
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Utilisation d’unique molécule fluorescente hybridation In Situ (FISH-SM) afin de quantifier et Localize ARNm dans les ovocytes murin
Dans cet article
Résumé
Pour compter reproductible les numéros des ARNm dans les ovocytes, in situ en fluorescence seule molécule RNA hybridation (RNA-poisson) a été optimisée pour les cellules non adhérentes. Ovocytes ont été recueillis, hybridaient avec des sondes spécifiques de transcription et quantifiées à l’aide d’un logiciel de quantification d’image.
Résumé
Cours méthodes couramment utilisées pour quantifier les ARNm d’ovocytes et d’embryons incluent réaction en chaîne de polymérase de transcription inverse numérique (dPCR), RT-PCR quantitative, en temps réel (RT-qPCR) et le séquençage de RNA. Lorsque ces techniques sont effectuées à l’aide d’un seul ovocyte ou embryon, ARNm de faibles copies n’est pas détectées avec fiabilité. Pour surmonter ce problème, des ovocytes ou des embryons peuvent être regroupés ensemble pour l’analyse ; Toutefois, cela conduit souvent à la grande variabilité entre les échantillons. Dans ce protocole, nous décrivons l’utilisation de l’hybridation in situ fluorescence (FISH) à l’aide de chimie de l’ADN ramifié. Cette technique identifie la répartition spatiale des ARNm dans les cellules individuelles. Lorsque la technique est associée à la recherche de Spot et logiciels de suivi, l’abondance des ARNm dans les cellules peut également être quantifiée. En utilisant cette technique, il existe une variabilité réduite au sein d’un groupe expérimental et moins des ovocytes et des embryons sont nécessaires pour détecter des différences significatives entre les groupes expérimentaux. Disponible dans le commerce ramifiée ADN-SM-kits de poissons ont été optimisés pour détecter les ARNm de coupes de tissus ou cellules adhérentes sur des lames. Cependant, les ovocytes n’adhèrent pas efficacement aux diapositives et certains réactifs de la trousse ont été trop sévères, aboutissant à la lyse de l’ovocyte. Pour éviter cette lyse, plusieurs modifications ont été apportées au kit de poissons. Plus précisément, tampons conçus pour l’immunofluorescence des ovocytes et des embryons de la perméabilisation et lavage ovocyte remplacé les tampons exclusifs. La perméabilisation, lavages et incubations avec amplificateur et sondes ont été réalisées en plaques 6 puits et ovocytes ont été placés sur des lames à la fin du protocole à l’aide de supports de montage. Ces modifications ont été en mesure de surmonter les limitations de la trousse disponible dans le commerce, en particulier, la lyse de l’ovocyte. Pour avec précision et de façon reproductible, compter le nombre des ARNm dans les ovocytes, logiciels a été utilisé. Ensemble, ce protocole représente une alternative à la PCR et séquençage de comparer l’expression des transcriptions précises dans des cellules individuelles.
Introduction
La transcriptase inverse amplification génique (PCR) a été l’étalon-or pour le dosage de l’ARNm. Deux essais, digital PCR (dPCR)1 et quantitative, real time PCR (qPCR)2 sont actuellement utilisés. De ces deux techniques PCR, dPCR a une plus grande sensibilité que qPCR suggérant qu’il pourrait être utilisé pour mesurer l’abondance d’ARNm dans les cellules. Toutefois, dans nos mains, dPCR analyse des ARNm de faible abondance dans les piscines de 5 à 10 ovocytes par chaque échantillon expérimental a produit des données avec faible reproductibilité et de la forte variation3. C’est probablement à cause de l’erreur expérimentale, associé à l’extraction de l’ARN et l’efficacité de la transcriptase inverse. Séquençage de RNA a également effectué à l’aide d’une seule souris et les ovocytes humains4,5. Cette technique requiert des étapes d’amplification de cDNA requis pour la génération de la bibliothèque qui augmente probablement la variabilité au sein d’un groupe expérimental. En outre, les transcriptions de faible abondance ne peuvent pas être détectables. Bien que les prix de séquençage ont baissé ces dernières années, il peut encore être coût prohibitif en raison du coût élevé des analyses bioinformatiques. Enfin, localisation de l’ARNm est un processus dynamique avec des changements spatiaux contribuant à la fonction de protéine6. Par conséquent, nous partîmes à adopter une technique qui produirait des mesures quantitatives précises et reproductibles et localisation des différents mRNAs dans les ovocytes unique.
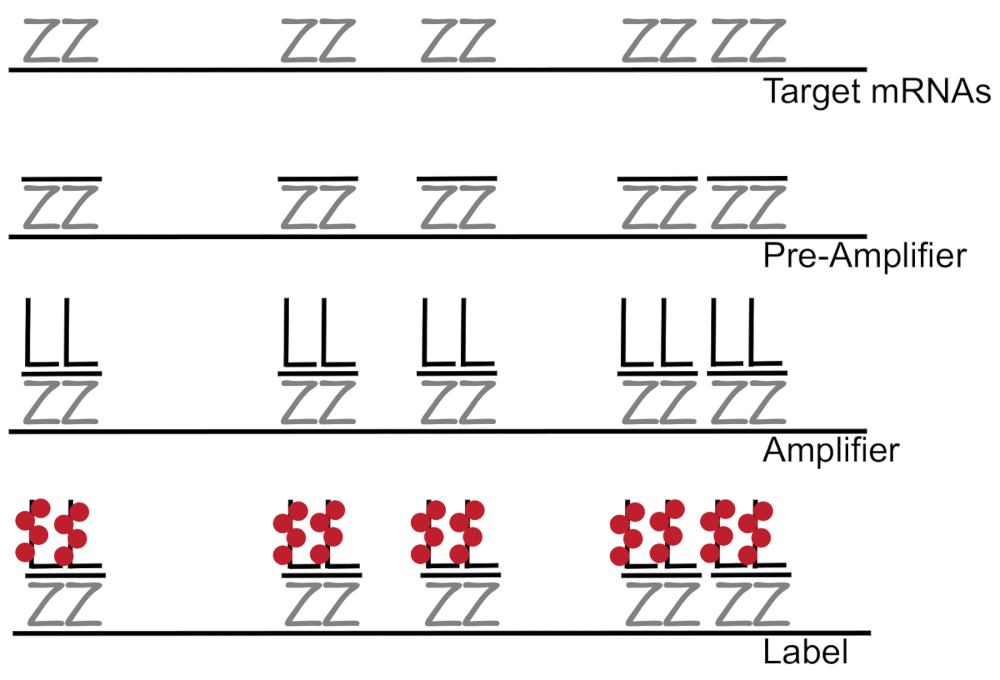
ADN ramifié couplé à l’hybridation in situ fluorescence amplifie le signal de fluorescence plutôt qu’amplification RNA/cDNA habilitante détection seul ARNm dans les cellules individuelles 7,8,9. L’analyse est réalisée à travers une série d’hybridation, amplification (en utilisant l’ADN ramifié) et fluorescence d’étiquetage afin d’amplifier le signal de fluorescence7étapes. La technique commence par liaison de paires de sonde oligonucléotide de 18 à 25 la base qui sont complémentaires à un8,3,d’ARNm spécifiques10. Quinze à vingt paires de sonde sont conçues pour chaque spécificité de veiller à ce que de transcription pour la transcription de la cible. L’hybridation de l’ARNm spécifique est suivie d’un préamplificateur et amplificateur des sondes qui forment une configuration ramifiée. Environ 400 étiquette fluorophores lier à chaque amplificateur, résultant en une 8000-fold augmentation de fluorescence permettant la détection des différents mRNAs (Figure 1)11.

Figure 1 : schéma du protocole SM-poisson. Séquentielle hybridation d’une sonde spécifique de transcription, ramifiée amplificateur ADN et fluorophore vers une cible apparaît à l’ARNm. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Des études antérieures utilisant des molécules simples fluorescence in situ hybridation (SM-FISH) localisée β-actine ARNm dans différents neurones12 et virus du papillome humain ADN dans le cancer du col utérin cellule lignes7. Les logiciels Spot trouver et Tracking Program identifie chaque signal fluorescent ponctuée et a été utilisé avec succès pour quantifier le nombre des ARNm dans chaque cellule3,13.
Selon les résultats de détection de l’ARNm dans les neurones12, nous avons supposé que SM-poisson s’avérera un outil utile pour quantifier les niveaux de transcription en murines ovocytes et d’embryons, y compris les ARNm de faible abondance. Cependant, la technique est optimisée pour une utilisation avec les cellules adhérentes fixes et formaldéhyde fixé paraffine intégrée des sections de tissu (FFIP). Ovocytes ne peuvent adhérer à une diapositive, même lorsqu’ils sont revêtus de la Poly-L-lysine. En outre, ils sont plus fragiles que les cellules somatiques et des sections de tissu ce qui entraîne la lyse des cellules lorsqu’elles sont soumises à certains des tampons exclusifs dans les kits disponibles dans le commerce3. Pour surmonter ces défis, les ovocytes ont été fixe et transférées manuellement entre les gouttes des tampons. En outre, tampons perméabilisation et de lavage dans les kits ont été remplacés pour réduire la lyse cellulaire. Sondes prédéfinis sont achetées aux côtés de la trousse de poissons ou des relevés de notes spécifiques peuvent être demandés. Chaque propriétaire de sonde est disponible dans l’un des trois canaux de fluorescence (C1, C2 et C3) permettant de multiplexage. Dans l’expérience actuelle, murins ovocytes étaient chiffrés à l’aide d’une sonde de C2 Nanog et une sonde de C3 Pou5f1 et double marquage. Ces sondes ont été choisis sur l’expression signalée de Nanog et Pou5f1 d’ovocytes et d’embryons. À l’issue de la procédure d’hybridation, ovocytes ont été placés en gouttes de supports de montage de l’anti-fondu pour application à histologiques. Images confocales ont été utilisées pour quantifier le nombre de signaux fluorescents ponctuées, qui représentent différents mRNAs. En plus de la quantification de l’ARNm, imagerie a également montré la distribution spatiale de l’ARNm spécifique dans la cellule, quelles autres méthodes de quantification ARN sont incapables d’atteindre. Cette technique s’est avérée pour avoir faible variabilité au sein d’un groupe expérimental permettant l’utilisation de plus petits nombres d’ovocytes dans chaque groupe expérimental afin d’identifier des différences significatives entre les groupes expérimentaux3.
Protocole
Procédures d’animaux ont été examinés et approuvés par le Comité de l’urbanisme à l’Université de Nebraska-Lincoln et d’institutionnels animalier et toutes les méthodes ont été effectuées conformément aux règlements et aux lignes directrices pertinentes. Pour cette étude, tel CD-1 souris avaient accès ad libitum à chow rongeur normaux et de l’eau ; Ils ont été maintenues dans un 12:12 sombres : light cycle.
1. préparation du média requis
- Pour les supports de base (OMM), ajouter 100 mM NaCl, KCl 5 mM, 0,5 mM KH2PO4et 1,7 mM CaCl2-2 H2O à 100 mL d’eau stérile.
NOTE : Support de l’OMM peut être stocké pendant environ 1 mois. - Pour les médias complètes (OMOPS), ajouter acide 3-morpholinopropane-1-sulfonique de 20 mM (MOPS), 1,2 mM MgSO4-7 H2O, glucose de 0,5 mM, 6 mM L-lactate, 1 mM ala-gln, taurine, 0,1 mM 1 x non essentiels des acides aminés (NEAA), (acide éthylènediaminetétraacétique) 0,01 mM EDTA), 10 µM acide alpha lipoïque, 10 gentamicine µg/mL dilué, 21 mM 1 M NaOH, 5 mM NaHCO3, 0,2 mM 0,5 mM le Citrate, le Pyruvate de 4 mg/mL FAF BSA pour un 01:10 dilution de l’OMM à l’eau stérile pour un volume total de 100 mL. Stériliser le milieu avec un filtre de 0,22 µM.
Remarque : Les OMOPS peuvent être stockés pendant 1 semaine. - Pour le support d’exploitation (HM) Ajoutez 5 % sérum de veau fœtal à OMOPS. Faire 2 mL HM par souris.
- Pour la solution de l’hyaluronidase ajouter 0,1 mg/mL d’hyaluronidase dérivé de testicules bovins, à 1 mL HM.
- Pour le tampon de fixation combiner paraformaldéhyde à 4 % dans 10 mL de solution de 1 PBS x 0,1 % embryon grade polyvinylpyrrolidone (PVP)14.
- 50 mL de tampon de lavage (WB), ajoutez surfactant non ionique de 0,1 % et 0,1 % PVP 1 x PBS14.
- Pour préparer 10 mL de tampon perméabilisation, ajouter 1 % non ioniques tensioactif de PBS 1 x14.
Remarque : Les tampons de lavage et perméabilisation décrits ci-dessus remplacent les tampons exclusifs dans les kits disponibles dans le commerce.
2. collecte des ovocytes ovulés des souris femelles
- Préparation :
- Stimuler les souris femelles à 5 à 8 semaines d’âge par une injection intrapéritonéale (IP) de 5 UI gonadotrophine chorionique équine (eCG) suivie de 5 UI gonadotrophine chorionique humaine (hCG) 44-48 h plus tard15,16.
- Garder 35 mm plats de pétri contenant 2 mL de HM sur une plaque chauffante de 37 ° C. Une pipette, déposer 100 µL de HM contenant dilué hyaluronidase suivie de trois gouttes de 50 µL de HM sans hyaluronidase dans un plat de pétri de 60 mm. Placer les plaques sur plaque de gouttes contenant sur le réchauffement de 37 ° C avant utilisation.
Remarque : Les gouttes de hyaluronidase il faudrait juste avant la dissection de chaque paire d’oviducte pour éviter l’évaporation et la concentration des composants du HM avec ou sans la hyaluronidase.
- Euthanasier souris, 16 h après l’injection Intrapéritonéale de l’hCG, à l’aide de surdosage isoflurane suivie de dislocation cervicale.
- Nettoyer la souris à l’aide d’éthanol à 70 %. Exposer la cavité abdominale et de visualiser l’appareil reproducteur féminin. Tenez l’ovaire avec une pince et enlever les ligaments utérins et l’excès de tissu adipeux d’autour de l’ovaire. Couper l’oviducte de l’utérus et placer la paire de l’ovaire-oviducte dans le HM chaud dans le plat de 35 mm.
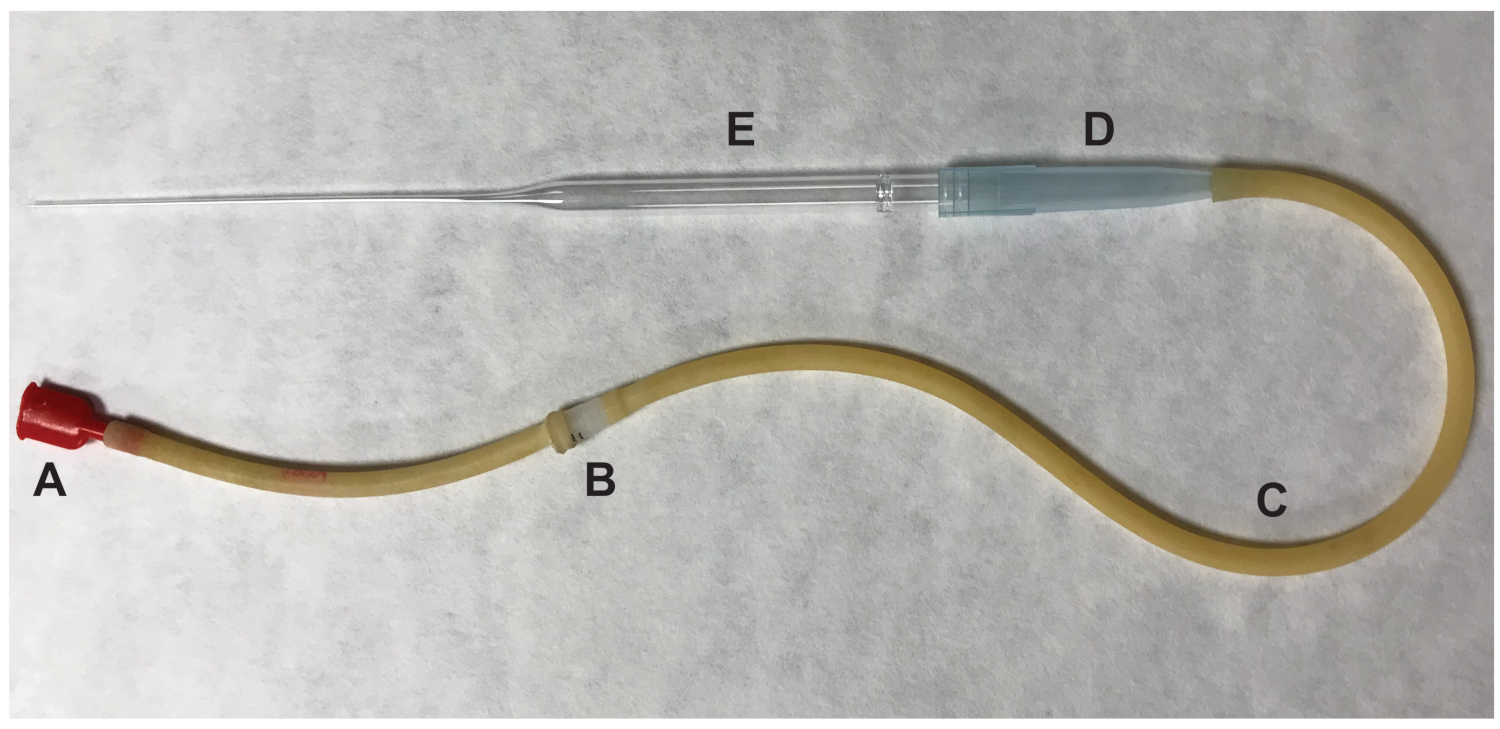
- Enlever l’ovaire et n’importe quel tissu adipeux environnant. Déchirer l’ampoule gonflée de l’oviducte à l’aide d’une aiguille de calibre 27 ½ pouce. Pousser l’oviducte à l’endroit de la déchirure et les complexes de cellule-ovocyte cumulus (COC) seront expulsés. Transférez les ovocytes ovulés, qui sont censés être en métaphase II (MII) de la méiose, la baisse de 100 μL contenant des médias HM avec hyaluronidase à l’aide d’une pipette de bouche (Figure 2).

Figure 2 : Certaines parties de la pipette de bouche utilisé pour transférer des ovocytes. (A) bouche totale (B) 0,22, euh, 4 mm (C) aspirateur tube (D) 1000 μL pipette extrémité du filtre (E) 9" pipette Pasteur. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
- Pipetter complexes cumulus-ovocytes cellule MII haut et en bas la hyaluronidase contenant HM avec la pipette de bouche pour déloger les cellules du cumulus. Transférer chaque ovocyte, une fois qu’ils sont dépourvus de cellules du cumulus à un lavage drop HM contenant uniquement à l’aide de la pipette de la bouche. Répétez cette opération pour chaque goutte de lavage. Ne transférez pas les ovocytes fragmenté ou transparent15.
Remarque : Il est important de transférer les ovocytes de chaque goutte dans le plat de 35 mm avec comme petite HM que possible. Cela est vrai pour chaque cession dans le protocole. Les ovocytes MII ne doivent pas rester dans l’hyaluronidase contenant le support HM pendant plus d’une minute.
3. poisson-SM la coloration des ovocytes
- Difficulté des ovocytes dans un puits individuel d’une plaque de 6 puits contenant 500 µL de tampon de Fixation. Plongez les 20 ovocytes ou moins dans le puits. Incuber pendant 20 min à température ambiante.
Remarque : Chaque étape de coloration SM-poisson se trouve dans un puits individuel dans une plaque 6 puits conique. S’assurer que les ovocytes sont complètement immergés dans des tampons et non flottante sur le dessus de la mémoire tampon. Chaque étape doit être effectuée avec 20 ovocytes ou moins dans chaque puits. - Transférez les ovocytes fixes à 500 μL de tampon de lavage (WB décrit à l’étape 1.6) pendant 10 min chaque. Répéter 2 fois plus.
- Incuber les ovocytes dans le tampon de la perméabilisation pendant 30 min à température ambiante.
NOTE : Le tampon de la perméabilisation décrit à l’étape 1.7 remplace le tampon de perméabilisation de bienséance.- Recueillir des ensembles de sonde et tourner rapidement vers le bas dans une micro-centrifugeuse. Réchauffer chaque sonde définie pendant 10 min dans un bain d’eau de 40 ° C ou de l’incubateur. Laisser refroidir à la température ambiante.
Remarque : Cette étape doit être effectuée au cours de l’incubation de la perméabilisation
- Recueillir des ensembles de sonde et tourner rapidement vers le bas dans une micro-centrifugeuse. Réchauffer chaque sonde définie pendant 10 min dans un bain d’eau de 40 ° C ou de l’incubateur. Laisser refroidir à la température ambiante.
- Laver les ovocytes dans 500 µL de WB pendant 10 min à température ambiante.
- Transférez les ovocytes dans 80 μL de protéase III tampon (disponible dans le kit), qui est dilué 1:8 à 1 X PBS, pendant 30 min à température ambiante.
Remarque : Le volume de 80 µL couvre adéquatement le fond d’un puits individuel dans une plaque 6 puits. - Laver les ovocytes dans 500 µL de WB pendant 10 min à température ambiante.
- Diluer les ensembles de sonde chauffée pour Nanog, Pou5f1 et DapB (un gène contrôle négatif), 01:50 dans le diluant de la sonde. Incuber les ovocytes dans 80 μL de la sonde de transcription spécifique pendant 2 heures à 40° C.
Remarque : Chaque jeu de sondes exclusives est disponible dans l’un des trois canaux de fluorescence (C1, C2 et C3). Les sondes Nanog et Pou5f1 ont été marqués avec C2 et C3, respectivement. - Réchauffer les propriétaires, 1 amplificateur (1 AMP), amplificateur 2 (AMP2), amplificateur 3 (AMP3) et amplificateur 4-fluorescence (AMP 4-FL) à température ambiante.
Remarque : Cette étape doit être effectuée au cours de l’incubation de la sonde de transcription spécifique de 2 heures. - Transférez les ovocytes dans 500 μL de WB et incuber pendant 10 min à température ambiante.
- Incuber les ovocytes séquentiellement dans des tampons de l’amplification.
- Incuber les ovocytes dans 80 µL d’AMP1 pendant 30 min à ovocytes transfert de 40° c à 500 µL de BM pendant 10 min à température ambiante.
- Incuber les ovocytes dans 80 µL de AMP2 pendant 15 min à 40 ° C. Transférez les ovocytes dans 500 µL de WB pendant 10 min à température ambiante.
- Incuber les ovocytes dans 80 µL de AMP3 pendant 30 min à 40 ° C. Transférez les ovocytes dans 500 µL de WB pendant 10 min à température ambiante.
NOTE : Le reste du protocole est effectué dans l’obscurité car AMP-FL contient le fluorophore. Lorsque vous travaillez sous le microscope à dissection, réduire autant que possible la lumière. - Ajouter des ovocytes à 80 μL de AMP4-FL pendant 15 min à 40° C.
Remarque : AMP4-FL est offert comme alternative tampon-A (Alt-A), Alt-B ou Alt-C. Sélectionnez le tampon AMP4-FL dépendant sur lequel les émissions de longueur d’onde est souhaitée.
- Laver les ovocytes dans 500 µL de WB pendant 10 min à température ambiante. Incuber les ovocytes dans 80 µL de DAPI pendant 20 min à température ambiante. Laver les ovocytes dans 500 µL de WB pendant 5 min à température ambiante.
- Distribuer 12 µL de supports de montage de l’anti-décoloration au centre d’une diapositive sans ajouter des bulles au réactif. Transférez les ovocytes avec comme petit WB que possible dans le milieu de montage et appliquez un lamelle couvre-objet.
- La lamelle à un angle d’inclinaison et délicatement placer au-dessus du liquide sur la diapositive. Évitez d’appuyer sur la lamelle couvre-objet trop dur pour empêcher toute déformation des ovocytes et introduction de bulles.
- Stocker les lames dans une boîte sombre sécher jusqu’au lendemain à la température ambiante. Enduisez les bords des lames en vernis à ongles transparent pour sceller la lamelle couvre-objet.
- Utiliser un microscope ordinaire pour rechercher les ovocytes sur la lame et le cercle avec un marqueur permanent.
Remarque : Cette étape n’est pas obligatoire mais améliore la localisation d’ovocytes sur la diapositive. Pour de meilleurs résultats, diapositives d’image dans les 1 à 5 jours comme le signal fluorescent commencera à s’estomper.
4. traitement de l’image
- Les ovocytes 3 dimensions, à l’aide de la microscopie confocale étape z de l’image.
Remarque : Pour analyser avec précision les images, chaque étape z serait de 1,0 µm/tranche. - Enregistrer les images confocales comme un nd2 compressé ou individuels. Fichiers TIFF pour chaque ovocyte. Les deux types d’images sont compatibles avec le programme de traitement d’image open source, Fidji.
- Téléchargez et installez le logiciel de Fidji de libre accès (https://imagej.net/Fiji/Downloads).
- Faire glisser des fichiers de nd2 à Fidji et choisissez hyperstack. Si les images confocales étaient enregistrés sous. Fichiers TIFF passez à l’étape 4.4.
Remarque : Lorsque le fichier nd2 est tomb6e Fidji la liste déroulante hyperstack devrait apparaître automatiquement. - Cliquez sur l’onglet Image , sélectionnez la couleuret cliquez sur Canaux Split pour séparer les canaux fluorescents du fichier nd2.
- Générer des individuels. Fichiers TIFF pour chaque tranche de z de l’ovocyte dans chaque canal fluorescent. Cliquez sur l’onglet Image , sélectionner les pileset cliquez sur la pile d’Images. Cliquez sur l’onglet Image , sélectionner le Typeet cliquez sur Couleur RVB pour convertir chaque tranche z en image RVB couleur séparée.
Remarque : La couleur RVB est artificielle et peuvent être choisis comme désiré pour chaque longueur d’onde d’émission. - Enregistrez chaque image convertie comme. Fichier TIFF. Placer les images d’un seul ovocyte pour chaque canal fluorescente dans un nouveau dossier pour éviter la confusion au cours de couture (étape 4.3).
- Faire glisser des fichiers de nd2 à Fidji et choisissez hyperstack. Si les images confocales étaient enregistrés sous. Fichiers TIFF passez à l’étape 4.4.
- Normaliser les uns. Image TIFF pour Pou5f1 et Nanog en utilisant des images de contrôle négatif (DapB).
NOTE : Normalisation s’effectue à l’aide d’un programme de retouche photo. Veillez à retirer les mêmes niveaux de fluorescence de fond de chaque image de contrôle. - Ouvert chaque normalisé. Fichier TIFF à Fidji pour assembler tous les z-tranches pour chaque ovocyte dans chaque longueur d’onde.
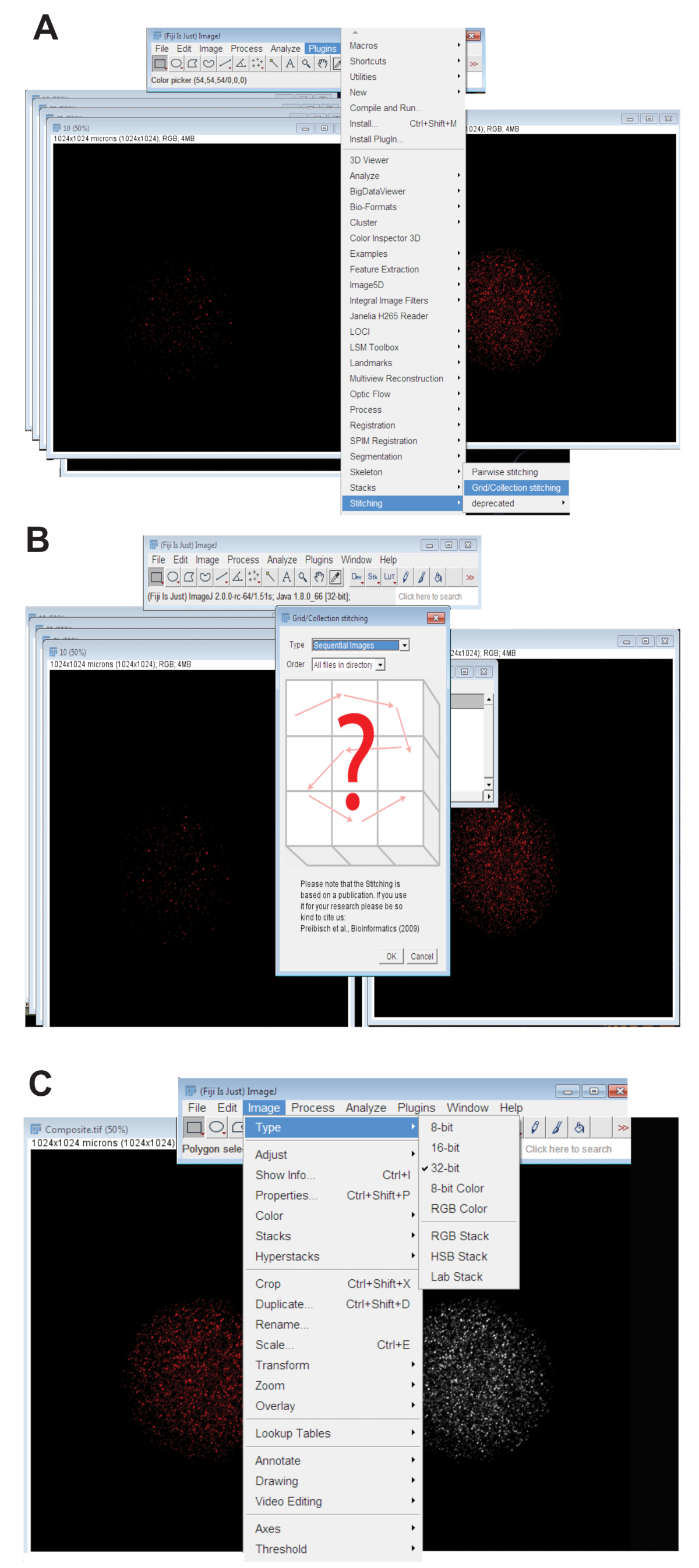
- Cliquez sur l’onglet Plugins , sélectionnez Stitchinget cliquez sur Grille/Collection (Figure 3 a). Sélectionnez Séquence d’Images dans le menu déroulant et cliquez sur OK (Figure 3 b).
- Parcourir le répertoire et sélectionnez le dossier contenant toutes les images de z-tranche pour un ovocyte individuel à une longueur d’onde (voir étape 4.3.4). Cliquez sur OK.
- Déplacez le curseur en bas de l’image cousu sur le canal de la couleur appropriée pour la longueur d’onde utilisée et créer l’image finale de cousu RGB en cliquant sur Image, sélectionner le Type, puis cliquez sur Couleur RVB.
NOTE : Cette image sera utilisée pour le dosage de fluorescence décrit à l’étape 4.6 ci-dessous.
- Convertir l’image cousu en image projetée maximale de 32 bits. Cliquez sur l’Image, sélectionnez le Typeet cliquez sur 32 bits (Figure 3). Enregistrez cette image sous un nouveau. Fichier TIFF.

Figure 3 : Assembler des images confocal z-série d’ovocytes. (A) capture d’écran montrant l’outil grille/Collection de plug-in dans les îles Fidji qui a été utilisé pour produire des images composites de l’ovocyte. (B) Images séquentielles utilise la fluorescence chevauchement entre séquentiel. Fichiers TIFF pour générer une image composite. (C) l’image composite a été enregistré comme un 32 bits. Fichier TIFF. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
- Téléchargez et installez la tache de trouver et de Tracking Program13, qui est disponible sur le site de D.R. Larson, un chercheur à la National instituts de santé National Cancer Institute (https://ccr.cancer.gov/Laboratory-of-Receptor-Biology-and-Gene-Expression/daniel-r-larson). Téléchargez et installez la machine virtuelle de libre accès pour le système d’exploitation interactive data language (IDL) est requis pour exécuter la recherche de Spot et Tracking Program (http://www.spacewx.com/pdf/idlvm.pdf).
- Ouvrez l’image 32 bits, cousu, qui a été généré à l’étape 4.6 (Figure 4 a), dans le programme de suivi et de trouver de la place. Sélectionnez la liste déroulante Localize et cliquez Localize (Figure 4 b), qui calcule les taches numéros trouvés dans l’image.
Remarque : Chaque point compté représente un ARNm individuels. Bande pass et photon seuil réglages sont indiqués dans la capture d’écran (Figure 4 b). Pour ce protocole, a été utilisé par défaut pour chaque paramètre de seuil. Représentant positif et taches d’arrière-plan sont affichent (Figure 4).

Figure 4 : Quantification des ARNm par Spot Finder et suivi. (A) images de z-séries individuelles ont été cousues ensemble tel que décrit à la Figure 3 et enregistrés sous un maximum de 32 bits projeté. Fichier TIFF. (B) image Composite a été ouvert en Spot Finder et suivi. Localiser a été utilisé pour compter les taches fluorescentes (zone rouge). Seuil de passage et le photon bande sont indiqués par la boîte bleue. (C) la flèche bleue pointe vers un signal positif (seuil). La flèche blanche montre une fluorescence spot au-dessous du seuil et, par conséquent, pas pris en compte. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Résultats
À la fin du protocole, le résultat sera des images individuelles de confocal z-series (Figure 4 a et Figure 5), images piquées (Figure 4), et l’ARNm comtes (Figure 4 b). Lorsque le multiplexage est effectuée, il y aura aussi des images fusionnées montrant l’étiquette pour deux différents ARNm (
Discussion
Une série de mesures mineures pendant le protocole assurera succès fluorescence et comptages précis des ARNm. Tout d’abord, le protocole doit être effectué immédiatement après la collecte et la fixation des ovocytes. Notez que le PVP est ajoutée au tampon de fixation paraformaldéhyde à 4 % pour empêcher des ovocytes de coller les uns aux autres. Nous avons constaté qu’il est nécessaire réaliser l’expérience immédiatement après la collecte et la fixation des ovocytes. Tout retard entraîne un beauco...
Déclarations de divulgation
Les auteurs n’ont rien à déclarer
Remerciements
Nous remercions le Dr Daniel R. Larson pour son aide généreuse avec l’installation et l’utilisation de la recherche de Spot et de Tracking Program 13 et l’appui technique de l’Université du Nebraska Lincoln microscopie Core pour l’imagerie de la microscopie confocale. Cette étude représente une contribution de l’Université du Nebraska Agricultural Research Division, Lincoln, Nebraska et a été soutenue par des fonds de Hatch UNL (NEB-26-206/numéro d’ordre-232435 et NEB-26-231/numéro d’ordre-1013511).
matériels
| Name | Company | Catalog Number | Comments |
| (±)-α-Lipoic acid | Sigma-Aldrich | T1395 | Alpha Lipoic Acid |
| Albumin, Bovine Serum, Low Fatty Acid | MP Biomedicals, LLC | 199899 | FAF BSA |
| BD 10mL TB Syringe | Becton, Dickinson and Company | 309659 | 10 mL syringe |
| BD PrecisionGlide Needle | Becton, Dickinson and Company | 305109 | 27 1/2 gauge needle |
| Calcium chloride dihydrate | Sigma-Aldrich | C7902 | CaCl2-2H2O |
| Citric acid | Sigma-Aldrich | C2404 | Citrate |
| D-(+)-Glucose | Sigma-Aldrich | G6152 | Glucose |
| Disodium phosphate | Na2HPO4 | ||
| Easy Grip Petri Dish | Falcon Corning | 351008 | 35 mm dish |
| Edetate Disodium | Avantor | 8994-01 | EDTA |
| Extra Fine Bonn Scissors | Fine Science Tools | 14084-08 | Straight, Sharp/Sharp, non-serrated, 13mm cutting edge scissors |
| Fetal Bovine Serum | Atlanta biologicals | S10250 | FBS |
| Gentamicin Reagent Solution | gibco | 15710-064 | Gentamicin |
| GlutaMAX-I (100X) | gibco | 35050-061 | Glutamax |
| Gold Seal Micro Slides | Gold Seal | 3039 | 25 x 75mm slides |
| Gonadotropin, From Pregnant Mares' Serum | Sigma | G4877 | eCG |
| hCG recombinant | NHPP | AFP8456A | hCG |
| Hyaluronidase, Type IV-S: From Bovine Testes | Sigma-Aldrich | H3884 | Hyaluronidase |
| Jewelers Style Forceps | Integra | 17-305X | Forceps 4-3/8", Style 5F, Straight, Micro Fine Jaw |
| L-(+)-Lactic Acid, free acid | MP Biomedicals, LLC | 190228 | L-Lactate |
| Magnesium sulfate heptahydrate | Sigma-Aldrich | M2773 | MgSO4-7H2O |
| MEM Nonessential Amino Acids | Corning | 25-025-Cl | NEAA |
| Microscope Cover Glass | Fisher Scientific | 12-542-C | 25 x 25x 0.15 mm cover slips |
| Mm-Nanog-O2-C2 RNAscope Probe | Advanced Cell Diagnostics | 501891-C2 | Nanog Probe |
| Mm-Pou5f1-O1-C3 RNAscope Probe | Advanced Cell Diagnostics | 501611-C3 | Pou5f1 Probe |
| MOPS | Sigma-Aldrich | M3183 | |
| Paraformaldehyde | Sigma-Aldrich | P6148 | Paraformaldehyde |
| PES 0.22 um Membrane -sterile | Millex-GP | SLGP033RS | 0.22 um filters |
| Polyvinylpyrrolidone | Sigma-Aldrich | P0930 | PVP |
| Potassium chloride | Sigma-Aldrich | 60128 | KCl |
| Potassium phosphate monobasic | Sigma-Aldrich | 60218 | KH2PO4 |
| Prolong Gold antifade reagent | invitrogen | P36934 | Antifade reagent without DAPI |
| RNAscope DAPI | Advanced Cell Diagnostics | 320858 | DAPI |
| RNAscope FL AMP 1 | Advanced Cell Diagnostics | 320852 | Amplifier 1 |
| RNAscope FL AMP 2 | Advanced Cell Diagnostics | 320853 | Amplifier 2 |
| RNAscope FL AMP 3 | Advanced Cell Diagnostics | 320854 | Amplifier 3 |
| RNAscope FL AMP 4 ALT A | Advanced Cell Diagnostics | 320855 | Amplifier 4 ALT A |
| RNAscope FL AMP 4 ALT B | Advanced Cell Diagnostics | 320856 | Amplifier 4 ALT B |
| RNAscope FL AMP 4 ALT C | Advanced Cell Diagnostics | 320857 | Amplifier 4 ALT C |
| RNAscope Fluorescent Multiplex Detection Reagents Kit | Advanced Cell Diagnostics | 320851 | FISH Reagent Kit |
| RNAscope Probe 3-plex Negative Control Probe | Advanced Cell Diagnostics | 320871 | Negative Control |
| RNAscope Probe 3-plex Positive Control | Advanced Cell Diagnostics | 320881 | Positive Control |
| RNAscope Probe Diluent | Advanced Cell Diagnostics | 300041 | Probe Diluent |
| RNAscope Protease III | Advanced Cell Diagnositics | 322337 | Protease III |
| RNAscope Protease III & IV Reagent Kit | Advanced Cell Diagnostics | 322340 | FISH Protease Kit |
| RNAscope Protease IV | Advanced Cell Diagnostics | 322336 | Protease IV |
| S/S Needle with Luer Hub 30G | Component Supply Co. | NE-301PL-50 | blunt 30 gauge needle |
| Sodium bicarbonate | Sigma-Aldrich | S6297 | NaHCO3 |
| Sodium chloride | Sigma-Aldrich | S6191 | NaCl |
| Sodium hydroxide | Sigma-Aldrich | 306576 | NaOH |
| Sodium pyruvate, >= 99% | Sigma-Aldrich | P5280 | Pyruvate |
| Solution 6 Well Dish | Agtechinc | D18 | 6 well dish |
| Taurine | Sigma-Aldrich | T8691 | Taurine |
| Tissue Culture Dish | Falcon Corning | 353002 | 60 mm dish |
| Triton X-100 | Sigma-Aldrich | X100 | Triton X-100 |
Références
- Vogelstein, B., Kinzler, K. W. Digital PCR. Proceedings of the National Academy of Sciences. 96 (16), 9236-9241 (1999).
- MacK, E. M., Smith, J. E., Kurz, S. G., Wood, J. R. CAMP-dependent regulation of ovulatory response genes is amplified by IGF1 due to synergistic effects on Akt phosphorylation and NF-kB transcription factors. Reproduction. 144 (5), 595-602 (2012).
- Xie, F., Timme, K. A., Wood, J. R. Using Single Molecule mRNA Fluorescent in Situ Hybridization (RNA-FISH) to Quantify mRNAs in Individual Murine Oocytes and Embryos. Scientific Reports. 8 (1), 7930 (2018).
- Ruebel, M. L., et al. Obesity modulates inflammation and lipidmetabolism oocyte gene expression: A single-cell transcriptome perspective. Journal of Clinical Endocrinology and Metabolism. 102 (6), 2029-2038 (2017).
- Borensztein, M., Syx, L., Servant, N., Heard, E. . Mouse Oocyte Development. 1818, 51-65 (2018).
- Jansova, D., Tetkova, A., Koncicka, M., Kubelka, M., Susor, A. Localization of RNA and translation in the mammalian oocyte and embryo. PLoS ONE. 13 (3), 1-25 (2018).
- Player, A. N., Shen, L. P., Kenny, D., Antao, V. P., Kolberg, J. A. Single-copy gene detection using branched DNA (bDNA) in situ hybridization. Journal of Histochemistry and Cytochemistry. 49 (5), 603-611 (2001).
- Wang, F., et al. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. Journal of Molecular Diagnostics. 14 (1), 22-29 (2012).
- Itzkovitz, S., van Oudenaarden, A. Validating transcripts with probes and imaging technology. Nature Methods. 8 (4), S12-S19 (2011).
- Derti, A., et al. ProbeDesigner: for the design of probesets for branched DNA (bDNA) signal amplification assays. Bioinformatics. 15 (5), 348-355 (1999).
- Larson, B., Malayter, D., Shure, M. Multiplexed Detection of Cytokine Cancer Biomarkers using Fluorescence RNA In Situ Hybridization and Cellular Imaging. BioTek Application Notes. , 1-5 (2016).
- Buxbaum, A. R., Wu, B., Singer, R. H. Single β-Actin mRNA Detection in Neurons Reveals a Mechanism for Regulating Its Translatability. Science. 343 (6169), 419-422 (2014).
- Thompson, R. E., Larson, D. R., Webb, W. W. Precise nanometer localization analysis for individual fluorescent probes. Biophysical Journal. 82 (5), 2775-2783 (2002).
- Herrick, J. R., Paik, T., Strauss, K. J., Schoolcraft, W. B., Krisher, R. L. Building a better mouse embryo assay: effects of mouse strain and in vitro maturation on sensitivity to contaminants of the culture environment. Journal of Assisted Reproduction and Genetics. 33 (2), 237-245 (2016).
- Pohlmeier, W. E., Xie, F., Kurz, S. G., Lu, N., Wood, J. R. Progressive obesity alters the steroidogenic response to ovulatory stimulation and increases the abundance of mRNAs stored in the ovulated oocyte. Molecular Reproduction and Development. 81 (8), 735-747 (2014).
- Xie, F., Anderson, C. L., Timme, K. R., Kurz, S. G., Fernando, S. C., Wood, J. R. Obesity-dependent increases in oocyte mRNAs are associated with increases in proinflammatory signaling and gut microbial abundance of lachnospiraceae in female mice. Endocrinology. 157 (4), 1630-1643 (2016).
- Hirao, Y., Yanagimachi, R. Detrimental effect of visible light on meiosis of mammalian eggs in vitro. Journal of Experimental Zoology. , (1978).
- Takenaka, M., Horiuchi, T., Yanagimachi, R. Effects of light on development of mammalian zygotes. Proceedings of the National Academy of Sciences. 104 (36), 14293-14293 (2007).
- Komminoth, P., Werner, M. Target and signal amplification: Approaches to increase the sensitivity of in situ hybridization. Histochemistry and Cell Biology. 108 (4-5), 325-333 (1997).
- Hornick, J. E., Duncan, F. E., Shea, L. D., Woodruff, T. K. Multiple follicle culture supports primary follicle growth through paracrine-acting signals. Reproduction. 145 (1), 19-32 (2013).
- Vlasova-St. Louis, I., Bohjanen, P. Feedback Regulation of Kinase Signaling Pathways by AREs and GREs. Cells. 5 (1), 4 (2016).
- Gilbert, C., Svejstrup, J. Q. RNA Immunoprecipitation for Determining RNA-Protein Associations In Vivo. Current Protocols in Molecular Biology. 75 (1), 27.4.1-27.4.11 (2006).
- Kwon, S., Chin, K., Nederlof, M., Gray, J. W. Quantitative, in situ analysis of mRNAs and proteins with subcellular resolution. Scientific Reports. 7 (1), 16459 (2017).
- Voigt, F., et al. Single-Molecule Quantification of Translation-Dependent Association of mRNAs with the Endoplasmic Reticulum. Cell Reports. 21 (13), 3740-3753 (2017).
- Halstead, J. M., Wilbertz, J. H., Wippich, F., Lionnet, T., Ephrussi, A., Chao, J. A. TRICK: A Single-Molecule Method for Imaging the First Round of Translation in Living Cells and Animals. Methods in Enzymology. 572, (2016).
- Cookson, W., Liang, L., Abecasis, G., Moffatt, M., Lathrop, M. Mapping complex disease traits with global gene expression. Nature Reviews Genetics. 10 (3), 184-194 (2009).
- Houle, D., Govindaraju, D. R., Omholt, S. Phenomics: the next challenge. Nature Reviews Genetics. 11, 855 (2010).
- Freimer, N., Sabatti, C. The Human Phenome Project. Nature Genetics. 34, 15 (2003).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.