Methods Article
Approche microfluidique pour résoudre la sécrétion simultanée et séquentielle de cytokines de cellules polyfonctionnelles individuelles
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Le protocole décrit une plate-forme microfluidique avancée pour mesurer quantitativement la dynamique de sécrétion de cytokines de cellules mononucléées du sang périphérique humain individuelles. La plateforme mesure jusqu’à trois cytokines en parallèle (IL-6, TNFα et IL-1β) pour chaque cellule individuelle stimulée par le lipopolysaccharide à titre d’exemple.
Résumé
Les infections, les maladies auto-immunes, les réponses immunologiques souhaitées et indésirables au traitement peuvent conduire à une réponse cytokinique complexe et dynamique in vivo. Cette réponse implique que de nombreuses cellules immunitaires sécrètent diverses cytokines pour orchestrer la réaction immunitaire. Cependant, la dynamique de sécrétion, les quantités et la cooccurrence des différentes cytokines par divers sous-types cellulaires restent mal comprises en raison d’un manque d’outils appropriés pour les étudier. Ici, nous décrivons un protocole utilisant une plateforme de gouttelettes microfluidiques qui permet la mesure quantitative résolue en temps de la dynamique de sécrétion de plusieurs cytokines en parallèle au niveau de la cellule unique. Ceci est rendu possible par l’encapsulation de cellules individuelles dans des gouttelettes microfluidiques ainsi qu’un immunodosage multiplexé pour la quantification parallèle des concentrations de cytokines, leur immobilisation pour l’imagerie fluorescente dynamique, et l’analyse des images respectives pour dériver les quantités et la dynamique sécrétées. Le protocole décrit la préparation de nanoparticules magnétiques fonctionnalisées, les expériences d’étalonnage, la préparation cellulaire et l’encapsulation des cellules et des nanoparticules en gouttelettes pour l’imagerie fluorescente et l’analyse ultérieure des images et des données en utilisant l’exemple des cellules mononucléées du sang périphérique humain stimulées par des lipopolysaccharides. La plateforme présentée a identifié un comportement distinct de sécrétion de cytokines pour les cellules uniques et co-sécrétrices, caractérisant l’hétérogénéité phénotypique attendue dans l’échantillon cellulaire mesuré. De plus, la nature modulaire du test permet son adaptation et son application à l’étude d’une variété de protéines, de cytokines et d’échantillons de cellules, ce qui pourrait conduire à une compréhension plus approfondie de l’interaction entre les différents types de cellules immunitaires et du rôle des différentes cytokines sécrétées dynamiquement pour façonner la réponse immunitaire étroitement régulée. Ces nouvelles connaissances pourraient être particulièrement intéressantes dans l’étude des dérèglements immunitaires ou dans l’identification de populations cibles dans le traitement et le développement de médicaments.
Introduction
Les infections provoquent souvent des réactions complexes de l’hôte impliquant les systèmes immunitaire inné et adaptatif 1,2. Lors de l’infection ou de la reconnaissance d’agents infectieux, les cellules hôtes peuvent produire une gamme variée de chimio- et de cytokines, qui sont de petites protéines connues sous le nom de communicateurs critiques et modulateurs du système immunitaire3. Les cytokines pro-inflammatoires sont libérées tôt après l’infection pour initier la réponse immunitaire, suivies plus tard par des cytokines anti-inflammatoires, qui sont essentielles pour prévenir les lésions tissulaires et les maladies chroniques ou auto-inflammatoires ultérieures. Cet équilibre entre l’élimination de la menace et la protection tissulaire se manifeste par un large répertoire de cytokines exerçant différentes fonctions pendant l’infection, permettant un ajustement fin de la réponse 4,5. Au sein de ce mélange, des signatures uniques peuvent être observées en fonction de l’agent pathogène et des signaux qu’il induit, de l’emplacement des tissus et des cellules immunitaires dont ils proviennent. Cependant, la libération de cytokines semble également constituer un processus biologique multifonctionnel propre à chaque population cellulaire, diversifié dans la dynamique de sécrétion et la réponse individuelle. Cette hétérogénéité est décrite dans la littérature depuis de nombreuses années, par exemple, parmi les sous-populations de lymphocytes T 6,7, où les investigations sur les maladies auto-inflammatoires et les infections graves à la COVID-19 ont montré une grande diversité fonctionnelle de marqueurs inflammatoires chez les patients et entre eux 8,9. Dernièrement, l’avènement du séquençage unicellulaire a mis en évidence la plasticité élevée et la diaphonie entre les sous-populations au sein des microenvironnements immunitaires qui n’étaient pas apparentes auparavant, indiquant que les méthodes unicellulaires sont nécessaires pour capturer cette hétérogénéité10,11. Bien que de nouvelles méthodes soient en cours de développement pour analyser le transcriptome, l’analyse phénotypique reste difficile, car elle nécessite des mesures simultanées, quantitatives et résolues en temps de la sécrétion de protéines au niveau d’une seule cellule. De telles mesures nous permettent d’étudier les identités, la dynamique et les schémas de sécrétion cellulaires sécrétrices (lent/rapide, précoce/tardif, simultané/séquentiel) pour un répertoire ou un panel de cytokines. En permettant d’étudier la dynamique de la libération de cytokines au cours d’une réponse immunitaire de manière quantitative et temporelle, les informations qui en résultent pourraient permettre de comprendre l’ensemble cellulaire et la réponse induite.
Dans les protocoles standard, les cytokines sont généralement détectées dans le surnageant des suspensions cellulaires et du sérum à l’aide d’un test immuno-enzymatique (ELISA), ce qui permet d’obtenir des quantités sécrétées en vrac. Les mesures en vrac ne permettent pas de quantifier les quantités de cytokines produites par chaque cellule, un problème particulièrement mis en évidence dans les échantillons de cellules hétérogènes. D’autres méthodes telles que la coloration intracellulaire des cytokines, le test ELISpot (Enzyme-linked Immunospot) ou les tests microgravés (par exemple, Isoplexis) détectent les cytokines exprimées par des cellules individuelles, mais ne fournissent des mesures finales que12,13. Cela signifie que la dynamique de la sécrétion et les changements qui peuvent se produire dans le modèle de sécrétion cellulaire au cours du temps d’incubation sont ignorés. De plus, les mesures du paramètre ne peuvent pas faire la différence entre la sécrétion simultanée et séquentielle de cytokines, de sorte que l’étendue réelle de la polyfonctionnalité simultanée des cellules immunitaires dans la sécrétion de cytokines reste incertaine à l’aide de ces méthodes.
La résolution d’une cellule unique peut être obtenue à l’aide de la microfluidique des gouttelettes pour générer et traiter des compartiments physiques de la taille d’un picoliter afin d’étudier les cellules immunitaires sur leurs phénotypes uniques de sécrétion de cytokines au niveau de la celluleunique 14,15. Ces compartiments sont constitués d’émulsions d’eau dans l’huile et peuvent être générés à l’aide de puces microfluidiques16,17. En effet, les tests microfluidiques à base de gouttelettes ont démontré une extrême polyvalence en permettant l’analyse de différents échantillons et répertoires biologiques au niveau de la cellule unique et leur intégration avec les processus en amont (traitement des cellules et des réactifs) et en aval (tri de cellules uniques, protéomique ou séquençage)18,19,20,21,22. En particulier, les configurations qui permettent l’immobilisation des gouttelettes permettent de mesurer la fonctionnalité d’une cellule unique dans le temps, ce qui est précieux pour l’analyse de la sécrétion de protéines18. De plus, l’intégration de tests quantitatifs multiplexés facilite des investigations supplémentaires dans des dimensions auparavant inaccessibles, dans des processus tels que la cosécrétion et l’identification de cellules immunitaires polyfonctionnelles23,24.
Dans ce protocole, nous décrivons un flux de travail unicellulaire basé sur des gouttelettes immobilisées pour détecter, quantifier et mesurer temporellement la sécrétion d’un maximum de trois cytokines en parallèle à partir de cellules individuelles17,23. La technologie offre la possibilité de surveiller les réponses cytokiniques de plus de 20 000 cellules en parallèle.
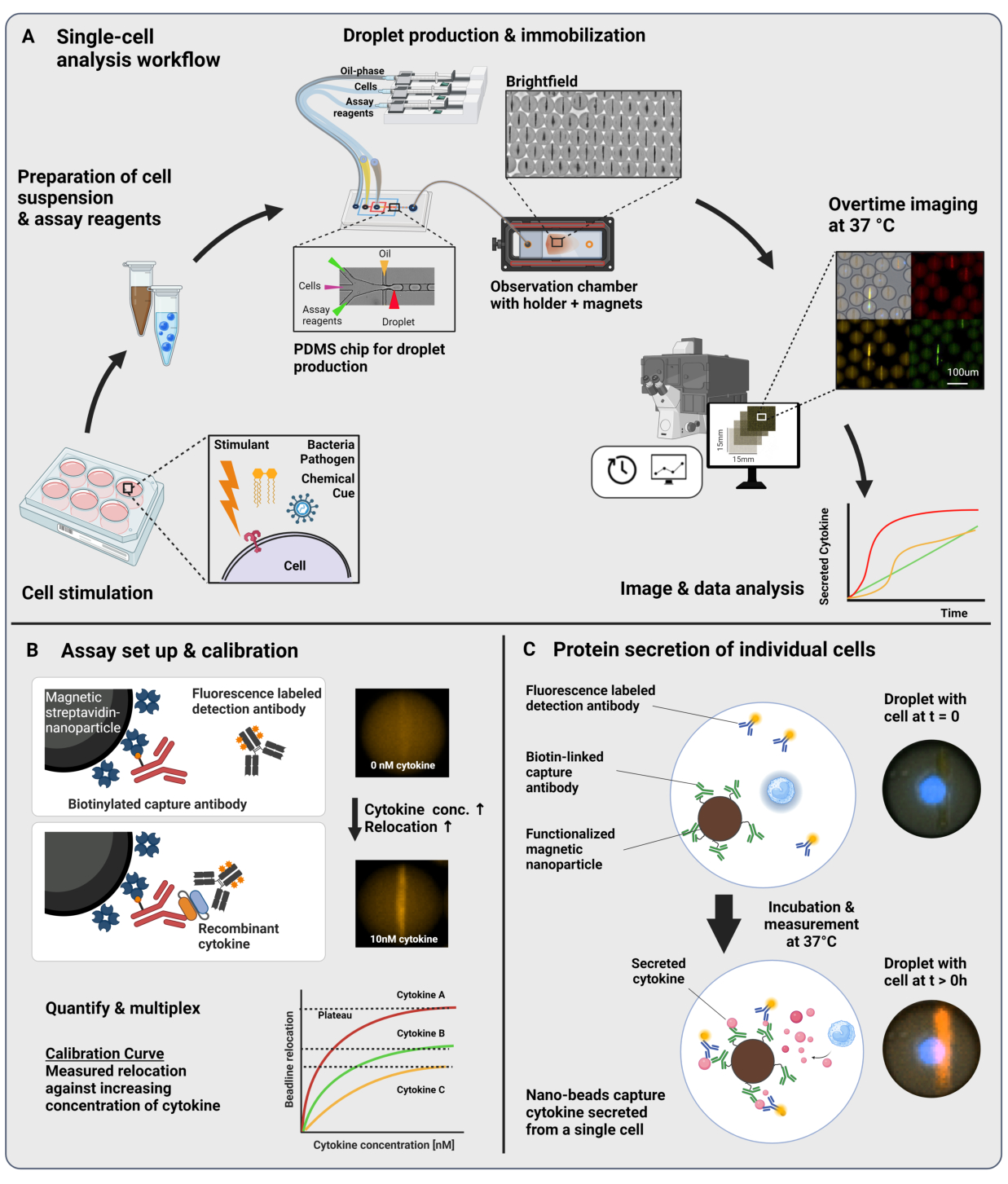
Le flux de travail présenté consiste en l’encapsulation microfluidique de cellules immunitaires uniques et de nanoparticules fonctionnalisées en gouttelettes d’eau dans l’huile de 60 pL. L’immobilisation de > 100 000 gouttelettes dans une chambre d’observation et la microscopie à fluorescence résolue en temps permettent de mesurer la dynamique de sécrétion de cytokines au sein de chaque gouttelette et de chaque cytokine (Figure 1A). Pour chaque cellule individuelle au sein d’une gouttelette, la sécrétion de cytokines est mesurée par un immunodosage sandwich, où des nanoparticules magnétiques fonctionnalisées avec un anticorps de capture spécifique se lient à la cytokine sécrétée, conduisant à la relocalisation et à la liaison ultérieures des anticorps de détection marqués par fluorescence (Figure 1B, C). Une ligne de perles est formée en alignant les nanoparticules magnétiques, vers lesquelles la relocalisation de la fluorescence peut être quantifiée en présence de cytokines. Ici, la relocalisation de la fluorescence est définie comme l’intensité moyenne de fluorescence trouvée sur la ligne de perle divisée par l’intensité moyenne de fluorescence de la gouttelette restante. Ce test peut être multiplexé pour plusieurs cytokines en mélangeant des lots de nanoparticules fonctionnalisés différemment et des anticorps de détection respectifs marqués dans différents canaux de fluorescence23, ce qui entraîne des relocalisations de fluorescence spécifiques dans les différents canaux. À l’aide d’un script d’analyse personnalisé, il est possible d’extraire les valeurs de relocalisation de la fluorescence et de convertir les images en profils dynamiques de sécrétion pour chaque cellule et cytokine individuelle. Par conséquent, les ensembles de données résultants produisent de nombreuses lectures, telles que la mesure quantitative de la sécrétion au fil du temps, l’identification des sous-populations co-sécrétrices et la distribution des cellules en fonction des quantités sécrétées, des taux et des combinaisons de cytokines.

Figure 1 : Flux de travail et principe de dosage. (A) Vue d’ensemble du flux de travail pour l’analyse des cellules sécrétant des cytokines après stimulation. Des suspensions unicellulaires et des nanoparticules magnétiques sont préparées et encapsulées dans des émulsions huile/eau de 60 pL (gouttelettes). Les gouttelettes sont immobilisées et les nanoparticules alignées à l’intérieur d’un champ magnétique avant d’être mesurées jusqu’à 4 h toutes les 30 min. Enfin, les images sont analysées et les paramètres de chaque gouttelette, point temporel et canal fluorescent sont extraits. Ce chiffre a été modifié au lieu de17. (B) Principe de l’essai biologique en sandwich de gouttelettes. Les nanoparticules fonctionnalisées se lient aux cytokines sécrétées, ce qui entraîne la relocalisation ultérieure des anticorps de détection marqués par fluorescence vers les nanoparticules. Cette relocalisation de la fluorescence est quantifiée et validée par des expériences d’étalonnage réalisées avec des cytokines recombinantes. Le mélange de différentes nanoparticules fonctionnalisées permet de mesurer simultanément jusqu’à trois cytokines. (C) Dans les expériences basées sur des cellules, les gouttelettes sont suivies pendant le temps de mesure et les cellules sécrétrices sont identifiées par une augmentation au fil du temps de la relocalisation de la fluorescence sur les nanoparticules. Les schémas ne sont pas à l’échelle. Figurine créée avec BioRender.com. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Protocole
Toutes les expériences ont été réalisées dans le cadre de l’accord d’éthique EK202-N-56 et approuvées par la commission d’éthique de l’ETH Zurich. La manipulation des cellules humaines a été effectuée dans une armoire à flux laminaire contenue dans un laboratoire de biosécurité de niveau 2.
REMARQUE : Les sections suivantes détaillent le protocole permettant de mesurer la sécrétion de cytokines résolues dans le temps au niveau d’une seule cellule. La procédure décrite ici est appliquée à la stimulation des cellules mononucléées du sang périphérique (PBMC) avec des lipopolysaccharides (LPS) et à la mesure parallèle des cytokines IL-6, TNFα et IL-1β. Cependant, si nécessaire, le protocole peut être adapté à d’autres types de cellules, de stimulants et de cytokines.
1. Fabrication de la chambre d’observation
REMARQUE : Pour éviter le mouvement des gouttelettes pendant l’imagerie, une chambre d’observation est préparée avec une hauteur d’environ 10% plus petite que le diamètre des gouttelettes.
- Préparation de la découpe du ruban adhésif double face et de la diapositive supérieure en verre
- Dessinez ou chargez le motif souhaité de la découpe de la chambre dans l’onglet Conception du logiciel de découpe. Pour les dimensions spécifiques utilisées ici, voir la figure 2E.
- Fixez le ruban adhésif double face de 32 μm d’épaisseur sur le tapis de découpe adhésif à l’aide de ruban adhésif et placez le tapis de découpe dans la machine de découpe automatique.
- Découpez la conception de la chambre dans le ruban, tout en faisant attention à couper les bords longs de la chambre dans la même direction pour faciliter le détachement à l’étape 1.3.
- Conservez les découpes de ruban à température ambiante pour un stockage à long terme. Pour un stockage à court terme, stockez-les à -20 °C et retirez-les peu de temps avant l’étape 1.3. pour une manipulation plus facile.
- Percez deux trous d’environ 1 mm de diamètre au milieu d’une lame de microscope standard (76 mm x 26 mm x 1 mm), avec une distance entre les deux trous d’environ 3,5 cm.
- Nettoyage et activation plasma des lames de verre
- Nettoyez une lame de verre avec des trous et une sans trous avec du savon. Rincez bien à l’eau distillée et séchez à l’aide de lingettes de précision non pelucheuses.
- Placez les lames de verre dans un nettoyeur plasma et traitez au plasma les surfaces supérieures à 55 W pendant 10 min. Retirez les lames de verre et passez à l’étape 1.3.
- Assemblage de la chambre
- Placez la lame de verre à trous sur une surface propre, le côté activé par plasma vers le haut, sans toucher la surface activée.
- Retirez la couche protectrice d’un côté du ruban adhésif double face, dans le même sens de la coupe. Sans toucher, alignez la découpe du ruban avec les bords de la glissière de verre et les trous percés, et mettez lentement le ruban en contact avec la glissière de verre en commençant par le bord court.
REMARQUE : Faites attention à ne pas générer d’étirements ou de plis dans le ruban car cela entraînerait des hauteurs de chambre incorrectes. Étant donné que cette étape est sujette aux erreurs et nécessite une certaine expérience pratique, il est conseillé de préparer plusieurs lames de verre en parallèle. - Retirez la deuxième couche protectrice du ruban, toujours dans le sens de la coupe, et placez la deuxième lame de verre sans trous avec la surface activée vers le bas. Appuyez sur toute la surface des deux lames de verre ensemble en plaçant une planche plate sur le dessus et en appuyant avec la force du haut du corps pendant environ 10 s.
- Après avoir assemblé les deux lames de verre, retournez la chambre de manière à ce que les deux trous soient face à vous. Collez les nanoports aux deux trous en mettant une petite quantité de colle durcissable aux UV dans l’anneau sous le port et en plaçant le port sur le trou dans la lame de verre. Ajoutez un anneau de colle durcissable aux UV autour de l’orifice et polymérisez la colle avec une lampe UV. La chambre devrait maintenant ressembler à celle illustrée à la figure 2E. Passez immédiatement à l’étape 1.4.
REMARQUE : La lumière UV peut endommager les yeux et la peau. Portez un équipement de protection approprié.
- Revêtement fluorophile de la surface de la chambre
REMARQUE : Cette étape doit avoir lieu dans l’heure qui suit le traitement au plasma des lames de verre (étape 1.2.2) pour assurer une bonne efficacité du revêtement.- Préparez fraîchement 1 mL de solution de fluorosilane à 1 % (1H, 1H, 2H, 2H-perfluorodécyltrichlorosilane) dans de l’huile fluorée (HFE-7500) et remplissez-la dans une seringue. Poussez la solution de revêtement à travers un filtre de seringue en PTFE et une aiguille de 27G x 0,75 pouce connectée à un microtube en PTFE de 0,3 mm x 0,76 mm, dans la chambre d’observation.
- Après 1 min d’incubation, rincez la solution d’enrobage hors de la chambre en utilisant la pression de l’azote sous une hotte. Rincez la chambre avec de l’huile fluorée (HFE-7500 uniquement) à l’aide d’une autre seringue.
- Stockez la chambre remplie d’huile fluorée avec des entrées fermées à température ambiante (RT). Après chaque expérience, lavez directement les cellules et les gouttelettes pour assurer une bonne conservation du revêtement.
REMARQUE : Le protocole peut être mis en pause ici, et les chambres peuvent être stockées et réutilisées pendant plusieurs mois.
- Support de chambre avec aimants
- Pour l’alignement des nanoparticules magnétiques, appliquez un champ magnétique statique à la chambre d’observation pendant l’encapsulation des gouttelettes et l’imagerie. Pour cela, placez la chambre dans un support de microscopie imprimé en 3D personnalisé (voir la figure 2D et le fichier trouvé dans Bounab et al.17 Données supplémentaires 4) qui contient deux aimants en néodyme le long des côtés longs de la chambre.
2. Fonctionnalisation des nanoparticules
REMARQUE : Le processus de fonctionnalisation des nanoparticules est similaire pour chaque cytokine, la seule différence étant l’ajout d’anticorps de capture spécifiques aux cytokines. La fonctionnalisation de chaque cytokine est effectuée dans différents tubes de réaction individuels en parallèle. Avant ce protocole, l’anticorps de capture TNFα et l’anticorps de détection de l’IL-1β étaient marqués en interne avec de la biotine et Alexa Fluor 647, respectivement. La conjugaison a été effectuée selon le protocole du fabricant trouvé sur le site Web du fournisseur (voir les liens dans la table des matériaux) et les anticorps ont été aliquote et stockés à -20 °C.
- Ajouter 50 μL de nanoparticules fonctionnalisées par la streptavidine (diamètre (Ø) 300 nm) dans le tube destiné à la détection du TNFα, 50 μL pour l’IL-1β et 100 μL pour l’IL-6. Diluer la solution de nanoparticules 1:1 (v/v) dans une solution saline tamponnée au phosphate (PBS).
- Ajouter dans chaque tube 1/20 (v/v) du volume respectif des anticorps de capture biotinylés (concentrations mères à 0,5 mg/mL) et incuber pendant 30 min à RT.
REMARQUE : Lorsque vous ajoutez de petits volumes à la solution de nanoparticules, déposez le volume en haut du tube et lavez-le plusieurs fois avec la majeure partie de la solution. Cela garantit un bon mélange et empêche la formation de granulats. - Ajouter 1/100 (v/v) de solution de D-biotine de 1 mM dans le tube et incuber pendant 5 min à RT. Il en résulte une concentration finale de biotine de 10 μM.
REMARQUE : L’excès de biotine bloque les côtés de liaison libre sur les nanoparticules et réduit la formation d’agrégats indésirables. - Collectez les particules en tenant un aimant en néodyme près du tube. Attendez que le surnageant soit clair et jetez-le.
REMARQUE : Les aimants utilisés tout au long du test présentent des forces d’attraction très fortes, qui peuvent causer des dommages physiques si deux aimants s’emboîtent accidentellement. - Pour réduire l’adsorption non spécifique à la surface des nanoparticules, remettez immédiatement les nanoparticules en suspension dans 0,5 fois le volume final de l’étape 2.1 de Pluronic F-127 (10 %) et 0,5 fois le volume PBS. Incuber la solution pendant 30 min à RT.
- Collectez les particules à l’aide de l’aimant, jetez le surnageant et mettez-les en suspension dans 1 fois le volume de la mémoire tampon de stockage (RPMI 1640, 5 % de remplacement sérique knock-out, 1 % Pen/Strep, 1 % d’albumine sérique humaine recombinante (HSA), 25 mM HEPES, 0,1 % Pluronic F-127). Incuber la solution pendant 30 min à RT.
REMARQUE : Le protocole peut être mis en pause ici, et les particules peuvent maintenant être stockées jusqu’à 1 semaine à 4 °C. - Immédiatement avant l’encapsulation, remettre les particules en suspension par pipetage et mélanger les nanoparticules conjuguées dans un rapport de 2:1:1 (v/v) pour IL-6 :TNFα :IL-1β, respectivement.
REMARQUE : Les différents ratios de nanoparticules fonctionnalisées dépendent de la paire d’anticorps utilisée pour chaque cytokine et ont été déterminés expérimentalement par des échantillons d’étalonnage pour obtenir une plage dynamique optimale. - Laver avec un support complet (RPMI 1640, 10 % FBS, 1 % Pen/Strep, 25 mM HEPES) en recueillant les particules avec l’aimant, en jetant le surnageant et en le remettant en suspension. Répétez cette étape, mais ne suspendez que dans 0,5 fois le volume du support complet de l’étape 2.7.
- Ajoutez à la solution les anticorps de détection IL-6, TNFα et IL-1β marqués différemment pour atteindre une concentration finale de 10 nM chacun. La solution est maintenant prête à être utilisée pour les expériences sur les gouttelettes.
3. Préparation des cellules
REMARQUE : Les PBMC ont été isolés à partir d’une couche leucocytaire reçue de la banque de sang de Zurich. Les cellules ont été congelées et stockées dans des flacons cryogéniques (1 x 107 cellules/flacon) dans de l’azote liquide pendant plusieurs mois.
- Décongélation cellulaire
- 1 h avant de commencer l’expérience, laisser le milieu complet et le tampon MACS (2 mM d’EDTA, 0,5 % BSA dans le DPBS, filtré stérilement) se réchauffer à RT. Préparez le tube contenant les cellules en ajoutant 9 mL de milieu complet dans un tube de 15 mL et en le maintenant dans le bain-marie à 37 °C.
- Récupérer un cryoflacon PBMC (contenant ~1 x 107 cellules) de son stockage dans de l’azote liquide. Faites tourner le cryotube dans le bain-marie à 37 °C jusqu’à ce qu’il ne reste qu’une petite quantité de glace.
- Essuyez le tube avec 70 % d’EtOH et transférez-le dans l’armoire à flux laminaire. Ajoutez 1 mL de milieu complet préchauffé dans le cryovial, mélangez doucement et transférez toutes les cellules dans le tube contenant le milieu complet chaud. Le cryoflacon peut être lavé avec 1 mL de milieu complet chaud pour récupérer le maximum de cellules.
- Faites tourner les cellules à 500 x g pendant 5 min à RT, jetez le surnageant et remettez doucement en suspension à l’aide d’une pipette la pastille cellulaire avec 1 mL de milieu complet. Ajouter 9 ml de support complet.
- Faites tourner les cellules à 500 x g pendant 5 min à RT. Jetez le surnageant et remettez-le en suspension comme précédemment dans 1 mL de milieu complet.
- Comptez les cellules à l’aide du compteur de cellules disponible. Dans ce cas, un compteur de cellules automatisé a été utilisé. Les cellules ont été comptées en mélangeant 10 μL de suspension cellulaire avec 10 μL de bleu de trypan et en transférant 10 μL du mélange dans la lame de comptage cellulaire.
- Coloration et blocage FcR
- Calculez le nombre total de cellules et le volume nécessaire pour remettre les cellules en suspension à 2 x 106 cellules vivantes/ml. Préparez la solution de coloration cellulaire (CellTrace Violet) en diluant la mère (5mM) 1000x dans du PBS (concentration de travail de 5 μM).
- Faites tourner les cellules à 500 x g pendant 5 min à RT. Jetez le surnageant et remettez les cellules en suspension dans le volume calculé de solution de coloration cellulaire préparé à l’étape 3.2.1. Incuber les cellules à 37 °C pendant 5 min.
- À la fin de l’incubation, trempez le colorant restant dans la solution en ajoutant un milieu complet (au moins 2 fois le volume de la solution de colorant). Faites tourner les cellules à 500 x g pendant 5 min à RT.
- Jetez le surnageant, remettez la pastille cellulaire en suspension dans 60 μL de tampon MACS et ajoutez 20 μL de bloc FcR humain par 1 x 107 cellules. Incuber les cellules pendant 10 min à RT.
- Remplissez le tube à 10 mL avec MACS Buffer et faites tourner les cellules à 500 x g pendant 5 min à RT.
- Jetez le surnageant et remettez les cellules en suspension dans 1 mL de milieu complet Comptez les cellules comme décrit à l’étape 3.1.6.
- Stimulation cellulaire avec LPS
- À l’aide du comptage cellulaire, diluez les cellules à 1 x 106 cellules/mL et transférez 2 mL de cellules dans chaque puits dans une plaque à 6 puits à très faible liaison.
- Diluer le LPS dans un milieu complet et l’ajouter dans le puits contenant les cellules pour une concentration finale de LPS de 1 μg/mL. Incuber les cellules pendant 6 h à 37 °C.
- Préparation pour l’encapsulation
- À la fin du temps de stimulation, transférez la suspension cellulaire dans un nouveau tube de 15 ml.
- Ajouter 1 mL de milieu complet dans le puits vide. À l’aide d’un grattoir à cellules, détachez les cellules restantes. Transférez les cellules dans un nouveau tube de 15 ml. Lavez le puits avec 1 ml de support complet et transférez-le dans un autre tube de 15 ml.
- Faites tourner les deux tubes à 500 x g pendant 5 min à l’heure RT et transférez 1 mL de la solution surnageante non diluée (du premier tube contenant les cellules non lavées) dans un nouveau tube pour une analyse plus approfondie si nécessaire (p. ex., ELISA).
- Jetez le reste des surnageants.
- Remettez les pastilles en suspension dans 0,5 mL de milieu complet, combinez les cellules du même puits et transférez-les dans un tube à centrifuger. Comptez les cellules comme décrit à l’étape 3.1.6.
- Faites tourner les cellules à 500 x g pendant 5 minutes à RT et jetez la majeure partie du surnageant (laissant environ 100 μL). Sans remettre la pastille en suspension, ajoutez très soigneusement 200 μL de média complet.
- Jetez le surnageant. Remettre les cellules en suspension dans un milieu complet à une concentration de 6,6 à 13,3 x 106 cellules/mL pour obtenir un nombre moyen de cellules par gouttelette de λ = 0,2-0,4 pour l’encapsulation, tel que défini à l’étape 8.6.
REMARQUE : Les étapes 3.4.6 et 3.4.7 doivent être effectuées immédiatement avant l’encapsulation afin d’éviter la sécrétion de cytokines dans le surnageant. Le nombre de cellules par gouttelette suit une distribution de Poisson : , où P indique la fraction de gouttelettes contenant X cellules et λ est le nombre moyen de cellules par gouttelette.
, où P indique la fraction de gouttelettes contenant X cellules et λ est le nombre moyen de cellules par gouttelette.
4. Encapsulation et production de gouttelettes
REMARQUE : L’encapsulation des cellules dans des gouttelettes est rendue possible par une puce génératrice de gouttelettes microfluidique, dont la fabrication est décrite en détail ailleurs17. Des alternatives sont disponibles dans le commerce (voir l’exemple dans la Table des matériaux). Une conception appropriée de puce de générateur de gouttelettes comporte deux entrées pour les phases aqueuses, une entrée pour la phase huileuse et une sortie pour les gouttelettes générées. De plus, une puce de générateur de gouttelettes commerciale appropriée devrait permettre de produire de l’eau dans des gouttelettes d’huile fluorée d’un volume de 40 à 60 pL. Le protocole décrit ici permet d’obtenir des émulsions eau/huile (gouttelettes) d’un diamètre de 50 μm. L’utilisation de diverses options pour modifier le protocole peut entraîner des gouttelettes plus grandes ou plus petites.
- Préparation du pousse-seringue (Figure 2A)
- Remplir une seringue de 1 mL avec 500 μL de phase continue composée de 2 % de tensioactif fluoré 008 dans de l’huile fluorée HFE-7500. Connectez une aiguille de 27 G x 0,75 pouce à un microtube en PTFE de 0,30 mm x 0,76 mm et montez l’ensemble sur la seringue, puis sur le pousse-seringue.
REMARQUE : Assurez-vous qu’il ne reste pas d’air dans la seringue ou la canule car cela empêche des débits constants. - Préparez deux connecteurs de pointe de pipette sur mesure pour les phases aqueuses (Figure 2B) : Percez un trou avec un poinçon de biopsie de Ø0,75 mm au milieu d’une découpe PDMS de ~5 mm de haut avec Ø6 mm. Tirez ~3 cm de tube en PTFE (0,56 mm de diamètre intérieur, 1,07 mm de diamètre extérieur) à travers le trou de la découpe PDMS et poussez l’ensemble dans le haut d’une pointe de pipette de 200 μL. Connectez l’autre côté du tube à une aiguille 23Gx 1,25 pouce. Scellez le connecteur en étalant de la colle durcissable aux UV sur le dessus de la pipette et polymérisez-le avec de la lumière UV.
REMARQUE : Étant donné que la lumière UV est nocive pour l’œil, portez des lunettes anti-UV pour vous protéger. - Remplissez deux seringues de 1 ml avec 500 μL d’huile minérale légère, fixez deux aiguilles 23G avec les embouts sur mesure et montez les deux sur le pousse-seringue.
- Aspirez 30 μL de nanoparticules et 30 μL de solution cellulaire dans les pointes de pipette des phases aqueuses à l’aide du logiciel de commande de la pompe à seringue.
- Préparez une chambre d’observation en nettoyant la surface avec de l’eau pour enlever la saleté et la poussière et séchez-la avec des lingettes de précision. Fixez la chambre dans le support de chambre imprimé équipé de deux aimants en néodyme.
REMARQUE : Assurez-vous que les aimants pointent dans la bonne direction (en s’attirant les uns les autres) pour former un agrégat allongé. - Inclinez légèrement la chambre (30°). Ouvrez les deux orifices et branchez une serviette en papier dans l’orifice supérieur pour absorber l’excès de phase extérieure pendant le remplissage.
- Remplir une seringue de 1 mL avec 500 μL de phase continue composée de 2 % de tensioactif fluoré 008 dans de l’huile fluorée HFE-7500. Connectez une aiguille de 27 G x 0,75 pouce à un microtube en PTFE de 0,30 mm x 0,76 mm et montez l’ensemble sur la seringue, puis sur le pousse-seringue.
- Production de gouttelettes et remplissage de la chambre
- Connectez la phase continue via un tube à l’entrée supérieure de la puce microfluidique (Figure 2A, C, F). Rincez la puce pendant environ 30 s en phase continue avec un débit de 1800 μL/h.
- Connectez les pointes de pipette des solutions aqueuses aux deux entrées centrales (Figure 2A, C, F).
- Démarrez l’écoulement de la solution aqueuse avec 200 μL/h chacun et laissez les canaux et la sortie être remplis de liquide. Lors de l’utilisation de nanoparticules magnétiques, une solution homogène de couleur brun-rouge doit s’écouler de la sortie de la puce.
- Une fois que le liquide apparaît à la sortie, démarrer le flux en phase d’huile fluorée à 800 μL/h et attendre qu’une production stable de gouttelettes s’établisse, confirmée par l’écoulement d’une solution homogène, grise et brillante à la sortie.
- Une fois qu’une production stable de gouttelettes est établie, collectez les gouttelettes produites en connectant des microtubes en PTFE (diamètre intérieur de 0,3 mm x diamètre extérieur de 0,76 mm) à l’orifice de sortie et dirigez-les dans une chambre d’observation en faisant passer le microtube à travers le module de virole d’un raccord monobloc étanche à la main (figure 2A).
- Si la production de gouttelettes est correcte, un liquide homogène et brillant doit remplir la chambre avec une façade droite de bas en haut.
- Une fois la chambre remplie, arrêtez l’écoulement et fermez les orifices avec des bouchons d’orifice en exerçant une pression serrée à la main.
REMARQUE : Faites attention à ne pas fermer la chambre trop hermétiquement. Le piégeage ou l’afflux d’air peut entraîner des mouvements des gouttelettes et ainsi compromettre le suivi pendant la mesure. - Après la production de gouttelettes, rincez la puce avec de l’huile fluorée et soufflez tous les rappels de liquide avec de l’azote pour préserver sa fonction. Les puces peuvent être réutilisées plusieurs fois et stockées pendant des mois tant qu’elles ne sont pas bouchées.

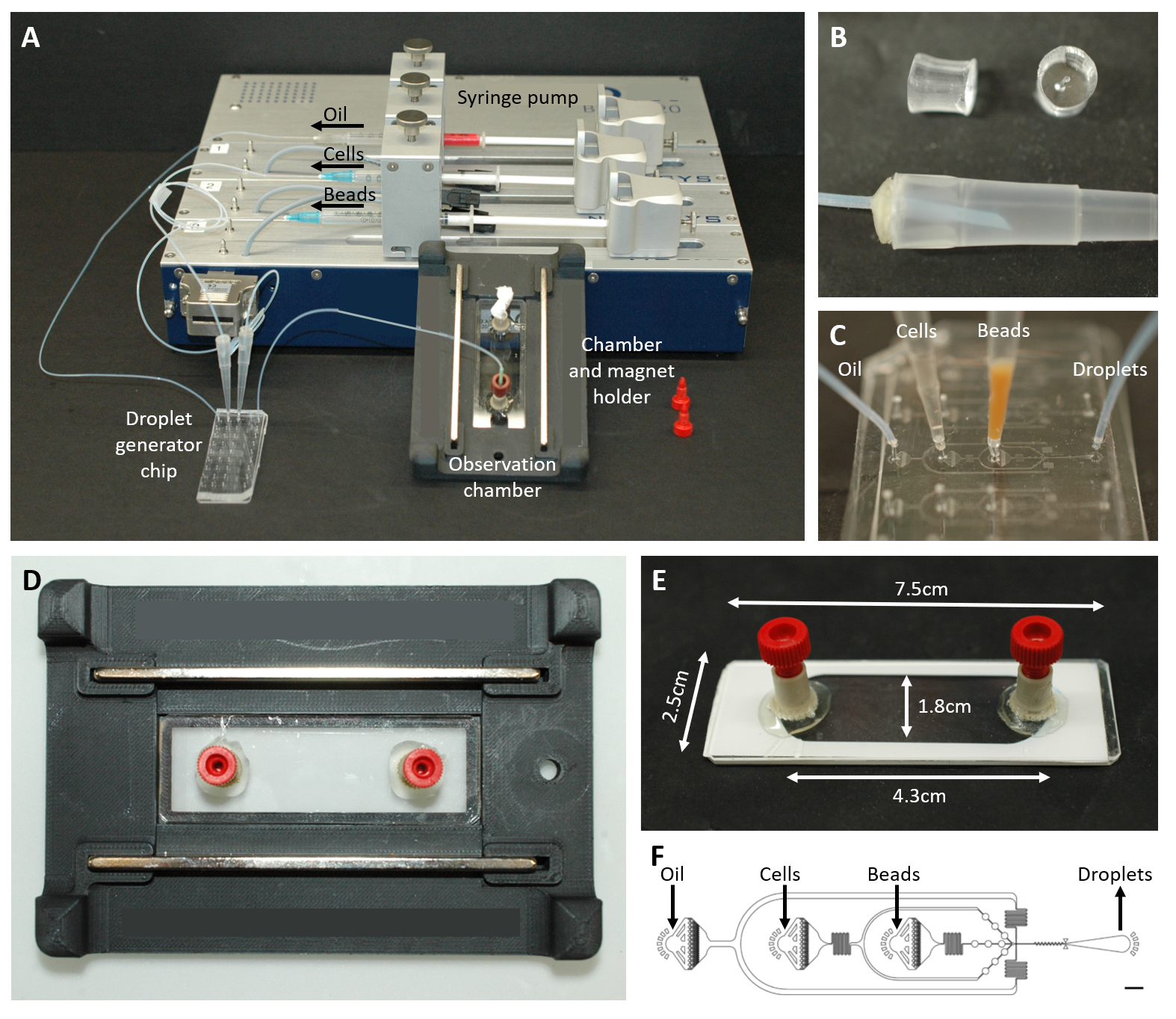
Figure 2 : Vue d’ensemble de la configuration microfluidique. (A) Configuration pour l’encapsulation des gouttelettes avec la pompe à seringue, la puce de génération de gouttelettes, la chambre d’observation et le support de microscope. (B) Image de la fiche PDMS perforée (en haut) pour former un connecteur à une pointe de pipette de 200 μL (en bas), comme décrit à l’étape 4.1.2 du protocole. (C) Images de la connexion des tubes et des pointes de pipette à la puce de génération de gouttelettes. (D) Photo de la chambre placée à l’intérieur du support de microscope imprimé en 3D avec deux aimants en haut et en bas. (E) Photo de la chambre d’observation (avec du ruban adhésif blanc pour l’illustration). (F) Disposition de la puce microfluidique pour la création de gouttelettes (barre d’échelle : 750 μm). Ce chiffre a été modifié au lieu de17. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
5. Acquisition et mesure d’images
REMARQUE : L’acquisition d’images est effectuée à l’aide d’un microscope à épifluorescence inversée standard enfermé dans un incubateur, permettant des mesures à 37 °C. Les paramètres décrits ici sont spécifiques à un microscope Nikon Eclipse Ti2 fonctionnant avec le logiciel NIS Elements (V. 5.30.04) équipé d’une caméra Orca Fusion, mais sont généralement adaptables à tous les autres microscopes et caméras à fluorescence.
- Réglage des paramètres de mesure
- Pour définir la taille de l’image, sélectionnez une taille de tableau de 10 x 10 images. Ce réseau contiendra environ 50 000 à 70 000 gouttelettes. Utilisez un chevauchement de 1 % et activez la fusion pour l’assemblage de l’image.
- Pour régler le nombre de canaux mesurés, sélectionnez le canal DAPI pour la détection de cellules, les canaux FITC, TRITC, Cy5 pour la détection des cytokines (lignes de perles) et le canal BF pour la détection des gouttelettes. Utilisez le regroupement de pixels 2 x 2 et 16 bits pour la profondeur de bits. Adaptez les paramètres de l’appareil photo pour obtenir des valeurs de pixels d’intensité en gouttelettes qui n’atteignent pas le maximum de l’appareil photo pour chaque canal de fluorescence.
REMARQUE : Les temps d’exposition et les intensités exactes de la lampe pour chaque canal dépendent du modèle utilisé et des réactifs et sont établis avant de générer les courbes d’étalonnage (étape 7). L’utilisation des mêmes paramètres d’acquisition pour l’étalonnage et les mesures de cellule est importante pour une quantification précise. - Pour configurer la mesure résolue dans le temps, sélectionnez une mesure toutes les 30 minutes pour 9 mesures au total.
REMARQUE : Les paramètres de mesure peuvent différer pour les cellules utilisées, les stimulants, les réactifs, les cytokines mesurées, la température d’incubation et les modèles de microscope.
- Démarrage de la mesure
- Montez le support de chambre sur le microscope à l’aide d’une platine au format plaque à puits (Figure 2D) et passez au canal en fond clair (BF) à l’aide de l’objectif 10x.
- Concentrez-vous sur les gouttelettes immobilisées dans BF et assurez-vous que l’ensemble est monté dans un plan parfait en se déplaçant et en ajustant si nécessaire. Déplacez-vous vers le milieu de la chambre pour les étapes suivantes.
- Activez le système de mise au point automatique (PFS) et réglez-le sur le plan de mesure optimal sur le canal BF afin que les bords des gouttelettes apparaissent sous forme de cercles noirs et nets qui peuvent être facilement distingués de la phase d’huile et de l’arrière-plan.
REMARQUE : Les mesures sont également possibles sans système de mise au point automatisé, mais si le microscope en est équipé, nous vous recommandons vivement de l’utiliser. Cela améliore la qualité de mesure des images de grande taille et fortement assemblées. - Passez en revue tous les canaux de fluorescence et définissez le plan de mesure optimal pour chacun d’eux. Pour les mesures de relocalisation sur les canaux FITC, TRITC et Cy5, assurez-vous que l’agrégat de nanoparticules est parfaitement mis au point, pour le canal DAPI, assurez-vous que les cellules sont nettes.
REMARQUE : Les plans focaux optimaux et les valeurs z peuvent différer pour tous les canaux mesurés. Assurez-vous d’enregistrer des décalages PFS individuels pour chaque canal. - Avant de commencer la mesure, passez en revue tous les canaux pour vérifier les foyers individuels et attendez 5 minutes pour équilibrer, car un mouvement peut se produire initialement pendant que les solutions se réchauffent.
- Commencez la mesure. Après avoir généré la première image, vérifiez qu’il n’y a pas d’irrégularités (mise au point, gouttelettes en mouvement, canaux incorrects, etc.). Redémarrez l’acquisition si nécessaire, ou dans le cas d’air, remplissez la chambre (commencez à partir de l’étape 4.1.4). Laissez l’assemblage imager les gouttelettes sur 4 h.
6. Analyse d’images
- Installez le logiciel d’analyse d’images (DropMap Analyzer App v 4.023) dans MatLab (https://github.com/ESPCI-LCMD/MiMB) et transférez le fichier .nd2 généré de l’expérience vers un ordinateur d’analyse.
- Ouvrez l’application. Sélectionnez les paramètres spécifiés, sinon laissez la valeur par défaut : CH1 : DAPI, WD (gouttelette entière) sélectionnée ; CH2 : FITC, BL (beadline) sélectionné ; CH3 : TRITC, BL sélectionné ; CH4 : Cy5, BL sélectionné ; Diamètre de chute maximum (μm) : 70 ; Détection de chute : Plein ; Suivi : Oui. Appuyez sur le bouton Démarrer (icône en forme de fruit) pour sélectionner l’emplacement du fichier .nd2 et lancer l’analyse.
- Après quelques minutes, le programme affichera une section d’exemple de l’image (Figure 3A). Appuyez sur Espace jusqu’à ce que vous en trouviez un adapté à la détection des gouttelettes, puis appuyez sur Entrée. Dans la même section d’image, dessinez un rectangle dans une zone représentative pour trouver des paramètres de seuillage afin de détecter les gouttelettes.
- Après quelques minutes, une autre fenêtre s’ouvrira montrant la distribution de l’intensité du canal DAPI. Faites glisser et déposez le curseur pour détecter uniquement le signal des cellules colorées et cliquez sur Terminé dans le coin supérieur droit.
- Après avoir segmenté l’image en gouttelettes uniques d’un diamètre inférieur au diamètre maximal de la goutte (μm), le programme effectuera maintenant les étapes suivantes pour chaque gouttelette, point temporel et canal de fluorescence sans autre intervention de l’utilisateur (voir Figure 3).
- Le logiciel calcule les pixels déplacés de la gouttelette entre les points temporels (les gouttelettes se déplaçant de plus de 40 pixels sont automatiquement exclues).
- Le logiciel mesure la valeur moyenne de fluorescence de l’ensemble de la gouttelette, détectant et mesurant l’intensité moyenne de la ligne de perle en trouvant le pixel le plus brillant sur une ligne horizontale et en faisant la moyenne de toutes les intensités de pixels sur une ligne verticale de haut en bas de la gouttelette. Cette opération est effectuée automatiquement et est utilisée pour calculer les valeurs moyennes de déplacement de la ligne de talon (Figure 3B) selon l’équation :

- Le logiciel calcule le pourcentage du nombre total de pixels dans la zone des gouttelettes au-dessus du seuil défini sur le canal DAPI.
- Le fichier .xslx résultant contiendra les colonnes suivantes d’intérêt pour une analyse plus approfondie : DropIdX (ID de la gouttelette suivie dans le temps), TrueCentroid_ t*2-1 et t+2 (coordonnées x et y, respectivement, du centre de la gouttelette pour le point temporel t), DiameterMicrons (diamètre de la gouttelette en μm), TrackingMove (nombre de pixels déplacés sur toute la durée de mesure), FluoChannel_BL_Ratio_t (valeur de relocalisation pour FluoChannel au point temporel t), DAPI_WD_PosPxlCount_t (nombre de pixels au-dessus du seuil dans l’ensemble de la gouttelette dans le canal DAPI au point t).

Figure 3 : Analyse d’image effectuée par le logiciel d’analyse d’image. (A) Les gouttelettes sont détectées dans le canal en fond clair (BF) à l’aide d’une transformation de Hough, en marquant chaque gouttelette d’un cercle rouge. Barres d’échelle : 200 μm. (B) À l’intérieur de chaque gouttelette, la ligne de perles de nanoparticules est identifiée par les pixels les plus brillants dans le plan horizontal et les intensités de fluorescence sont moyennées pour tous les pixels s’étendant de haut en bas de la gouttelette. De plus, la cellule est identifiée par un pourcentage de pixels >0 au-dessus du seuil pour l’ensemble de la zone des gouttelettes. Barre d’échelle : 20 μm. (C) Le logiciel de l’analyseur compare l’intensité de fluorescence sur les nanoparticules au fond des gouttelettes pour les canaux FITC, TRITC et Cy5 sur tous les points temporels mesurés pour chaque gouttelette individuelle. Les points de temps 0, 4 (120 min) et 9 (240 min) sont affichés. Pour vérifier manuellement la détection correcte des gouttelettes et des cellules, les canaux DAPI et BF sont également affichés. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
7. Étalonnage
REMARQUE : Pour une lecture quantitative, l’étalonnage des concentrations de cytokines en fonction des valeurs de relocalisation de fluorescence doit être effectué une seule fois, car des différences entre différentes configurations expérimentales peuvent survenir. Toutes les étapes requises sont détaillées dans les sections précédentes du protocole, comme indiqué.
- Préparez les nanoparticules comme décrit à l’étape 2.
- Reconstituer les protéines recombinantes humaines IL-6, TNFα et IL-1β selon les instructions du fabricant.
REMARQUE : Assurez-vous que les aliquotes congelées ne sont décongelées qu’une seule fois et utilisées rapidement. - Préparez une série de dilutions en 2 fois pour les trois protéines en utilisant des milieux complets (10 % FBS, 1 % Pen/Strep, 25 mM HEPES) avec une concentration initiale de 80 nM jusqu’à 0,625 nM.
- Effectuez l’encapsulation comme décrit à l’étape 4 avec des nanoparticules fonctionnalisées dans la première et des RPMI uniquement dans la deuxième phase aqueuse. Cette mesure sert de blanc et l’écart-type mesuré est utilisé ultérieurement pour l’analyse des données.
- Attendez 5 min et imagez les gouttelettes comme décrit à l’étape 5. Prenez 3 images avec une taille de matrice de 2 x 2 dans les canaux de fluorescence respectifs.
- Répétez les étapes 7.4 et 7.5 avec toutes les solutions d’étalonnage préparées, en commençant par la plus faible et en terminant par la concentration la plus élevée.
- Analysez les images comme décrit à l’étape 6. N’utilisez pas l’option WD pour le canal DAPI et réglez le suivi sur Non.
- L’analyse produit des valeurs de relocalisation de fluorescence pour chaque gouttelette mesurée dans une image. Extrayez la médiane et l’écart-type pour chaque canal de fluorescence. Faites la moyenne de la médiane et de l’écart-type pour chaque image mesurée par concentration.
- Générez une courbe d’étalonnage en traçant la relocalisation médiane moyenne par rapport aux concentrations mesurées de chaque protéine recombinante.
- Ajustez les courbes à l’aide d’une association monophasée :
 ,
,
avec Y = déplacement à x, Y0 = déplacement de la mesure à blanc et x la concentration utilisée. La courbe d’étalonnage obtenue est utilisée pour quantifier les valeurs de déplacement comme décrit à l’étape 8.
REMARQUE : N’ajustez que les valeurs jusqu’au déplacement mesuré le plus élevé et excluez les valeurs des concentrations plus élevées avec un déplacement mesuré plus faible. Une diminution des valeurs de relocalisation mesurées à des concentrations plus élevées est attendue et se produit en raison de l’effet crochet et de la capacité de liaison limitée des nanoparticules.
8. Analyse des données
- Excluez les gouttelettes dont la valeur TrackingMove est supérieure à 10, c’est-à-dire qui se sont déplacées de plus de 10 pixels au cours de la mesure.
- Identifiez les gouttelettes contenant des cellules colorées (canal DAPI) dans le premier point temporel en triant les gouttelettes dont les valeurs sont supérieures à 0 dans la colonne DAPI_WD_PosPxlPercent_1.
- Identifiez les gouttelettes contenant des cellules sécrétrices en appliquant les 3 critères suivants à la relocalisation de fluorescence de chaque canal de fluorescence (colonnes de FluoChannel_BL_Ratio_t).
- Identification des gouttelettes avec des valeurs de relocalisation croissantes, en triant pour une pente positive sur le temps de mesure.
- Identification des gouttelettes dont les valeurs de relocalisation atteignent la limite de détection (LOD). Une gouttelette est sélectionnée lorsque la relocalisation maximale de la fluorescence sur le temps de mesure est supérieure à la LD, calculée comme décrit ailleurs25 :
 , où μRelocalisation t0 est la médiane de toutes les valeurs de relocalisation au point temporel 0 et σBLK l’écart type du blanc mesuré lors de l’étalonnage, chacune étant spécifique à la cytokine.
, où μRelocalisation t0 est la médiane de toutes les valeurs de relocalisation au point temporel 0 et σBLK l’écart type du blanc mesuré lors de l’étalonnage, chacune étant spécifique à la cytokine. - Vérifier que l’augmentation de la valeur de relocalisation est significative en vérifiant que la variation entre la relocalisation maximale et minimale de la fluorescence mesurée au cours de la durée de mesure est supérieure à :
 .
.
- Identifier les cellules co-sécrétrices en répondant aux critères décrits à l’étape 8.3. pour plus d’un canal de fluorescence simultanément.
- Répétez l’étape 8.3. pour toutes les gouttelettes ne contenant aucune cellule (DAPI_WD_PosPxlPercent_1 = 0). Utilisez ces gouttelettes pour calculer le pourcentage de faux positifs.
- Déterminez la valeur λ précise de la mesure en sélectionnant de manière aléatoire 200 à 500 gouttelettes et en les inspectant à l’aide de la fonction de vérification et de tri du logiciel d’analyse d’images. Comptez le nombre de cellules dans ces gouttelettes et calculez :
λ = nombre de cellules comptées / nombre de gouttelettes analysées - Calculez le nombre total de cellules encapsulées pour la mesure en procédant comme suit :
Nombre total de cellules = λ × nombre de gouttelettes analysées - Calculez le pourcentage de cellules sécrétrices à l’aide du nombre de cellules déterminé. De plus, calculez le pourcentage de faux positifs pour chaque cytokine (généralement inférieur à 3 % à 5 % par rapport au nombre de vrais positifs par canal) et utilisez-les comme contrôle interne pour la cohérence et la reproductibilité expérimentales.
- Pour calculer les concentrations de cytokines sécrétées, convertissez les valeurs de relocalisation en concentration à l’aide des équations d’étalonnage établies à l’étape 7.10.
- Calculez le taux de sécrétion (SR) entre les points temporels à l’aide de l’équation suivante.

- Calculez le taux de sécrétion moyen sur la mesure en faisant la moyenne des taux de sécrétion individuels entre les points temporels. Si le déplacement maximal mesurable a été atteint avant la fin de la mesure, réglez la concentration sur la concentration maximale mesurable (cette valeur est spécifique aux cytokines et correspond à la concentration maximale mesurée et utilisée dans la courbe d’étalonnage à l’étape 6.10) et ne calculez pas d’autre concentration. Calculez le taux de sécrétion et faites la moyenne uniquement jusqu’à ce point de temps.
REMARQUE : Si moins de 50 cellules sécrétrices sont détectées dans un canal de fluorescence, les gouttelettes doivent être examinées visuellement à l’aide de la fonction de vérification et de tri et les gouttelettes ayant des agrégats de fluorescence ou de nanoparticules peuvent être exclues de l’analyse. - Pour extraire d’autres paramètres de la courbe de sécrétion de chaque cellule, effectuez un ajustement des moindres carrés à la courbe temps-concentration pour chaque cellule et cytokine à l’aide d’un script Python personnalisé (disponible sur demande). La fonction ajustée est une courbe sigmoïdale suivant la formule décrite ci-dessous (les ajustements avec R2<0,95 sont exclus des étapes suivantes) :

où C correspond au plateau de concentration [nM], t50 au décalage du demi-maximum [s], et m à la pente de Hill [min-1]. À partir de ces paramètres, les descripteurs de courbe suivants sont extraits comme décrit ci-dessous.- Cmax [nM] : Concentration maximale mesurée.
 : Le temps de sécrétion commence, où la crise atteint 10% de C.
: Le temps de sécrétion commence, où la crise atteint 10% de C. : taux de sécrétion comme la pente linéaire approximative de la courbe entre 10 % et 90 % de la courbe temps-concentration.
: taux de sécrétion comme la pente linéaire approximative de la courbe entre 10 % et 90 % de la courbe temps-concentration.
Résultats
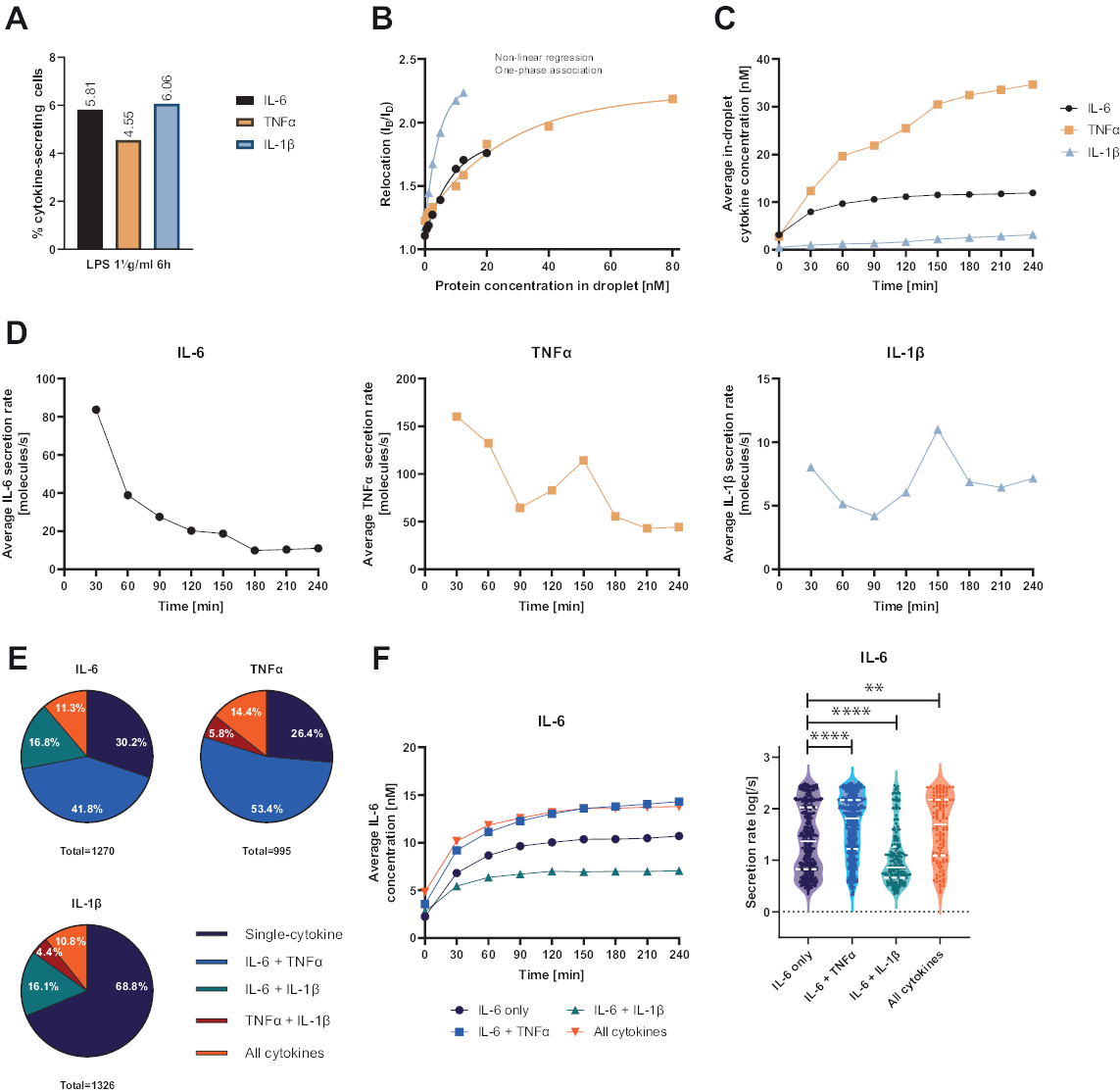
La plate-forme monocellulaire fonctionnelle présentée a permis de mesurer plusieurs paramètres. Tout d’abord, et à l’instar des techniques standard, la fréquence des cellules sécrétrices est représentée à la fin de la mesure (Figure 4A). Suite à la stimulation avec 1 μg/mL de lipopolysaccharide (LPS) pendant 6 h de cellules mononucléées du sang périphérique (PBMC), 5,81 % des cellules ont sécrété de l’IL-6 (n = 1270), 4,55 % du TNFα (n = 995) et 6,06 % de l’IL-1β (n = 1326).
Pour quantifier la sécrétion de cytokines, des courbes d’étalonnage ont été générées avec des concentrations connues de cytokines recombinantes (Figure 4B). Ces courbes d’étalonnage permettent de quantifier les concentrations de cytokines en gouttelettes dans le temps. À titre d’exemple, la concentration moyenne d’IL-6 en gouttelettes a atteint un plateau après 90 minutes pour les PBMC stimulées par le LPS, tandis que la concentration moyenne d’IL-1β en gouttelettes a augmenté plus rapidement à partir de 90 minutes, montrant la résolution dynamique de la plateforme et la possibilité d’extraire des sous-populations cellulaires sécrétant des cytokines spécifiques (Figure 4C). Comme la concentration change entre les points de mesure, il est possible de calculer les taux de sécrétion dynamiques par cytokine. Si l’on considère le taux de sécrétion moyen de chaque cytokine (Figure 4D), les cellules sécrétant l’IL-6 ont montré une diminution constante du taux de sécrétion moyen, tandis que les cellules sécrétant le TNFα et l’IL-1β ont toutes deux montré une augmentation du taux de sécrétion après 90 minutes de temps de mesure et une seconde diminution après 150 min.
De plus, il est possible de regrouper les cellules en sous-populations en fonction des cytokines sécrétées et co-sécrétées (Figure 4E). Ici, l’IL-6 et le TNFα sont sécrétés par 30,2 % et 26,4 % des cellules sécrétant respectivement de l’IL-6 ou du TNFα, tandis que les cellules IL-1β à sécrétion unique représentaient 68,8 % de toutes les cellules sécrétrices d’IL-1β. De plus, les effets de la cosécrétion sur les concentrations et les taux de sécrétion peuvent être résolus (figure 4F). En examinant les cellules sécrétant de l’IL-6, différentes quantités d’IL-6 ont été sécrétées si les cellules produisaient en outre du TNFα ou de l’IL-1β. De même, la distribution des taux de sécrétion moyens sur la mesure différait statistiquement entre les cellules sécrétant uniquement de l’IL-6 ou de l’IL-6 avec le TNFα (taux de sécrétion plus élevés) et l’IL-1β (taux de sécrétion d’IL-6 plus faibles).

Figure 4 : Résultats représentatifs de l’IL-6, du TNFα et de l’IL-1β sécrétant de la PBMC après 6 h de stimulation avec 1 μg/mL de LPS. (A) Pourcentage de PBMC sécrétant de l’IL-6, du TNFα et de l’IL-1β à la fin de la mesure de 4 h. (B) Des courbes d’étalonnage de cytokines multiplexées sont générées à partir de concentrations connues de cytokines recombinantes. Cela permet de quantifier les expériences cellulaires en calculant à partir de la valeur de relocalisation la concentration de cytokines dans la gouttelette. Les points ont été ajustés à l’aide d’un ajustement de courbe d’association monophasé non linéaire, r2 = 0,9926 (IL-6), 0,9901 (TNFα), 0,9990 (IL-1β). (C) Concentrations moyennes sécrétées d’IL-6, de TNFα et d’IL-1β libérées par la sécrétion de PBMC sur la durée de mesure de 4 h. (D) Taux moyens de sécrétion d’IL-6, de TNFα et d’IL-1β sur la durée de mesure de 4 h. (E) Pourcentage relatif de cellules cosécrétrices d’IL-6, de TNFα ou d’IL-1β et combinaisons de ceux-ci. Normalisé à l’ensemble des cellules sécrétrices détectées pour chaque cytokine. (F) Concentrations moyennes d’IL-6 sur le temps de mesure et distributions du taux de sécrétion moyen (log) pour les cellules sécrétrices d’IL-6 avec résolution de co-sécrétion (n = 383 pour l’IL-6 seulement, n = 531 pour l’IL-6 + TNFα, n = 213 pour l’IL-6 + IL-1β et n = 143 pour l’IL-6 + TNFα + IL-1β). Les différences statistiques dans les distributions du taux de sécrétion ont été évaluées à l’aide de tests de Kolmogorov-Smirnov bifaces, non appariés, non paramétriques, avec un niveau de confiance de 95 %, la valeur p est représentée. ** (p <0,002) et **** (p <0,0001). La ligne pleine représente la médiane et la ligne pointillée les quartiles. ncellules au total = 21 866. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Pour extraire des informations supplémentaires au niveau de la cellule unique, une fonction sigmoïde peut être ajustée aux points de concentration-temps de chaque cellule et cytokine (Figure 5). La figure 5A illustre un exemple de concentration dans le temps d’un ensemble de données pour une cellule et l’ajustement sigmoïdal correspondant. Ici, la procédure d’ajustement des moindres carrés donne les paramètres suivants : C, correspondant à la valeur du plateau supérieur de la courbe, t50 quantifiant le décalage temporel de la courbe par rapport à zéro, et la pente de Hill m, décrivant la pente de la partie montante de la courbe sigmoïde avec des valeurs de concentration de 10 % et 90 % atteintes tout au long de la mesure. À partir de ces paramètres d’ajustement, certains descripteurs de courbe peuvent être extraits comme expliqué à l’étape 7.12. ce qui donne le Cmax, la valeur de concentration la plus élevée des données, tstart, le moment de début de la sécrétion, défini comme atteignant 10 % de la valeur de concentration du plateau supérieur, et SRlin, le taux de sécrétion pendant la partie montante de la courbe.
Pour classer les sous-populations cellulaires, les descripteurs de courbe obtenus à partir de tous les ajustements de cellules uniques ont été classés en trois catégories chacune : les valeurs Cmax ont été regroupées en faible, moyenne et élevée pour tcommencer en précoce, moyen et tardif et SRlin en sécrétions lentes, moyennes et rapides. Pour illustrer cette classification, quatre courbes de sécrétion unicellulaire exemplaires et leurs descripteurs de courbe correspondants sont présentés (Figure 5A-D), où la courbe A présente les caractéristiques d’un sécréteur précoce faible de débit moyen, la courbe B est un sécréteur précoce, lent et élevé, la courbe C un sécréteur précoce et rapide et la courbe D montre une sécrétion tardive faible. Il est important de noter que les seuils pour ces critères sont spécifiques aux cellules, aux cytokines et aux paramètres de dosage, et doivent être adaptés à chaque question de recherche. De plus, seule la sécrétion de PBMC par l’IL-6 après une stimulation LPS de 1 μg/mL pendant 6 h a été prise en compte ici, ce qui signifie que la plupart des cellules étaient des sécrétrices précoces et élevées avec 80 % et 79 %, respectivement (Figure 5E-F). En ce qui concerne le taux de sécrétion, une réponse bipolaire a été observée avec 55% des cellules sécrétrices d’IL-6 sont des sécrétrices lentes et 39% des sécrétrices rapides (Figure 5G).
Pour caractériser davantage le comportement de la sécrétion, les descripteurs de courbe de chaque cellule ont été tracés les uns par rapport aux autres et différents groupes ont été extraits (Figure 5H-J). Il n’y a pas de corrélation claire entre tstart et Cmax (Figure 5H) : les deux plus grandes populations étaient des sécréteurs précoces faibles et des sécrétions élevées indépendamment du début de la sécrétion. Si l’on considère la relation entre tstart etSR lin (Figure 5I), la plupart des cellules étaient des sécréteurs précoces lents avec une population claire de sécréteurs précoces élevés et peu de sécréteurs lents/moyens à tardifs. En ce qui concerne les corrélations RSlin et Cmax (Figure 5J), il n’y avait presque pas de sécréteurs rapides faibles à moyens, avec seulement une plus grande population de sécréteurs rapides faibles. De plus, il y avait une grande population de sécréteurs rapides qui ne dépendaient pas de la concentration maximale mesurée, et deux populations de sécréteurs élevés sécrétaient soit lentement, soit rapidement. En résumé, on peut conclure que l’étude de la relation entre les descripteurs de courbe pour des cellules individuelles donne une analyse beaucoup plus détaillée et peut potentiellement extraire de nouvelles découvertes biologiques à partir de mesures de sécrétion unicellulaire.
Avec l’analyse présentée ci-dessus, nous avons extrait la dynamique de sécrétion des cellules co-sécrétrices (Figure 6). Deux exemples de courbes montrent une dynamique différente de co-sécrétion d’IL-6 et de TNFα à partir de deux cellules uniques avec un démarrage simultané des deux cytokines (Figure 6A), ou un démarrage séquentiel de la sécrétion, l’IL-6 étant sécrétée en premier (Figure 6B). Pour classer toutes les cellules co-sécrétrices, un délai de sécrétion de 60 min a été défini, où toutes les cellules commençant à sécréter dans cette plage sont considérées comme des sécréteurs simultanés et toutes les cellules avec des délais plus longs sont considérées comme des sécréteurs séquentiels. Cette analyse a également permis d’observer quelle cytokine était sécrétée en premier. Pour l’IL-6 et le TNFα, une cosécrétion principalement simultanée a été observée dans 76 % des cellules (Figure 6C), tandis que pour l’IL-6 et l’IL-1β, une cosécrétion séquentielle a été observée dans 86 % des cellules, l’IL-6 étant la première cytokine à être sécrétée dans la plupart des cas (Figure 6D).
En examinant l’heure de début de la sécrétion des différentes cytokines pour toutes les cellules co-sécrétrices individuelles, aucune corrélation claire entre les heures de début de la sécrétion n’a été observée dans les expériences réalisées. Pour la co-sécrétion d’IL-6 et de TNFα (Figure 6E), un amas vertical plus grand autour de 0 min était présent, correspondant aux cellules co-sécrétrices plus prévalentes à partir de l’IL-6. Pour la co-sécrétion d’IL-6 et d’IL-1β (Figure 6F), la plupart des cellules ont commencé à sécréter de l’IL-6 au début de la mesure, tandis que l’IL-1β a été principalement sécrétée plus tard. En résumé, l’analyse présentée ici a permis d’identifier différentes sous-populations sécrétrices et des dynamiques complexes de cosécrétion de cytokines.

Figure 5 : Analyse détaillée de différents modèles dynamiques de sécrétion pour les courbes de cellules sécrétant uniques d’IL-6. (A) Données représentatives de concentration de cytokines sur cellule unique sur le temps de mesure avec la courbe sigmoïde ajustée et les paramètres extraits. (B-D) Trois courbes exemplaires de concentration de cytokines unicellulaires pour les différents types de sécréteurs de cytokines trouvés pour la sécrétion d’IL-6 après stimulation LPS. (E-G) Pourcentages de cellules sécrétrices d’IL-6 qui sont classées dans les différents types de sécréteurs avec les critères suivants (n=633) : E. Cmax : faible <5 nM, élevé >19,5 nM, F. tdébut : précoce <30 min, fin >120 min, G. SRlin : lent <250 molécules/s, rapide >750 molécules/s. (H-J) Relation entre les trois descripteurs de la courbe de sécrétion Cmax, tstart et SRlin pour chaque cellule individuelle (n = 633). La grande population à Cmax=20nM résulte de l’atteinte de la limite supérieure de détection du test. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 6 : Extraction des modèles de cosécrétion à partir de courbes de concentration de cellules uniques. (A-B) Courbes de concentration représentatives pour les cellules uniques co-sécrétant l’IL-6 et le TNFα (A) simultanément et (B) séquentiellement, respectivement. (C-D) Pourcentage de cellules présentant une cosécrétion simultanée et séquentielle d’IL-6 et de TNFα (n = 249), ou d’IL-6 et d’IL-1β (n = 72), respectivement. La sécrétion séquentielle est définie par le délai de plus de 60 minutes entre les débuts de la sécrétion de cytokines. Les couleurs indiquent quelles cytokines ont commencé à sécréter en premier. (E-F) Relation entre les heures de début de sécrétion des différentes cytokines pour chaque cellule sécrétrice (nIL6-TNFα=249, nIL6-IL1β=72). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Discussion
La libération et la sécrétion de cytokines sont fréquemment étudiées en immunologie et en médecine clinique3. Une sécrétion déséquilibrée de cytokines peut entraîner des effets néfastes pour les patients souffrant d’infections, mais aussi de maladies neurologiques, d’inflammation ou de cancer 26,27,28. Même si leur importance dans la santé et la maladie est bien établie, l’étude des cytokines et de leurs cellules sécrétrices reste difficile, car les méthodologies actuelles ne sont pas capables de détecter et de quantifier avec précision les cytokines provenant d’une seule cellule de manière résolue dans le temps. Pour le flux de travail présenté ici, un protocole de stimulation établi avec des PBMC a été utilisé et leur sécrétion d’IL-6, de TNF-α et d’IL-1β a été mesurée. Le choix d’utiliser des PBMC plutôt que des sous-populations individuelles purifiées découle de la demande précédente visant à étudier les syndromes de libération de cytokines (CRS)23, une affection caractérisée par des concentrations plasmatiques très élevées de cytokines pro-inflammatoires, notamment l’IL-6, le TNF-α et l’IL-1β29. Comme le SRC n’est généralement pas lié à une seule population, nous avons utilisé des PBMC car ils seraient présents in vivo. Cependant, les sous-populations cellulaires peuvent être purifiées et évaluées individuellement, si la question scientifique l’exige. Le temps d’incubation, les conditions de stimulation et les plages de dosage dynamique ont été optimisés pour mesurer la sécrétion des trois cytokines d’intérêt. Le flux de travail et les données présentés ici montrent comment configurer, calibrer, quantifier, mesurer et analyser la sécrétion unicellulaire résolue en temps de plusieurs cytokines. Ce protocole fournit un plan sur la façon dont l’analyse multifonctionnelle de la sécrétion de cytokines pourrait permettre la grande diversité fonctionnelle et dynamique des cytokines sécrétées chez les patients.
Plusieurs aspects cruciaux du protocole de test décrit permettent une lecture biologique unique. Tout d’abord, l’encapsulation d’une seule cellule dans des gouttelettes microfluidiques a permis d’extraire des données pour chaque cellule individuelle. Les événements de plusieurs encapsulations de cellules peuvent être détectés et triés par l’analyse d’image, en fonction de la question de recherche. Deuxièmement, l’inclusion de plusieurs immunoessais fluorescents indépendants en gouttelettes et l’alignement des nanoparticules fonctionnalisées ont permis de mesurer quantitativement jusqu’à trois concentrations de cytokines en parallèle. Ce multiplexage a permis d’analyser les modèles de cosécrétion de cytokines au niveau d’une seule cellule. Troisièmement, l’immobilisation des gouttelettes a permis de mesurer et de corréler dans le temps la sécrétion de cytokines pour chaque cellule sécrétrice et a permis de distinguer la sécrétion co-occurrente de la sécrétion séquentielle. La résolution temporelle a fourni des données uniques sur les modèles de sécrétion et les sous-populations de différents types de sécréteurs. Enfin, l’analyse d’images parallélisées a permis d’extraire et de suivre efficacement de grandes quantités de données à partir de mesures portant sur plus de 20 000 cellules individuelles. L’extraction à partir de courbes de sécrétion uniques a permis de découvrir des sous-populations et des fonctionnalités phénotypiques.
Outre sa lecture unique, le test présente également des avantages techniques par rapport à l’analyse standard des cytokines. Grâce à la petite taille des compartiments d’encapsulation d’environ 60 pL, des quantités absolues de cytokines sécrétées peuvent être détectées directement à partir de la source biologique, avec des limites de détection adaptées à la sécrétion cellulaire. La miniaturisation du test utilise également de plus petites quantités de bio-réactifs coûteux. De plus, la configuration nécessite très peu d’équipement spécialisé, qui est souvent déjà disponible dans les laboratoires de biologie et de bio-ingénierie. Les microscopes à fluorescence sont largement disponibles, et les pousse-seringues sont fréquemment utilisés dans les laboratoires de bio-ingénierie ou peuvent être achetés à un coût relativement faible. S’il s’agit d’une culture cellulaire, le coût de l’équipement complet nécessaire au déroulement des expériences est d’environ 148 000 euros, la majorité étant fournie par le microscope épifluorescent automatisé (130 000 euros). Cependant, un tel instrument peut souvent être trouvé dans les laboratoires biologiques, et le reste du coût est distribué à la pompe à seringue (13 000 euros, mais des alternatives moins chères sont disponibles) et à des équipements plus petits. La fabrication de la puce à gouttelettes et de la chambre d’observation est très bien décrite17 et peut être réalisée en dehors d’un environnement de salle blanche avec l’infrastructure nécessaire, telle que les fours et les nettoyeurs plasma présents dans la plupart des laboratoires de bio-ingénierie. Alternativement, différents fournisseurs sont disponibles pour fournir aux laboratoires intéressés des puces génératrices de gouttelettes. En raison des petits volumes nécessaires, le test est rentable et simple à mettre en place.
Afin d’assurer le plus haut degré de reproductibilité, nous avons identifié certaines étapes critiques pour le succès du protocole. Un problème courant pour les nouveaux utilisateurs est le mouvement des gouttelettes pendant la mesure. Bien que le logiciel d’analyse puisse suivre les gouttelettes individuelles dans une certaine mesure, un mouvement excessif entraîne une perte de résolution de cellule unique et des résultats inexacts. Il est possible d’éviter les mouvements en utilisant des chambres de mesure correctement étanches à l’air, une taille de gouttelette et de chambre correctes, une courte période d’équilibrage avant de commencer la mesure et une concentration appropriée de tensioactif. Une autre étape critique est la mise au point précise avant de commencer la mesure. Une mise au point incorrecte entraîne une baisse significative des valeurs de relocalisation de la fluorescence et une sous-estimation de la quantité de cytokines sécrétées. Enfin, en fonction de la question et du protocole à l’étude, le bon timing entre les différentes étapes est de la plus haute importance pour la reproductibilité. En particulier, le temps d’attente entre le remplissage de la chambre et le début de la mesure doit être cohérent, sinon la fenêtre de mesure des cytokines sécrétées pourrait être manquée.
Les limites de la technologie présentée comprennent la capacité limitée de manipuler davantage les cellules après encapsulation. Il n’est donc actuellement pas possible d’ajouter ou de supprimer des stimulants, des anticorps ou des réactifs supplémentaires. De plus, comme les cellules sont encapsulées dans leur bioréacteur isolé, aucune interaction entre les cellules (signalisation par contact ou paracrine) ne peut avoir lieu pendant la mesure. Cette limitation peut être partiellement surmontée par des incubations en vrac au préalable. En outre, des effets autocrines accrus des cytokines sécrétées sont également possibles et ces effets ne peuvent pas être quantifiés ou exclus avec certitude, car seules les cytokines sécrétées détectées par des anticorps sont mesurées. Ainsi, la vue isolée de la sécrétion de cytokines doit toujours être décrite dans le contexte de la question et de l’application correspondantes. Cependant, cette limitation pourrait également être utilisée pour l’étude détaillée des multiples, des doublets et des triplets encapsulés si cela vous intéresse. Cela fournirait une configuration intéressante utile pour étudier le contact cellule-cellule ou les questions basées sur le paracrine. Enfin, la plage dynamique du test est également limitée et doit être adaptée à l’application spécifique. Ici, nous avons adapté la plage dynamique du test à la quantité sécrétée attendue des cytokines mesurées.
Afin de faire progresser les capacités et l’applicabilité du test, plusieurs développements pourraient être abordés à l’avenir, dans les aspects biologiques, techniques et d’analyse des données. Sur le plan biologique, la mesure de cytokines supplémentaires, d’autres protéines sécrétées, de marqueurs métaboliques ou de surface cellulaire pourrait être intégrée en adaptant le dosage. De plus, ce test pourrait être intégré dans un flux de travail aux côtés d’autres tests cellulaires afin d’élargir les lectures (par exemple, la coloration ou le séquençage par cytométrie en flux). De plus, la facilité d’utilisation du test pourrait être simplifiée, par exemple en créant une puce microfluidique intégrée pour la création et l’observation de gouttelettes, permettant ainsi une application plus large en dehors des laboratoires de bio-ingénierie dans un cadre clinique. En ce qui concerne l’analyse des données, l’extraction et le suivi d’informations à partir d’images pourraient être étendus en améliorant l’automatisation et en utilisant des approches d’apprentissage automatique, par exemple, pour détecter la présence et la position de la ou des cellules et de la ligne de perle dans chaque gouttelette sans marquage fluorescent. Cela ouvrirait des canaux fluorescents supplémentaires qui pourraient être utilisés pour les immunoessais, ce qui permettrait de mesurer encore plus de cytokines en parallèle.
Le test présenté et les protocoles et analyses associés peuvent être appliqués à divers cas d’utilisation potentiels liés à la dynamique de sécrétion de cytokines. Plus précisément, le test pourrait potentiellement aborder des questions immunologiques fondamentales telles que l’identification des profils de sécrétion de cytokines spécifiques au type de cellule et à l’activation, la polyfonctionnalité des cellules sécrétant des cytokines ou la temporalité et les mécanismes de maintien des équilibres de cytokines. De plus, dans les applications cliniques, la plateforme pourrait permettre de démêler le rôle des cytokines lors des réponses inflammatoires actives ou chroniques, comme observé dans le COVID-1930, ou fournir un outil pour stratifier les patients et personnaliser les traitements en fonction de signatures uniques comme dans l’auto-inflammation31. En conclusion, l’évaluation quantitative résolue en temps de la sécrétion de cytokines à partir de cellules uniques est une méthode indispensable car elle permet d’élucider comment un médicament particulier, une infection, une altération génétique ou une stimulation ex vivo induit une réponse particulière.
Déclarations de divulgation
Des aspects spécifiques tels que les mesures de la ligne de perle des cellules ont été brevetés.
Remerciements
Ce projet a été soutenu par la subvention #2021-349 du domaine d’intervention stratégique Santé personnalisée et technologies connexes (PHRT) du Domaine des EPF (écoles polytechniques fédérales suisses), la subvention de démarrage du Conseil européen de la recherche (subvention #803,336) et le Fonds national suisse de la recherche scientifique (subvention #310030_197619). De plus, nous remercions Guilhem Chenon et Jean Baudry pour leur travail et le développement de l’analyseur initial DropMap.
matériels
| Name | Company | Catalog Number | Comments |
| 008-FluoroSurfactant | RAN Biotechnologies | 008-FluoroSurfactant-10G | |
| 2-Stream flow-focusing droplet maker, 30 µm nozzle, PFOS hydrophobic surface treatment | Wunderli chips | ||
| Alexa Fluor 647 NHS Ester | ThermoFisher | A20006 | https://www.thermofisher.com/ch/en/home/references/protocols/cell-and-tissue-analysis/labeling-chemistry-protocols/fluorescent-amine-reactive-alexa-fluor-dye-labeling-of-igm-antibodies.html |
| Anti-Human IL-1β (Monoclonal Mouse), AF647 labelled in-house | PeproTech | 500-M01B | |
| ARcare92524 double-sided adhesive tape | Adhesvies Reasearch | ARcare92524 | |
| Bio-Adembeads Streptavidin plus 300nm | Ademtech | Cat#03233 | |
| Biotinylated Goat Anti-Human IL-1β | PeproTech | 500-P21BGBT | |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A3059 | |
| Cell Scraper | TPP | 99002 | |
| CellTrace Violet Cell Proliferation Kit | Invitrogen | C34557 | Cell staining solution |
| Chromafil Xtra PTFE-45/25 syringe filters | Macherey-Nagel | 729205 | |
| Costar 6-well Clear Flat Bottom Ultra-Low Attachment | Corning | 3471 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10283 | |
| D-Biotin | Fluorochem | M02926 | |
| DPBS, no calcium, no magnesium | Gibco | 14196-094 | |
| epT.I.P.S. Standard 2-200 µl | Eppendorf | 30000889 | |
| Ethylenediaminetetraacetic acid disodium salt solution | Sigma-Aldrich | 3690 | |
| EZ-LINK-NHS-PEG4-Biotin | ThermoFisher | A39259 | https://www.thermofisher.com/order/catalog/product/20217 |
| FcR Blocking Reagent, human | Miltenyi Biotec | 130-059-901 | |
| Fetal Bovine Serum | Gibco | 10270-106 | |
| Handy dish soap | Migros | 5.01002E+11 | |
| HEPES (1 M) | Gibco | 15630-080 | |
| HFE-7500 Oil 3M TM Novec | Fluorochem | B40045191 | |
| Idex F-120 Fingertight One-Piece Fitting, Standard Knurl, Natural PEEK, 1/16" OD Tubing, 10-32 Coned | Cole-Parmer | GZ-02014-15 | |
| IL-6 Monoclonal Antibody (MQ2-13A5 - Rat), FITC | ThermoFisher | 11-7069-81 | |
| IL-6 Monoclonal Antibody (MQ2-39C3), Biotin | ThermoFisher | 13-7068-85 | |
| KnockOut Serum Replacement | ThermoFisher | 10828-010 | |
| Loctite AA 3491 curable UV glue | Henkel AG & Co | 3491 | |
| Microscope slides (76x26x1mm, clear white) | Menzel Gläser | ||
| Mineral oil light | Sigma-Aldrich | 330779 | |
| NanoPort Assembly Headless, 10-32 Coned, for 1/16" OD | Idex | N-333 | |
| Neodymium block magnet | K&J Magnetics | BZX082 | |
| Omnifix-F Spritze, 1 ml, LS | Braun | 9161406V | |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | 15140-122 | |
| Phosphate buffered saline | Sigma-Aldrich | P4417 | |
| Pluronic F-127, 0.2 µm filtered (10% Solution in Water) | ThermoFisher | P6866 | |
| Precision wipes | Kimtech Science | 5511 | |
| PTFE microtubing 0.30 × 0.76 mm | FisherScientific | 1191-9445 | |
| PTFE microtubing 0.56 × 1.07 mm | FisherScientific | 1192-9445 | |
| Recombinant Human IL-1β | Peprotech | Cat#200-01B | |
| Recombinant Human IL-6 | Peprotech | Cat#200-06 | |
| Recombinant human serum albumine (HSA) | Sigma-Aldrich | A9731 | |
| Recombinant Human TNF-α | Peprotech | Cat#300-01A | |
| Reusable biopsy punch diameter 0.75 mm and 6 mm | Stiefel | 504529 and 504532 | |
| RPMI 1640 Medium, no phenol red | Gibco | 11835-030 | |
| Standard LPS, E. coli K12 | InvivoGen | tlrl-eklps | |
| Sterican needles 23 G for 0.56 mm diameter microtubing | FisherScientific | 15351547 | |
| Sterican needles 27 G for 0.30mm diameter microtubing | FisherScientific | 15341557 | |
| TNF alpha Monoclonal Antibody (MAb11), PE | ThermoFisher | 12-7349-81 | |
| TNF-alpha Monoclonal Antibody (MAb1), biotinylated in-house | ThermoFisher | 14-7348-85 | |
| Trypan Blue Stain (0.4%) for use with the Countess Automated Cell Counter | Invitrogen | T10282 | |
| Vacuum Filtration "rapid"-Filtermax | TPP | 99500 | |
| Devices | |||
| Cameo 4 automatic cutting machine | Silhouette | ||
| Cetoni Base 120 + 3x NEMESYS Low Pressure Syringe Pumps | Cetoni | NEM-B101-03 A | |
| Countess II Automated Cell Counter | ThermoFisher | ||
| Inverted Epi-fluorescence microscope Ti2 | Nikon | ECLIPSE Ti2-E, Ti2-E/B*1 | |
| OKO Lab Cage Incubator, dark panels | OKO Lab | ||
| ORCA-Fusion Digital CMOS camera | Hammatsu | C14440 | |
| SOLA Light Engine | Lumencor | sola 80-10247 |
Références
- Chen, L., et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 9 (6), 7204-7218 (2017).
- Cicchese, J. M., et al. Dynamic balance of pro- and anti-inflammatory signals controls disease and limits pathology. Immunol Rev. 285 (1), 147-167 (2018).
- Liu, C., et al. Cytokines: From clinical significance to quantification. Adv Sci. 8 (15), e2004433 (2021).
- Rojas, J. M., Avia, M., Martín, V., Sevilla, N. IL-10: A multifunctional cytokine in viral infections. J Immunol Res. 2017, 6104054 (2017).
- Kohanawa, Y. M. A regulatory effect of the balance between TNF-α and IL-6 in the granulomatous and inflammatory response to Rhodococcus aurantiacus infection in mice. J Immunol. 177 (1), 642-650 (2006).
- Geginat, J., et al. Plasticity of human CD4 T cell subsets. Front Immunol. 5, 630 (2014).
- Sallusto, F. Heterogeneity of human CD4+ T cells against microbes. Ann Rev Immunol. 34 (1), 317-334 (2016).
- Chetaille Nézondet, A. L., Poubelle, P. E., Pelletier, M. The evaluation of cytokines to help establish diagnosis and guide treatment of autoinflammatory and autoimmune diseases. J Leukocyte Biol. 108 (2), 647-657 (2020).
- Sims, J. T., et al. Characterization of the cytokine storm reflects hyperinflammatory endothelial dysfunction in COVID-19. J Allergy Clin Immunol. 147 (1), 107-111 (2021).
- Yasen, A., et al. Single-cell RNA sequencing reveals the heterogeneity of infiltrating immune cell profiles in the hepatic cystic echinococcosis microenvironment. Infection and Immunity. 89 (12), (2021).
- Jiang, Y., et al. Single-cell RNA sequencing highlights intratumor heterogeneity and intercellular network featured in adamantinomatous craniopharyngioma. Sci Adv. 9 (15), (2023).
- Tanguay, S., Killion, J. J. Direct comparison of ELISPOT and ELISA-based assays for detection of individual cytokine-secreting cells. Lymphokine Cytokine Res. 13 (4), 259-263 (1994).
- Bucheli, O. T. M., Sigvaldadóttir, I., Eyer, K. Measuring single-cell protein secretion in immunology: Technologies, advances, and applications. Eur J Immunol. 51 (6), 1334-1347 (2021).
- Brower, K. K., et al. Double emulsion flow cytometry with high-throughput single droplet isolation and nucleic acid recovery. Lab Chip. 20 (12), 2062-2074 (2020).
- Brower, K. K., et al. Double emulsion picoreactors for high-throughput single-cell encapsulation and phenotyping via FACS. Anal Chem. 92 (19), 13262-13270 (2020).
- Luo, X., Chen, J. Y., Ataei, M., Lee, A. Microfluidic compartmentalization platforms for single cell analysis. Biosensors. 12 (2), 58 (2022).
- Bounab, Y., et al. Dynamic single-cell phenotyping of immune cells using the microfluidic platform DropMap. Nat Protoc. 15 (9), 2920-2955 (2020).
- Eyer, K., et al. Single-cell deep phenotyping of IgG-secreting cells for high-resolution immune monitoring. Nat Biotechnol. 35 (10), 977-982 (2017).
- Gaa, R., et al. Versatile and rapid microfluidics-assisted antibody discovery. mAbs. 13 (1), 198130 (2021).
- Gerard, A., et al. High-throughput single-cell activity-based screening and sequencing of antibodies using droplet microfluidics. Nat Biotechnol. 38 (6), 715-721 (2020).
- De Jonghe, J., et al. spinDrop: a droplet microfluidic platform to maximise single-cell sequencing information content. Nat Comm. 14 (1), 4788 (2023).
- Wheeler, M. A., et al. Droplet-based forward genetic screening of astrocyte-microglia cross-talk. Science. 379 (6636), 1023-1030 (2023).
- Portmann, K., Linder, A., Oelgarth, N., Eyer, K. Single-cell deep phenotyping of cytokine release unmasks stimulation-specific biological signatures and distinct secretion dynamics. Cell Rep Meth. 3 (7), 100502 (2023).
- Portmann, K., Linder, A., Eyer, K. Stimulation-induced cytokine polyfunctionality as a dynamic concept. eLife. 12, 89781 (2023).
- Armbruster, D. A., Pry, T. Limit of blank, limit of detection and limit of quantitation. Clin Biochem Rev. 29, S49-S52 (2008).
- Yang, J., et al. New insight into neurological degeneration: Inflammatory cytokines and blood-brain barrier. Front Mol Neurosci. 15, 1013933 (2022).
- Kim, P. S., Ahmed, R. Features of responding T cells in cancer and chronic infection. Curr Opin Immunol. 22 (2), 223-230 (2010).
- Becher, B., Spath, S., Goverman, J. Cytokine networks in neuroinflammation. Nat Rev Immunol. 17 (1), 49-59 (2017).
- Cosenza, M., Sacchi, S., Pozzi, S. Cytokine release syndrome associated with T-cell-based therapies for hematological malignancies: Pathophysiology, clinical presentation, and treatment. Int J Mol Sci. 22 (14), 7652 (2021).
- Hu, B., Huang, S., Yin, L. The cytokine storm and COVID-19. J Med Virol. 93 (1), 250-256 (2021).
- Marcuzzi, A., et al. Autoinflammatory diseases and cytokine storms-Imbalances of innate and adaptative immunity. Int J Mol Sci. 22 (20), 11241 (2021).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.