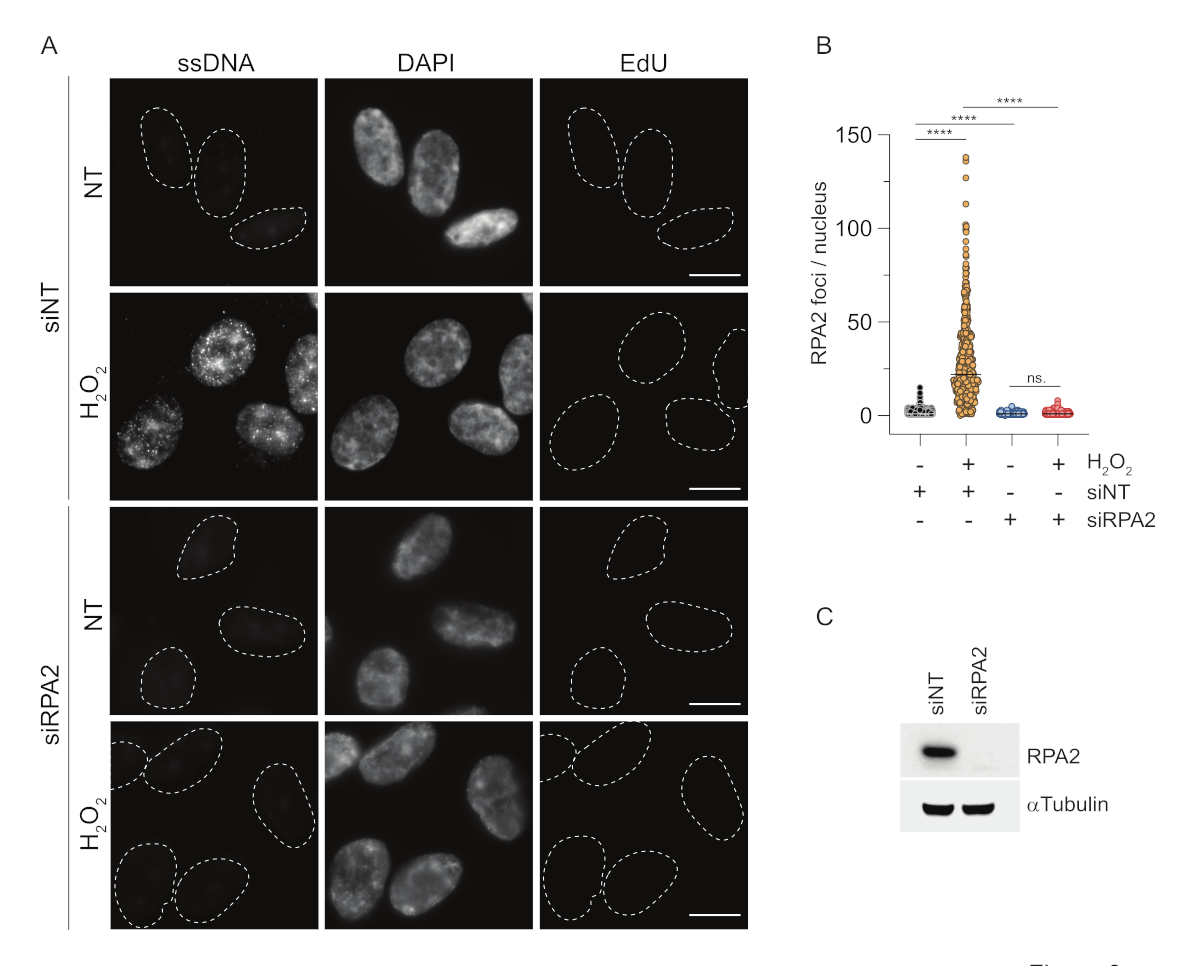
En revanche, la coloration RPA2 montre la formation de foyers dépendants de NCS et H2O2 non seulement dans la phase S mais aussi dans d’autres phases du cycle cellulaire (Figure 2B). En tant que témoin, nous avons également traité les cellules avec de l’HU, qui ne provoque que l’accumulation d’ADNss dans les cellules en cours de réplication. Comme prévu, nous n’avons détecté qu’une augmentation du signal lors du traitement par HU avec l’anticorps RPA2 dans les cellules EdU-positives, ce qui souligne la spécificité de cette approche. L’anticorps RPA2 peut également détecter la formation naturelle d’ADNss pendant la réplication en l’absence de stress génotoxique exogène (Figure 2B). La nature très sensible de l’anticorps RPA2 nous a incités à essayer de l’utiliser dans la phase G1 où la coloration BrdU conventionnelle n’a pas permis de détecter de signal lors d’un stress génotoxique (Figure supplémentaire S2C). La figure 3A montre que la formation de foyers d’ADNss lors du traitement par H2O2 était détectable lors de l’utilisation d’un anticorps anti-RPA2, même en G1. Il y a eu une augmentation significative du nombre de foyers de RPA2 dans ces noyaux lors du traitement par H2O2 (Figure 3B). Ces foyers étaient spécifiques à la RPA2 car le silence de la RPA2 abolissait le signal IF (Figure 3A,B). La figure 3C et la figure supplémentaire S1C montrent l’efficacité de l’inactivation de la RPA2 dans ces cellules. Par rapport aux méthodes conventionnelles, la détection de l’ADNss basée sur la RPA2 est très sensible, et son application peut donc être étendue aux cellules de la phase G1.

Figure 1 : Efficacité de synchronisation des cellules RPE1 après privation de sérum. (A) Les immunoblots montrent les niveaux de protéines indiqués dans les cellules RPE1 synchronisées en phase asynchrone, G1 et S. (B) Des images représentatives montrent des cellules RPE1 asynchrones, G1 et synchronisées en phase S qui ont été exposées à 10 μM EdU pendant 30 min avant la fixation et visualisées par la réaction Click-IT. Le DAPI a été utilisé pour contre-colorer l’ADN nucléaire. Barres d’échelle = 50 μm. (C) Le graphique montre le pourcentage de cellules EdU positives par rapport à la population cellulaire totale évaluée par DAPI. La barre d’erreur représente l’erreur type de la moyenne, et le nombre de noyaux analysés était le suivant : AS n = 219, G1 n = 630, S n = 437. Abréviations : RPE1 = cellules épithéliales pigmentaires rétiniennes immortalisées par hTERT ; AS = asynchrone ; EdU = 5-éthynyl-2'-désoxyuridine ; DAPI = 4',6-diamidino-2-phénylindole. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Détection de l’ADNss avec l’anticorps BrdU ou l’anticorps RPA2 lors d’une lésion de l’ADN. (A) Des images représentatives illustrent les foyers d’ADNss utilisant αBrdU (vert), les cellules de la phase S sont mises en évidence par EdU (violet) et le DAPI a été utilisé pour contre-colorer l’ADN nucléaire (bleu). Les cellules RPE1 ont été conservées dans 10 μM BrdU pendant 48 h avant tout traitement supplémentaire. Après 48 h, les cellules ont été pulsées avec 10 μM EdU pendant 30 min, suivies d’un traitement par H2O2 (250 μM) pendant 1 h ou par néocarzinosatine (0,5 μg/mL) pendant 4 h. Les cellules ont été fixées après la pré-extraction de CSK. Une ligne pointillée blanche indique la bordure de chaque noyau. Barre d’échelle = 5 μm. Les panneaux de droite sont des images agrandies des noyaux de phase S ou non S indiqués. (B) Des images représentatives illustrent les foyers d’ADNss utilisant l’anticorps αRPA2 (vert). Les cellules de la phase S sont mises en évidence par EdU (violet), et le DAPI a été utilisé pour contre-colorer l’ADN nucléaire (bleu). Les cellules RPE1 ont été pulsées avec 10 μM d’EdU pendant 30 min, suivies de 1 hH 2O2 (250 μM), 4 h d’hydroxyurée (2 mM) ou 4 h de NCS (0,5 μg/mL). Les cellules ont été fixées après la pré-extraction de CSK. Une ligne pointillée blanche indique la bordure de chaque noyau. Barre d’échelle = 10 μm. Les panneaux de droite sont des images agrandies des noyaux de phase S ou non S indiqués. Abréviations : ssDNA = ADN simple brin ; BrdU = 5-bromo-2'-désoxyuridine ; DAPI = 4',6-diamidino-2-phénylindole ; RPE1 = cellules épithéliales pigmentaires rétiniennes immortalisées par hTERT ; EdU = 5-éthynyl-2'-désoxyuridine ; NCS = Néocarzinostatine ; HU = hydroxyurée. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Détection de foyers d’ADNss en phase G1 à l’aide d’un anticorps RPA2. (A) Les cellules RPE1 ont été transfectées avec des siRNA ciblant RPA2 ou un siRNA témoin non ciblant, puis synchronisées dans G1 et marquées par impulsions avec 10 μM EdU pendant 30 min avant de les traiter avec H2O2 (250 μM) pendant 1 h là où cela était indiqué. Le DAPI a été utilisé pour contre-colorer l’ADN nucléaire. Les cellules ont été fixées après la pré-extraction de CSK. Une ligne pointillée blanche indique la bordure de chaque noyau. Barre d’échelle = 5 μm. (B) Les mesures du nombre de foyers/noyaux de RPA2 ont été effectuées à partir de deux expériences indépendantes. Seules les cellules EdU négatives ont été prises en compte lors de l’analyse. Les lignes représentent la valeur moyenne sur les tracés. Un test ANOVA non paramétrique (Kruskal-Wallis) a été réalisé pour l’analyse statistique. Les étoiles indiquent P < 0,0001. Le nombre de noyaux analysés était le suivant : siNT no H2O2 n = 513, siNT H2O2 n = 603, siRPA2 no H2O2 n = 266, siRPA2 H2O2 n = 536. (C) L’efficacité de l’inhibition de l’ARNi est démontrée dans l’immunotransfert. Abréviations : siNT = contrôle de l’ARNi non ciblant ; BrdU = 5-bromo-2'-désoxyuridine ; DAPI = 4',6-diamidino-2-phénylindole ; RPE1 = cellules épithéliales pigmentaires rétiniennes immortalisées par hTERT ; EdU = 5-éthynyl-2'-désoxyuridine. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Quantification des foyers d’ADNss à l’aide des îles Fidji. Étapes détaillées aux Fidji montrant comment évaluer le nombre de foyers RPA2 dans le noyau. (A-E) La création d’un masque nucléaire à l’aide du canal DAPI. (F-H) Seuillage pour identifier les foyers individuels d’ADNss nucléaire à partir du signal de fond. Abréviations : ssDNA = ADN simple brin ; DAPI = 4',6-diamidino-2-phénylindole. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
| Tampon cytosquelettique (CSK) | |
| TUYAUX pH 7,0 | 10 mM |
| NaCl | 100 mM |
| EDTA pH 8 | 1 mM |
| MgCl2 | 3 mM |
| D-saccharose | 300 mètres d’épaisseur |
| Triton X-100 | 0.20% |
| Cocktail d’inhibiteurs de phosphatase | 1 comprimé par 10 mL |
| Cocktail d’inhibiteurs de protéase | 1 comprimé par 10 mL |
| dilué dans du ddH2O | n/a |
| Tampon de lavage | |
| Triton X-100 | 0.05% |
| dilué dans du PBS | n/a |
| Tampon de perméabilisation | |
| Triton X-100 | 0.50% |
| dilué dans du PBS | n/a |
| Solution de fixation | |
| Paraformaldéhyde | 3.60% |
| Triton X-100 | 0.05% |
| dilué dans du PBS | n/a |
| Tampon de blocage | |
| Albumine sérique bovine (BSA) | 5% |
| Triton X-100 | 0.10% |
| dilué dans du PBS | n/a |
| Cocktail de réaction Click-iT Plus | |
| 1x tampon de réaction Click-iT | 435 mL |
| Solution PCA Alexa Fluor | 5 ml |
| Prémélange protecteur CuSO4-cuivre | 10 ml |
| 1x additif tampon Click-iT | 50 ml |
| Volume total | 500 ml |
Tableau 1 : Composition des tampons utilisés dans ce protocole.
Figure supplémentaire S1. (A) Les cellules RPE1 ont été synchronisées à la phase G0 en utilisant la privation de sérum pendant 72 h, puis libérées dans différentes phases du cycle cellulaire en réintroduisant le sérum. Les diagrammes à points montrent les cellules en phases G0/G1, S ou G2/M, où les heures indiquent le temps qui suit la ré-addition de sérum après la famine sérique. Le graphique de droite montre le pourcentage de cellules G0/G1, S et G2/M dans chaque condition. L’analyse FACS a été réalisée à l’aide d’un kit de prolifération cellulaire disponible dans le commerce utilisant de l’EdU et de l’iodure de propidium selon les recommandations du fabricant. (B) Balayage par transfert Western non recadré pour la figure 1. Les chiffres montrent les marqueurs de poids moléculaire en kDa. PARP1 a été utilisé comme contrôle de charge et chargé sur le gel qui a également été développé contre CCNA2, p27 (encore dénudé pour le PCNA) et pH3 (S10) (encore dépouillé pour H3) en coupant la membrane. Le CCNB1 et le RPA2 ont été chargés sur un gel séparé, en utilisant la même quantité de lysat de protéines pour assurer la comparabilité. (C) Balayage par transfert Western non recadré pour la figure 3. Les chiffres montrent les marqueurs de poids moléculaire en kDa. Abréviation : EdU = 5-éthynyl-2'-désoxyuridine. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire S2 : (A) Des images représentatives illustrent les foyers d’ADNss à l’aide de l’anticorps BrdU (vert) ; Les cellules de la phase S sont mises en évidence par la cycline A2 (rouge) ; et le DAPI a été utilisé pour contrer la coloration de l’ADN nucléaire (bleu). Les cellules RPE1 ont été conservées dans 10 μM BrdU pendant 48 h avant un traitement ultérieur. Après 48 h, les cellules ont été traitées avec H2O2 (250 μM) pendant 1 h ou avec de la néocarzinosatine (0,5 μg/mL) pendant 4 h avant fixation. Une ligne pointillée blanche indique la bordure de chaque noyau. Barre d’échelle = 5 μm. (B) Coloration BrdU des cellules RPE1 avec et sans condition de dénaturation. Les cellules RPE1 asynchrones ont été prétraitées avec 10 μM BrdU pendant 48 h. Barre d’échelle = 10 μm. (C) Les mesures du nombre de foyers/noyaux BrdU ont été effectuées à partir de deux expériences indépendantes dans des cellules RPE1 synchronisées G1. Seules les cellules EdU négatives ont été prises en compte lors de l’analyse. Les lignes représentent la valeur moyenne sur les tracés. Un test ANOVA non paramétrique (Kruskal-Wallis) a été réalisé pour l’analyse statistique. Le « ns » indique une différence non significative. Le nombre de noyaux analysés était le suivant : NT n = 52, NCS n = 105, H2O2 n = 82. Abréviations : siNT = contrôle de l’ARNi non ciblant ; BrdU = 5-bromo-2'-désoxyuridine ; DAPI = 4',6-diamidino-2-phénylindole ; RPE1 = cellules épithéliales pigmentaires rétiniennes immortalisées par hTERT ; NCS = Néocarzinostatine. Veuillez cliquer ici pour télécharger ce fichier.
Vidéo supplémentaire S1 : Enregistrement d’écran de l’analyse des foyers RPA2 basée aux Fidji. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Le maintien d’une culture cellulaire saine et exempte de mycoplasmes est essentiel pour toutes les expériences décrites ci-dessus. Les cellules RPE1 ont une forte fixation aux articles en plastique traités par culture tissulaire dans des milieux de culture normaux ; Cependant, leurs caractéristiques de liaison diminuent considérablement lorsqu’elles sont conservées dans des conditions sans sérum. De plus, pour capturer des images haute résolution des foyers d’ADNss au microscope, les cellules doivent être plaquées sur un verre de protection de 0,17 mm d’épaisseur, qui n’est pas suffisamment hydrophile pour permettre une fixation correcte des cellules RPE1. Sans cellules correctement aplaties et uniformément réparties, il est très difficile de visualiser les foyers individuels d’ADNss. Par conséquent, il est essentiel de choisir le matériau de revêtement approprié (par exemple, la vitronectine) et de laisser suffisamment de temps (6 à 12 h) pour que les cellules se propagent et se fixent après les avoir libérées en phase G1.
Une partie difficile du protocole consiste à obtenir des cellules RPE1 homogènes synchronisées avec G1. Cela nécessite deux étapes essentielles. Tout d’abord, pour une famine sérique efficace, les cellules doivent être trypsinisées, soigneusement lavées avec du PBS et directement ensemencées sur de nouvelles boîtes de culture tissulaire à l’aide d’un milieu sans sérum. Le lavage des cellules directement dans des boîtes de culture tissulaire pour éliminer le sérum ne donnera pas une synchronisation efficace de G0. Deuxièmement, lors de la libération des cellules en phase G1, les cellules doivent être trypsinisées à nouveau et ensemencées sur des plaques de culture de tissus frais. De même, le simple fait de changer le milieu et d’ajouter un milieu de culture contenant du sérum aux cellules n’entraînera pas une entrée synchrone de G1. De plus, pour une entrée correcte de G1, la densité d’ensemencement des cellules sur les verres de protection enduits doit être à certains niveaux de confluence. Bien qu’une synchronisation cellulaire parfaite soit généralement impossible, ce protocole de synchronisation décrit ici donne une population G1 pure de ~97%. La densité de semis recommandée pour RPE1 sur une lamelle de 12 mm de diamètre est de ~4 × 104 afin d’obtenir un champ de vision homogène pour l’imagerie, avec une confluence d’environ 70 %. Une densité d’ensemencement plus élevée provoque le détachement et le « décollement » des cellules après l’extraction de CSK et entraîne un signal de fond plus élevé lors de l’acquisition de l’image.
Pour réduire tout signal de fond et obtenir un rapport signal/bruit favorable, un lavage minutieux après l’incubation des anticorps primaires et secondaires est essentiel. Étant donné que de nombreuses étapes de lavage doivent être appliquées, il est également essentiel d’éviter que le puits ne se dessèche à chaque étape de lavage. Nous minimisons cet artefact en appliquant un minimum de 0,05 % de Triton X-100 dans toutes les étapes de lavage et d’incubation. Une fois les puits asséchés, les cellules ont affiché un rapport signal/bruit altéré ; Cela conduit à un motif en forme de mosaïque au microscope et pourrait interférer avec l’évaluation. L’acquisition d’images Z-stack combinée à la déconvolution peut aider à capturer des foyers dans différents plans focaux pour améliorer l’analyse.
Les méthodes conventionnelles reposent sur la détection du BrdU incorporé dans des conditions non dénaturantes. Ces méthodes, cependant, dépendent du prétraitement des cellules avec des doses élevées de BrdU pendant au moins 1 à 2 jours (ou un temps équivalent à un cycle cellulaire complet dans la lignée cellulaire utilisée) pour assurer une incorporation génomique uniforme. Il n’est pas souhaitable qu’une incorporation étendue de BrdU puisse provoquer des interférences dans le cycle cellulaire36. Pour remédier à ces limitations, cette méthode utilise la RPA2 endogène pour détecter les foyers d’ADNss. Cette approche ne nécessite pas d’incorporation BrdU pilotée par réplication, elle peut également être utilisée dans les cellules post-mitotiques. Étant donné qu’il n’est pas nécessaire d’intégrer BrdU à grande échelle, cela permet de gagner du temps et de réduire la complexité expérimentale. En utilisant la coloration RPA2 pour visualiser l’ADNss, nous pouvons utiliser la 2′-désoxy-5-éthynyluridine (EdU) et la chimie click pour marquer la réplication de l’ADN tout en évitant une éventuelle réactivité croisée des anticorps BrdU contre EdU 27,37,38. Des précautions particulières doivent être prises pour masquer correctement l’EdU incorporé lors de la réaction de clic afin que les anticorps BrdU ne réagissent pas de manière croisée avec l’EdU27,39.
Enfin, un avantage important de l’utilisation de RPA2 au lieu de BrdU est simplement d’avoir un rapport signal/bruit supérieur par rapport à la coloration BrdU en dehors de la phase S. Nous avons constaté que la coloration BrdU non dénaturante et sa capacité à visualiser l’ADNss sont limitées à la phase S, même dans les cellules en réplication (Figure 2). L’anticorps BrdU ne se lie qu’au BrdU suffisamment exposé dans les étirements de l’ADNss. La liaison des protéines de réparation, y compris RPA2, aux étirements de l’ADNss peut supprimer ou entraver l’exposition suffisante de BrdU dans l’ADNss. Nous avons également constaté que la pré-extraction CSK est nécessaire pour la visualisation de l’ADNss à l’aide de l’anticorps BrdU. Cela est possible parce que les traces d’ADNss ne sont pas accessibles pour l’anticorps sans en retirer les composants protéiques légèrement liés.
Néanmoins, il existe certaines limitations associées à ce protocole. L’une des limites de l’utilisation de RPA2 pour la détection de l’ADNss est la nécessité d’optimiser l’étape de pré-extraction de la CSK. Non lié, l’excès de RPA2 doit être éliminé de l’ADN avant de fixer les cellules. D’une part, la sous-extraction conduit à un bruit de fond élevé en raison de la fraction protéique RPA2 qui n’est pas liée à l’ADNss. D’autre part, une surextraction entraînera une perte de signal. Pour la détection de BrdU, il ne s’agit pas d’une variable puisque BrdU est incorporé de manière stable dans l’ADN et ne peut pas être éliminé par pré-extraction. Par conséquent, le moment de la pré-extraction CSK, la quantité de Triton X-100 dans le tampon, le volume et la température à laquelle la pré-extraction est effectuée doivent être soigneusement pris en compte. La pré-extraction CSK limite également l’utilisation de la taille du noyau pour distinguer les cellules G0/G1 des cellules S/G2.
De plus, nous ne pouvons pas exclure la possibilité qu’une partie du signal provenant de RPA2 provienne de sa liaison à d’autres interacteurs protéiques de liaison à la chromatine. Il faut également tenir compte de la spécificité de l’espèce de l’anticorps RPA2. L’anticorps utilisé dans ce protocole peut reconnaître l’APR2 humaine, la souris, le rat, le hamster et le singe. Une autre limite de cette approche est que toutes les lignées cellulaires ne peuvent pas être privées de sérum pour la synchronisation G0. La plupart des lignées cellulaires cancéreuses peuvent contourner les points de contrôle du cycle cellulaire et proliférer même dans les milieux privés de sérum. Bien que la privation de sérum soit bénéfique, car elle ne cause pas de dommages à l’ADN, il faut surveiller attentivement l’efficacité de la synchronisation cellulaire pour s’assurer que l’enrichissement de phase du cycle cellulaire est approprié. Pour les cellules qui ne répondent pas à la privation sérique, d’autres méthodes de synchronisation cellulaire doivent être envisagées (par exemple, la secousse mitotique, l’inhibition de CDK1 pour l’arrêt de G2 ou des techniques non invasives telles que l’élutriation centrifuge). Une autre méthode possible consiste à utiliser l’imagerie à haut contenu pour mesurer l’EdU et la teneur en ADN nucléaire pour le profilage du cycle cellulaire des cellules asynchrones31. Il faut tenir compte des implications de l’utilisation d’autres méthodes de synchronisation pour éviter les interférences avec l’analyse en aval. Par exemple, l’utilisation d’un double bloc de thymidine ou d’aphidicoline, souvent utilisée dans la littérature, entraînera un stress de réplication et des dommages à l’ADN40.
L’étude des mécanismes de réparation de l’ADN continue d’être un point central de discussion dans les domaines du cancer et de la biologie cellulaire. Le protocole présenté ici offre une approche précieuse pour la préparation des cellules, permettant la visualisation et l’analyse quantitative de l’ADNss lors de l’exposition à des agents endommageant l’ADN. Notamment, ce protocole met en évidence l’utilisation de la protéine de liaison à l’ADNss, RPA2, démontrant sa grande spécificité pour visualiser de petites quantités de foyers d’ADNss tout en évitant une réactivité croisée indésirable dans toutes les phases du cycle cellulaire. L’utilisation de la RPA2 présente de nombreux avantages, notamment pour les chercheurs qui souhaitent analyser des cellules en phase G1 du cycle cellulaire. Ce protocole tient compte de plusieurs limitations et répond aux préoccupations liées aux interférences de signal, au bruit de fond indésirable et à la réactivité croisée lors de l’utilisation de la coloration RPA2 ou BrdU pour détecter l’ADNss.
Déclarations de divulgation
Les auteurs n’ont pas d’intérêts concurrents à déclarer.
Remerciements
Les auteurs remercient Michele Pagano pour son soutien et ses idées utiles, Ashley Chui et Sharon Kaisari pour leur lecture critique du manuscrit, et Jeffrey Estrada et Vilma Diaz pour leur soutien continu. Ce travail a été soutenu par un supplément de diversité au GM136250 de subvention des National Institutes of Health.
matériels
| Name | Company | Catalog Number | Comments |
| Alpha-tubulin antibody | Sigma-Aldrich | T6074 | primary antibody (1:5,000) |
| Axio Observer Inverted Microscope | Zeiss | na | microscope |
| Bis-Tris Plus Mini Protein Gels, 4-12% | Invitrogen | NW04127BOX | Western Blot |
| Bovine Serum Albumin | Jackson ImmunoResearch | 001-000-162 | blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Sigma-Aldrich | B5002-100MG | nucleotide analogue |
| BrdU antibody BU1/75 | Abcam | ab6326 | primary antibody (1:500) |
| CellAdhere Dilution Buffer | Stemcell Technologies | 07183 | coating reagent |
| Click-iT Plus EdU Flow Cytometry Assay Kits | Invitrogen | C10632 | flow cytomery |
| Click-iT Plus EdU Cell Proliferation Kit for Imaging, Alexa Fluor 647 dye | Thermo Fisher Scientific | C10640 | click-reaction kit |
| cOmplete ULTRA Protease inhibitor tablets | Sigma-Aldrich | 5892791001 | reagent |
| Countess 3 Automated cell counter | Thermo Scientific | AMQAX2000 | cell counter |
| Coverslip | neuVitro | GG12PRE | tissue culture |
| Cyclin A2 antibody | Santa Cruz Biotechnology | sc-271682 | primary antibody (1:1,000) for IF and WB |
| Cyclin B1 antibody | Santa Cruz Biotechnology | sc-245 | primary antibody (1:5,000) |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D2650-100ML | vehicle control |
| DMEM, high glucose, with HEPES | Gibco | 12430051 | cell culture medium for RPE cells |
| DPBS, no calcium, no magnesium | Gibco | 14190144 | the PBS used throughout the protocol |
| D-Sucrose | Thermo Fisher Scientific | bp220-1 | reagent |
| Eclipse Ti2 Series Epifluorescent Microscope | Nikon | na | microscope |
| EdU (5-Ethynyl-2'-deoxyuridine) | Thermo Fisher Scientific | C10637 | nucleotide analogue |
| Falcon 24-well plate | Corning | 351147 | tissue culture |
| Falcon Cell Culture Dishes 100 mm | Corning | 353003 | tissue culture |
| Fetal Bovine Serum, heat inactivated | Gibco | 16140071 | media supplement |
| Fiji (ImageJ) | NIH | version 1.54f | software and algorithms |
| FxCycle PI/RNase Staining Solution | Invitrogen | F10797 | PI staining |
| Goat anti-mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 555 | Thermo Fisher Scientific | A21422 | secondary antibody (1:1,000) |
| Goat anti-rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A48262 | secondary antibody (1:1,000) |
| Histone H3 antibody | Abcam | ab1791 | primary antibody (1:10,000) |
| hTERT RPE1 | ATCC | CRL-3216 | cell line |
| Hydrochloric acid | Sigma-Aldrich | H1758-100ML | reagent |
| Hydrogen peroxide 30% soultion | Sigma-Aldrich | H1009-100ML | reagent |
| Hydroxyurea,98% powder | Sigma-Aldrich | H8627-5G | reagent |
| Invitrogen Ultra Pure 0.5 M EDTA pH 8.0 | Thermo Fisher Scientific | 15-575-020 | reagent |
| Lipfectamine RNAiMAX Transfection Reagent | Invitrogen | 13778150 | transfection reagent |
| Magnesium chloride solution 1 M | Sigma-Aldrich | M1028-100ML | reagent |
| MycoFluor | Thermo Fisher | M7006 | Mycoplasma Detection Kit |
| Neocarzinostatin from Streptomyces carzinostaticus | Sigma-Aldrich | N9162-100UG | reagent |
| NuPage MES SDS Running Buffer (20x) | Invitrogen | NP0002 | Western Blot |
| onTARGETplus Human RPA2 siRNA | Dharmacon | L-017058-01-0005 | siRNA |
| p27 antibody | BD Biosciences | 610241 | primary antibody (1:1,000) |
| Paraformaldehyde aqueous solution (32%) | Electron Microscopy Sciences | 50-980-494 | fixative |
| PARP1 antibody | Cell Signaling Technology | 9542S | primary antibody (1:1,000) |
| PCNA antibody | Cell Signaling Technology | 13110S | primary antibody (1:2,000) |
| Penicillin-Streptomycin | Gibco | 15140163 | media supplement |
| pH3 antibody | Cell Signaling Technology | 3377S | primary antibody (1:2,000) |
| PhosSTOP phosphatase inhibitor tablets | Sigma-Aldrich | 4906837001 | reagent |
| PIPES Buffer 0.5 M solution, pH 7.0 | Bioworld | 41620034-1 | reagent |
| Precision Plus Protein Kaleidoscope Prestained Protein Standards | Bio-Rad | 1610395 | Western Blot |
| Prism | GraphPad | version 10 | statistical analysis and graph |
| ProLong Diamond Antifade Mountant | Thermo Scientific | P36961 | mounting media |
| Reduced serum media (Opti-MEM) | Gibco | 31985070 | used for transfection |
| Rpa32/rpa2 antibody (mouse) | EMD Millipore | NA19L | primary antibody (1:1,000) for WB |
| Rpa32/rpa2 antibody (rat) | Cell Signaling Technology | 2208S | primary antibody (1:1,000) for IF |
| Sodium Chloride solution (5 M) | Sigma-Aldrich | S5150 | reagent |
| Sodium Pyruvate (100 mM) | Gibco | 11360070 | media supplement |
| Sodium tetraborate decahydrate | Sigma-Aldrich | B3535-500G | reagent |
| Thermo Scientific Pierce DAPI Nuclear Counterstain | Thermo Scientific | 62248 | nucleic acid stain |
| Thymidine,powder | Sigma-Aldrich | T1985-1G | reagent |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | detergent |
| Trypsin-EDTA (0.5%), no phenol red | Gibco | 1540054 | cell dissociation agent |
| Vitronectin XF | Stemcell Technologies | 07180 | coating reagent |
| ZE5 Cell Analyzer | Bio-Rad | na | flow cytomery |
Références
- Hakem, R. DNA-damage repair; the good, the bad, and ugly. EMBO J. 27 (4), 589-605 (2008).
- Gutierrez, R., O'Connor, T. R. DNA direct reversal repair and alkylating agent drug resistance. Cancer Drug Resist. 4 (2), 414-423 (2021).
- Krokan, H. E., Bjoras, M. Base excision repair. Cold Spring Harb Perspect Biol. 5 (4), a012583 (2013).
- Marteijn, J. A., Lans, H., Vermeulen, W., Hoeijmakers, J. H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 15 (7), 465-481 (2014).
- Li, G. M. Mechanisms and functions of DNA mismatch repair. Cell Res. 18 (1), 85-98 (2008).
- Hustedt, N., Durocher, D. The control of DNA repair by the cell cycle. Nat Cell Biol. 19 (1), 1-9 (2016).
- Yang, W., Gao, Y. Translesion and repair DNA polymerases: diverse structure and mechanism. Annu Rev Biochem. 87, 239-261 (2018).
- Bhat, D. S., et al. Therapeutic disruption of RAD52-ssDNA complexation via novel drug-like inhibitors. NAR Cancer. 5 (2), zcad018 (2023).
- Gupta, P., Saha, B., Chattopadhyay, S., Patro, B. S. Pharmacological targeting of differential DNA repair, radio-sensitizes WRN-deficient cancer cells in vitro and in vivo. Biochem Pharmacol. 186, 114450 (2021).
- Pena-Diaz, J., et al. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol Cell. 47 (5), 669-680 (2012).
- Schroering, A. G., Edelbrock, M. A., Richards, T. J., Williams, K. J. The cell cycle and DNA mismatch repair. Exp Cell Res. 313 (2), 292-304 (2007).
- Scully, R., Panday, A., Elango, R., Willis, N. A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 20 (11), 698-714 (2019).
- Escribano-Diaz, C., et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 49 (5), 872-883 (2013).
- Genschel, J., Modrich, P. Mechanism of 5'-directed excision in human mismatch repair. Mol Cell. 12 (5), 1077-1086 (2003).
- Hu, J., et al. Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J Biol Chem. 288 (29), 20918-20926 (2013).
- Huertas, P., Jackson, S. P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 284 (14), 9558-9565 (2009).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in DNA replication with putative roles in cancer. Int J Mol Sci. 20 (1), 74 (2018).
- Symington, L. S. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol. 6 (8), a016436 (2014).
- Liu, Y., et al. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem. 280 (5), 3665-3674 (2005).
- Wold, M. S., Kelly, T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc Natl Acad Sci U S A. 85 (8), 2523-2527 (1988).
- Wienholz, F., Vermeulen, W., Marteijn, J. A. Amplification of unscheduled DNA synthesis signal enables fluorescence-based single cell quantification of transcription-coupled nucleotide excision repair. Nucleic Acids Res. 45 (9), e68 (2017).
- Wold, M. S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 66, 61-92 (1997).
- Chen, R., Wold, M. S. Replication protein A: single-stranded DNA's first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays. 36 (12), 1156-1161 (2014).
- Kang, Y., et al. Alteration of replication protein A binding mode on single-stranded DNA by NSMF potentiates RPA phosphorylation by ATR kinase. Nucleic Acids Res. 51 (15), 7936-7950 (2023).
- Kilgas, S., Kiltie, A. E., Ramadan, K. Immunofluorescence microscopy-based detection of ssDNA foci by BrdU in mammalian cells. STAR Protoc. 2 (4), 100978 (2021).
- Madabhushi, R., Pan, L., Tsai, L. H. DNA damage and its links to neurodegeneration. Neuron. 83 (2), 266-282 (2014).
- Liboska, R., Ligasova, A., Strunin, D., Rosenberg, I., Koberna, K. Most anti-BrdU antibodies react with 2'-deoxy-5-ethynyluridine -- the method for the effective suppression of this cross-reactivity. PLoS One. 7 (12), e51679 (2012).
- Biehs, R., et al. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol Cell. 65 (4), 671-684.e5 (2017).
- Cruz-Garcia, A., Lopez-Saavedra, A., Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 9 (2), 451-459 (2014).
- Ercilla, A., et al. Physiological tolerance to ssDNA enables strand uncoupling during DNA replication. Cell Rep. 30 (7), 2416-2429.e7 (2020).
- Lezaja, A., et al. RPA shields inherited DNA lesions for post-mitotic DNA synthesis. Nat Commun. 12 (1), 3827 (2021).
- Mukherjee, B., Tomimatsu, N., Burma, S. Immunofluorescence-based methods to monitor DNA end resection. Methods Mol Biol. 1292, 67-75 (2015).
- Ochs, F., et al. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol. 23 (8), 714-721 (2016).
- Raderschall, E., Golub, E. I., Haaf, T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad Sci U S A. 96 (5), 1921-1926 (1999).
- Forment, J. V., Walker, R. V., Jackson, S. P. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytometry A. 81 (10), 922-928 (2012).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Sci Rep. 6, 19567 (2016).
- Aten, J. A., Bakker, P. J., Stap, J., Boschman, G. A., Veenhof, C. H. DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem J. 24 (5), 251-259 (1992).
- Podgorny, O., Peunova, N., Park, J. H., Enikolopov, G. Triple S-phase labeling of dividing stem cells. Stem Cell Reports. 10 (2), 615-626 (2018).
- Cappella, P., Gasparri, F., Pulici, M., Moll, J. Cell proliferation method: click chemistry based on BrdU coupling for multiplex antibody staining. Curr Protoc Cytom. Chapter 7, (2008).
- Ligasova, A., Koberna, K. Strengths and weaknesses of cell synchronization protocols based on inhibition of DNA synthesis. Int J Mol Sci. 22 (19), 10759 (2021).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.