
Method Article
Sequenziamento dell'intero genoma per una rapida caratterizzazione del virus della rabbia utilizzando la tecnologia Nanopore
In questo articolo
Riepilogo
Qui, presentiamo un flusso di lavoro rapido ed economico per la caratterizzazione dei genomi del virus della rabbia (RABV) utilizzando la tecnologia dei nanopori. Il flusso di lavoro ha lo scopo di supportare la sorveglianza basata sulla genomica a livello locale, fornendo informazioni sui lignaggi RABV circolanti e sul loro posizionamento all'interno delle filogenesi regionali per guidare le misure di controllo della rabbia.
Abstract
I dati genomici possono essere utilizzati per tracciare la trasmissione e la diffusione geografica delle malattie infettive. Tuttavia, la capacità di sequenziamento richiesta per la sorveglianza genomica rimane limitata in molti paesi a basso e medio reddito (LMIC), dove la rabbia mediata dai cani e/o la rabbia trasmessa da animali selvatici come i pipistrelli vampiro pongono gravi preoccupazioni per la salute pubblica e l'economia. Presentiamo qui un flusso di lavoro rapido e conveniente dal campione alla sequenza all'interpretazione utilizzando la tecnologia dei nanopori. Vengono brevemente descritti i protocolli per la raccolta dei campioni e la diagnosi della rabbia, seguiti dai dettagli del flusso di lavoro ottimizzato per il sequenziamento dell'intero genoma, tra cui la progettazione e l'ottimizzazione del primer per la reazione a catena della polimerasi multiplex (PCR), una preparazione modificata e a basso costo della libreria di sequenziamento, il sequenziamento con chiamata di base dal vivo e offline, la designazione del lignaggio genetico e l'analisi filogenetica. Viene dimostrata l'implementazione del flusso di lavoro e vengono evidenziati i passaggi critici per l'implementazione locale, come la convalida della pipeline, l'ottimizzazione del primer, l'inclusione di controlli negativi e l'uso di dati disponibili pubblicamente e strumenti genomici (GLUE, MADDOG) per la classificazione e il posizionamento all'interno di filogenesi regionali e globali. Il tempo di consegna per il flusso di lavoro è di 2-3 giorni e il costo varia da $ 25 per campione per un'esecuzione di 96 campioni a $ 80 per campione per un'esecuzione di 12 campioni. Concludiamo che l'istituzione di una sorveglianza genomica del virus della rabbia nei paesi a basso e medio reddito è fattibile e può supportare i progressi verso l'obiettivo globale di zero decessi per rabbia umana mediata dai cani entro il 2030, nonché un migliore monitoraggio della diffusione della rabbia nella fauna selvatica. Inoltre, la piattaforma può essere adattata ad altri agenti patogeni, contribuendo a costruire una capacità genomica versatile che contribuisce alla preparazione alle epidemie e alle pandemie.
Introduzione
Il virus della rabbia (RABV) è un lyssavirus della famiglia Rhabdoviridae che causa una malattia neurologica fatale nei mammiferi1. Sebbene la rabbia sia prevenibile al 100% con la vaccinazione, rimane una delle principali preoccupazioni per la salute pubblica e l'economia nei paesi endemici. Dei 60.000 decessi per rabbia umana che si stima si verifichino ogni anno, oltre il 95% si verifica in Africa e in Asia, dove i cani sono il serbatoio primario. Al contrario, la vaccinazione dei cani ha portato all'eliminazione della rabbia mediata dai cani in tutta l'Europa occidentale, il Nord America e gran parte dell'America Latina. In queste regioni, le riserve di rabbia sono ora limitate alla fauna selvatica, come pipistrelli, procioni, puzzole e canidi selvatici3. In tutta l'America Latina, il pipistrello vampiro comune è una fonte problematica di rabbia a causa della regolare trasmissione di spillover dai pipistrelli sia agli esseri umani che al bestiame durante l'alimentazione notturnadel sangue. L'impatto economico globale annuale della rabbia è stimato in 8,6 miliardi di dollari, con perdite di bestiame che rappresentano il 6%5.
I dati di sequenza dei patogeni virali, combinati con i metadati sulla tempistica e l'origine delle infezioni, possono fornire solide informazioni epidemiologiche6. Per il RABV, il sequenziamento è stato utilizzato per indagare l'origine dei focolai7,8, identificare le associazioni dell'ospite con la fauna selvatica o i cani domestici 8,9,10,11,12 e rintracciare le fonti dei casi umani 13,14. Le indagini sull'epidemia che utilizzano l'analisi filogenetica hanno indicato che la rabbia è emersa nella provincia di Bali, in Indonesia, precedentemente libera dalla rabbia, attraverso una singola introduzione dalle vicine aree endemiche di Kalimantan o Sulawesi15. Nel frattempo, nelle Filippine, è stato dimostrato che un focolaio sull'isola di Tablas, nella provincia di Romblon, è stato introdotto dall'isola principale di Luzon16. I dati genomici virali sono stati utilizzati anche per comprendere meglio le dinamiche di trasmissione dei patogeni necessarie per indirizzare geograficamente le misure di controllo. Ad esempio, la caratterizzazione genomica di RABV illustra il raggruppamento geografico dei cladi 17,18,19, la co-circolazione dei lignaggi 20,21,22, il movimento virale mediato dall'uomo17,23,24 e le dinamiche di metapopolazione 25,26.
Il monitoraggio della malattia è un'importante funzione della sorveglianza genomica che è stata potenziata con l'aumento globale della capacità di sequenziamento in risposta alla pandemia di SARS-CoV-2. La sorveglianza genomica ha supportato il tracciamento in tempo reale delle varianti di SARS-COV-2 che destano preoccupazione27,28 e le relative contromisure6. I progressi nella tecnologia di sequenziamento accessibile, come la tecnologia dei nanopori, hanno portato a protocolli migliori e più convenienti per il sequenziamento rapido di patogeniumani 29,30,31,32 e animali33,34,35. Tuttavia, in molti paesi in cui la rabbia è endemica, ci sono ancora ostacoli all'operatività della sorveglianza genomica dei patogeni, come dimostrato dalle disparità globali nella capacità di sequenziamento del SARS-CoV-236. I limiti dell'infrastruttura di laboratorio, delle catene di approvvigionamento e delle conoscenze tecniche rendono difficile l'istituzione e la routinizzazione della sorveglianza genomica. In questo articolo, dimostriamo come un flusso di lavoro ottimizzato, rapido e conveniente per il sequenziamento dell'intero genoma possa essere implementato per la sorveglianza RABV in contesti con risorse limitate.
Protocollo
Lo studio è stato approvato dal Comitato di Coordinamento della Ricerca Medica dell'Istituto Nazionale per la Ricerca Medica (NIMR/HQ/R.8a/vol. IX/2788), il Ministero dell'Amministrazione Regionale e del Governo Locale (AB.81/288/01) e l'Ifakara Health Institute Institutional Review Board (IHI/IRB/No:22-2014) in Tanzania; l'Istituto di Malattie Tropicali e Infettive dell'Università di Nairobi (P947/11/2019) e l'Istituto di Ricerca Medica del Kenya (KEMRI-SERU; protocollo n. 3268) in Kenya; e l'Istituto di ricerca per la medicina tropicale (RITM), Dipartimento della salute (2019-023) nelle Filippine. Il sequenziamento dei campioni provenienti dalla Nigeria è stato effettuato su materiale diagnostico archiviato raccolto nell'ambito della sorveglianza nazionale.
NOTA: Le sezioni 1-4 sono prerequisiti. La sezione 5-16 descrive il flusso di lavoro dal campione alla sequenza all'interpretazione per il sequenziamento dei nanopori RABV (Figura 1). Per le fasi successive del protocollo che richiedono la centrifugazione a impulsi, centrifugare a 10-15.000 x g per 5-15 s.
1. Configurazione dell'ambiente computazionale per il sequenziamento e l'analisi dei dati
- Apri il sito web di Oxford Nanopore Technology (ONT)37 e crea un account per accedere a risorse specifiche per i nanopori.
- Accedere e installare il software di sequenziamento e chiamata di baseONT 38.
- Apri GitHub39 e crea un account.
- Accedere ai repository artic-rabv40 e MADDOG41 e seguire le istruzioni di installazione.
2. Progettare o aggiornare lo schema di primer multiplex
NOTA: gli schemi RABV esistenti sono disponibili nell'archivio artic-rabv40. Quando ci si rivolge a una nuova area geografica, è necessario progettare un nuovo sistema o modificare un sistema esistente per incorporare una maggiore diversità.
- Scegliere un set di riferimento del genoma per rappresentare la diversità nell'area di studio; si tratta in genere di un insieme di sequenze disponibili al pubblico (ad esempio, da NCBI GenBank) o di dati interni preliminari. Seguire il passaggio 2.1.1 per utilizzare RABV-GLUE42, una risorsa di dati di sequenza RABV, per filtrare e scaricare le sequenze NCBI e i metadati associati.
NOTA: Scegliere sequenze di riferimento con genomi completi (cioè senza lacune e basi mascherate). Si consiglia di scegliere fino a 10 sequenze come set di riferimento per la progettazione del primer. Se i dati di sequenza disponibili sono incompleti o non rappresentativi dell'area di studio, fare riferimento ai consigli43,44,45 nel file supplementare 1.- Passare alla pagina NCBI RABV Sequences by Clade dal menu a discesa Sequence Data (Dati sequenza ) in RABV-GLUE. Fare clic sul collegamento Rabies Virus (RABV) per accedere a tutti i dati disponibili o selezionare un particolare clade di interesse. Utilizzare l'opzione filtro per aggiungere filtri che soddisfano i criteri desiderati (ad esempio, paese di origine, lunghezza della sequenza). Scarica sequenze e metadati.
- Generare uno schema di primer per la reazione a catena della polimerasi multiplex (PCR) seguendo le istruzioni fornite da Primal Scheme46. Si consiglia uno schema a 400 bp con una sovrapposizione di 50 bp per sequenziare campioni di bassa qualità. Scaricare e salvare tutti gli output (non modificare i nomi dei file o delle prime applicazioni).
NOTA: Lo schema sarà indicizzato alla prima sequenza nella fasta di input, d'ora in poi denominata "riferimento all'indice" (Figura 2). Vedere File supplementare 1 per le opzioni per ottimizzare le prestazioni del primer.
3. Impostare le pipeline di bioinformatica RAMPART e ARTIC
- Fare riferimento al File supplementare 2 per impostare una struttura di directory per gestire i file di input/output per RAMPART e la pipeline bioinformatica ARTIC.
4. Biosicurezza e configurazione del laboratorio
- Gestire campioni potenzialmente positivi alla rabbia in condizioni di livello di biosicurezza (BSL) 2 o 3.
- Assicurarsi che il personale di laboratorio abbia completato la vaccinazione antirabbica prima dell'esposizione e si sottoponga al monitoraggio dell'immunità secondo le raccomandazioni dell'Organizzazione Mondiale della Sanità (OMS)3.
- Garantire che per il laboratorio siano in atto procedure operative standard dedicate e valutazioni dei rischi, seguendo le linee guida nazionali o internazionali.
- Configurazione del laboratorio richiesta: ridurre al minimo la contaminazione mantenendo la separazione fisica tra le aree pre e post-PCR. Nei laboratori con spazio limitato o in ambienti di laboratorio sul campo, utilizzare scatole a guanti portatili o stazioni di laboratorio improvvisate per ridurre al minimo la contaminazione.
- In questo protocollo, assicurarsi di designare aree separate per:
- Estrazione del campione: allestire un armadio/vano portaoggetti BSL2/3 per gestire il materiale biologico ed eseguire l'inattivazione e l'estrazione dell'RNA.
- Area del modello: allestire un armadio/vano portaoggetti BSL1 per l'aggiunta di stampo (RNA/cDNA) alla miscela master di reazione pre-preparata.
- Area master mix: Allestire un'area pulita designata (armadio/vano portaoggetti BSL1) per la preparazione delle miscele master dei reagenti. Non dovrebbe esserci alcun modello in quest'area.
- Area post-PCR: allestire un'area separata per il lavoro sugli ampliconi e sulla preparazione della libreria di sequenziamento.
NOTA: Tutte le aree devono essere pulite con un decontaminante per superfici e sterilizzate con raggi ultravioletti (UV) prima e dopo l'uso.
5. Raccolta e diagnosi dei campioni sul campo
NOTA: I campioni devono essere raccolti da personale addestrato e immunizzato che indossi dispositivi di protezione individuale e seguendo le procedure standarddi riferimento 47,48,49.
- Raccogliere il campione attraverso il forame magno (cioè la via occipitale), come descritto in dettaglio in Mauti et al.50.
- Diagnosticare la rabbia sul campo con test diagnostici rapidi e confermare in laboratorio utilizzando le procedure raccomandate47, come il test degli anticorpi fluorescenti diretti (DFA), il test immunoistochimico rapido diretto (DRIT)51,52 o la trascrizione inversa in tempo reale (RT)-PCR 53.
- Utilizzare campioni cerebrali positivi confermati per l'estrazione dell'RNA o conservarli in un congelatore a -20 °C per 2-3 mesi o -80 °C per periodi più lunghi. Conservare l'RNA per la conservazione e il trasporto utilizzando un mezzo di stabilizzazione del DNA/RNA adatto.
6. Preparazione del campione ed estrazione dell'RNA (3 ore)
NOTA: Utilizzare un kit di estrazione dell'RNA virale basato su colonna di rotazione adatto al tipo di campione.
- Preparare due provette di microsfere di ceramica riempiendo una provetta PCR da 2 mL con una provetta da circa 200 μL piena di perline di ceramica da 1,4 mm ed etichettare la provetta.
- Aggiungere il volume raccomandato di tampone di lisi fornito nel kit di estrazione dell'RNA alla provetta PCR etichettata.
- Prelevare circa un cubo di 3 mm dal campione di cervello confermato con infezione da rabbia utilizzando un applicatore di legno e metterlo in una provetta etichettata con l'ID del campione e 100 μL di acqua priva di nucleasi nella provetta etichettata con controllo negativo.
NOTA: Utilizzare l'omogeneizzazione a microsfere a tubo chiuso per limitare l'esposizione del campione. Se non è possibile, utilizzare altri disgregatori meccanici idonei (ad es. a rotore) o un micropestello manuale. Tuttavia, questi possono essere meno efficaci del battitura delle perline su una superficie dura per distruggere il tessuto (i campioni di tessuto possono indurirsi in alcuni mezzi di conservazione). - Distruggi manualmente il tessuto cerebrale utilizzando un bastoncino applicatore di legno e poi vortica alla massima velocità fino a raggiungere la completa omogeneizzazione del tessuto.
- Centrifugare il lisato secondo le istruzioni del produttore e utilizzare una pipetta per trasferire il surnatante in una nuova provetta per microcentrifuga etichettata. Utilizzare questo surnatante solo nei passaggi successivi.
- Seguire le istruzioni della colonna di rotazione del kit di estrazione dell'RNA per ottenere RNA purificato.
- Includi qui un controllo di estrazione negativo (NEC) e procedi fino alla fase di sequenziamento.
7. preparazione del cDNA (20 min)
- Nell'area del master mix, preparare un master mix per la sintesi del cDNA del primo filamento in base al numero di campioni e controlli da processare (con un volume in eccesso del 10% per garantire un reagente adeguato; Tabella 1). In questa fase è necessario includere un controllo senza modello (NTC).
- Etichettare le provette con striscia PCR da 0,2 mL e aliquotare 5 μL della miscela master nelle provette.
- Portare le provette preparate nell'area del modello. Aggiungere 5 μL di RNA in ciascuna provetta marcata, incluso il NEC. Aggiungere 5 μL di acqua priva di nucleasi (NFW) all'NTC.
- Incubare in un termociclatore seguendo le condizioni indicate nella Tabella 1.
NOTA: Punto di pausa opzionale: il cDNA può essere conservato a -20 °C per un massimo di 1 mese, se necessario, ma è preferibile procedere alla PCR.
8. Preparazione del materiale di base del primer (1 h)
NOTA: Questo passaggio è necessario solo se si creano nuovi materiali da singoli primer, dopodiché è possibile utilizzare soluzioni madre pre-preparate.
- Preparare un pool di primer di 100 μM nell'area della miscela principale.
- Risospendere i primer liofilizzati in 1x tampone tris-EDTA (TE) o NFW a una concentrazione di 100 μM ciascuno. Vorticare accuratamente e ruotare verso il basso.
NOTA: Nei passaggi seguenti, i singoli primer vengono separati in due pool di primer, numerati dispari (denominati Pool A) e pari (denominati Pool B), per evitare interazioni tra i primer che fiancheggiano le sovrapposizioni degli ampliconi. Questi pool di primer generano ampliconi sovrapposti di 400 bp che coprono il genoma bersaglio. - Disporre tutti i primer con numero dispari in un rack per provette. Generare un pool di primer aggiungendo 5 μL da ciascun primer a una provetta per microcentrifuga da 1,5 mL etichettata come "nome dello schema di primer - Pool A (100 μM)".
- Ripetere il processo per tutti i primer pari ed etichettare come "nome dello schema di innesco - Pool B (100 μM)".
- Diluire ogni pool di primer 1:10 in acqua di grado molecolare per generare 10 μM di primer.
NOTA: Effettuare aliquote multiple di diluizioni di primer da 10 μM e congelarle in caso di degradazione o contaminazione.
9. PCR multiplex (5 ore)
- Preparare due master mix PCR, uno per ogni pool di primer nell'area del master mix.
- Utilizzare una concentrazione finale di 0,015 μM per primer. Calcolare il volume del pool di primer richiesto per la reazione PCR (Tabella 2) utilizzando la seguente formula:
Volume del pool di primer = numero di primer x volume di reazione x 0,015/concentrazione (μM) di primer
- Utilizzare una concentrazione finale di 0,015 μM per primer. Calcolare il volume del pool di primer richiesto per la reazione PCR (Tabella 2) utilizzando la seguente formula:
- Aliquotare 10 μL ciascuno della miscela master del pool A e della miscela master del pool B alle provette della striscia PCR etichettate nell'area del modello. Per ogni campione, aggiungere 2,5 μL di cDNA (dal passaggio 3) a ciascuna delle corrispondenti reazioni del pool A e B del primer marcato. Il cDNA in eccesso può essere conservato a -20 °C.
- Mescolare agitando delicatamente e centrifugare a impulsi.
- Incubare i campioni con le condizioni menzionate nella Tabella 2 su una macchina PCR.
NOTA: Il programma non include una fase di estensione specifica a causa del lungo tempo di ricottura di 5 min (richiesto a causa dell'elevato numero di primer) e della breve lunghezza degli ampliconi (400 bp) che è sufficiente per l'estensione.
10. Pulizia e quantificazione della PCR (3,5 ore)
- Eseguire tutto il lavoro da questo punto in poi nell'area post-PCR.
- Le microsfere di immobilizzazione reversibile in fase solida (SPRI) aliquote vengono inserite in provette per microcentrifuga dal flacone principale. Conservare a 4 °C.
- Riscaldare un'aliquota di microsfere SPRI a temperatura ambiente (RT; ~20 °C) e vorticare accuratamente fino a quando le perle non sono completamente risospese nella soluzione.
- In provette da 1,5 mL, combinare i prodotti primer Pool A e primer Pool B PCR per ciascun campione. Se necessario, aggiungere acqua per portare il volume a 25 μL.
- Aggiungere 25 μL di microsfere SPRI a ciascun campione (rapporto perline:campione 1:1). Miscelare pipettando su e giù o picchiettando delicatamente la provetta.
- Incubare a RT per 10 minuti, invertendo o muovendo di tanto in tanto le provette.
- Posizionare su una griglia magnetica fino a quando le perline e la soluzione non si sono completamente separate. Rimuovere e gettare il surnatante, facendo attenzione a non disturbare il pellet di perline.
- Lavare due volte con etanolo all'80% (riscaldato a RT).
- Aggiungere 200 μL di etanolo al pellet. Attendere 30 secondi per assicurarsi che le perline siano lavate correttamente.
- Rimuovere con cautela e gettare il surnatante, cercando di non toccare il pellet di perline.
- Ripetere i passaggi 10.8.1-10.8.2 per lavare il pellet una seconda volta.
- Rimuovere tutte le tracce di etanolo utilizzando una punta da 10 μL. Asciugare all'aria fino a quando le tracce di etanolo non sono evaporate (con piccole perle questo accade rapidamente, ~30 s); Quando ciò accade, il pellet dovrebbe passare da lucido a opaco. Fare attenzione a non asciugare eccessivamente (se il pellet si sta rompendo, è troppo secco), poiché ciò influirà sul recupero del DNA.
- Risospendere le perle in 15 μL di NFW e incubare a RT (fuori dal rack magnetico) per 10 minuti.
- Rimettere nel rack magnetico e trasferire il surnatante (prodotto pulito) in una nuova provetta da 1,5 mL.
- Preparare una diluizione 1:10 di ciascun campione in una provetta separata (2 μL di prodotto + 18 μL di NFW).
NOTA: Prestare molta attenzione in questa fase per evitare la contaminazione incrociata. Tenere aperto un solo tubo dell'amplicone alla volta. Introdurre prima 18 μL di acqua nelle provette (in un'area di miscelazione master pulita). - Misurare la concentrazione di DNA di ciascun campione diluito utilizzando un fluorimetro altamente sensibile e specifico, come descritto al punto protocols.io 54,55.
11. Normalizzazione (30 min)
- Utilizzare il modello di normalizzazione (file supplementare 3) e la concentrazione di DNA (ng/μL) di ciascun campione per calcolare il volume di campione diluito (o puro) richiesto per 200 fmol di ciascun campione in un volume totale di 5 μL.
- Etichettare le nuove provette PCR e aggiungere volumi calcolati di NFW e campione per ottenere DNA normalizzato.
- Utilizzare il volume calcolato per i campioni non diluiti (puliti) se sono necessari più di 5 μL del campione diluito per ottenere 200 fmol.
NOTA: Punto di pausa opzionale: A questo punto, il prodotto PCR purificato può essere conservato a 4 °C per un massimo di 1 settimana o posto a -20 °C per una conservazione a lungo termine, se necessario.
12. End-prep e codice a barre (1,5 ore)
NOTA: I passaggi successivi presuppongono l'uso di reagenti specifici da kit di codifica a barre e sequenziamento della legatura specifici per nanopori (vedere la tabella dei materiali per i dettagli). Il protocollo è trasferibile tra diverse versioni chimiche, ma gli utenti devono fare attenzione a utilizzare kit compatibili, secondo le istruzioni del produttore.
- Riparazione finale e dA-tailing
- Impostare la reazione di fine preparazione per ciascun campione menzionato nella Tabella 3. Preparare una miscela master in base al numero di campioni (più il 10% in eccesso). Prestare attenzione durante il pipettaggio poiché i reagenti sono viscosi.
- Aggiungere 5 μL di master mix in ciascuna provetta di DNA normalizzato (5 μL). La miscela di reazione totale deve essere di 10 μL. Cambiare le punte ogni volta e tenere aperta solo una provetta alla volta.
- Incubare in un termociclatore nelle condizioni indicate nella Tabella 3.
- Codici a barre
- Aliquotare i codici a barre dal kit di codifica a barre alle provette a strisce PCR a 1,25 μL/provetta e registrare il codice a barre assegnato a ciascun campione.
- Aggiungere 0,75 μL del campione preparato all'aliquota del codice a barre assegnato.
- Preparare una miscela master di legatura in base al numero di campioni (più il 10% in eccesso) (Tabella 4).
- Aggiungere 8 μL di miscela master di legatura al campione preparato alla fine + codici a barre, ottenendo una reazione totale di 10 μL.
- Incubare in un termociclatore utilizzando le condizioni indicate nella Tabella 4.
- Pulizia delle microsfere SPRI e quantificazione del DNA
- Scongelare il tampone a frammenti corti (SFB) a RT, mescolare mediante vortex, centrifugare a impulsi e posizionare sul ghiaccio.
- Raggruppare tutti i campioni con codice a barre in una provetta per microcentrifuga lobind da 1,5 mL. Per non rendere il volume di pulizia troppo grande da utilizzare, raggruppare 12-24 campioni (10 μL/campione), fino a 48 campioni (5 μL/campione) o fino a 96 campioni (2,5 μL/campione) da ciascuna reazione di codice a barre nativa.
- Aggiungere un volume di 0,4 volte di perline SPRI al pool con codice a barre. Mescolare delicatamente (flicking o pipettaggio) e incubare a RT per 5 min.
- Posizionare i campioni su un magnete fino a quando le perle non si sono pellettate e il surnatante è completamente limpido (~2 min). Rimuovere ed eliminare il surnatante. Fare attenzione a non disturbare le perline.
- Lavare due volte con 250 μL di SFB.
- Rimuovere il tubo dal magnete e risospendere completamente il pellet in 250 μL di SFB. Incubare per 30 s, centrifugare a impulsi e tornare al magnete.
- Rimuovere il surnatante e gettarlo.
- Ripetere il passaggio 12.3.5 per eseguire un secondo lavaggio SFB.
- Centrifugare a impulsi e rimuovere eventuali residui di SFB.
- Aggiungere 200 μL di etanolo all'80% (RT) per bagnare il pellet. Rimuovere e gettare l'etanolo, facendo attenzione a non disturbare il pellet di perline. Asciugare all'aria per 30 secondi o fino a quando il pellet non ha perso la sua lucentezza.
- Risospendere in 22 μL di NFW a RT per 10 min.
- Posizionare sul magnete, lasciare riposare per ~2 minuti, quindi rimuovere con cautela la soluzione e trasferirla in una provetta per microcentrifuga pulita da 1,5 mL.
- Utilizzare 1 μL per ottenere la concentrazione di DNA, come descritto in precedenza (passaggio 10.13).
NOTA: Punto di pausa opzionale: A questo punto, la libreria può essere conservata a 4 °C per un massimo di 1 settimana o a -20 °C per una conservazione a lungo termine, ma è preferibile continuare con la legatura e il sequenziamento dell'adattatore.
13. Sequenziamento (massimo 48 ore)
- Preparare il computer (fare riferimento anche alle sezioni 1-4 dei prerequisiti).
- Verificare che ci sia spazio sufficiente per archiviare i nuovi dati (minimo 150 GB), che venga eseguito il backup/spostamento dei dati delle vecchie esecuzioni su un server prima dell'eliminazione e che sia installata l'ultima versione di MinKNOW.
- Rimuovere la cella di flusso immagazzinata dal frigorifero e lasciarla raggiungere RT.
- Legatura dell'adattatore (1 h)
- Centrifugare a impulsi la miscela adattatore e la ligasi e metterla sul ghiaccio.
- Scongelare il tampone di eluizione (EB), l'SFB e il tampone di legatura a RT. Miscelare mediante vortex, centrifuga a impulsi e mettere su ghiaccio.
- Preparare la miscela master di legatura dell'adattatore (Tabella 5), combinando i reagenti nell'ordine specificato in una provetta a basso legame.
NOTA: A seconda della disponibilità presso il laboratorio, è possibile utilizzare alternative per i reagenti della miscela master di legatura dell'adattatore (Tabella 5). Vedere il file supplementare 3 e l'indice dei materiali per un elenco di alternative. Usa il calcolo nel foglio di lavoro File supplementare 3 per ottenere il volume della libreria di DNA equivalente a 200 fmol. Se viene calcolato un valore inferiore a 20 μL, aggiungere NFW per ottenere fino a 20 μL. - Mescolare con un leggero battito e centrifuga a impulsi. Incubare a RT per 20 min.
NOTA: Durante l'incubazione, iniziare a preparare la cella di flusso (paragrafo 13.5).
- Pulire utilizzando le perle SPRI (non utilizzare etanolo come nelle pulizie precedenti).
- Aggiungere un volume 0,4x di microsfere SPRI (RT) ai campioni. Incubare a RT per 10 minuti, agitare delicatamente a intermittenza per favorire la miscelazione.
- Posizionare sul magnete fino a quando le perle e la soluzione non si sono completamente separate (~5 min). Rimuovere e scartare il surnatante; Fare attenzione a non disturbare il pellet di perline.
- Lavare due volte con 125 μL di SFB.
- Risospendere completamente il pellet con 125 μL di SFB mescolando con una pipetta. Lasciare incubare per 30 s.
- Centrifuga a impulsi per raccogliere il liquido alla base della provetta e posizionarlo sul magnete. Rimuovere il surnatante e gettarlo.
- Ripetere i passaggi 13.4.4-13.4.5 per lavare il pellet una seconda volta.
- Centrifugare a impulsi e rimuovere l'SFB in eccesso.
- Risospendere in 15 μL di EB e incubare per 10 minuti a RT.
- Tornare al magnete per ~2 minuti e poi trasferire con cautela la soluzione in una provetta per microcentrifuga pulita da 1,5 mL.
- Quantificare 1 μL della libreria eluita, come descritto in precedenza al punto 10.13
NOTA: Per ottenere i migliori risultati, procedere direttamente al sequenziamento MinION; tuttavia, la libreria finale può essere conservata in EB a 4 °C per un massimo di 1 settimana, se necessario.
- Eseguire un controllo di qualità della cella di flusso.
- Collegare il dispositivo di sequenziamento a un laptop e aprire il software di sequenziamento.
- Selezionare il tipo di cella di flusso, quindi fare clic su Controlla cella di flusso e su Avvia test.
- Una volta completato, verrà visualizzato il numero totale di pori attivi (cioè vitali). Una nuova cella di flusso dovrebbe avere >800 pori attivi; In caso contrario, contattare il produttore per la sostituzione.
- Adescamento e caricamento della cella di flusso (20 min)
- Scongelare i seguenti reagenti a RT e quindi posizionare il tampone di sequenziamento, un cavo di lavaggio, un tampone di lavaggio e le perle di caricamento sul ghiaccio.
- Vorticare il tampone di sequenziamento e il tampone di lavaggio, centrifugare a impulsi e posizionare sul ghiaccio.
- Centrifugare a impulsi il cavo di lavaggio e miscelare mediante pipettaggio; quindi mettere sul ghiaccio.
- Preparare la miscela di adescamento della cella di flusso aggiungendo 30 μL di cavo di lavaggio direttamente alla provetta di tampone di lavaggio da un kit di adescamento della cella di flusso e miscelare mediante pipettaggio.
- Miscelare le perle di caricamento pipettando immediatamente prima dell'uso, poiché si depositano rapidamente.
- In una nuova provetta, preparare la diluizione finale della libreria per il sequenziamento, come indicato nella Tabella 5.
NOTA: Utilizzare il calcolo nel foglio di lavoro File supplementare 3 per ottenere il volume della libreria di DNA equivalente a 50 fmol. Se viene calcolato un valore inferiore a 12 μL, aggiungere EB per ottenere fino a 12 μL. - Capovolgere il coperchio del dispositivo di sequenziamento e far scorrere il coperchio della porta di adescamento in senso orario in modo che la porta di adescamento sia visibile (Figura 3)
- Rimuovere con cautela le bolle d'aria impostando una pipetta P1000 su 200 μL, inserire il puntale nella porta di adescamento e ruotare la rotella fino a quando non si vede un piccolo volume che entra nel puntale della pipetta (rotazione massima a 230 μL).
- Caricare 800 μL di miscela di adescamento della cella di flusso nella cella di flusso attraverso la porta di adescamento, facendo attenzione a evitare la formazione di bolle.
- Lasciare agire per 5 min.
- Sollevare delicatamente il coperchio della porta del campione e caricare 200 μL di miscela di adescamento nella cella di flusso attraverso la porta di adescamento utilizzando una pipetta P1000.
- Pipettare la miscela della libreria su e giù prima del caricamento, assicurandosi che le perle di caricamento nella miscela principale siano risospese prima del caricamento.
- Caricare 75 μL di miscela di libreria nella cella di flusso attraverso la porta del campione in modo dropwise. Assicurarsi che ogni gocciolamento fluisca nella porta prima di aggiungere il successivo.
- Riposizionare delicatamente il coperchio della porta del campione, assicurandosi che il tappo entri nella porta del campione.
- Chiudere la porta di adescamento e riposizionare il coperchio del dispositivo di sequenziamento.
- Esecuzione del sequenziamento (massimo 48 ore)
- Collegare il dispositivo di sequenziamento al laptop e aprire il software di sequenziamento.
- Fare clic su Start , quindi su Avvia sequenziazione.
- Fare clic su Nuovo esperimento e seguire il flusso di lavoro dell'interfaccia utente grafica (GUI) del software di sequenziazione per impostare i parametri per l'esecuzione.
- Digita il nome dell'esperimento e l'ID del campione (ad esempio, rabv_run1) e scegli il tipo di cella di flusso dal menu a discesa.
- Continuare con la selezione del kit e scegliere il kit di sequenziamento della legatura pertinente e il kit di codici a barre nativi utilizzati.
- Continuare con le opzioni Esegui . Mantenere le impostazioni predefinite, a meno che non si desideri che l'esecuzione si interrompa automaticamente dopo un certo numero di ore (le esecuzioni possono essere interrotte manualmente in qualsiasi momento).
- Continuare con Basecalling. Scegliere di attivare o disattivare Basecalling in base alle risorse di elaborazione (vedere Configurazione del computer). Scegli Opzioni di modifica sotto il codice a barre e assicurati che l'opzione Codice a barre su entrambe le estremità sia attivata. Salva e continua con la sezione di output.
- Accettare le impostazioni predefinite e passare alla revisione finale, controllare le impostazioni e registrare i dettagli nel foglio di lavoro (File supplementare 3). Fare clic su Start.
NOTA: Se la cella di flusso viene riutilizzata, regolare la tensione di avviamento (nella sezione avanzata delle opzioni di funzionamento), come indicato dallo schema nel file supplementare 3. - Registrare i canali attivi iniziali: se questo è significativamente inferiore al controllo di qualità (QC), riavviare il software di sequenziamento. Se è ancora inferiore, riavviare il computer.
- Registrare i canali iniziali nel filamento rispetto al poro singolo per ottenere un'occupazione approssimativa dei pori. Questo numero fluttuerà, quindi fornisci un'approssimazione.
- Monitorare l'esecuzione man mano che procede.
14. Chiamate di base dal vivo e offline
NOTA: queste istruzioni presuppongono che sia stata seguita la struttura di directory preesistente fornita nel repository artic-rabv e che siano state seguite le sezioni 1 e 3 dei prerequisiti.
- Nel file system locale, creare una nuova directory denominata analysis, in cui verranno archiviati tutti gli output dell'analisi. Per organizzare ulteriormente: creare una sottodirectory con il nome del progetto e all'interno di essa una nuova directory per l'esecuzione, utilizzando l'ID di esempio fornito a MinKNOW come run_name. Eseguire questa operazione con un unico comando come segue:
mkdir -p
analisi/project_name/run_name
Quindi vai alla sua posizione:
CD
percorso/analisi/project_name/run_name - Chiamate in tempo reale
NOTA: Per eseguire la chiamata di base dei nanopori in tempo reale, i laptop richiedono un'unità di elaborazione grafica (GPU) compatibile con NVIDIA CUDA. Assicurarsi che le istruzioni per la configurazione della chiamata di base della GPU siano state eseguite utilizzando il protocollo guppy56.- Durante la configurazione dell'esecuzione, attiva le chiamate di base dal vivo.
- Utilizzare RAMPART per monitorare la copertura del sequenziamento in tempo reale, come indicato di seguito.
- Nel terminale del computer, attivare l'ambiente artic-rabv conda:
Conda attiva ARTIC-RABV - Creare una nuova directory per l'output rampart all'interno della directory run_name e navigare al suo interno:
cd /percorso/analisi/project_name/run_name
mkdir rampart_output
CD rampart_output - Creare un file barcodes.csv per accoppiare i codici a barre e i nomi dei campioni. Deve avere una riga per codice a barre e specificare solo i codici a barre presenti nella libreria, con le intestazioni "codice a barre" e "campione". Seguire l'esempio nella directory artic-rabv:
analisi/example_project/example_run/rampart_output/codici a barre.csv - Avviare RAMPART fornendo la cartella del protocollo pertinente e il percorso della cartella fastq_pass nell'output MinKNOW per l'esecuzione:
rampart --protocol /path/rampart/scheme_name_V1_protocol - basecalledPath - Aprire una finestra del browser e passare a localhost:3000 nella casella URL. Attendere che vengano chiamati dati sufficienti prima che i risultati vengano visualizzati sullo schermo.
- Chiamata di base offline (eseguita dopo l'esecuzione)
- Se la chiamata di base in tempo reale non è stata impostata, l'output di MinKNOW sarà costituito da dati di segnale grezzi (file fast5). Non sarà possibile utilizzare RAMPART durante l'esecuzione. Convertire i file fast5 in dati basecalled (file fastq) dopo l'esecuzione usando guppy (vedere l'installazione in Prerequisiti passaggio 1.1.1.). Eseguire RAMPART post-hoc sulla basedati chiamati.
- Esegui il basecaller guppy:
guppy_basecaller -c dna_r9.4.1_450bps_fast.cfg -i /percorso/della/lettura/fast5_* -s /percorso/analisi/project_name/run_name -x auto -r
-c è il file di configurazione per specificare il modello di chiamata di base, -i è il percorso di input, -s è il percorso di salvataggio, -x specifica la chiamata di base da parte del dispositivo GPU (escludere se si utilizza la versione per computer di guppy) e -r specifica di cercare i file di input in modo ricorsivo.
NOTA: Il file di configurazione (.cfg) può essere modificato in un basecaller ad alta precisione sostituendo _fast con _hac, anche se questa operazione richiederà molto più tempo.
15. Lavaggio delle celle di flusso
- Le celle di flusso possono essere lavate e riutilizzate per sequenziare nuove librerie se i pori sono ancora vitali. Vedere le istruzioni per il lavaggio al protocollo di lavaggio della cella di flusso ONT57.
16. Analisi e interpretazione
- Generazione di sequenze di consenso con la pipeline bioinformatica ARTIC
- Segui le istruzioni dettagliate nel repository GitHub artic-rabv40 nella cartella rabv_protocols per generare sequenze di consenso da file fast5 raw o basecalled fastq.
NOTA: Fare riferimento a Tubazione Artica - Pipelineprincipale 58 per ulteriori indicazioni.
- Segui le istruzioni dettagliate nel repository GitHub artic-rabv40 nella cartella rabv_protocols per generare sequenze di consenso da file fast5 raw o basecalled fastq.
- Facoltativo: analizza la profondità di lettura media per amplicone.
- Adattare gli script disponibili dal repository artic-rabv, facendo riferimento al File Supplementare 1. In breve, le statistiche approfondite sono generate utilizzando SAMtools59 e la copertura per nucleotide tracciata in R.
- Analisi filogenetica con GLUE
- A partire da RABV_GLUE42, selezionare la scheda Analisi > genotipizzazione e interpretazione > Aggiungi file, selezionando il file fasta delle sequenze di consenso.
- Fare clic su Invia e attendere. Una volta completate le analisi, sarà possibile fare clic sul pulsante Mostra analisi , che mostra le assegnazioni di clade e sotto cladi, la copertura per gene, la variazione rispetto alle sequenze di riferimento e il parente più prossimo.
- Le sequenze contestuali rilevanti possono essere identificate anche nella sezione Dati di sequenza > Sequenze NCBI per clade .
- Selezionare il clade identificato o fare clic su Rabies Virus (RABV) per visualizzare tutte le sequenze disponibili.
- Filtra per sequenze rilevanti (ad esempio, paese di origine).
- Scarica queste sequenze e i metadati corrispondenti per l'analisi e il confronto.
- Assegnazione del lignaggio con MADDOG41
- Estrai il repository MADDOG da GitHub per assicurarti di lavorare con la versione più aggiornata.
- Creare una cartella di assegnazione all'interno del repository MADDOG locale (creato in precedenza nella sezione Prerequisiti) denominata nome dell'esecuzione.
- All'interno della cartella, aggiungere il file fasta contenente le sequenze di consenso.
- Aggiungere un file di metadati alla cartella.
NOTA: questo file deve essere un file csv con 4 colonne denominate "ID", "paese", "anno" e "assegnazione", che dettagliano gli ID della sequenza, il paese di campionamento e l'anno di raccolta del campione, mentre la colonna "assegnazione" deve essere vuota. - Nell'interfaccia della riga di comando, attivare l'ambiente conda: conda activate MADDOG.
- Nell'interfaccia della riga di comando, passare alla cartella del repository MADDOG.
- Inizialmente, eseguire l'assegnazione del lignaggio sulle sequenze per verificare la presenza di eventuali anomalie potenziali e per identificare se l'esecuzione del passaggio di designazione del lignaggio più lungo sarebbe appropriato. Per questo, digita questo nella riga di comando: sh assignment.sh.
- Quando richiesto, immettere Y per indicare che è stato eseguito il pull del repository e che si sta lavorando con la versione più aggiornata di MADDOG.
- Quando richiesto, immettere il nome della cartella all'interno della cartella del repository MADDOG che contiene il file fasta.
- Al termine dell'assegnazione della derivazione, controllare il file di output nella cartella. Se l'output è quello previsto e sono presenti più sequenze assegnate alla stessa derivazione, eseguire la designazione della derivazione.
- Se si esegue la designazione di derivazione, eliminare il file di output dell'assegnazione appena creato.
- Nel terminale, all'interno della cartella del repository MADDOG, eseguire il comando sh designation.sh.
- Quando richiesto, immettere Y per indicare che è stato eseguito il pull del repository e che si sta lavorando con la versione più aggiornata di MADDOG.
- Quando richiesto, immettere il nome della cartella all'interno della cartella del repository MADDOG contenente il file fasta e i metadati. Questo produce informazioni sul lignaggio su ciascuna sequenza, una filogenesi delle sequenze precedenti nuove e rilevanti (da 16.3.6), informazioni gerarchiche sui lignaggi e dettagli sui lignaggi potenzialmente emergenti e aree di sottocampionamento.
NOTA: I dettagli completi del protocollo, dell'utilizzo e delle uscite sono disponibili in Campbell et al.60. - Una volta completata l'analisi iniziale, se viene richiesto di verificare anche le linee di derivazione emergenti e sottocampionate, immettere Y, se necessario. In caso contrario, immettere N.
- Se viene richiesto di confermare le derivazioni appena trovate, immettere Y e seguire le istruzioni nel file NEXT_STEPS.eml risultante. In caso contrario, immettere N.
Risultati
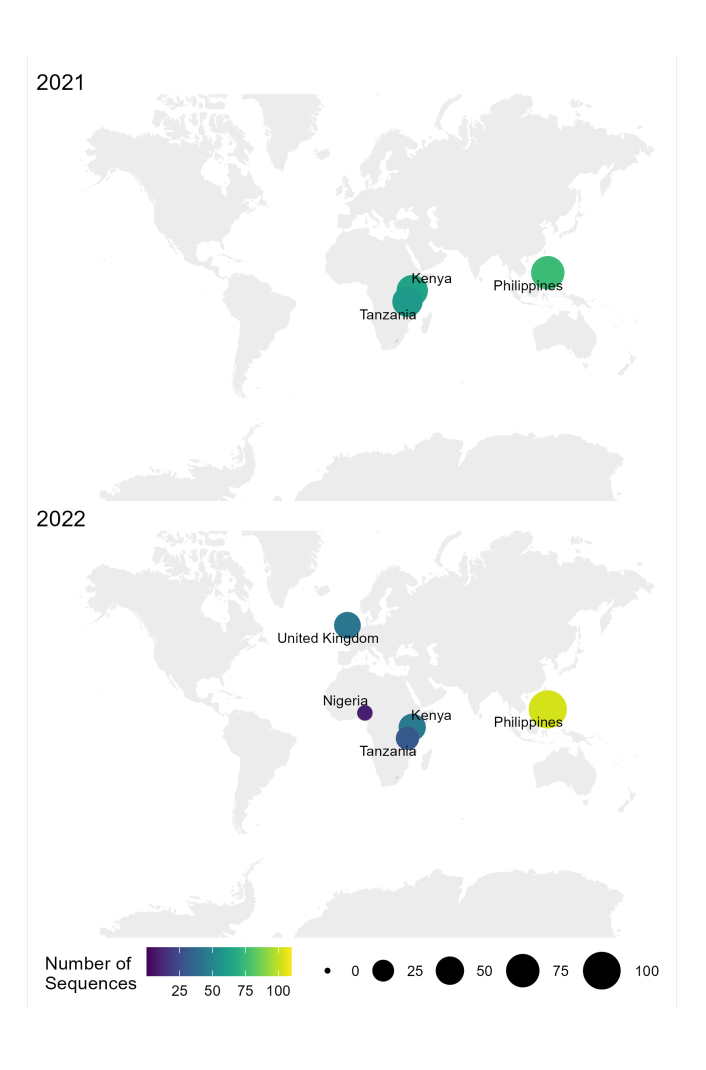
Il flusso di lavoro dal campione alla sequenza e all'interpretazione per RABV descritto in questo protocollo è stato utilizzato con successo in diverse condizioni di laboratorio in paesi endemici, come Tanzania, Kenya, Nigeria e Filippine (Figura 4). Il protocollo è stato utilizzato su diversi tipi di campioni e condizioni (Tabella 6): tessuto cerebrale fresco e congelato, estratti di cDNA e RNA da tessuto cerebrale trasportato sotto catena del freddo per periodi prolungati e schede FTA con strisci di tessuto cerebrale.
Le chiamate in tempo reale con RAMPART (Figura 5) mostrano la generazione quasi in tempo reale delle letture e la percentuale di copertura per campione. Ciò è particolarmente utile per decidere quando interrompere la corsa e salvare la cella di flusso per il riutilizzo. È stata osservata una variazione nel tempo di esecuzione, con alcuni finiti in 2 ore, mentre altri potrebbero richiedere più di 12 ore per raggiungere un'adeguata profondità di copertura (x100). Possiamo anche visualizzare regioni con scarsa amplificazione; ad esempio, la Figura 6 mostra un'istantanea di un'esecuzione di sequenziamento in cui i profili di copertura mostrano alcuni ampliconi con un'amplificazione molto bassa, indicando primer potenzialmente problematici. Studiando più a fondo queste regioni scarsamente amplificanti, siamo stati in grado di identificare le discrepanze dei primer, che ci consentiranno di riprogettare e migliorare i singoli primer. Alcuni schemi di primer hanno mostrato più discrepanze di altri. Ciò si osserva nel programma di primer per l'Africa orientale, rispetto alle Filippine, in linea con la diversità mirata, poiché il programma per l'Africa orientale mira a catturare una diversità molto più ampia.
RABV-GLUE42, una risorsa generica per la gestione dei dati del genoma RABV, e MADDOG60, un sistema di classificazione e nomenclatura del lignaggio, sono stati utilizzati per compilare e interpretare le sequenze RABV risultanti. La Tabella 7 mostra i cladi maggiori e minori che circolano in ciascun paese assegnato utilizzando RABV-GLUE. Viene inoltre mostrata una classificazione a risoluzione più elevata dei lignaggi locali dopo l'assegnazione di MADDOG.

Figura 1: Flusso di lavoro dal campione alla sequenza all'interpretazione per RABV. Vengono mostrati i passaggi riassuntivi per (A) la preparazione del campione, (B) la PCR e la preparazione della libreria, e (C) il sequenziamento e la bioinformatica fino all'analisi e all'interpretazione. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Schema dello schema di base. Posizioni di ricottura lungo il "genoma di riferimento indice" (viola scuro) per coppie di primer diretti e inversi (mezze frecce), che sono assegnati in due pool separati: A (rosso) e B (verde). Le coppie di primer generano ampliconi sovrapposti di 400 bp (blu) che sono numerati in sequenza lungo il genoma di riferimento dell'indice nel formato 'scheme_name_X_DIRECTION', dove 'X' è un numero che si riferisce all'amplicone generato dal primer e 'DIRECTION' è 'LEFT' o 'RIGHT', che descrive rispettivamente l'avanti o l'indietro. I valori pari o dispari di 'X' determinano il pool (A o B, rispettivamente). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Cella di flusso a nanopori48. Le etichette blu illustrano le diverse parti della cella di flusso, tra cui il coperchio della porta di adescamento che copre la porta di adescamento in cui viene aggiunta la soluzione di adescamento, il coperchio della porta del campione SpotON che copre la porta del campione in cui il campione viene aggiunto a goccia, le porte di scarico 1 e 2 e l'ID della cella di flusso. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Mappa che mostra la posizione in cui è stato condotto il sequenziamento RABV utilizzando il flusso di lavoro ottimizzato nel 2021 e nel 2022. La dimensione e il colore delle bolle corrispondono al numero di sequenze per posizione, dove più piccole e più scure sono meno, mentre più grandi e più chiare sono di più. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Screenshot della visualizzazione di RAMPART nel browser web. I nomi dei codici a barre vengono sostituiti dai nomi dei campioni in base alla configurazione bioinformatica. I tre pannelli superiori mostrano grafici riepilogativi per l'intera esecuzione: profondità di copertura delle letture mappate per ogni codice a barre per posizione nucleotidica sul genoma di riferimento dell'indice (in alto a sinistra, colorato per codice a barre), letture mappate sommate da tutti i codici a barre nel tempo (in alto al centro) e letture mappate per codice a barre (in alto a destra, colorate per codice a barre). I pannelli inferiori mostrano le righe di grafici per codice a barre. Da sinistra a destra: la profondità di copertura delle letture mappate per posizione nucleotidica sul genoma di riferimento dell'indice (a sinistra), la distribuzione della lunghezza delle letture mappate (al centro) e la proporzione delle posizioni nucleotidiche sul genoma di riferimento dell'indice che hanno ottenuto una copertura di 10x, 100x e 1.000x delle letture mappate nel tempo (a destra). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 6: Un esempio di copertura di lettura attraverso il genoma per un campione di virus della rabbia proveniente dalle Filippine sequenziato utilizzando il protocollo. Viene mostrata la copertura di lettura in corrispondenza di ciascuna posizione nucleotidica nel genoma, insieme alla posizione degli ampliconi sovrapposti (1-41) utilizzati per generare la libreria. I picchi nella profondità di copertura corrispondono alle aree di sovrapposizione dell'amplicone. Gli ampliconi con una bassa profondità di copertura corrispondono alle aree di sovrapposizione degli ampliconi. Gli ampliconi con una bassa profondità di copertura sono evidenziati in rosso, indicando le regioni problematiche che potrebbero richiedere un'ottimizzazione. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Tabella 1: Condizioni della miscela principale e del termociclatore per la preparazione del cDNA. Clicca qui per scaricare questa tabella.
Tabella 2: Condizioni della miscela master e del termociclatore per la PCR multiplex. Clicca qui per scaricare questa tabella.
Tabella 3: Condizioni della miscela principale e del termociclatore per la reazione di fine preparazione. Clicca qui per scaricare questa tabella.
Tabella 4: Condizioni della miscela principale e del termociclatore per la codifica a barre. Clicca qui per scaricare questa tabella.
Tabella 5: Miscela master di legatura dell'adattatore e diluizione finale della libreria per il sequenziamento. Clicca qui per scaricare questa tabella.
Tabella 6: Il numero di sequenze dell'intero genoma del virus della rabbia generate e il tipo di campioni utilizzati nei diversi paesi utilizzando il flusso di lavoro dal campione alla sequenza all'interpretazione. Clicca qui per scaricare questa tabella.
Tabella 7: Assegnazioni di clade maggiori e minori da RABV-GLUE e assegnazioni di derivazione da MADDOG per le sequenze generate utilizzando il flusso di lavoro. Clicca qui per scaricare questa tabella.
File supplementare 1: Progettazione e ottimizzazione dello schema di primer e analisi della profondità di lettura dell'amplicone. Fare clic qui per scaricare il file.
File supplementare 2: Setup computazionale Fare clic qui per scaricare questo file.
File supplementare 3: Foglio di lavoro del protocollo RABV WGS Fare clic qui per scaricare questo file.
Discussione
Brunker et al.61 ha sviluppato un flusso di lavoro accessibile per il sequenziamento dell'intero genoma RABV, basato su nanopori, utilizzando le risorse della rete ARTIC46. Qui presentiamo un flusso di lavoro aggiornato, con passaggi completi dal campione alla sequenza all'interpretazione. Il flusso di lavoro descrive in dettaglio la preparazione di campioni di tessuto cerebrale per il sequenziamento dell'intero genoma, presenta una pipeline bioinformatica per elaborare le letture e generare sequenze di consenso ed evidenzia due strumenti specifici per la rabbia per automatizzare l'assegnazione del lignaggio e determinare il contesto filogenetico. Il flusso di lavoro aggiornato fornisce anche istruzioni complete per la configurazione di aree di lavoro computazionali e di laboratorio appropriate, con considerazioni per l'implementazione in contesti diversi (incluse le impostazioni con risorse limitate). Abbiamo dimostrato il successo dell'implementazione del flusso di lavoro sia in ambito accademico che di istituto di ricerca in quattro LMIC endemici RABV con capacità di sorveglianza genomica nulla o limitata. Il flusso di lavoro si è dimostrato resiliente all'applicazione in diversi contesti e comprensibile da utenti con competenze diverse.
Questo flusso di lavoro per il sequenziamento RABV è il protocollo più completo disponibile al pubblico (che copre le fasi dal campione alla sequenza fino all'interpretazione) e specificamente adattato per ridurre i costi di avvio e di gestione. Il tempo e i costi necessari per la preparazione e il sequenziamento della libreria con la tecnologia dei nanopori sono notevolmente ridotti rispetto ad altre piattaforme, come Illumina61, e i continui sviluppi tecnologici stanno migliorando la qualità e l'accuratezza della sequenza per essere paragonabili a Illumina62.
Questo protocollo è progettato per essere resiliente in diversi contesti con risorse limitate. Facendo riferimento alle linee guida per la risoluzione dei problemi e le modifiche fornite insieme al protocollo di base, gli utenti sono supportati per adattare il flusso di lavoro alle proprie esigenze. L'aggiunta di strumenti bioinformatici di facile utilizzo al flusso di lavoro costituisce un importante sviluppo del protocollo originale, fornendo metodi rapidi e standardizzati che possono essere applicati da utenti con una minima esperienza bioinformatica precedente per interpretare i dati di sequenza in contesti locali. La capacità di farlo in situ è spesso limitata dalla necessità di avere competenze specifiche di programmazione e filogenetiche, che richiedono un investimento intensivo e a lungo termine nella formazione delle competenze. Sebbene questo set di competenze sia importante per interpretare a fondo i dati di sequenza, gli strumenti di interpretazione di base e accessibili sono altrettanto auspicabili al fine di capacitare i "campioni di sequenziamento" locali, la cui competenza principale può essere basata sul wet lab, consentendo loro di interpretare e assumere la proprietà dei propri dati.
Poiché il protocollo è stato intrapreso per un certo numero di anni in diversi paesi, ora possiamo fornire indicazioni su come ottimizzare gli schemi di primer multiplex per migliorare la copertura e gestire la diversità accumulata. Sono stati compiuti sforzi anche per aiutare gli utilizzatori a migliorare l'efficacia in termini di costi o per facilitare l'approvvigionamento in una determinata regione, il che rappresenta in genere una sfida per la sostenibilità degli approcci molecolari63. Ad esempio, in Africa (Tanzania, Kenya e Nigeria), abbiamo optato per la miscela master di ligasi blunt/TA nella fase di legatura dell'adattatore, che era più facilmente disponibile presso i fornitori locali e un'alternativa più economica ad altri reagenti di legatura.
In base all'esperienza, esistono diversi modi per ridurre il costo per campione e per esecuzione. La riduzione del numero di campioni per ciclo (ad esempio, da 24 a 12 campioni) può prolungare la durata delle celle di flusso su più cicli, mentre l'aumento del numero di campioni per ciclo massimizza il tempo e i reagenti. Nelle nostre mani, siamo stati in grado di lavare e riutilizzare le celle di flusso per un ogni tre cicli di sequenziamento, consentendo di sequenziare altri 55 campioni. Il lavaggio della cella di flusso immediatamente dopo l'uso o, se non possibile, la rimozione del fluido di scarto dal canale di scarico dopo ogni corsa, sembrava preservare il numero di pori disponibili per una seconda corsa. Prendendo in considerazione il numero iniziale di pori disponibili in una cella di flusso, è anche possibile ottimizzare un'esecuzione per pianificare il numero di campioni da eseguire in una particolare cella di flusso.
Sebbene il flusso di lavoro miri a essere il più completo possibile, con l'aggiunta di una guida dettagliata e di risorse segnalate, la procedura è ancora complessa e può essere scoraggiante per un nuovo utente. L'utente è incoraggiato a cercare formazione e supporto di persona, idealmente localmente, o in alternativa tramite collaboratori esterni. Nelle Filippine, ad esempio, un progetto sullo sviluppo delle capacità all'interno dei laboratori regionali per la sorveglianza genomica del SARS-CoV-2 utilizzando l'ONT ha sviluppato competenze di base tra i diagnostici sanitari che sono prontamente trasferibili al sequenziamento RABV. Passaggi importanti, come la pulizia del cordone SPRI, possono essere difficili da padroneggiare senza una formazione pratica e una pulizia inefficace può danneggiare la cella di flusso e compromettere la corsa. La contaminazione del campione è sempre una delle principali preoccupazioni quando gli ampliconi vengono processati in laboratorio e può essere difficile da eliminare. In particolare, la contaminazione incrociata tra i campioni è estremamente difficile da rilevare durante la bioinformatica post-esecuzione. Buone tecniche e pratiche di laboratorio, come il mantenimento di superfici di lavoro pulite, la separazione delle aree pre e post-PCR e l'incorporazione di controlli negativi, sono indispensabili per garantire il controllo della qualità. Il ritmo veloce degli sviluppi del sequenziamento dei nanopori è sia un vantaggio che uno svantaggio per la sorveglianza genomica di routine del RABV. I continui miglioramenti all'accuratezza, all'accessibilità e al repertorio di protocolli di nanopore ampliano e migliorano la portata della sua applicazione. Tuttavia, gli stessi sviluppi rendono difficile mantenere le procedure operative standard e le pipeline bioinformatiche. In questo protocollo, forniamo un documento che assiste la transizione dai vecchi agli attuali kit di preparazione della libreria di nanopori (Tabella dei materiali).
Un ostacolo comune al sequenziamento nei LMIC è l'accessibilità, che include non solo il costo, ma anche la capacità di procurarsi i materiali di consumo in modo tempestivo (in particolare i reagenti di sequenziamento, che sono relativamente nuovi per i team di approvvigionamento e i fornitori) e le risorse computazionali, oltre ad avere semplicemente accesso a un'alimentazione stabile e a Internet. L'utilizzo della tecnologia di sequenziamento dei nanopori portatili come base di questo flusso di lavoro aiuta a risolvere molti di questi problemi di accessibilità e abbiamo dimostrato l'uso del nostro protocollo in una vasta gamma di impostazioni, conducendo l'intero protocollo e l'analisi nel paese. Certo, l'approvvigionamento tempestivo delle attrezzature e il sequenziamento dei materiali di consumo rimane una sfida e, in molti casi, siamo stati costretti a trasportare o spedire reagenti dal Regno Unito. Tuttavia, in alcune aree, siamo stati in grado di fare affidamento interamente sulle rotte di approvvigionamento locali per i reagenti, beneficiando degli investimenti nel sequenziamento del SARS-CoV-2 (ad esempio, nelle Filippine) che hanno semplificato i processi di approvvigionamento e iniziato a normalizzare l'applicazione della genomica dei patogeni.
La necessità di una connessione Internet stabile è ridotta al minimo dalle installazioni una tantum; ad esempio, i repository GitHub, il download di software e lo stesso sequenziamento dei nanopori richiedono solo l'accesso a Internet per avviare l'esecuzione (non per tutto il tempo) o possono essere eseguiti completamente offline con l'accordo dell'azienda. Se i dati mobili sono disponibili, è possibile utilizzare un telefono come hotspot per il laptop per iniziare l'esecuzione della sequenza, prima di disconnettersi per la durata dell'esecuzione. Quando si elaborano regolarmente i campioni, i requisiti di archiviazione dei dati possono crescere rapidamente e, idealmente, i dati dovrebbero essere archiviati su un server. Per il resto, i dischi rigidi con unità a stato solido (SSD) sono relativamente economici da reperire.
Pur riconoscendo che ci sono ancora barriere alla sorveglianza genomica nei paesi a basso e medio reddito, l'aumento degli investimenti nella costruzione di accessibilità e competenze genomiche (ad esempio, l'Africa Pathogen Genomics Initiative [Africa PGI])64 suggerisce che questa situazione migliorerà. La sorveglianza genomica è fondamentale per la preparazione alle pandemie6 e la capacità può essere stabilita attraverso la routinizzazione della sorveglianza genomica di patogeni endemici come il RABV. Le disparità globali nelle capacità di sequenziamento evidenziate durante la pandemia di SARS-CoV-2 dovrebbero essere un motore di cambiamento catalizzatore per affrontare queste disuguaglianze strutturali.
Questo flusso di lavoro dal campione alla sequenza all'interpretazione per RABV, compresi gli strumenti bioinformatici accessibili, ha il potenziale per essere utilizzato per guidare le misure di controllo mirate all'obiettivo di zero morti umane per rabbia mediata dai cani entro il 2030 e, in ultima analisi, per l'eliminazione delle varianti di RABV. In combinazione con i metadati pertinenti, i dati genomici generati da questo protocollo facilitano la rapida caratterizzazione del RABV durante le indagini sui focolai e nell'identificazione dei lignaggi circolanti in un paese o in una regione60,61,65. Illustriamo la nostra pipeline utilizzando principalmente esempi di rabbia mediata dai cani; Tuttavia, il flusso di lavoro è direttamente applicabile alla rabbia della fauna selvatica. Questa trasferibilità e il basso costo riducono al minimo le sfide nel rendere facilmente disponibile il sequenziamento di routine, non solo per la rabbia ma anche per altri agenti patogeni 46,66,67, per migliorare la gestione e il controllo della malattia.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Questo lavoro è stato sostenuto da Wellcome [207569/Z/17/Z, 224670/Z/21/Z], dal finanziamento Newton del Medical Research Council [MR/R025649/1] e dal Dipartimento di Scienza e Tecnologia delle Filippine (DOST), dallo sforzo globale di ricerca e innovazione del Regno Unito su COVID-19 [MR/V035444/1], dal Fondo di supporto strategico istituzionale dell'Università di Glasgow [204820], dal Medical Research Council New Investigator Award (KB) [MR/X002047/1], e Fondo per lo sviluppo del partenariato internazionale, una borsa di studio DOST British Council-Philippines (CB), una borsa di studio GemVi dell'Istituto nazionale per la ricerca sanitaria [17/63/82] (GJ) e borse di studio dell'Università di Glasgow dal MVLS DTP (KC) [125638-06], dall'EPSRC DTP (RD) [EP/T517896/1] e dal Wellcome IIB DTP (HF) [218518/Z/19/Z]. Siamo grati ai colleghi e collaboratori che hanno sostenuto questo lavoro: Daniel Streicker, Alice Broos, Elizabeth Miranda, DVM, Daria Manalo, DVM, Thumbi Mwangi, Kennedy Lushasi, Charles Kayuki, Jude Karlo Bolivar, Jeromir Bondoc, Esteven Balbin, Ronnel Tongohan, Agatha Ukande, Davis Kuchaka, Mumbua Mutunga, Lwitiko Sikana e Anna Czupryna.
Materiali
| Name | Company | Catalog Number | Comments |
| Brand name | |||
| Software | |||
| Sequencing software (MinKnow) | Oxford Nanopore Technologies | https://community.nanoporetech.com/downloads | |
| Bioinformatics tool kit (Guppy) | Oxford Nanopore Technologies | https://community.nanoporetech.com/docs/prepare/library_prep_protocols/Guppy-protocol/v/gpb_2003_v1_revao_14dec2018 | |
| Equipment | |||
| Thermal cycler (miniPCR™ mini16 thermal cycler) | Cambio | MP-QP-1016-01 | |
| Homogenizer (Precellys Evolution Touch Homogenizer) | Bertin Instruments | EQ02520-300 | |
| Cold Racks (0.2-0.5mL) (PCR Mini-cooler with transparent lid) | BRAND | 781260 | |
| Pipettor | |||
| (Pipetman L Fixed F1000L, 1000 uL) | Gilson | SKU: FA10030 | |
| (Pipetman L Fixed F100L, 100 uL) | Gilson | SKU: FA10024 | |
| (Pipetman L Fixed F10L, 10 uL) | Gilson | SKU: FA10020 | |
| (Pipetman L Fixed F1L, 1 uL) | Gilson | SKU: FA10025 | |
| (Pipetman L Fixed F20L, 20 uL) | Gilson | SKU: FA10021 | |
| (Pipetman L Fixed F250L, 250 uL) | Gilson | SKU: FA10026 | |
| Fluorometer (Qubit 4 Fluorometer) | Thermofisher scientific/Fisher scientific | Q33238 | |
| Laptop (Any brand with ~2 GB of drive space, minimum of 512 GB storage space, msi installer [GPU]) | |||
| Microcentrifuge (Refrigerated centrifuge) | Thermofisher scientific/Fisher scientific | 75004081 | |
| Vortex mixer (Basic vortex mixer) | Thermofisher scientific/Fisher scientific | 88882011 | |
| Magnetic rack (DynaMag -2 Magnet) | Thermofisher scientific/Fisher scientific | 12321D | |
| Sequencing device (MinION) | Oxford Nanopore Technologies | MinION Mk1B | |
| RNA Extraction | |||
| RNA extraction kit (Qiagen RNEasy Mini Kit 250) | Qiagen | 74106 | |
| RNA stabilizing reagents | |||
| (RNA later) | Invitrogen | AM7020 | |
| (DNA/RNA Shield) | Zymo Research | R1100-50 | |
| PCR | |||
| Nuclease-free Water (Nuclease-free Water [not DEPC-treated]) | Thermofisher scientific/Fisher scientific | AM9937 | |
| Master mix for first strand cDNA synthesis (LunaScript RT SuperMix Kit) | New England Biolabs | E3010S | |
| DNA amplification master mix (Q5® Hot Start High-Fidelity 2X Master Mix [NEB]) | New England Biolabs | M0494L | |
| Primer (Scheme) (Custom DNA oligos) | Invitrogen | ||
| SPRI Bead Clean-up | |||
| SPRI beads (Aline Biosciences PCR Clean DX ) | Cambio | AL-AC1003-50 | |
| Ethanol, Pure Absolute, >99.8% (GC) [Riedel-De Haen] | Merck | 818760 | |
| Short Fragment buffer (SFB expansion pack) | Oxford Nanopore Technologies | EXP-SFB001 | |
| DNA Quantification | |||
| DNA quantification kit (Qubit® dsDNA HS Assay Kit) | Thermofisher scientific/Fisher scientific | Q32854 | |
| DNA quantification assay tubes (Qubit™ Assay Tubes) | Thermofisher scientific/Fisher scientific | Q32856 | |
| End Prep and barcoding (Qubit™ Assay Tubes) | |||
| End Prep master mix (NEBNext Ultra End Repair/dA-Tailing Module) | New England Biolabs | E7546L | |
| Barcoding kit | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 | |
| (Native Barcoding Expansion 1-12) | EXP-NBD104 | ||
| (Native Barcoding Expansion 13-24) | EXP-NBD114 | ||
| (Native Barcoding Expansion 96) | EXP-NBD196 | ||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 | |
| (not compatible) | (not compatible) | ||
| (Native Barcoding Kit 24 V14) | SQK-NBD114.24 | ||
| (Native Barcoding Kit 96 V14) | SQK-NBD114.96 | ||
| Ligation mastermix (Blunt/TA Ligase Master Mix) | New England Biolabs | M0367S | |
| Adapter Ligation | |||
| Adapter ligation master mix | |||
| (NEBNext Quick Ligation Module) | New England Biolabs | E6056S | |
| (NEBNext Ultra II Ligation Module) | New England Biolabs | E7595S | |
| (Blunt/TA Ligase Master Mix) | New England Biolabs | M0367S | |
| Adapter mix | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 EXP-AMII001 | |
| (Adapter Mix II [AMII]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 EXP-NBA114 | |
| (Native adapter [NA]) | |||
| Sequencing | |||
| Flowcell priming kit | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 EXP-FLP002 | |
| (Flush Buffer [FB]) | |||
| (Flush Tether [FT]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 EXP-FLP004 | |
| (Flow Cell Flush [FCF]) | |||
| (Flow Cell Tether [FCT]) | |||
| Ligation Sequencing Kit | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 SQK-LSK109 | |
| Adapter Mix (Adapter Mix [AMX]) | |||
| Ligation Buffer (Ligation buffer [LNB]) | |||
| Short Fragment Buffer (Short Fragment buffer [SFB]) | |||
| Sequencing Buffer (Sequencing Buffer [SQB]) | |||
| Elution Buffer (Elution buffer [EB]) | |||
| Loading Beads (Loading Beads [LB]) | |||
| Sequencing Tether (Sequencing Tether [SQT]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 SQK-LSK114 | |
| Adapter Mix (Ligation Adapter [LA]) | |||
| Ligation Buffer (Ligation buffer [LNB]) | |||
| Short Fragment Buffer (Short Fragment buffer [SFB]) | |||
| Sequencing Buffer (Sequencing Buffer [SB]) | |||
| Elution Buffer (Elution buffer [EB]) | |||
| Loading Beads (Loading Beads [LIB]) | |||
| Sequencing Tether (Flow Cell Tether [FCT]) | |||
| Library solution (Library solution [LIS]) | |||
| Flush buffer (Flow Cell Flush [FCF]) | |||
| Flow Cell | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 FLO-MIN106D | |
| (Flow Cell [R9.4.1]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 FLO-MIN114 | |
| (Flow Cell [R10.4.1]) | |||
| Flow Cell wash | |||
| Flowcell wash kit (Flow cell wash kit) | Oxford Nanopore Technologies | EXP-WSH004 | |
| Consummables | |||
| Surface decontaminant | |||
| (DNA Away Surface Decontaminant, Squeeze Bottle [Molecular Bio]) | Thermofisher scientific/Fisher scientific | 7010PK | |
| (RNase Away Surface Decontaminant, Bottle [Molecular Bio]) | Thermofisher scientific/Fisher scientific | 7002PK | |
| PCR 8-Tube Strip 0.2ml, individual cap (PCR 8-Tube Strip 0.2ml, with Individual attached Flat Caps, Sterile, DNAse/RNAse, Pyrogen Free,Natural [Greiner]) | Greiner | 608281 | |
| PCR Tube 0.2ml (PCR Tube 0.2ml, Natural [Domed Cap] Bagged in 500s, Non-Sterile [Greiner]) | Greiner | 671201 | |
| 1000µL Filter Tips (500) (Stacked 1000µL Filter Tips [500]) | Thermofisher scientific/Fisher scientific | 11977724 | |
| 100µL Filter Tips (1000) | Thermofisher scientific/Fisher scientific | 11947724 | |
| 10µL Filter Tips (1000) (Stacked 100µL Filter Tips [1000]) | Thermofisher scientific/Fisher scientific | 11907724 | |
| Reinforced tubes tubes (2ml) with screw caps and o-rings (Fisherbrand™ Bulk tubes) | Thermofisher scientific/Fisher scientific | 15545809 | |
| Microcentrifuge tube (1.5ml) (1.5 ml Eppendorf Tubes [500]) | Eppendorf | 1229888 | |
| DNA LoBind Tubes (1.5ml) (DNA LoBind Tubes) | Thermofisher scientific/Fisher scientific | 10051232 | |
| Cryobabies labels | |||
| Gloves (S/M/L) | |||
| Paper towel |
Riferimenti
- Rupprecht, C. E. Rhabdoviruses: rabies virus. Medical Microbiology. , University of Texas Medical Branch. Galveston, TX. (1996).
- Rabies. World Health Organization. , Available from: https://www.who.int/news-room/fact-sheets/detail/rabies (2023).
- Health Organization, W. orld WHO Expert Consultation on Rabies: WHO TRS N°1012. World Health Organization. , Available from: https://www.who.int/publications-detail-redirect/WHO-TRS-1012 (2018).
- Benavides, J. A., et al. Defining new pathways to manage the ongoing emergence of bat rabies in Latin America. Viruses. 12 (9), 1002(2020).
- Hampson, K. Estimating the global burden of endemic canine rabies. PLoS Neglected Tropical Diseases. 9 (4), e0003709(2015).
- Global genomic surveillance strategy for pathogens with pandemic and epidemic potential, 2022-2032. World Health Organization. , Available from: https://www.who.int/publications-detail-redirect/978924004679 (2022).
- Tsai, K. J., et al. Emergence of a sylvatic enzootic formosan ferret badger-associated rabies in Taiwan and the geographical separation of two phylogenetic groups of rabies viruses. Veterinary Microbiology. 182, 28-34 (2016).
- Chiou, H. -Y., et al. Molecular characterization of cryptically circulating rabies virus from ferret badgers, Taiwan. Emerging Infectious Diseases. 20 (5), 790-798 (2014).
- Sabeta, C. T., Mansfield, K. L., McElhinney, L. M., Fooks, A. R., Nel, L. H. Molecular epidemiology of rabies in bat-eared foxes (Otocyon megalotis) in South Africa. Virus Research. 129 (1), 1-10 (2007).
- Scott, T. P. Complete genome and molecular epidemiological data infer the maintenance of rabies among kudu (Tragelaphus strepsiceros) in Namibia. PLoS One. 8 (3), e58739(2013).
- Lembo, T., et al. Exploring reservoir dynamics: a case study of rabies in the Serengeti ecosystem. The Journal of Applied Ecology. 45 (4), 1246-1257 (2008).
- Coetzee, P., Nel, L. H. Emerging epidemic dog rabies in coastal South Africa: a molecular epidemiological analysis. Virus Research. 126 (1-2), 186-195 (2007).
- Ou de Munnink, B. B. First molecular analysis of rabies virus in Qatar and clinical cases imported into Qatar, a case report. International Journal of Infectious Diseases. 96, 323-326 (2020).
- Smith, J., et al. Case report: Rapid ante-mortem diagnosis of a human case of rabies imported into the UK from the Philippines. Journal of Medical Virology. 69, 150-155 (2003).
- Mahardika, G. N. K., et al. Phylogenetic analysis and victim contact tracing of rabies virus from humans and dogs in Bali, Indonesia. Epidemiology and Infection. 142 (6), 1146-1154 (2014).
- Tohma, K., et al. Molecular and mathematical modeling analyses of inter-island transmission of rabies into a previously rabies-free island in the Philippines. Infection, Genetics and Evolution. 38, 22-28 (2016).
- Tohma, K., et al. Phylogeographic analysis of rabies viruses in the Philippines. Infection, Genetics and Evolution. 23, 86-94 (2014).
- Saito, M., et al. Genetic diversity and geographic distribution of genetically distinct rabies viruses in the Philippines. PLoS Neglected Tropical Diseases. 7 (4), e2144(2013).
- Biek, R., Henderson, J. C., Waller, L. A., Rupprecht, C. E., Real, L. A. A high-resolution genetic signature of demographic and spatial expansion in epizootic rabies virus. Proceedings of the National Academy of Sciences. 104 (19), 7993-7998 (2007).
- Reddy, G. B., et al. Molecular characterization of Indian rabies virus isolates by partial sequencing of nucleoprotein (N) and phosphoprotein (P) genes. Virus Genes. 43, 13-17 (2011).
- David, D., Dveres, N., Yakobson, B. A., Davidson, I. Emergence of dog rabies in the northern region of Israel. Epidemiology and Infection. 137 (4), 544-548 (2009).
- Benjathummarak, S. Molecular genetic characterization of rabies virus glycoprotein gene sequences from rabid dogs in Bangkok and neighboring provinces in Thailand, 2013-2014. Archives of Virology. 161 (5), 1261-1271 (2016).
- Denduangboripant, J., et al. Transmission dynamics of rabies virus in Thailand: implications for disease control. BMC Infectious Diseases. 5, 52(2005).
- Talbi, C., et al. Phylodynamics and human-mediated dispersal of a zoonotic virus. PLoS Pathogens. 6 (10), e1001166(2010).
- Bourhy, H., et al. Revealing the micro-scale signature of endemic zoonotic disease transmission in an African urban setting. PLoS Pathogens. 12 (4), e1005525(2016).
- Zinsstag, J., et al. Vaccination of dogs in an African city interrupts rabies transmission and reduces human exposure. Science Translational Medicine. 9 (421), (2017).
- Yakovleva, A., et al. Tracking SARS-COV-2 variants using Nanopore sequencing in Ukraine in 2021. Scientific Reports. 12, 15749(2022).
- Mannsverk, S., et al. SARS-CoV-2 variants of concern and spike protein mutational dynamics in a Swedish cohort during 2021, studied by Nanopore sequencing. Virology Journal. 19, 164(2022).
- Soufi, M., et al. Fast and easy nanopore sequencing workflow for rapid genetic testing of familial Hypercholesterolemia. Frontiers in Genetics. 13, 836231(2022).
- Cabibbe, A. M. Application of targeted next-generation sequencing assay on a portable sequencing platform for culture-free detection of drug-resistant tuberculosis from clinical samples. Journal of Clinical Microbiology. 58 (10), 00632(2020).
- Xu, Y., et al. Nanopore metagenomic sequencing of influenza virus directly from respiratory samples: diagnosis, drug resistance and nosocomial transmission, United Kingdom, 2018/19 influenza season. Euro Surveillance. 26 (27), 2000004(2021).
- Stubbs, S. C. B., et al. Assessment of a multiplex PCR and Nanopore-based method for dengue virus sequencing in Indonesia. Virology Journal. 17, 24(2020).
- Croville, G., et al. Rapid whole-genome based typing and surveillance of avipoxviruses using nanopore sequencing. Journal of Virological Methods. 261, 34-39 (2018).
- Theuns, S., et al. Nanopore sequencing as a revolutionary diagnostic tool for porcine viral enteric disease complexes identifies porcine kobuvirus as an important enteric virus. Scientific Reports. 8, 9830(2018).
- O'Donnell, V. K., et al. Rapid sequence-based characterization of African swine fever virus by use of the Oxford Nanopore MinION sequence sensing device and a companion analysis software tool. Journal of Clinical Microbiology. 58, 01104-01119 (2019).
- Brito, A. F. Global disparities in SARS-CoV-2 genomic surveillance. Nature Communications. 13, 7003(2022).
- ONT login/register. Oxford Nanopore Technology. , Available from: https://nanoporetech.com/login-register (2023).
- Software Downloads. Oxford Nanopore Technology. , Available from: https://community.nanoporetech.com/downloads (2023).
- GitHub. , Available from: https://github.com/ (2023).
- Brunker, K. Artic-rabv. , Available from: https://github.com/kirstyn/artic-rabv (2022).
- Campbell, K. MADDOG: Method for Assignment, Definition and Designation of Global Lineages. , Available from: https://github.com/KathrynCampbell/MADDOG (2022).
- Campbell, K. RABV-GLUE. Centre for Virus Research. , Available from: https://github.com/KathrynCampbell/MADDOG (2022).
- Itokawa, K., Sekizuka, T., Hashino, M., Tanaka, R., Kuroda, M. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS ONE. 15 (9), e0239403(2020).
- Davis, M. W., Jorgensen, E. M. ApE, A plasmid editor: A freely available DNA manipulation and visualization program. Frontiers in Bioinformatics. 2, 818619(2022).
- Döring, M., Pfeifer, N. openPrimeR: Multiplex PCR primer design and analysis. , (2023).
- Quick, J. Multiplex PCR method for MiniON and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nature Protocols. 12 (6), 1261-1276 (2017).
- Laboratory Techniques in Rabies. World Health Organization. 1, Available from: https://apps.who.int/iris/bitstream/handle/10665/310836/9789241515153-eng.pdf (2018).
- Lembo, T. Partners for Rabies Prevention. The blueprint for rabies prevention and control: a novel operational toolkit for rabies elimination. PLoS Neglected Tropical Diseases. 6 (2), e1388(2012).
- Terrestrial Manual Online Access. World Organization for Animal Health. , Available from: https://www.woah.org/en/what-we-do/standards/codes-and-manuals/terrestrial-manual-online-access/ (2023).
- Mauti, S. Field postmortem rabies rapid immunochromatographic diagnostic test for resource-limited settings with further molecular applications. Journal of Visualized Experiments. (160), e60008(2020).
- Patrick, E., et al. Enhanced rabies surveillance using a direct rapid immunohistochemical test. Journal of Visualized Experiments. (146), e59416(2019).
- Lembo, T., et al. Evaluation of a direct, rapid immunohistochemical test for rabies diagnosis. Emerging Infectious Diseases. 12 (2), 310-313 (2006).
- Marston, D. A., et al. Pan-lyssavirus real time RT-PCR for rabies diagnosis. Journal of Visualized Experiments. (149), e59709(2019).
- Brunker, K. DNA quantification using the Qubit fluorometer. , Available from: https://www.protocols.io/view/dna-quantification-using-the-qubit-fluorometer-bc6vize6 (2020).
- Quick, J. DNA quantification using the Quantus fluorometer. , Available from: https://www.protocols.io/view/dna-quantification-using-the-quantus-fluorometer-7pzhmp6 (2020).
- Guppy protocol. Nanopore Community. , Available from: https://community.nanoporetech.com/protocols/Guppy-protocol/v/gpb_2003_v1_revaq_14dec2018 (2023).
- Flow Cell Wash Kit (EXP-WSH004). Nanopore Community. , Available from: https://community.nanoporetech.com/protocols/flow-cell-wash-kit-exp-wsh004/v/wfc_9120_v1_revh_08dec2020 (2023).
- Core Pipeline - arctic pipeline. , Available from: https://artic.readthedocs.io/en/latest/minion/ (2023).
- Samtools. , Available from: https://www.htslib.org (2023).
- Campbell, K., et al. Making genomic surveillance deliver: A lineage classification and nomenclature system to inform rabies elimination. PLoS Pathogens. 18 (5), e1010023(2022).
- Brunker, K., et al. Rapid in-country sequencing of whole virus genomes to inform rabies elimination programmes. Wellcome Open Research. 5, 3(2020).
- Bull, R. A., et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nature Communications. 11, 6272(2020).
- Okeke, I. N., Ihekweazu, C. The importance of molecular diagnostics for infectious diseases in low-resource settings. Nature Reviews. Microbiology. 19 (9), 547-548 (2021).
- Inzaule, S. C., Tessema, S. K., Kebede, Y., Ouma, A. E. O., Nkengasong, J. N. Genomic-informed pathogen surveillance in Africa: opportunities and challenges. The Lancet Infectious Diseases. 21 (9), 281-289 (2021).
- Kennedy, L. Integrating contact tracing and whole-genome sequencing to track the elimination of dog-mediated rabies: an observational and genomic study. eLife. , (2023).
- Pallerla, S. R. Diagnosis of pathogens causing bacterial meningitis using Nanopore sequencing in a resource-limited setting. Annals of Clinical Microbiology and Antimicrobials. 21, 39(2022).
- Quick, J. Real-time, portable genome sequencing for Ebola surveillance. Nature. 530 (7589), 228-232 (2016).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati