
Method Article
Séquençage du génome entier pour la caractérisation rapide du virus de la rage à l’aide de la technologie des nanopores
Dans cet article
Résumé
Nous présentons ici un flux de travail rapide et rentable pour caractériser les génomes du virus de la rage (RABV) à l’aide de la technologie nanopore. Le flux de travail vise à soutenir la surveillance fondée sur la génomique à l’échelle locale, en fournissant des informations sur les lignées de RABV en circulation et leur placement dans les phylogénies régionales afin d’orienter les mesures de lutte contre la rage.
Résumé
Les données génomiques peuvent être utilisées pour suivre la transmission et la propagation géographique des maladies infectieuses. Cependant, la capacité de séquençage requise pour la surveillance génomique reste limitée dans de nombreux pays à revenu faible et intermédiaire (PRFI), où la rage transmise par les chiens et/ou la rage par des animaux sauvages tels que les chauves-souris vampires posent des problèmes de santé publique et économiques majeurs. Nous présentons ici un flux de travail rapide et abordable de l’échantillon, de la séquence à l’interprétation utilisant la technologie nanopore. Les protocoles de collecte d’échantillons et de diagnostic de la rage sont brièvement décrits, suivis de détails sur le flux de travail optimisé du séquençage du génome entier, y compris la conception et l’optimisation de l’amorce pour la réaction en chaîne par polymérase (PCR) multiplexe, la préparation d’une bibliothèque de séquençage modifiée et peu coûteuse, le séquençage avec appel de base en direct et hors ligne, la désignation de lignées génétiques et l’analyse phylogénétique. La mise en œuvre du flux de travail est démontrée et les étapes critiques sont mises en évidence pour le déploiement local, telles que la validation du pipeline, l’optimisation de l’amorce, l’inclusion de contrôles négatifs et l’utilisation de données et d’outils génomiques accessibles au public (GLUE, MADDOG) pour la classification et le placement dans les phylogénies régionales et mondiales. Le délai d’exécution du flux de travail est de 2 à 3 jours et le coût varie de 25 $ par échantillon pour un cycle de 96 échantillons à 80 $ par échantillon pour un cycle de 12 échantillons. Nous concluons que la mise en place d’une surveillance génomique du virus de la rage dans les PRFI est réalisable et peut contribuer à l’atteinte de l’objectif mondial de zéro décès humain dus à la rage chez l’homme d’ici 2030, ainsi qu’à une surveillance accrue de la propagation de la rage chez les animaux sauvages. De plus, la plateforme peut être adaptée à d’autres agents pathogènes, ce qui permet de construire une capacité génomique polyvalente qui contribue à la préparation aux épidémies et aux pandémies.
Introduction
Le virus de la rage (RABV) est un lyssavirus de la famille des Rhabdoviridae qui provoque une maladie neurologique mortelle chez les mammifères1. Bien que la rage soit évitable à 100 % par la vaccination, elle reste une préoccupation majeure en matière de santé publique et d’économie dans les pays endémiques. Sur les 60 000 décès humains dus à la rage estimés chaque année, plus de 95 % se produisent en Afrique et en Asie, où les chiens constituent le principal réservoir2. En revanche, la vaccination des chiens a conduit à l’élimination de la rage transmise par les chiens en Europe occidentale, en Amérique du Nord et dans une grande partie de l’Amérique latine. Dans ces régions, les réservoirs de la rage sont maintenant limités à la faune, comme les chauves-souris, les ratons laveurs, les mouffettes et les canidés sauvages3. Dans toute l’Amérique latine, la chauve-souris vampire commune est une source problématique de rage en raison de la transmission régulière des chauves-souris aux humains et au bétail lors de l’alimentation nocturne du sang4. L’impact économique mondial annuel de la rage est estimé à 8,6 milliards de dollars, les pertes de bétail représentant 6 %5.
Les données de séquençage des agents pathogènes viraux, combinées à des métadonnées sur le moment et la source des infections, peuvent fournir des informations épidémiologiques solides6. Dans le cas du RABV, le séquençage a été utilisé pour étudier l’origine des éclosions7,8, identifier les associations de l’hôte avec des animaux sauvages ou des chiens domestiques 8,9,10,11,12 et retracer les sources des cas humains 13,14. Des enquêtes sur les épidémies à l’aide d’analyses phylogénétiques ont indiqué que la rage est apparue dans la province de Bali, en Indonésie, autrefois exempte de rage, par une seule introduction en provenance des zones endémiques voisines de Kalimantan ou de Sulawesi15. Pendant ce temps, aux Philippines, il a été prouvé qu’une épidémie sur l’île de Tablas, dans la province de Romblon, avait été introduite à partir de l’île principale de Luçon16. Les données génomiques virales ont également été utilisées pour mieux comprendre la dynamique de transmission des agents pathogènes nécessaire au ciblage géographique des mesures de contrôle. Par exemple, la caractérisation génomique du RABV illustre le regroupement géographique des clades 17,18,19, la co-circulation des lignées 20,21,22, le mouvement viral médié par l’homme17,23,24 et la dynamique des métapopulations 25,26.
La surveillance des maladies est une fonction importante de la surveillance génomique qui a été renforcée par l’augmentation mondiale de la capacité de séquençage en réponse à la pandémie de SRAS-CoV-2. La surveillance génomique a permis de suivre en temps réel les variants préoccupants du SRAS-COV-227,28 et les contre-mesures associées6. Les progrès réalisés dans le domaine des technologies de séquençage accessibles, telles que la technologie des nanopores, ont permis de mettre au point des protocoles améliorés et plus abordables pour le séquençage rapide des agents pathogènes humains 29,30,31,32 et animaux 33,34,35. Cependant, dans de nombreux pays où la rage est endémique, il existe encore des obstacles à l’opérationnalisation de la surveillance génomique des agents pathogènes, comme le montrent les disparités mondiales dans la capacité de séquençage du SRAS-CoV-236. Les limites de l’infrastructure des laboratoires, des chaînes d’approvisionnement et des connaissances techniques compliquent la mise en place et la banalisation de la surveillance génomique. Dans cet article, nous démontrons comment un flux de travail optimisé, rapide et abordable de séquençage du génome entier peut être déployé pour la surveillance du RABV dans des contextes aux ressources limitées.
Protocole
L’étude a été approuvée par le Comité de coordination de la recherche médicale de l’Institut national de recherche médicale (NIMR/HQ/R.8a/vol. IX/2788), le Ministère de l’administration régionale et des collectivités locales (AB.81/288/01) et le Conseil d’examen institutionnel de l’Institut de santé d’Ifakara (IHI/IRB/No :22-2014) en Tanzanie ; l’Institut des maladies tropicales et infectieuses de l’Université de Nairobi (P947/11/2019) et l’Institut de recherche médicale du Kenya (KEMRI-SERU ; protocole n° 3268) au Kenya ; et l’Institut de recherche en médecine tropicale (RITM), ministère de la Santé (2019-023) aux Philippines. Le séquençage d’échantillons provenant du Nigeria a été entrepris à partir de matériel de diagnostic archivé collecté dans le cadre de la surveillance nationale.
REMARQUE : Les sections 1 à 4 sont des conditions préalables. Les sections 5 à 16 décrivent le processus de séquençage des nanopores de l’échantillon, de la séquence et de l’interprétation (Figure 1). Pour les étapes suivantes du protocole qui nécessitent une centrifugation par impulsions, centrifuger à 10-15 000 x g pendant 5-15 s.
1. Configuration de l’environnement informatique pour le séquençage et l’analyse des données
- Ouvrez le site Web d’Oxford Nanopore Technology (ONT)37 et créez un compte pour accéder à des ressources spécifiques aux nanopores.
- Connectez-vous et installez le logiciel de séquençage et d’appel de baseONT 38.
- Ouvrez GitHub39 et créez un compte.
- Allez dans les dépôts artic-rabv40 et MADDOG41 et suivez les instructions d’installation.
2. Concevoir ou mettre à jour le schéma d’amorçage multiplex
NOTE : Les schémas RABV existants sont disponibles dans le référentiel artic-rabv40. Lorsque l’on cible une nouvelle zone géographique, il convient de concevoir un nouveau système ou de modifier un système existant pour y intégrer une diversité supplémentaire.
- Choisir un ensemble de référence du génome pour représenter la diversité dans la zone d’étude ; il s’agit généralement d’un ensemble de séquences accessibles au public (par exemple, à partir de NCBI GenBank) ou de données internes préliminaires. Suivez l’étape 2.1.1 pour utiliser RABV-GLUE42, une ressource de données de séquence RABV, afin de filtrer et de télécharger les séquences NCBI et les métadonnées associées.
REMARQUE : Choisissez des séquences de référence avec des génomes complets (c’est-à-dire sans lacunes ni bases masquées). Il est recommandé de choisir jusqu’à 10 séquences comme jeu de référence pour la conception de l’apprêt. Si les données de séquence disponibles sont incomplètes ou non représentatives de la zone d’étude, se référer à l’avis43,44,45 dans le fichier supplémentaire 1.- Accédez à la page NCBI RABV Sequences by Clade (Séquences RABV Sequences by Clade ) dans le menu déroulant Sequence Data (Données de séquence ) de RABV-GLUE. Cliquez sur le lien Virus de la rage (RABV) pour accéder à toutes les données disponibles ou sélectionnez un clade d’intérêt particulier. Utilisez l’option de filtre pour ajouter des filtres qui correspondent aux critères souhaités (par exemple, le pays d’origine, la longueur de la séquence). Télécharger les séquences et les métadonnées.
- Générer un schéma d’amorce pour la réaction en chaîne par polymérase multiplex (PCR) en suivant les instructions fournies par Primal Scheme46. Un schéma de 400 pb avec un chevauchement de 50 pb est recommandé pour séquencer des échantillons de faible qualité. Téléchargez et enregistrez toutes les sorties (ne modifiez pas les noms des fichiers ou des amorces).
NOTE : Le schéma sera indexé sur la première séquence de la fasta d’entrée, ci-après appelée « référence d’index » (Figure 2). Reportez-vous au fichier supplémentaire 1 pour connaître les options permettant d’optimiser les performances de l’apprêt.
3. Mettre en place les pipelines bioinformatiques RAMPART et ARTIC
- Reportez-vous au fichier supplémentaire 2 pour configurer une structure de répertoires afin de gérer les fichiers d’entrée/sortie de RAMPART et du pipeline bioinformatique ARTIC.
4. Biosécurité et configuration du laboratoire
- Manipuler des échantillons potentiellement positifs pour la rage dans des conditions de niveau de biosécurité (BSL) 2 ou 3.
- S’assurer que le personnel de laboratoire a terminé la vaccination pré-exposition contre la rage et qu’il se soumet à une surveillance de l’immunité conformément aux recommandations de l’Organisation mondiale de la santé (OMS)3.
- S’assurer que des procédures opérationnelles normalisées et des évaluations des risques sont en place pour le laboratoire, conformément aux directives nationales ou internationales.
- Configuration de laboratoire requise : Minimisez la contamination en maintenant une séparation physique entre les zones pré et post-PCR. Dans les laboratoires où l’espace est limité ou dans les laboratoires sur le terrain, utilisez des boîtes à gants portatives ou des postes de laboratoire de fortune pour minimiser la contamination.
- Dans ce protocole, assurez-vous de désigner des zones distinctes pour :
- Extraction d’échantillons : Installez une armoire/boîte à gants BSL2/3 pour manipuler le matériel biologique et effectuer l’inactivation et l’extraction de l’ARN.
- Zone du gabarit : Installez une armoire/boîte à gants BSL1 pour l’ajout du gabarit (ARN/ADNc) au mélange maître de réaction pré-préparé.
- Zone de mélange maître : Mettez en place une zone de nettoyage désignée (armoire BSL1/boîte à gants) pour la préparation des mélanges maîtres de réactifs. Il ne devrait pas y avoir de modèle dans cette zone.
- Zone post-PCR : Mettez en place une zone distincte pour le travail sur les amplicons et la préparation de la bibliothèque de séquençage.
REMARQUE : Toutes les zones doivent être nettoyées avec un décontaminant de surface et stérilisé aux ultraviolets (UV) avant et après utilisation.
5. Prélèvement d’échantillons sur le terrain et diagnostic
REMARQUE : Les échantillons doivent être prélevés par du personnel formé et immunisé, portant de l’équipement de protection individuelle et suivant les procédures normalisées mentionnéesaux articles 47, 48 et 49.
- Prélever l’échantillon par le foramen magnum (c’est-à-dire la voie occipitale), comme décrit en détail dans Mauti et al.50.
- Diagnostiquer la rage sur le terrain à l’aide de tests de diagnostic rapide et confirmer en laboratoire à l’aide des procédures recommandées47, telles que le test d’anticorps fluorescents directs (DFA), le test immunohistochimique rapide direct (DRIT)51,52 ou la transcription inverse (RT)-PCR en temps réel 53.
- Utiliser des échantillons de cerveau confirmés positifs pour l’extraction de l’ARN ou les conserver au congélateur à -20 °C pendant 2 à 3 mois ou à -80 °C pendant de plus longues périodes. Conserver l’ARN pour le stockage et le transport à l’aide d’un milieu de stabilisation ADN/ARN approprié.
6. Préparation de l’échantillon et extraction de l’ARN (3 h)
REMARQUE : Utilisez un kit d’extraction d’ARN viral basé sur une colonne de spin adapté au type d’échantillon.
- Préparez deux tubes de billes de céramique en remplissant un tube PCR de 2 mL avec un tube d’environ 200 μL rempli de billes de céramique de 1,4 mm et étiquetez le tube.
- Ajoutez le volume recommandé de tampon de lyse fourni dans le kit d’extraction d’ARN dans le tube PCR étiqueté.
- Prélever un cube d’environ 3 mm de l’échantillon de cerveau confirmé avec une infection par la rage à l’aide d’un applicateur en bois et le mettre dans un tube étiqueté avec ID de l’échantillon et 100 μL d’eau sans nucléase dans le tube étiqueté témoin négatif.
REMARQUE : Utiliser l’homogénéisation à base de billes à tube fermé pour limiter l’exposition de l’échantillon. Si ce n’est pas possible, utiliser d’autres perturbateurs mécaniques appropriés (p. ex., à rotor) ou un micropilon manuel. Cependant, ceux-ci peuvent être moins efficaces que le battage de billes sur une surface dure pour perturber les tissus (les échantillons de tissus peuvent durcir dans certains milieux de stockage). - Perturbez manuellement le tissu cérébral à l’aide d’un bâtonnet applicateur en bois, puis tourbillonnez à la vitesse maximale jusqu’à ce que l’homogénéisation complète des tissus soit atteinte.
- Centrifugez le lysat selon les instructions du fabricant et utilisez une pipette pour transférer le surnageant dans un nouveau tube de microcentrifugation étiqueté. N’utilisez ce surnageant que dans les étapes suivantes.
- Suivez les instructions de la colonne de spin du kit d’extraction d’ARN pour obtenir de l’ARN purifié.
- Incluez ici un contrôle d’extraction négatif (NEC) et allez jusqu’à l’étape du séquençage.
7. Préparation de l’ADNc (20 min)
- Dans la zone du mélange maître, préparer un mélange maître pour la synthèse de l’ADNc du premier brin en fonction du nombre d’échantillons et de témoins à traiter (avec un volume excédentaire de 10 % pour assurer un réactif adéquat ; Tableau 1). Un contrôle sans modèle (NTC) doit être inclus à ce stade.
- Étiqueter les tubes de 0,2 mL de bandelettes PCR et aliquote 5 μL du mélange maître dans les tubes.
- Amenez les tubes préparés dans la zone du modèle. Ajouter 5 μL d’ARN dans chaque tube marqué, y compris le NEC. Ajouter 5 μL d’eau sans nucléase (NFW) au NTC.
- Incuber dans un thermocycleur en suivant les conditions mentionnées dans le tableau 1.
REMARQUE : Point de pause facultatif : l’ADNc peut être conservé à -20 °C jusqu’à 1 mois si nécessaire, mais il est préférable de procéder à la PCR.
8. Préparation du stock de piscine d’apprêt (1 h)
REMARQUE : Cette étape n’est nécessaire que si vous fabriquez de nouvelles souches à partir d’apprêts individuels, après quoi des solutions mères pré-préparées peuvent être utilisées.
- Préparez un pool d’apprêt de 100 μM dans la zone du mélange principal.
- Remettre en suspension les amorces lyophilisées dans 1x tampon tris-EDTA (TE) ou NFW à une concentration de 100 μM chacune. Tourbillonnez soigneusement et essorez vers le bas.
REMARQUE : Dans les étapes suivantes, les amorces individuelles sont séparées en deux pools d’amorces - impaires (nommés groupe A) et pairs (nommés groupe B) - pour éviter les interactions entre les amorces flanquant les chevauchements d’amplicons. Ces pools d’amorces génèrent des amplicons de 400 pb qui se chevauchent et s’étendent sur le génome cible. - Disposez toutes les amorces impaires dans un support à tubes. Générer un stock d’amorces en ajoutant 5 μL de chaque amorce à un tube de microcentrifugation de 1,5 mL étiqueté « nom du schéma d’amorce - Pool A (100 μM) ».
- Répétez le processus pour toutes les amorces paires et étiquetez-les comme « nom du schéma d’amorce - Pool B (100 μM) ».
- Diluer chaque pool d’apprêt 1 :10 dans de l’eau de qualité moléculaire pour générer des stocks d’apprêt de 10 μM.
REMARQUE : Faire plusieurs aliquotes de dilutions d’amorce de 10 μM et les congeler en cas de dégradation ou de contamination.
9. PCR multiplex (5 h)
- Préparez deux mélanges maîtres PCR, un pour chaque pool d’apprêt dans la zone du mélange maître.
- Utiliser une concentration finale de 0,015 μM par apprêt. Calculer le volume requis de la piscine d’amorce pour la réaction PCR (tableau 2) à l’aide de la formule suivante :
Volume du pool d’amorces = nombre d’amorces x volume de réaction x 0,015/concentration (μM) du stock d’apprêt
- Utiliser une concentration finale de 0,015 μM par apprêt. Calculer le volume requis de la piscine d’amorce pour la réaction PCR (tableau 2) à l’aide de la formule suivante :
- Aliquote 10 μL chacun du mélange maître du groupe A et du mélange maître du groupe B sur des tubes de bandelettes PCR étiquetés dans la zone du modèle. Pour chaque échantillon, ajoutez 2,5 μL d’ADNc (à partir de l’étape 3) à chacune des réactions d’amorce marquées correspondantes des groupes A et B. L’excès d’ADNc peut être stocké à -20 °C.
- Mélanger en agitant doucement et en centrifugeant par impulsions.
- Incuber les échantillons dans les conditions mentionnées dans le tableau 2 sur un appareil de PCR.
REMARQUE : Le programme n’inclut pas d’étape d’extension spécifique en raison du long temps de recuit de 5 min (nécessaire en raison du nombre élevé d’amorces) et de la courte longueur des amplicons (400 bp) qui est suffisante pour l’extension.
10. Nettoyage et quantification de la PCR (3,5 h)
- Effectuez tous les travaux à partir de ce point dans la zone post-PCR.
- Les billes d’immobilisation réversible en phase solide (SPRI) aliquote sont insérées dans des tubes de microcentrifugation à partir de la bouteille principale. Conserver à 4 °C.
- Réchauffez une aliquote de billes SPRI à température ambiante (RT ; ~20 °C) et tourbillonnez soigneusement jusqu’à ce que les billes soient complètement remises en suspension dans la solution.
- Dans des tubes de 1,5 mL, mélanger les produits PCR de l’amorce du groupe A et de l’amorce du groupe B pour chaque échantillon. Si nécessaire, ajoutez de l’eau pour porter le volume à 25 μL.
- Ajouter 25 μL de billes SPRI à chaque échantillon (rapport 1 :1 bille/échantillon). Mélangez en pipetant de haut en bas ou en tapotant doucement le tube.
- Incuber à RT pendant 10 minutes, en inversant ou en faisant tourner les tubes de temps en temps.
- Placez-les sur une grille magnétique jusqu’à ce que les billes et la solution se soient complètement séparées. Retirez et jetez le surnageant en prenant soin de ne pas déranger la pastille de billes.
- Laver deux fois avec de l’éthanol à 80 % (réchauffé à RT).
- Ajoutez 200 μL d’éthanol à la pastille. Attendez 30 s pour vous assurer que les perles sont correctement lavées.
- Retirez et jetez soigneusement le surnageant en essayant de ne pas toucher la pastille de bille.
- Répétez les étapes 10.8.1 à 10.8.2 pour laver la pastille une deuxième fois.
- Éliminez toute trace d’éthanol à l’aide d’une douille de 10 μL. Sécher à l’air libre jusqu’à ce que l’éthanol à l’état de traces se soit évaporé (avec de petites billes, cela se produit rapidement, ~30 s) ; Lorsque cela se produit, la pastille doit passer de brillante à mate. Veillez à ne pas trop sécher (si la pastille se fissure, elle est trop sèche), car cela affectera la récupération de l’ADN.
- Remettre les billes en suspension dans 15 μL de NFW et incuber à RT (hors grille magnétique) pendant 10 min.
- Remettre dans la grille magnétique et transférer le surnageant (produit nettoyé) dans un nouveau tube de 1,5 ml.
- Préparer une dilution de 1 :10 de chaque échantillon dans un tube séparé (2 μL de produit + 18 μL de NFW).
REMARQUE : Soyez très prudent à ce stade pour éviter la contamination croisée. N’ouvrez qu’un seul tube d’amplicon à la fois. Aliquote 18 μL d’eau dans les tubes d’abord (dans une zone de master mix propre). - Mesurer la concentration d’ADN de chaque échantillon dilué à l’aide d’un fluorimètre très sensible et spécifique, tel que décrit aux protocols.io54,55.
11. Normalisation (30 min)
- Utiliser le modèle de normalisation (fichier supplémentaire 3) et la concentration d’ADN (ng/μL) de chaque échantillon pour calculer le volume d’échantillon dilué (ou net) requis pour 200 fmol de chaque échantillon dans un volume total de 5 μL.
- Étiqueter les nouveaux tubes PCR et ajouter les volumes calculés de NFW et d’échantillon pour obtenir de l’ADN normalisé.
- Utiliser le volume calculé pour les échantillons non dilués (purs) s’il faut plus de 5 μL de l’échantillon dilué pour obtenir 200 fmol.
REMARQUE : Point de pause facultatif : À ce stade, le produit PCR nettoyé peut être stocké à 4 °C jusqu’à 1 semaine ou placé à -20 °C pour un stockage à plus long terme si nécessaire.
12. Préparation finale et code-barres (1,5 h)
REMARQUE : Les étapes suivantes supposent l’utilisation de réactifs spécifiques provenant de kits de séquençage de la ligature et des codes-barres spécifiques aux nanopores (voir le tableau des matériaux pour plus de détails). Le protocole est transférable entre différentes versions de chimie, mais les utilisateurs doivent veiller à utiliser des kits compatibles, conformément aux instructions du fabricant.
- Réparation d’extrémités et dA-tailing
- Réglez la réaction de préparation finale pour chaque échantillon mentionné dans le tableau 3. Préparez un master mix en fonction du nombre d’échantillons (plus 10% d’excédent). Soyez prudent lors du pipetage car les réactifs sont visqueux.
- Ajouter 5 μL de mélange maître dans chaque tube d’ADN normalisé (5 μL). Le mélange réactionnel total doit être de 10 μL. Changez les embouts à chaque fois et n’ouvrez qu’un seul tube à la fois.
- Incuber dans un thermocycleur dans les conditions mentionnées dans le tableau 3.
- Code-barres
- Aliquotez les codes-barres du kit de codes-barres sur les tubes à bandelettes PCR à 1,25 μL/tube, et enregistrez le code-barres attribué à chaque échantillon.
- Ajouter 0,75 μL de l’échantillon préparé aux extrémités à l’aliquote de code à barres qui lui est attribuée.
- Préparer un mélange maître de ligature en fonction du nombre d’échantillons (plus 10 % d’excédent) (tableau 4).
- Ajouter 8 μL de mélange maître de ligature à l’échantillon préparé en fin de match + codes-barres, ce qui donne une réaction totale de 10 μL.
- Incuber dans un thermocycleur en utilisant les conditions mentionnées dans le tableau 4.
- Nettoyage des billes SPRI et quantification de l’ADN
- Décongeler le tampon à fragments courts (SFB) à RT, mélanger par vortex, centrifugeuse à impulsions et placer sur de la glace.
- Regroupez tous les échantillons à code-barres dans un tube de microcentrifugation lobind de 1,5 ml. Afin de ne pas rendre le volume de nettoyage trop important pour être utilisé, regroupez 12 à 24 échantillons (10 μL/échantillon), jusqu’à 48 échantillons (5 μL/échantillon) ou jusqu’à 96 échantillons (2,5 μL/échantillon) de chaque réaction de codage à barres native.
- Ajoutez un volume de 0,4x de perles SPRI à la piscine à code-barres. Mélanger délicatement (effleurement ou pipetage) et incuber à RT pendant 5 min.
- Placez les échantillons sur un aimant jusqu’à ce que les billes se soient agglomérées et que le surnageant soit complètement clair (~2 min). Retirez et jetez le surnageant. Veillez à ne pas déranger les perles.
- Laver deux fois avec 250 μL de SFB.
- Retirez le tube de l’aimant et remettez complètement la pastille en suspension dans 250 μL de SFB. Incuber pendant 30 s, centrifuger par impulsions et revenir à l’aimant.
- Retirez le surnageant et jetez-le.
- Répétez l’étape 12.3.5 pour effectuer un deuxième lavage SFB.
- Centrifuger à impulsions et éliminer tout SFB résiduel.
- Ajouter 200 μL d’éthanol à 80 % (RT) pour baigner la pastille. Retirez et jetez l’éthanol, en prenant soin de ne pas déranger la pastille de bille. Sécher à l’air libre pendant 30 s ou jusqu’à ce que le granulé ait perdu de son éclat.
- Remettre en suspension dans 22 μL de NFW à RT pendant 10 min.
- Placez sur l’aimant, laissez reposer pendant ~2 min, puis retirez délicatement la solution et transférez-la dans un tube de microcentrifugation propre de 1,5 ml.
- Utilisez 1 μL pour obtenir la concentration d’ADN, comme décrit précédemment (étape 10.13).
REMARQUE : Point de pause facultatif : À ce stade, la bibliothèque peut être stockée à 4 °C jusqu’à 1 semaine ou à -20 °C pour un stockage à plus long terme, mais il est préférable de continuer avec la ligature et le séquençage de l’adaptateur.
13. Séquençage (48 h maximum)
- Préparez l’ordinateur (reportez-vous également aux sections 1 à 4 des conditions préalables).
- Vérifiez qu’il y a suffisamment d’espace pour stocker les nouvelles données (min 150 Go), que les données des anciennes exécutions sont sauvegardées/déplacées vers un serveur avant d’être supprimées et que la dernière version de MinKNOW est installée.
- Retirez la cellule d’écoulement stockée du réfrigérateur et laissez-la atteindre RT.
- Ligature de l’adaptateur (1 h)
- Centrifuger à impulsions le mélange adaptateur et la ligase et placer sur de la glace.
- Dégeler le tampon d’élution (EB), le SFB et le tampon de ligature à RT. Mélanger par vortex, centrifugeuse à impulsions et placer sur de la glace.
- Préparez le mélange maître de ligature de l’adaptateur (tableau 5), en combinant les réactifs dans l’ordre spécifié dans un tube à faible liaison.
REMARQUE : Des solutions de rechange peuvent être utilisées pour les réactifs de mélange maître pour la ligature par adaptateur (tableau 5) en fonction de la disponibilité au laboratoire. Voir le dossier supplémentaire 3 et le tableau des matériaux pour une liste des solutions de rechange. Utilisez le calcul de la feuille de calcul Fichier supplémentaire 3 pour obtenir le volume de la bibliothèque d’ADN équivalent à 200 fmol. Si moins de 20 μL sont calculés, ajoutez NFW pour obtenir jusqu’à 20 μL. - Mélanger par effleurement doux et centrifugeuse à impulsions. Incuber à RT pendant 20 min.
REMARQUE : Pendant l’incubation, commencer à préparer la cellule d’écoulement (section 13.5).
- Nettoyez à l’aide de billes SPRI (n’utilisez pas d’éthanol comme dans les nettoyages précédents).
- Ajoutez un volume de 0,4 fois les billes SPRI (RT) aux échantillons. Incuber à RT pendant 10 min, effleurer doucement par intermittence pour faciliter le mélange.
- Placez sur l’aimant jusqu’à ce que les billes et la solution se soient complètement séparées (~5 min). Retirer et jeter le surnageant ; Veillez à ne pas déranger la pastille de perles.
- Laver deux fois avec 125 μL de SFB.
- Remettre complètement la pastille en suspension avec 125 μL de SFB en mélangeant à l’aide d’une pipette. Laisser incuber 30 s.
- Centrifugeuse à impulsions pour recueillir le liquide à la base du tube et le placer sur l’aimant. Retirez le surnageant et jetez-le.
- Répétez les étapes 13.4.4 à 13.4.5 pour laver le granulé une deuxième fois.
- Centrifuger à impulsions et éliminer l’excès de SFB.
- Remettre en suspension dans 15 μL d’EB et incuber pendant 10 min à RT.
- Retournez à l’aimant pendant ~2 minutes, puis transférez délicatement la solution dans un tube de microcentrifugation propre de 1,5 mL.
- Quantifier 1 μL de la bibliothèque éluée, comme décrit précédemment à l’étape 10.13
REMARQUE : Pour de meilleurs résultats, passez directement au séquençage MinION ; cependant, la bibliothèque finale peut être stockée dans EB à 4 °C jusqu’à 1 semaine si nécessaire.
- Exécutez un contrôle de la qualité de la cellule d’écoulement.
- Connectez l’appareil de séquençage à un ordinateur portable et ouvrez le logiciel de séquençage.
- Sélectionnez le type de cellule de flux, puis cliquez sur Vérifier la cellule de flux et démarrer le test.
- Une fois terminé, le nombre total de pores actifs (c’est-à-dire viables) sera affiché. Une nouvelle cellule d’écoulement doit avoir > 800 pores actifs ; Si ce n’est pas le cas, contactez le fabricant pour un remplacement.
- Amorçage et chargement de la cellule d’écoulement (20 min)
- Décongelez les réactifs suivants à RT, puis placez le tampon de séquençage, une attache de rinçage, un tampon de rinçage et les billes de chargement sur de la glace.
- Vortex le tampon de séquençage et le tampon de rinçage, la centrifugeuse d’impulsions et placez-le sur de la glace.
- Centrifuger par impulsions l’attache affleurante et mélanger par pipetage ; puis placez-les sur de la glace.
- Préparez le mélange d’amorçage de la cellule d’écoulement en ajoutant 30 μL d’attache de rinçage directement dans le tube du tampon de rinçage à partir d’un kit d’amorçage de la cellule d’écoulement et mélangez par pipetage.
- Mélangez les billes de chargement par pipetage immédiatement avant l’utilisation car elles se déposent rapidement.
- Dans un tube frais, préparer la dilution finale de la bibliothèque pour le séquençage, comme indiqué dans le tableau 5.
REMARQUE : Utilisez le calcul de la feuille de calcul du fichier supplémentaire 3 pour obtenir le volume de la bibliothèque d’ADN équivalent à 50 fmol. Si moins de 12 μL sont calculés, ajoutez EB pour obtenir jusqu’à 12 μL. - Retournez le couvercle du dispositif de séquençage et faites glisser le couvercle du port d’amorçage dans le sens des aiguilles d’une montre afin que le port d’amorçage soit visible (Figure 3)
- Éliminez soigneusement les bulles d’air en réglant une pipette P1000 à 200 μL, insérez l’embout dans l’orifice d’amorçage et tournez la roue jusqu’à ce qu’un petit volume entrant dans l’embout de la pipette soit visible (tourner maximum à 230 μL).
- Chargez 800 μL de mélange d’amorçage de la cellule d’écoulement dans la cellule d’écoulement via l’orifice d’amorçage, en prenant soin d’éviter les bulles.
- Laisser poser 5 min.
- Soulevez doucement le couvercle de l’orifice d’échantillonnage et chargez 200 μL de mélange d’amorçage dans la cellule d’écoulement via l’orifice d’amorçage à l’aide d’une pipette P1000.
- Pipetez le mélange de la bibliothèque de haut en bas avant le chargement, en veillant à ce que les billes de chargement dans le mélange maître soient remises en suspension avant le chargement.
- Chargez 75 μL de mélange de bibliothèque dans la cellule d’écoulement via l’orifice d’échantillonnage de manière goutte à goutte. Assurez-vous que chaque goutte s’écoule dans le port avant d’ajouter la suivante.
- Replacez doucement le couvercle de l’orifice d’échantillonnage, en vous assurant que la bonde pénètre dans l’orifice d’échantillonnage.
- Fermez l’orifice d’amorçage et replacez le couvercle du dispositif de séquençage.
- Séquençage (48 h maximum)
- Connectez le dispositif de séquençage à l’ordinateur portable et ouvrez le logiciel de séquençage.
- Cliquez sur Démarrer , puis sur Démarrer le séquençage.
- Cliquez sur Nouvelle expérience et suivez le processus de l’interface utilisateur graphique (GUI) du logiciel de séquençage pour configurer les paramètres de l’exécution.
- Saisissez le nom de l’expérience et l’ID de l’échantillon (par exemple, rabv_run1), puis choisissez le type de cellule de flux dans le menu déroulant.
- Passez à la sélection du kit et choisissez le kit de séquençage de ligature approprié et le(s) kit(s) de code-barres natif utilisé(s).
- Continuez à exécuter les options. Conservez les valeurs par défaut, sauf si vous souhaitez que l’exécution s’arrête automatiquement après un certain nombre d’heures (les exécutions peuvent être arrêtées manuellement à tout moment).
- Passez à Basecalling. Choisissez d’activer ou de désactiver l’appel de base en fonction des ressources informatiques (voir configuration de l’ordinateur). Choisissez Options de modification sous Code-barres et assurez-vous que l’option Code-barres aux deux extrémités est activée. Enregistrez et passez à la section de sortie.
- Acceptez les valeurs par défaut et passez à l’examen final, vérifiez les paramètres et enregistrez les détails dans la feuille de calcul (fichier supplémentaire 3). Cliquez sur Démarrer.
REMARQUE : Si la cellule d’écoulement est réutilisée, ajustez la tension de démarrage (dans la section avancée des options de fonctionnement), comme indiqué par le schéma dans le fichier supplémentaire 3. - Enregistrez les canaux actifs initiaux - si cela est nettement inférieur à la vérification du contrôle de la qualité (CQ), redémarrez le logiciel de séquençage. Si le niveau est encore inférieur, redémarrez l’ordinateur.
- Enregistrez les canaux initiaux dans le brin par rapport au pore unique pour donner une occupation approximative des pores. Ce nombre fluctuera, alors donnez une approximation.
- Surveillez l’exécution au fur et à mesure de sa progression.
14. Appels de base en direct et hors ligne
NOTE : Ces instructions supposent que la structure de répertoires préexistante fournie dans le dépôt artic-rabv et que les sections 1 et 3 du protocole ont été suivies.
- Sur votre système de fichiers local, créez un répertoire appelé analyse, dans lequel vous stockerez toutes vos sorties d’analyse. Pour aller plus loin : créez un sous-répertoire avec le nom de votre projet et à l’intérieur de celui-ci un nouveau répertoire pour l’exécution, en utilisant l’exemple d’ID fourni à MinKNOW comme run_name. Faites-le en une seule commande comme suit :
mkdir -p
analyse/project_name/run_name
Naviguez ensuite jusqu’à son emplacement :
CD
chemin/analyse/project_name/run_name - Appels de base en direct
REMARQUE : Pour effectuer des appels de base nanoporeux en temps réel, les ordinateurs portables nécessitent une unité de traitement graphique (GPU) compatible NVIDIA CUDA. Assurez-vous que les instructions pour la configuration de l’appel de base GPU ont été exécutées à l’aide du protocole guppy56.- Lors de la configuration de l’exécution, activez les appels de base en direct.
- Utilisez RAMPART pour surveiller la couverture du séquençage en temps réel, conformément aux instructions ci-dessous.
- Dans le terminal de l’ordinateur, activez l’environnement artic-rabv conda :
conda activer artic-rabv - Créez un nouveau répertoire pour la sortie du rempart à l’intérieur du répertoire run_name et naviguez-y :
cd /chemin/analyse/project_name/run_name
mkdir rampart_output
rampart_output de CD - Créez un fichier codes-barres.csv pour associer les codes-barres et les exemples de noms. Il doit comporter une ligne par code-barres et ne spécifier que les codes-barres présents dans la bibliothèque, avec les en-têtes « code-barres » et « échantillon ». Suivez l’exemple dans le répertoire artic-rabv :
analyse/example_project/example_run/rampart_output/codes-barres.csv - Démarrez RAMPART en fournissant le dossier de protocole approprié et le chemin d’accès au dossier fastq_pass dans la sortie MinKNOW pour l’exécution :
rampart --protocol /path/rampart/scheme_name_V1_protocol - basecalledPath - Ouvrez une fenêtre de navigateur et accédez à localhost :3000 dans la zone URL. Attendez que suffisamment de données soient appelées pour que les résultats apparaissent à l’écran.
- Appel de base hors ligne (effectué après l’exécution)
- Si l’appel de base en direct n’a pas été défini, la sortie de MinKNOW sera constituée de données de signal brutes (fichiers fast5). Il ne sera pas possible d’utiliser RAMPART pendant la course. Convertissez les fichiers fast5 en données de base (fichiers fastq) après l’exécution à l’aide de guppy (voir la configuration dans Prérequis étape 1.1.1.). Exécutez RAMPART post-hoc sur les données de base.
- Exécutez le basecaller guppy :
guppy_basecaller -c dna_r9.4.1_450bps_fast.cfg -i /chemin/vers/lit/fast5_* -s /chemin/analyse/project_name/run_name -x auto -r
-c est le fichier de configuration pour spécifier le modèle d’appel de base, -i est le chemin d’entrée, -s est le chemin d’enregistrement, -x spécifie l’appel de base par le périphérique GPU (à exclure si vous utilisez la version ordinateur de guppy), et -r spécifie de rechercher les fichiers d’entrée de manière récursive.
REMARQUE : Le fichier de configuration (.cfg) peut être remplacé par un appel de base de haute précision en remplaçant _fast par _hac, bien que cela prenne beaucoup plus de temps.
15. Lavage des cellules d’écoulement
- Les cellules d’écoulement peuvent être lavées et réutilisées pour séquencer de nouvelles banques si les pores sont encore viables. Voir les instructions de lavage au protocole de lavage de la cellule d’écoulement ONT57.
16. Analyse et interprétation
- Génération de séquences de consensus avec le pipeline bioinformatique ARTIC
- Suivez les instructions détaillées dans le référentiel GitHub artic-rabv40 dans le dossier rabv_protocols pour générer des séquences de consensus à partir de fichiers fast5 bruts ou de fichiers fastq de base.
NOTA : Se reporter à la section Gazoduc Artic - Pipeline principal58 pour obtenir de plus amples renseignements.
- Suivez les instructions détaillées dans le référentiel GitHub artic-rabv40 dans le dossier rabv_protocols pour générer des séquences de consensus à partir de fichiers fast5 bruts ou de fichiers fastq de base.
- Facultatif : Analysez la profondeur de lecture moyenne par amplicon.
- Adapter les scripts disponibles dans le dépôt artic-rabv, en se référant au fichier supplémentaire 1. En bref, des statistiques détaillées sont générées à l’aide de SAMtools59 et de la couverture par nucléotide tracée dans R.
- Analyse phylogénétique à l’aide de GLUE
- À partir de RABV_GLUE42, sélectionnez l’onglet Analyse > génotypage et interprétation > Ajouter des fichiers, en sélectionnant le fichier fasta des séquences de consensus.
- Cliquez sur Soumettre et attendez. Une fois les analyses terminées, vous pourrez cliquer sur le bouton Afficher l’analyse , indiquant les affectations de clades et de sous-clades, la couverture par gène, la variation par rapport aux séquences de référence et le parent le plus proche.
- Les séquences contextuelles pertinentes peuvent également être identifiées dans la section Données de séquence > Séquences NCBI par clade .
- Sélectionnez le clade identifié ou cliquez sur Virus de la rage (RABV) pour voir toutes les séquences disponibles.
- Filtrez les séquences pertinentes (p. ex., pays d’origine).
- Téléchargez ces séquences et les métadonnées correspondantes à des fins d’analyse et de comparaison.
- Assignation de lignées à l’aide de MADDOG41
- Extrayez le dépôt MADDOG de GitHub pour vous assurer que vous travaillez avec la version la plus récente.
- Créez un dossier d’affectation dans le référentiel MADDOG local (précédemment créé dans la section Conditions préalables) appelé le nom de l’exécution.
- À l’intérieur du dossier, ajoutez le fichier fasta contenant les séquences de consensus.
- Ajoutez un fichier de métadonnées au dossier.
REMARQUE : Ce fichier doit être au format csv avec 4 colonnes intitulées « ID », « pays », « année » et « affectation », détaillant les ID de séquence, le pays d’échantillonnage et l’année de prélèvement de l’échantillon, tandis que la colonne « affectation » doit être vide. - Dans l’interface de ligne de commande, activez l’environnement conda : conda activate MADDOG.
- Dans l’interface de ligne de commande, accédez au dossier du référentiel MADDOG.
- Dans un premier temps, entreprenez l’affectation de lignées sur les séquences afin de vérifier toute anomalie potentielle et de déterminer s’il serait approprié d’exécuter l’étape de désignation de lignée la plus longue. Pour cela, tapez ceci dans la ligne de commande : sh assignment.sh.
- Lorsque vous y êtes invité, entrez Y pour indiquer que vous avez extrait le référentiel et que vous travaillez avec la version la plus récente de MADDOG.
- Lorsque vous y êtes invité, entrez le nom du dossier dans le dossier du référentiel MADDOG qui contient le fichier fasta.
- Une fois l’affectation de lignage terminée, vérifiez le fichier de sortie dans votre dossier. Si la sortie est conforme aux attentes et que plusieurs séquences sont affectées à la même lignée, exécutez la désignation de lignage.
- Si vous exécutez la désignation de lignage, supprimez le fichier de sortie d’affectation que vous venez de créer.
- Dans le terminal, à l’intérieur du dossier du référentiel MADDOG, exécutez la commande sh designation.sh.
- Lorsque vous y êtes invité, entrez Y pour indiquer que vous avez extrait le dépôt et que vous travaillez avec la version la plus récente de MADDOG.
- Lorsque vous y êtes invité, entrez le nom du dossier dans le dossier du référentiel MADDOG contenant le fichier fasta et les métadonnées. Cela produit des informations sur les lignées sur chaque séquence, une phylogénie des séquences précédentes nouvelles et pertinentes (à partir de la version 16.3.6), des informations hiérarchiques sur les lignées et des détails sur les lignées potentiellement émergentes et les zones de sous-échantillonnage.
NOTE : Tous les détails du protocole, de l’utilisation et des résultats peuvent être trouvés dans Campbell et al.60. - Une fois l’analyse initiale terminée, si vous êtes invité à tester également les lignées émergentes et sous-échantillonnées, entrez Y, si cela est nécessaire. Dans le cas contraire, entrez N.
- Si vous êtes invité à confirmer les lignées nouvellement trouvées, entrez Y et suivez les instructions dans le fichier NEXT_STEPS.eml résultant. Dans le cas contraire, entrez N.
Résultats
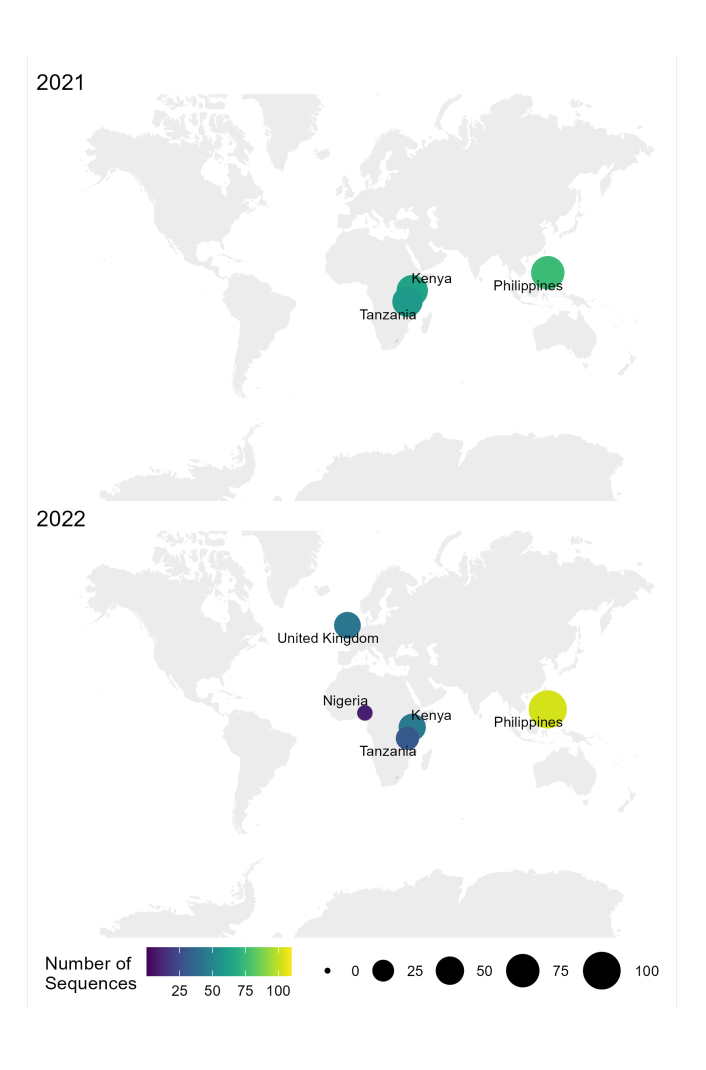
Le flux de travail de l’échantillon, de la séquence et de l’interprétation du RABV décrit dans ce protocole a été utilisé avec succès dans différentes conditions de laboratoire dans des pays endémiques, tels que la Tanzanie, le Kenya, le Nigeria et les Philippines (Figure 4). Le protocole a été utilisé sur différents types d’échantillons et conditions (tableau 6) : tissus cérébraux frais et congelés, extraits d’ADNc et d’ARN de tissus cérébraux transportés sous chaîne du froid pendant de longues périodes, et cartes FTA avec frottis de tissus cérébraux.
L’appel de base en direct à l’aide de RAMPART (Figure 5) montre la génération presque en temps réel des lectures et le pourcentage de couverture par échantillon. Ceci est particulièrement utile pour décider quand arrêter le cycle et enregistrer la cellule d’écoulement pour la réutiliser. Des variations dans le temps d’exécution ont été observées, certains terminant en 2 h, tandis que d’autres pouvaient prendre plus de 12 h pour atteindre une profondeur de couverture adéquate (x100). Nous pouvons également voir des régions avec une faible amplification ; par exemple, la figure 6 montre un instantané d’un cycle de séquençage où les profils de couverture montrent certains amplicons avec une très faible amplification, ce qui indique des amorces potentiellement problématiques. En étudiant plus en profondeur ces régions faiblement amplificatrices, nous avons été en mesure d’identifier les incompatibilités d’amorces, ce qui nous permettra de redessiner et d’améliorer les amorces individuelles. Certains schémas d’amorces ont montré plus d’incohérences que d’autres. C’est ce que l’on observe dans le programme d’introduction de l’Afrique de l’Est, par rapport aux Philippines, conformément à la diversité ciblée, car le programme de l’Afrique de l’Est vise à capturer une diversité beaucoup plus large.
RABV-GLUE42, une ressource polyvalente pour la gestion des données génomiques du RABV, et MADDOG60, un système de classification et de nomenclature des lignages, ont été utilisés pour compiler et interpréter les séquences RABV résultantes. Le tableau 7 montre les clades majeurs et mineurs circulant dans chaque pays assigné à l’aide de RABV-GLUE. On voit également une classification à plus haute résolution des lignées locales suite à l’affectation MADDOG.

Figure 1 : Flux de travail de l’échantillon, de la séquence à l’interprétation pour le RABV. Les étapes résumées sont indiquées pour (A) la préparation des échantillons, (B) la PCR et la préparation des bibliothèques, et (C) le séquençage et la bio-informatique jusqu’à l’analyse et l’interprétation. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Schéma du schéma d’amorçage. Positions de recuit le long du « génome de référence index » (violet foncé) pour les paires d’amorces directes et inverses (demi-flèches), qui sont attribuées dans deux pools distincts : A (rouge) et B (vert). Les paires d’amorces génèrent 400 pb d’amplicons superposés (bleus) qui sont numérotés séquentiellement le long du génome de référence de l’indice au format 'scheme_name_X_DIRECTION', où 'X' est un nombre se référant à l’amplicon généré par l’amorce et 'DIRECTION' est soit 'GAUCHE', soit 'DROITE', décrivant respectivement l’avant ou l’arrière. Les valeurs paires ou impaires de 'X' déterminent le pool (A ou B, respectivement). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Cellule d’écoulement nanoporeux48. Les étiquettes bleues illustrent les différentes parties de la cellule d’écoulement, y compris le couvercle de l’orifice d’amorçage qui recouvre l’orifice d’amorçage où la solution d’amorçage est ajoutée, le couvercle de l’orifice d’échantillonnage SpotON couvrant l’orifice d’échantillonnage où l’échantillon est ajouté goutte à goutte, les orifices d’évacuation 1 et 2 et l’ID de la cellule d’écoulement. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Carte montrant l’endroit où le séquençage du RABV a été effectué à l’aide du flux de travail optimisé en 2021 et 2022. La taille et la couleur des bulles correspondent au nombre de séquences par emplacement, où plus petit et plus foncé est moins nombreux, tandis que plus grand et plus clair est plus. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 5 : Capture d’écran de la visualisation de RAMPART dans un navigateur Web. Les noms de codes-barres sont remplacés par des noms d’échantillons en fonction de la configuration bioinformatique. Les trois panneaux du haut montrent des graphiques récapitulatifs pour l’ensemble de l’exécution : profondeur de couverture des lectures cartographiées pour chaque code-barres par position nucléotidique sur le génome de référence de l’index (en haut à gauche, coloré par code-barres), lectures mappées additionnées de tous les codes-barres au fil du temps (en haut au milieu) et lectures cartographiées par code-barres (en haut à droite, colorées par code-barres). Les panneaux inférieurs affichent les rangées de tracés par code-barres. De gauche à droite : la profondeur de couverture des lectures cartographiées par position nucléotidique sur le génome de référence de l’indice (à gauche), la distribution de longueur des lectures cartographiées (au milieu) et la proportion de positions de nucléotides sur le génome de référence de l’indice qui ont obtenu une couverture de 10x, 100x et 1 000x des lectures cartographiées au fil du temps (à droite). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 6 : Exemple de couverture de lecture sur le génome d’un échantillon de virus de la rage des Philippines séquencé à l’aide du protocole. La couverture de lecture à chaque position nucléotidique dans le génome est indiquée, ainsi que la position des amplicons superposés (1-41) utilisés pour générer la bibliothèque. Les pics de profondeur de couverture correspondent à des zones de chevauchement d’amplicons. Les amplicons avec une faible profondeur de couverture correspondent à des zones de chevauchement d’amplicons. Les amplicons avec une faible profondeur de couverture sont surlignés en rouge pour indiquer les zones problématiques qui peuvent nécessiter une optimisation. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Tableau 1 : Conditions du mélange maître et du thermocycleur pour la préparation de l’ADNc. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 2 : Conditions du mélange maître et du thermocycleur pour la PCR multiplex. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 3 : Conditions du mélange maître et du thermocycleur pour la réaction de préparation finale. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 4 : Conditions du mélange principal et du thermocycleur pour le codage à barres. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 5 : Mélange maître de ligature par adaptateur et dilution finale de la bibliothèque pour le séquençage. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 6 : Le nombre de séquences du génome entier du virus de la rage générées et le type d’échantillons utilisés dans différents pays à l’aide du flux de travail de l’échantillon à la séquence et à l’interprétation. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 7 : Affectations de clades majeurs et mineurs à partir de RABV-GLUE et affectations de lignées à partir de MADDOG pour les séquences générées à l’aide du flux de travail. Veuillez cliquer ici pour télécharger ce tableau.
Fichier supplémentaire 1 : Conception et optimisation du schéma d’amorce, et analyse de la profondeur de lecture de l’amplocon. Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 2 : Configuration du calcul Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 3 : Feuille de travail sur le protocole RABV WGS Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Brunker et al.61 ont mis au point un processus de séquençage du génome entier accessible par RABV, basé sur des nanopores, en utilisant les ressources du réseau ARTIC46. Nous présentons ici un flux de travail mis à jour, avec des étapes complètes de l’échantillon, de la séquence à l’interprétation. Le flux de travail détaille la préparation d’échantillons de tissus cérébraux pour le séquençage du génome entier, présente un pipeline bioinformatique pour traiter les lectures et générer des séquences consensuelles, et met en évidence deux outils spécifiques à la rage pour automatiser l’assignation des lignées et déterminer le contexte phylogénétique. Le flux de travail mis à jour fournit également des instructions complètes pour la configuration des espaces de travail de calcul et de laboratoire appropriés, avec des considérations pour la mise en œuvre dans différents contextes (y compris les environnements à faibles ressources). Nous avons démontré la mise en œuvre réussie du flux de travail dans les milieux universitaires et les instituts de recherche dans quatre PRFI endémiques du RABV avec une capacité de surveillance génomique nulle ou limitée. Le flux de travail s’est avéré résilient à l’application dans divers contextes et compréhensible par des utilisateurs ayant des expertises variées.
Ce flux de travail pour le séquençage RABV est le protocole le plus complet accessible au public (couvrant les étapes de l’échantillon à la séquence et à l’interprétation) et spécialement adapté pour réduire les coûts de démarrage et d’exploitation. Le temps et le coût requis pour la préparation et le séquençage de la bibliothèque avec la technologie des nanopores sont considérablement réduits par rapport à d’autres plates-formes, telles qu’Illumina61, et les développements technologiques continus améliorent la qualité et la précision de la séquence pour être comparables à Illumina62.
Ce protocole est conçu pour être résilient dans divers contextes à faibles ressources. En se référant aux conseils de dépannage et de modification fournis avec le protocole de base, les utilisateurs sont aidés à adapter le flux de travail à leurs besoins. L’ajout d’outils bioinformatiques conviviaux au flux de travail constitue une évolution majeure du protocole original, fournissant des méthodes rapides et standardisées qui peuvent être appliquées par des utilisateurs ayant une expérience préalable minimale en bioinformatique pour interpréter les données de séquence dans des contextes locaux. La capacité de le faire in situ est souvent limitée par la nécessité d’avoir des compétences spécifiques en programmation et en phylogénétique, ce qui nécessite un investissement intensif et à long terme dans la formation professionnelle. Bien que cet ensemble de compétences soit important pour interpréter en profondeur les données de séquence, des outils d’interprétation de base et accessibles sont tout aussi souhaitables afin de renforcer les capacités des « champions du séquençage » locaux, dont l’expertise de base peut être basée sur les laboratoires humides, leur permettant d’interpréter et de s’approprier leurs données.
Comme le protocole a été mis en œuvre pendant un certain nombre d’années dans plusieurs pays, nous pouvons maintenant fournir des conseils sur la façon d’optimiser les schémas d’amorces multiplex afin d’améliorer la couverture et de faire face à la diversité accumulée. Des efforts ont également été faits pour aider les utilisateurs à améliorer le rapport coût-efficacité ou pour faciliter l’approvisionnement dans une région donnée, ce qui constitue généralement un défi pour la durabilité des approches moléculaires63. Par exemple, en Afrique (Tanzanie, Kenya et Nigeria), nous avons opté pour un mélange maître blunt/TA ligase à l’étape de la ligase par adaptateur, qui était plus facilement disponible auprès des fournisseurs locaux et constituait une alternative moins chère aux autres réactifs de ligaturation.
D’expérience, il existe plusieurs façons de réduire le coût par échantillon et par cycle. La réduction du nombre d’échantillons par cycle (par exemple, de 24 à 12 échantillons) peut prolonger la durée de vie des cellules d’écoulement sur plusieurs essais, tandis que l’augmentation du nombre d’échantillons par cycle maximise le temps et les réactifs. Entre nos mains, nous avons pu laver et réutiliser les cellules d’écoulement pour un cycle de séquençage sur trois, ce qui a permis de séquencer 55 échantillons supplémentaires. Le lavage de la cellule d’écoulement immédiatement après utilisation, ou si ce n’est pas possible, l’élimination du fluide résiduaire du canal d’évacuation après chaque passage, semblait préserver le nombre de pores disponibles pour un deuxième cycle. En tenant compte du nombre initial de pores disponibles dans une cellule d’écoulement, un cycle peut également être optimisé pour planifier le nombre d’échantillons à traiter dans une cellule d’écoulement particulière.
Bien que le flux de travail vise à être aussi complet que possible, avec l’ajout de conseils détaillés et de ressources balisées, la procédure reste complexe et peut être intimidante pour un nouvel utilisateur. L’utilisateur est encouragé à rechercher une formation et un soutien en personne, idéalement localement, ou par l’intermédiaire de collaborateurs externes. Aux Philippines, par exemple, un projet de renforcement des capacités au sein des laboratoires régionaux pour la surveillance génomique du SRAS-CoV-2 à l’aide de l’ONT a permis aux diagnostiqueurs de soins de santé de développer des compétences de base qui sont facilement transférables au séquençage du RABV. Les étapes importantes, telles que le nettoyage du cordon SPRI, peuvent être difficiles à maîtriser sans formation pratique, et un nettoyage inefficace peut endommager la cellule d’écoulement et compromettre le fonctionnement. La contamination des échantillons est toujours une préoccupation majeure lorsque les amplicons sont traités en laboratoire et peut être difficile à éliminer. En particulier, la contamination croisée entre les échantillons est extrêmement difficile à détecter lors de la bio-informatique post-analyse. De bonnes techniques et pratiques de laboratoire, telles que le maintien de surfaces de travail propres, la séparation des zones pré et post-PCR et l’intégration de contrôles négatifs, sont impératives pour assurer le contrôle de la qualité. Le rythme rapide des développements de séquençage des nanopores est à la fois un avantage et un inconvénient pour la surveillance génomique de routine du RABV. L’amélioration continue de la précision, de l’accessibilité et du répertoire de protocoles de nanopore élargit et améliore le champ d’application de son application. Cependant, les mêmes développements rendent difficile le maintien de procédures opérationnelles standard et de pipelines bioinformatiques. Dans ce protocole, nous fournissons un document d’aide à la transition entre les anciens et les actuels kits de préparation de bibliothèques de nanopores (Table of Materials).
L’un des obstacles courants au séquençage dans les PRFI est l’accessibilité, y compris non seulement le coût, mais aussi la capacité à se procurer des consommables en temps opportun (en particulier les réactifs de séquençage, qui sont relativement nouveaux pour les équipes d’approvisionnement et les fournisseurs) et des ressources informatiques, ainsi que le simple fait d’avoir accès à une alimentation stable et à Internet. L’utilisation de la technologie de séquençage portable des nanopores comme base de ce flux de travail permet de résoudre bon nombre de ces problèmes d’accessibilité, et nous avons démontré l’utilisation de notre protocole dans divers contextes, en effectuant le protocole complet et l’analyse dans le pays. Certes, l’approvisionnement en équipement et le séquençage des consommables en temps opportun restent un défi et, dans de nombreux cas, nous avons été contraints de transporter ou d’expédier des réactifs depuis le Royaume-Uni. Cependant, dans certaines régions, nous avons pu compter entièrement sur les voies d’approvisionnement locales pour les réactifs, bénéficiant d’investissements dans le séquençage du SRAS-CoV-2 (par exemple, aux Philippines) qui ont rationalisé les processus d’approvisionnement et commencé à normaliser l’application de la génomique des agents pathogènes.
Le besoin d’une connexion Internet stable est minimisé par des installations uniques ; par exemple, les dépôts GitHub, le téléchargement de logiciels et le séquençage nanopore lui-même ne nécessitent qu’un accès à Internet pour démarrer l’exécution (et non tout au long) ou peuvent être effectués complètement hors ligne avec l’accord de l’entreprise. Si des données mobiles sont disponibles, un téléphone peut être utilisé comme point d’accès à l’ordinateur portable pour commencer l’exécution du séquençage, avant de se déconnecter pendant la durée de l’exécution. Lors du traitement de routine d’échantillons, les besoins en stockage de données peuvent augmenter rapidement et, idéalement, les données devraient être stockées sur un serveur. Sinon, les disques durs à semi-conducteurs (SSD) sont relativement bon marché à l’approvisionnement.
Bien que nous reconnaissions qu’il existe encore des obstacles à la surveillance génomique dans les PRFI, l’augmentation des investissements dans le renforcement de l’accessibilité et de l’expertise en génomique (par exemple, l’Initiative africaine de génomique des agents pathogènes [IGP Afrique])64 suggère que cette situation va s’améliorer. La surveillance génomique est essentielle pour la préparation aux pandémies6, et la capacité peut être établie en routinisant la surveillance génomique des agents pathogènes endémiques tels que le RABV. Les disparités mondiales dans les capacités de séquençage mises en évidence pendant la pandémie de SRAS-CoV-2 devraient être un moteur de changement catalytique pour remédier à ces inégalités structurelles.
Ce flux de travail de l’échantillon à la séquence et à l’interprétation pour le RABV, y compris les outils bioinformatiques accessibles, a le potentiel d’être utilisé pour guider les mesures de contrôle visant l’objectif de zéro décès humain dû à la rage transmise par le chien d’ici 2030 et, en fin de compte, pour l’élimination des variants du RABV. Combinées aux métadonnées pertinentes, les données génomiques générées à partir de ce protocole facilitent la caractérisation rapide du RABV lors des enquêtes sur les épidémies et dans l’identification des lignées circulantes dans un pays ou une région60,61,65. Nous illustrons notre pipeline principalement à l’aide d’exemples de rage transmise par les chiens ; Cependant, le flux de travail est directement applicable à la rage de la faune. Cette transférabilité et ce faible coût minimisent les difficultés liées à la facilité d’accès au séquençage de routine, non seulement pour la rage, mais aussi pour d’autres agents pathogènes46,66,67, afin d’améliorer la gestion et le contrôle de la maladie.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Ce travail a été soutenu par Wellcome [207569/Z/17/Z, 224670/Z/21/Z], un financement Newton du Medical Research Council [MR/R025649/1] et du Département de la science et de la technologie des Philippines (DOST), l’effort mondial de recherche et d’innovation du Royaume-Uni sur le COVID-19 [MR/V035444/1], le Fonds de soutien stratégique institutionnel de l’Université de Glasgow [204820], le Medical Research Council New Investigator Award (KB) [MR/X002047/1], et le Fonds de développement des partenariats internationaux, une bourse d’études du DOST British Council-Philippines (CB), une bourse GemVi (GJ) de l’Institut national de recherche en santé [17/63/82] et des bourses d’études de l’Université de Glasgow du MVLS DTP (KC) [125638-06], de l’EPSRC DTP (RD) [EP/T517896/1] et du Wellcome IIB DTP (HF) [218518/Z/19/Z]. Nous sommes reconnaissants envers les collègues et collaborateurs qui ont soutenu ce travail : Daniel Streicker, Alice Broos, Elizabeth Miranda, DVM, Daria Manalo, DVM, Thumbi Mwangi, Kennedy Lushasi, Charles Kayuki, Jude Karlo Bolivar, Jeromir Bondoc, Esteven Balbin, Ronnel Tongohan, Agatha Ukande, Davis Kuchaka, Mumbua Mutunga, Lwitiko Sikana et Anna Czupryna.
matériels
| Name | Company | Catalog Number | Comments |
| Brand name | |||
| Software | |||
| Sequencing software (MinKnow) | Oxford Nanopore Technologies | https://community.nanoporetech.com/downloads | |
| Bioinformatics tool kit (Guppy) | Oxford Nanopore Technologies | https://community.nanoporetech.com/docs/prepare/library_prep_protocols/Guppy-protocol/v/gpb_2003_v1_revao_14dec2018 | |
| Equipment | |||
| Thermal cycler (miniPCR™ mini16 thermal cycler) | Cambio | MP-QP-1016-01 | |
| Homogenizer (Precellys Evolution Touch Homogenizer) | Bertin Instruments | EQ02520-300 | |
| Cold Racks (0.2-0.5mL) (PCR Mini-cooler with transparent lid) | BRAND | 781260 | |
| Pipettor | |||
| (Pipetman L Fixed F1000L, 1000 uL) | Gilson | SKU: FA10030 | |
| (Pipetman L Fixed F100L, 100 uL) | Gilson | SKU: FA10024 | |
| (Pipetman L Fixed F10L, 10 uL) | Gilson | SKU: FA10020 | |
| (Pipetman L Fixed F1L, 1 uL) | Gilson | SKU: FA10025 | |
| (Pipetman L Fixed F20L, 20 uL) | Gilson | SKU: FA10021 | |
| (Pipetman L Fixed F250L, 250 uL) | Gilson | SKU: FA10026 | |
| Fluorometer (Qubit 4 Fluorometer) | Thermofisher scientific/Fisher scientific | Q33238 | |
| Laptop (Any brand with ~2 GB of drive space, minimum of 512 GB storage space, msi installer [GPU]) | |||
| Microcentrifuge (Refrigerated centrifuge) | Thermofisher scientific/Fisher scientific | 75004081 | |
| Vortex mixer (Basic vortex mixer) | Thermofisher scientific/Fisher scientific | 88882011 | |
| Magnetic rack (DynaMag -2 Magnet) | Thermofisher scientific/Fisher scientific | 12321D | |
| Sequencing device (MinION) | Oxford Nanopore Technologies | MinION Mk1B | |
| RNA Extraction | |||
| RNA extraction kit (Qiagen RNEasy Mini Kit 250) | Qiagen | 74106 | |
| RNA stabilizing reagents | |||
| (RNA later) | Invitrogen | AM7020 | |
| (DNA/RNA Shield) | Zymo Research | R1100-50 | |
| PCR | |||
| Nuclease-free Water (Nuclease-free Water [not DEPC-treated]) | Thermofisher scientific/Fisher scientific | AM9937 | |
| Master mix for first strand cDNA synthesis (LunaScript RT SuperMix Kit) | New England Biolabs | E3010S | |
| DNA amplification master mix (Q5® Hot Start High-Fidelity 2X Master Mix [NEB]) | New England Biolabs | M0494L | |
| Primer (Scheme) (Custom DNA oligos) | Invitrogen | ||
| SPRI Bead Clean-up | |||
| SPRI beads (Aline Biosciences PCR Clean DX ) | Cambio | AL-AC1003-50 | |
| Ethanol, Pure Absolute, >99.8% (GC) [Riedel-De Haen] | Merck | 818760 | |
| Short Fragment buffer (SFB expansion pack) | Oxford Nanopore Technologies | EXP-SFB001 | |
| DNA Quantification | |||
| DNA quantification kit (Qubit® dsDNA HS Assay Kit) | Thermofisher scientific/Fisher scientific | Q32854 | |
| DNA quantification assay tubes (Qubit™ Assay Tubes) | Thermofisher scientific/Fisher scientific | Q32856 | |
| End Prep and barcoding (Qubit™ Assay Tubes) | |||
| End Prep master mix (NEBNext Ultra End Repair/dA-Tailing Module) | New England Biolabs | E7546L | |
| Barcoding kit | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 | |
| (Native Barcoding Expansion 1-12) | EXP-NBD104 | ||
| (Native Barcoding Expansion 13-24) | EXP-NBD114 | ||
| (Native Barcoding Expansion 96) | EXP-NBD196 | ||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 | |
| (not compatible) | (not compatible) | ||
| (Native Barcoding Kit 24 V14) | SQK-NBD114.24 | ||
| (Native Barcoding Kit 96 V14) | SQK-NBD114.96 | ||
| Ligation mastermix (Blunt/TA Ligase Master Mix) | New England Biolabs | M0367S | |
| Adapter Ligation | |||
| Adapter ligation master mix | |||
| (NEBNext Quick Ligation Module) | New England Biolabs | E6056S | |
| (NEBNext Ultra II Ligation Module) | New England Biolabs | E7595S | |
| (Blunt/TA Ligase Master Mix) | New England Biolabs | M0367S | |
| Adapter mix | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 EXP-AMII001 | |
| (Adapter Mix II [AMII]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 EXP-NBA114 | |
| (Native adapter [NA]) | |||
| Sequencing | |||
| Flowcell priming kit | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 EXP-FLP002 | |
| (Flush Buffer [FB]) | |||
| (Flush Tether [FT]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 EXP-FLP004 | |
| (Flow Cell Flush [FCF]) | |||
| (Flow Cell Tether [FCT]) | |||
| Ligation Sequencing Kit | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 SQK-LSK109 | |
| Adapter Mix (Adapter Mix [AMX]) | |||
| Ligation Buffer (Ligation buffer [LNB]) | |||
| Short Fragment Buffer (Short Fragment buffer [SFB]) | |||
| Sequencing Buffer (Sequencing Buffer [SQB]) | |||
| Elution Buffer (Elution buffer [EB]) | |||
| Loading Beads (Loading Beads [LB]) | |||
| Sequencing Tether (Sequencing Tether [SQT]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 SQK-LSK114 | |
| Adapter Mix (Ligation Adapter [LA]) | |||
| Ligation Buffer (Ligation buffer [LNB]) | |||
| Short Fragment Buffer (Short Fragment buffer [SFB]) | |||
| Sequencing Buffer (Sequencing Buffer [SB]) | |||
| Elution Buffer (Elution buffer [EB]) | |||
| Loading Beads (Loading Beads [LIB]) | |||
| Sequencing Tether (Flow Cell Tether [FCT]) | |||
| Library solution (Library solution [LIS]) | |||
| Flush buffer (Flow Cell Flush [FCF]) | |||
| Flow Cell | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 FLO-MIN106D | |
| (Flow Cell [R9.4.1]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 FLO-MIN114 | |
| (Flow Cell [R10.4.1]) | |||
| Flow Cell wash | |||
| Flowcell wash kit (Flow cell wash kit) | Oxford Nanopore Technologies | EXP-WSH004 | |
| Consummables | |||
| Surface decontaminant | |||
| (DNA Away Surface Decontaminant, Squeeze Bottle [Molecular Bio]) | Thermofisher scientific/Fisher scientific | 7010PK | |
| (RNase Away Surface Decontaminant, Bottle [Molecular Bio]) | Thermofisher scientific/Fisher scientific | 7002PK | |
| PCR 8-Tube Strip 0.2ml, individual cap (PCR 8-Tube Strip 0.2ml, with Individual attached Flat Caps, Sterile, DNAse/RNAse, Pyrogen Free,Natural [Greiner]) | Greiner | 608281 | |
| PCR Tube 0.2ml (PCR Tube 0.2ml, Natural [Domed Cap] Bagged in 500s, Non-Sterile [Greiner]) | Greiner | 671201 | |
| 1000µL Filter Tips (500) (Stacked 1000µL Filter Tips [500]) | Thermofisher scientific/Fisher scientific | 11977724 | |
| 100µL Filter Tips (1000) | Thermofisher scientific/Fisher scientific | 11947724 | |
| 10µL Filter Tips (1000) (Stacked 100µL Filter Tips [1000]) | Thermofisher scientific/Fisher scientific | 11907724 | |
| Reinforced tubes tubes (2ml) with screw caps and o-rings (Fisherbrand™ Bulk tubes) | Thermofisher scientific/Fisher scientific | 15545809 | |
| Microcentrifuge tube (1.5ml) (1.5 ml Eppendorf Tubes [500]) | Eppendorf | 1229888 | |
| DNA LoBind Tubes (1.5ml) (DNA LoBind Tubes) | Thermofisher scientific/Fisher scientific | 10051232 | |
| Cryobabies labels | |||
| Gloves (S/M/L) | |||
| Paper towel |
Références
- Rupprecht, C. E. Rhabdoviruses: rabies virus. Medical Microbiology. , University of Texas Medical Branch. Galveston, TX. (1996).
- Rabies. World Health Organization. , Available from: https://www.who.int/news-room/fact-sheets/detail/rabies (2023).
- Health Organization, W. orld WHO Expert Consultation on Rabies: WHO TRS N°1012. World Health Organization. , Available from: https://www.who.int/publications-detail-redirect/WHO-TRS-1012 (2018).
- Benavides, J. A., et al. Defining new pathways to manage the ongoing emergence of bat rabies in Latin America. Viruses. 12 (9), 1002(2020).
- Hampson, K. Estimating the global burden of endemic canine rabies. PLoS Neglected Tropical Diseases. 9 (4), e0003709(2015).
- Global genomic surveillance strategy for pathogens with pandemic and epidemic potential, 2022-2032. World Health Organization. , Available from: https://www.who.int/publications-detail-redirect/978924004679 (2022).
- Tsai, K. J., et al. Emergence of a sylvatic enzootic formosan ferret badger-associated rabies in Taiwan and the geographical separation of two phylogenetic groups of rabies viruses. Veterinary Microbiology. 182, 28-34 (2016).
- Chiou, H. -Y., et al. Molecular characterization of cryptically circulating rabies virus from ferret badgers, Taiwan. Emerging Infectious Diseases. 20 (5), 790-798 (2014).
- Sabeta, C. T., Mansfield, K. L., McElhinney, L. M., Fooks, A. R., Nel, L. H. Molecular epidemiology of rabies in bat-eared foxes (Otocyon megalotis) in South Africa. Virus Research. 129 (1), 1-10 (2007).
- Scott, T. P. Complete genome and molecular epidemiological data infer the maintenance of rabies among kudu (Tragelaphus strepsiceros) in Namibia. PLoS One. 8 (3), e58739(2013).
- Lembo, T., et al. Exploring reservoir dynamics: a case study of rabies in the Serengeti ecosystem. The Journal of Applied Ecology. 45 (4), 1246-1257 (2008).
- Coetzee, P., Nel, L. H. Emerging epidemic dog rabies in coastal South Africa: a molecular epidemiological analysis. Virus Research. 126 (1-2), 186-195 (2007).
- Ou de Munnink, B. B. First molecular analysis of rabies virus in Qatar and clinical cases imported into Qatar, a case report. International Journal of Infectious Diseases. 96, 323-326 (2020).
- Smith, J., et al. Case report: Rapid ante-mortem diagnosis of a human case of rabies imported into the UK from the Philippines. Journal of Medical Virology. 69, 150-155 (2003).
- Mahardika, G. N. K., et al. Phylogenetic analysis and victim contact tracing of rabies virus from humans and dogs in Bali, Indonesia. Epidemiology and Infection. 142 (6), 1146-1154 (2014).
- Tohma, K., et al. Molecular and mathematical modeling analyses of inter-island transmission of rabies into a previously rabies-free island in the Philippines. Infection, Genetics and Evolution. 38, 22-28 (2016).
- Tohma, K., et al. Phylogeographic analysis of rabies viruses in the Philippines. Infection, Genetics and Evolution. 23, 86-94 (2014).
- Saito, M., et al. Genetic diversity and geographic distribution of genetically distinct rabies viruses in the Philippines. PLoS Neglected Tropical Diseases. 7 (4), e2144(2013).
- Biek, R., Henderson, J. C., Waller, L. A., Rupprecht, C. E., Real, L. A. A high-resolution genetic signature of demographic and spatial expansion in epizootic rabies virus. Proceedings of the National Academy of Sciences. 104 (19), 7993-7998 (2007).
- Reddy, G. B., et al. Molecular characterization of Indian rabies virus isolates by partial sequencing of nucleoprotein (N) and phosphoprotein (P) genes. Virus Genes. 43, 13-17 (2011).
- David, D., Dveres, N., Yakobson, B. A., Davidson, I. Emergence of dog rabies in the northern region of Israel. Epidemiology and Infection. 137 (4), 544-548 (2009).
- Benjathummarak, S. Molecular genetic characterization of rabies virus glycoprotein gene sequences from rabid dogs in Bangkok and neighboring provinces in Thailand, 2013-2014. Archives of Virology. 161 (5), 1261-1271 (2016).
- Denduangboripant, J., et al. Transmission dynamics of rabies virus in Thailand: implications for disease control. BMC Infectious Diseases. 5, 52(2005).
- Talbi, C., et al. Phylodynamics and human-mediated dispersal of a zoonotic virus. PLoS Pathogens. 6 (10), e1001166(2010).
- Bourhy, H., et al. Revealing the micro-scale signature of endemic zoonotic disease transmission in an African urban setting. PLoS Pathogens. 12 (4), e1005525(2016).
- Zinsstag, J., et al. Vaccination of dogs in an African city interrupts rabies transmission and reduces human exposure. Science Translational Medicine. 9 (421), (2017).
- Yakovleva, A., et al. Tracking SARS-COV-2 variants using Nanopore sequencing in Ukraine in 2021. Scientific Reports. 12, 15749(2022).
- Mannsverk, S., et al. SARS-CoV-2 variants of concern and spike protein mutational dynamics in a Swedish cohort during 2021, studied by Nanopore sequencing. Virology Journal. 19, 164(2022).
- Soufi, M., et al. Fast and easy nanopore sequencing workflow for rapid genetic testing of familial Hypercholesterolemia. Frontiers in Genetics. 13, 836231(2022).
- Cabibbe, A. M. Application of targeted next-generation sequencing assay on a portable sequencing platform for culture-free detection of drug-resistant tuberculosis from clinical samples. Journal of Clinical Microbiology. 58 (10), 00632(2020).
- Xu, Y., et al. Nanopore metagenomic sequencing of influenza virus directly from respiratory samples: diagnosis, drug resistance and nosocomial transmission, United Kingdom, 2018/19 influenza season. Euro Surveillance. 26 (27), 2000004(2021).
- Stubbs, S. C. B., et al. Assessment of a multiplex PCR and Nanopore-based method for dengue virus sequencing in Indonesia. Virology Journal. 17, 24(2020).
- Croville, G., et al. Rapid whole-genome based typing and surveillance of avipoxviruses using nanopore sequencing. Journal of Virological Methods. 261, 34-39 (2018).
- Theuns, S., et al. Nanopore sequencing as a revolutionary diagnostic tool for porcine viral enteric disease complexes identifies porcine kobuvirus as an important enteric virus. Scientific Reports. 8, 9830(2018).
- O'Donnell, V. K., et al. Rapid sequence-based characterization of African swine fever virus by use of the Oxford Nanopore MinION sequence sensing device and a companion analysis software tool. Journal of Clinical Microbiology. 58, 01104-01119 (2019).
- Brito, A. F. Global disparities in SARS-CoV-2 genomic surveillance. Nature Communications. 13, 7003(2022).
- ONT login/register. Oxford Nanopore Technology. , Available from: https://nanoporetech.com/login-register (2023).
- Software Downloads. Oxford Nanopore Technology. , Available from: https://community.nanoporetech.com/downloads (2023).
- GitHub. , Available from: https://github.com/ (2023).
- Brunker, K. Artic-rabv. , Available from: https://github.com/kirstyn/artic-rabv (2022).
- Campbell, K. MADDOG: Method for Assignment, Definition and Designation of Global Lineages. , Available from: https://github.com/KathrynCampbell/MADDOG (2022).
- Campbell, K. RABV-GLUE. Centre for Virus Research. , Available from: https://github.com/KathrynCampbell/MADDOG (2022).
- Itokawa, K., Sekizuka, T., Hashino, M., Tanaka, R., Kuroda, M. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS ONE. 15 (9), e0239403(2020).
- Davis, M. W., Jorgensen, E. M. ApE, A plasmid editor: A freely available DNA manipulation and visualization program. Frontiers in Bioinformatics. 2, 818619(2022).
- Döring, M., Pfeifer, N. openPrimeR: Multiplex PCR primer design and analysis. , (2023).
- Quick, J. Multiplex PCR method for MiniON and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nature Protocols. 12 (6), 1261-1276 (2017).
- Laboratory Techniques in Rabies. World Health Organization. 1, Available from: https://apps.who.int/iris/bitstream/handle/10665/310836/9789241515153-eng.pdf (2018).
- Lembo, T. Partners for Rabies Prevention. The blueprint for rabies prevention and control: a novel operational toolkit for rabies elimination. PLoS Neglected Tropical Diseases. 6 (2), e1388(2012).
- Terrestrial Manual Online Access. World Organization for Animal Health. , Available from: https://www.woah.org/en/what-we-do/standards/codes-and-manuals/terrestrial-manual-online-access/ (2023).
- Mauti, S. Field postmortem rabies rapid immunochromatographic diagnostic test for resource-limited settings with further molecular applications. Journal of Visualized Experiments. (160), e60008(2020).
- Patrick, E., et al. Enhanced rabies surveillance using a direct rapid immunohistochemical test. Journal of Visualized Experiments. (146), e59416(2019).
- Lembo, T., et al. Evaluation of a direct, rapid immunohistochemical test for rabies diagnosis. Emerging Infectious Diseases. 12 (2), 310-313 (2006).
- Marston, D. A., et al. Pan-lyssavirus real time RT-PCR for rabies diagnosis. Journal of Visualized Experiments. (149), e59709(2019).
- Brunker, K. DNA quantification using the Qubit fluorometer. , Available from: https://www.protocols.io/view/dna-quantification-using-the-qubit-fluorometer-bc6vize6 (2020).
- Quick, J. DNA quantification using the Quantus fluorometer. , Available from: https://www.protocols.io/view/dna-quantification-using-the-quantus-fluorometer-7pzhmp6 (2020).
- Guppy protocol. Nanopore Community. , Available from: https://community.nanoporetech.com/protocols/Guppy-protocol/v/gpb_2003_v1_revaq_14dec2018 (2023).
- Flow Cell Wash Kit (EXP-WSH004). Nanopore Community. , Available from: https://community.nanoporetech.com/protocols/flow-cell-wash-kit-exp-wsh004/v/wfc_9120_v1_revh_08dec2020 (2023).
- Core Pipeline - arctic pipeline. , Available from: https://artic.readthedocs.io/en/latest/minion/ (2023).
- Samtools. , Available from: https://www.htslib.org (2023).
- Campbell, K., et al. Making genomic surveillance deliver: A lineage classification and nomenclature system to inform rabies elimination. PLoS Pathogens. 18 (5), e1010023(2022).
- Brunker, K., et al. Rapid in-country sequencing of whole virus genomes to inform rabies elimination programmes. Wellcome Open Research. 5, 3(2020).
- Bull, R. A., et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nature Communications. 11, 6272(2020).
- Okeke, I. N., Ihekweazu, C. The importance of molecular diagnostics for infectious diseases in low-resource settings. Nature Reviews. Microbiology. 19 (9), 547-548 (2021).
- Inzaule, S. C., Tessema, S. K., Kebede, Y., Ouma, A. E. O., Nkengasong, J. N. Genomic-informed pathogen surveillance in Africa: opportunities and challenges. The Lancet Infectious Diseases. 21 (9), 281-289 (2021).
- Kennedy, L. Integrating contact tracing and whole-genome sequencing to track the elimination of dog-mediated rabies: an observational and genomic study. eLife. , (2023).
- Pallerla, S. R. Diagnosis of pathogens causing bacterial meningitis using Nanopore sequencing in a resource-limited setting. Annals of Clinical Microbiology and Antimicrobials. 21, 39(2022).
- Quick, J. Real-time, portable genome sequencing for Ebola surveillance. Nature. 530 (7589), 228-232 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.