Method Article
Calcio dei periciti cerebrali e imaging emodinamico nei topi transgenici in vivo
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Questo protocollo presenta passaggi per acquisire e analizzare immagini fluorescenti di calcio da periciti che riscaldano il cervello e dati sul flusso sanguigno dai vasi sanguigni vicini in topi anestetizzati. Queste tecniche sono utili per gli studi di fisiologia delle cellule murali e possono essere adattate per studiare i transitori di calcio in qualsiasi tipo di cellula.
Abstract
I recenti progressi nella biologia delle proteine e nella genetica dei topi hanno permesso di misurare le fluttuazioni intracellulari del calcio delle cellule cerebrali in vivo e di correlarle con l'emodinamica locale. Questo protocollo utilizza topi transgenici che sono stati preparati con una finestra cranica cronica ed esprimono l'indicatore di calcio geneticamente codificato, RCaMP1.07, sotto il promotore dell'actina muscolare α-liscia per etichettare specificamente le cellule murali, come le cellule muscolari lisce vascolari e i periciti che riscaldano. I passaggi sono delineati su come preparare un catetere della vena della coda per l'iniezione endovenosa di coloranti fluorescenti per tracciare il flusso sanguigno, nonché su come misurare il calcio dei periciti cerebrali e l'emodinamica dei vasi sanguigni locali (diametro, velocità dei globuli rossi, ecc.) mediante due microscopi a fotoni in vivo attraverso la finestra cranica in topi anestetizzati con ketamina / xilazina. Infine, vengono forniti dettagli per l'analisi delle fluttuazioni del calcio e dei filmati del flusso sanguigno tramite gli algoritmi di elaborazione delle immagini sviluppati da Barrett et al. 2018, con particolare attenzione a come questi processi possono essere adattati ad altri dati di imaging cellulare.
Introduzione
La vascolarizzazione del sistema nervoso centrale è costituita da arteriole penetranti, capillari e venule ascendenti. All'interno di questa rete, le cellule murali come le cellule muscolari lisce vascolari encase arteriole e periciti estendono i processi cellulari lungo i primi rami e capillari arterioli1. I periciti sembrano avere diversi ruoli all'interno del cervello tra cui il mantenimento della barriera emato-encefalica1,2,la migrazione e la motilità3,le potenziali proprietà delle cellule staminali e la regolazione del flusso sanguigno cerebrale4,5,6. Molti dei ruoli funzionali dei periciti sono stati collegati a fluttuazioni del calcio intracellulare che possono regolare la dilatazione o la contrazione di queste cellule4,5,6.
Diversi studi recenti hanno stabilito criteri per identificare diversi tipi di periciti cerebrali7,8. Le cellule murali all'interno dei primi 4 rami delle arteriole penetranti sono periciti avvolgenti in base alla loro espressione della proteina contrattile α-liscia dell'actina muscolare (αSMA) e del loro somata ovoidale sporgente con processi che avvolgono i vasi7,8,9. Per visualizzare le fluttuazioni del calcio nei periciti ensheathing, questo protocollo utilizza una nuova linea di topo transgenico, Acta2-RCaMP1.07, nota anche come Tg(RP23-370F21-RCaMP1.07)B3-3Mik/J10. Questi topi esprimono l'indicatore rosso del calcio geneticamente codificato, RCaMP1.07, nelle cellule che esprimono αSMA (cellule muscolari lisce vascolari e periciti che si riscaldano). Le colonie riproduttive vengono mantenute incrociando animali non aerei con emizigoti. RCaMP1.07 è una proteina fluorescente rossa con un dominio di legame calmodulina, che aumenta la fluorescenza quando si lega al calcio intracellulare10,11. Questo protocollo delinea i passaggi per l'imaging combinato del calcio dei periciti insanguinati e le misurazioni del flusso sanguigno mediante microscopia a due fotoni, comprese le procedure per l'iniezione della vena caudale di coloranti fluorescenti, l'acquisizione di immagini al microscopio in topi anestetizzati e l'analisi dei dati con piattaforme di programmazione (Figura 1). Queste tecniche sono utili per affrontare domande sulla fisiologia delle cellule murali, ma possono essere adattate per studiare i transitori di calcio in qualsiasi tipo di cellula nel cervello o in altri sistemi di organi.
Un topo Acta2-RCaMP1.07 femmina di 10 mesi è stato utilizzato per l'esperimento presentato in questo articolo. Il topo è stato sottoposto a un intervento chirurgico per la finestra cranica cronica e l'impianto post testa due mesi prima. I dettagli per il protocollo chirurgico sono discussi in studi precedenti12,13 e procedure simili sono state eseguite in altri protocolli precedentemente pubblicati14,15. La vascolarizzazione è etichettata con fluoresceina-destrano verde (70.000 MW, soluzione anionica, 2,5% p/v) iniettata per via endovenosa. Questo colorante è economico e facilmente disponibile da fonti commerciali, ma ha uno spettro di emissione più ampio che può sovrapporsi all'emissione RCaMP e sanguinare durante l'acquisizione di immagini al microscopio. I passaggi per l'unmixing spettrale sono descritti nella Sezione 4 di seguito per aggirare questo, ma possono essere utilizzati anche altri coloranti verdi con spettri di emissione più stretti, come quelli basati su EGFP.
Protocollo
Tutte le procedure che coinvolgono animali da esperimento descritte di seguito sono state approvate dal Comitato per la cura degli animali dell'Università di Manitoba, che è governato dal Canadian Council on Animal Care.
1. Impostazione e preparazione della procedura
NOTA: I seguenti elementi sono necessari per l'iniezione di catetere della vena della coda: siringhe da insulina, un pezzo di tubo PE10 da 15 cm, aghi da 30 G, garza, soluzione salina, pinza, colorante destrone alla fluoresceina verde, pinze e forbici. Inoltre, avere un ago pronto con ketamina / xilazina anestesia che verrà iniettato prima della sessione di imaging.
CRITICO: Tutti i materiali e le attrezzature nelle fasi 1 e 2 devono essere sterilizzati prima dell'uso in autoclave o risciacquo con etanolo al 70%. Se le pinze non possono essere adeguatamente sterilizzate, si raccomanda l'uso di un paio di portaaghi di grandi dimensioni. Il montaggio del catetere deve essere fatto con pinze e pinze per evitare punture accidentali dell'ago.
- Taglio di circa 15-20 cm di tubi in polietilene, PE10 (I.D. 28 mm; O.D. 61 mm).
- Riempire una siringa da insulina da 27 G con soluzione salina allo 0,9% e allacciare l'ago della siringa nella punta del tubo di polietilene. Spingere la soluzione salina attraverso il tubo, assicurandosi che non ci siano perdite.
- Usando le pinze, piegare un ago da 30 G (0,3 mm x 25 mm) avanti e indietro fino a quando non si rompe dal mozzo. L'ago deve essere pulito senza pieghe.
- Tenendo l'ago con una pinna, inserire con attenzione l'ago nell'estremità del tubo PE10 che è attaccato alla siringa riempita di soluzione salina e rimuovere le bolle d'aria. Questo è il catetere per iniezione.
- Filtrare 30 μL di 2,5% (p/v) fluoresceina destrano attraverso un filtro da 13-25 μm prima dell'iniezione.
- Riempire un'altra siringa da insulina con l'aliquota di 30 μL del destro e assicurarsi che non vi siano bolle nella siringa riempita.
2. Iniezione della vena della coda
- Anestetizzare il topo con isoflurano (4% di induzione, 1,5% di mantenimento) o ketamina/xilazina (60 mg/kg; 10 mg/kg; i.p.) e applicare gel lubrificante per gli occhi. Ketamina/xilazina è raccomandata per misurazioni del flusso sanguigno più stabili durante l'imaging e la dose può essere aumentata a 90 mg/kg; 10 mg/kg di ketamina/xilazina per sessioni di imaging più lunghe.
- Quando il topo è sul piano chirurgico dell'anestesia, posizionare un guanto pieno di acqua tiepida sulla coda per dilatare la vena laterale.
- Rimuovere il guanto dopo 30 s e pulire la coda con etanolo.
- Posiziona la coda tra il pollice e il dito medio. Fornire pressione con l'indice sulla coda per dilatare la vena. Con l'altra mano, raccogliere l'ago del catetere con una pinetta orientando la lunetta verso l'alto verso il soffitto.
- Dopo aver pulito la coda con etanolo al 70%, inserire agevolmente l'ago nella vena con un angolo di 0 ° e iniettare delicatamente soluzione salina attraverso il catetere per garantire che l'ago sia posizionato correttamente.
NOTA: Se non c'è resistenza sullo stantuffo e nessun gonfiore della coda, allora l'ago è nella vena. Se c'è una resistenza o gonfiore significativi, l'ago deve essere rimosso. I passaggi 2.2-2.5 possono essere ripetuti fino a 3 volte su ciascun lato della coda, sostituendo l'ago 30G alla fine del catetere ogni secondo prova fino a quando il posizionamento non è corretto. - Una volta che l'ago è nella vena, commutare la siringa salina all'estremità del catetere con la siringa contenente fluoresceina destrano (Fase 1.6). Iniettare lentamente il destro nel tubo del catetere, assicurandosi che non entrino bolle nel tubo. Se è evidente una bolla d'aria, tagliare il tubo contenente la bolla per rimuoverlo e riattaccare la siringa.
- Quando tutto il destrano (aliquota 30 μL) è stato iniettato, rimuovere la siringa e sostituirla con la siringa salina. Iniettare il destro residuo dal tubo nel mouse fino a quando non rimane alcun colorante nel tubo.
- Rimuovere l'ago dalla coda e fornire pressione con una garza per 10-30 s fino a quando il sanguinamento si ferma.
CRITICO: Se dopo 6 tentativi l'iniezione della vena della coda non ha successo, l'animale deve essere ripreso in un'altra sessione. Inoltre, il volume totale (soluzione salina e destrano) iniettato nel topo non deve superare i 100 μL.
3. Microscopia a due fotoni
- Concentrarsi sulla finestra cranica
NOTA: Utilizzare l'anestesia ketamina/xilazina durante l'acquisizione dei dati perché ha meno effetti vascolari (vasodilatazione) rispetto all'isoflurano. Se si utilizza isoflurano nei passaggi precedenti, iniettare nel mouse ketamina/xilazina i.p. (dose raccomandata sopra descritta) prima dell'imaging.- Fissare il mouse con una vite attraverso il suo palo di testa a una piattaforma con una piastra riscaldante sotto il microscopio.
- Applicare lubrificante per gli occhi sugli occhi del mouse.
- Pulire la finestra cranica con applicatori dentali umidi. Assicurarsi che non rimangano particelle che potrebbero interferire con il processo di imaging.
- Applicare il gel ad ultrasuoni sulla finestra.
- Metti a fuoco attraverso l'obiettivo del microscopio a due fotone fino a quando i vasi sanguigni piali possono essere visti sotto la finestra.
- Controllare la respirazione del mouse e assicurarsi che la piastra riscaldante fornisca un supporto di temperatura sufficiente.
- Acquisizione di immagini
NOTA: Il microscopio a due fotone utilizzato in questo esperimento ha un laser Ti-Sapphire sintonizzabile per l'eccitazione della fluorescenza con una cella Pockel che controlla la quantità di laser che raggiunge il campione. La luce emessa viene divisa da un dicroico a passaggio lungo 565 a due tubi fotomoltiplicatori GaAsP (PMT) con filtro passa banda 595/50 (rosso) e filtro passa banda 525/70 (verde) per il rilevamento.
Le procedure descritte nei passaggi da 3.2 a 3.4 vengono eseguite utilizzando software specifici del microscopio a due fotone di questo protocollo (vedi Tabella dei materiali). Questi passaggi possono essere adattati ad altri software e apparecchiature per microscopi.- Con le luci della stanza spente, impostare la lunghezza d'onda desiderata nel software del microscopio su 990 nm per eccitare sia RCaMP che fluoresceina-destrano facendo clic sulla casella Laser 2-P.
- Impostare la potenza del laser facendo clic sulla sezione Potenza/Guadagno /Laser e regolando la tensione della cella Pockels 1 al 30% o un valore di 300 su una scala di 1000. La potenza laser che raggiunge il campione a questa impostazione è stata precedentemente determinata come ~ 30 mW.
- Impostare la sensibilità del rilevatore PMT facendo clic sulla sezione Power/Gain box/PMTs e regolando il valore su 700-800.
NOTA: questi valori possono essere regolati in relazione all'intensità del campione fluorescente e devono essere impostati su zero prima di accendere le luci nella stanza. - Vai alla sezione Risoluzione immagine e fai clic sulla risoluzione 512 x 512 per una dimensione dell'immagine più grande.
- Fare clic su 2-P Laser/Open per aprire l'otturatore laser 2-P.
- Vai alla sezione Scansione e fai clic sul pulsante Live Scan.
NOTA: scansione dal vivo con questi parametri e risoluzione più elevata, è possibile vedere le cellule murali positive RCaMP e il plasma sanguigno marcato fluorescentemente. Se il segnale è debole, il valore di pockel può essere aumentato fino a quando l'immagine non è chiara.
CRITICO: Negli strati di tessuto superficiale, la potenza del laser non deve superare i 50 mW, che è di circa 600 nelle impostazioni delle celle di Pockels in questo esempio.
- Acquisizione di una pila di profondità delle celle murali e della rete vascolare
NOTA: si consiglia l'acquisizione di una pila di profondità per localizzare correttamente i periciti nella rete vascolare. I periciti ensheathing si trovano sul primo al quarto ramo dall'arteriola penetrante7,8,9. Il software del microscopio utilizzato in questo protocollo si riferisce alle pile di profondità come "serie Z".- Spostando l'obiettivo del microscopio nel piano X, Y e Z, localizzare una grande arteria sulla superficie del cervello in base all'etichettatura delle cellule muscolari lisce di RCaMP.
- Fare clic sulla casella Z-Series.
- Focalizzati nella parte superiore del tessuto vicino ai vasi piali, impostalo come punto zero e parte superiore dello stack della serie Z facendo clic sulla sezione Current Z-series/ Start position [μm]. Clicca sulla casella con quattro strisce nere e una striscia rossa in alto.
- Focalizzatevi verso il basso nel tessuto alla profondità desiderata e impostatela come parte inferiore della pila facendo clic sulla sezione Current Z-series/Stop position [μm]. Fai clic sulla casella con quattro strisce nere e una striscia rossa sul fondo.
- Impostare lo spessore di ciascun piano dell'immagine (dimensione del passo) su 1-2 μm digitando il valore desiderato nella casella sotto il pulsante"Dimensione passo"(il pulsanteDimensione passo è localizzato nella sezione Serie Z/Serie Z corrente). Questo definirà il numero di immagini acquisite nello stack.
- Impostare la potenza del laser in modo che aumenti esponenzialmente man mano che il microscopio si sposta più in profondità attraverso la pila facendo clic sulla casella Compensazione laser/PMT e selezionando Relativo (gradiente esponenziale).
- Assegna un nome al file, scegli una cartella per salvarlo e fai clic su Avvia serie Z.
- Dopo l'acquisizione, aprire la serie Z nel software di elaborazione delle immagini.
- Unisci i due canali come immagini colorate e scansiona la pila alla ricerca di periciti e vasi sanguigni di interesse facendo clic nella casella Immagine | | colore Canali divisi; | immagine | colore Unisci canali.
- Selezionare le regioni di interesse (ROI) che contengono periciti e salvare le posizioni per aiutare a localizzare nuovamente questi punti nelle future sessioni di imaging.
- Acquisizione di filmati di imaging di calcio serie T (tempo)
- Utilizzando la pila di profondità e i ROI dall'alto come riferimento, spostare l'obiettivo del microscopio nell'asse X, Y e Z durante la modalità di scansione dal vivo fino a trovare un pericita di interesse.
- Per raccogliere un filmato di eventi di calcio pericitario, aumentare il frame rate di acquisizione (>10 fotogrammi al secondo) andando alla sezione Risoluzione immagine e facendo clic sulla casella 128x128.
- Impostare la durata dell'imaging su 60 s facendo clic sulla casella della serie T e inserendo l'ora nella casella della durata.
- Accanto alla casella Salva percorso, fare clic sul pulsante con tre punti per aggiornare il percorso di salvataggio con un nome di file univoco.
- Zoom ottico sul vaso per tenere conto della risoluzione inferiore e per ottenere una visione più ravvicinata del pericita regolando il valore nella sezione Zoom ottico [mag].
- Acquisire la serie T facendo clic su Avvia serie T.
- Misure emodinamiche con kymografi (scansioni di linea)
- Concentrati sulla nave di interesse con una risoluzione di 512 x 512pixel in modalità Live Scan.
- Per misurare il diametro dei vasi sanguigni e la velocità dei globuli rossi, fare clic sulla scansione della linea per avviare una scansione unidimensionale con il microscopio.
- Impostare la durata della scansione (30-60 s) in millisecondi.
- Disegna una linea che taglia in due la nave di interesse e si muove parallelamente lungo la nave. Questo genererà un chimografo del diametro del vaso a sinistra e delle striature dei globuli rossi che si muovono attraverso il vaso a destra.
NOTA: Più vasi sanguigni possono essere misurati con la stessa linea purché si trovino sullo stesso piano di imaging. - Assegna un nome al file e fai clic su Start Linescan(s) per acquisire i dati.
4. Analisi delle immagini
- Analisi del film di calcio.
NOTA: questo protocollo illustra i passaggi per l'unmixing spettrale (Figura 2) e due diversi metodi per analizzare gli eventi di calcio dei periciti che riscaldano il corpo utilizzando ROI manuali selezionati a mano (Figura 3) e la selezione automatizzata del ROI basata sulle attività (Figura 4)16,17. Al fine di rilevare e classificare i picchi di segnale con la traccia di calcio normalizzata da ciascun ROI, i dati sono filtrati long-pass e band-pass che aiutano a smussare i dati per le stime di ampiezza e larghezza e anche a identificare picchi di forme diverse: picchi singoli, multi-picchi e altipiani (Figura 3B). I parametri per questa analisi possono essere ottimizzati per rilevare diversi tipi di segnali cellulari dinamici. I passaggi seguenti richiederanno l'uso di software di elaborazione delle immagini e software di programmazione con pacchetti di elaborazione delle immagini che contengono codici diversi per analizzare i film di calcio come menzionato sopra. Si prega di consultare la tabella dei materiali per un elenco completo dei programmi e dei pacchetti utilizzati in questo protocollo. I dati di imaging di diversi tipi di microscopi possono essere importati con questi pacchetti mantenendo i metadati dalle immagini.
NOTA: i passaggi 4.1.1-4.1.7 descrivono come selezionare manualmente il ROI nel software di elaborazione delle immagini per il successivo utilizzo nel metodo di analisi manuale del calcio (passaggio 4.1.16)- Caricare la serie T di imaging del calcio nel software di elaborazione delle immagini, trascinando il file .xml sulla barra degli strumenti del software. Fare clic sulla casella OK.
- Prendi la media dello stack (la media dello stack è etichettata come "proiezione Z" dal software di elaborazione delle immagini). Questo può essere fatto facendo clic su Immagine | Stack | Proiezione Z nella barra degli strumenti.
- Creare un'immagine colorata da entrambi i canali come nel passaggio 3.3.9.
- Aprire la finestra roi manager facendo clic nella casella Analizza | Strumenti | ROI Manager,o semplicemente premendo la lettera"T"nella tastiera.
- Selezionare lo strumento poligono facendo clic sulla forma del poligono sulla barra degli strumenti del software di elaborazione delle immagini e delineare le strutture dei periciti di rivestimento visibili, come il soma e i processi.
- Fare clic sul pulsante Aggiungi situato nella finestra ROI manager per aggiungere i ROI selezionati nel ROI manager.
- Assegna a ciascuna regione di interesse un nome univoco facendo clic sul pulsante Rinomina e salvali come cartella zip che può essere caricata in un secondo momento nel software di programmazione facendo clic su Altro>> | Salva.
NOTA: I passaggi 4.1.8-4.1.14 descrivono come importare la serie T di calcio nella piattaforma di programmazione e come smisare i diversi fluorofori rilevati dal microscopio PMT in canali diversi (Figura 2). - Aprire il software di programmazione e assicurarsi che le cartelle per i pacchetti di elaborazione delle immagini siano sul percorso (vedere Tabella dei materiali).
- Importare la serie T di calcio nel software di programmazione chiamando la funzione BioFormats nella finestra di comando della piattaforma di programmazione, che apre automaticamente la finestra di selezione dei file.
- Definisci cosa c'è su ciascun canale inserendo il numero desiderato. In questi dati di esempio, Canale 1 risposta =6 (cellular_signal), Canale 2 risposta =1 (blood_plasma).
- Traccia i dati come un film all'interno del software di programmazione per facilitare la visualizzazione chiamando la funzione di trama.
- Per rimuovere la fluorescenza verde dalla fluoresceina-destrano che sanguina nel canale rosso RCaMP, deselezionare i canali nel pacchetto di elaborazione delle immagini chiamando la funzione unmix_chs nella finestra di comando della piattaforma di programmazione.
- Selezionare una regione che contiene solo fluorescenza da questo fluoroforo nel canale 1, ad esempio RCaMP in questo caso.
- Selezionare una regione che contiene solo fluorescenza dal fluoroforo 2, ad esempio la fluoresceina nel plasma sanguigno in questo esempio.
- Selezionare un'area di sfondo che non abbia fluorescenza da nessuno dei due fluorofori. Questo genera una matrice di contributo spettrale che viene applicata a ciascun pixel in ciascun canale. Migliora significativamente la localizzazione del segnale RCaMP che migliorerà il rilevamento di eventi di calcio in queste strutture.
NOTA: come accennato in precedenza, esistono diversi modi in cui i dati di imaging del calcio possono essere analizzati all'interno dei pacchetti di elaborazione delle immagini. I passaggi 4.1.16-4.1.23 descrivono il metodo per analizzare gli eventi di calcio dei periciti che riscaldano utilizzando ROI manuali selezionati a mano. - Eseguire l'analisi della segnalazione cellulare sul filmato di calcio non mixato chiamando la funzione CellScan nella finestra di comando della piattaforma di programmazione.
- Il codice chiederà "Quale metodo di rilevamento del ROI si desidera utilizzare?". Digitare il numero 2 per caricare i ROI selezionati manualmente sulla piattaforma di programmazione.
- Caricare le regioni di interesse dalla cartella zip che sono state selezionate dai periciti a mano in precedenza (Passaggio 4.1.6).
- Il codice chiederà "Qual è il fattore di scala?". Determinare il fattore di scala per i ROI selezionati manualmente rispetto alla serie di immagini analizzata e digitare il numero della scala. In questo esempio, il fattore di scala è 1 perché non è necessario ridimensionare i ROI poiché sono stati selezionati su immagini con 128x128 pixel, la stessa risoluzione del filmato di calcio originale.
- Genera grafici di ciascun ROI e delle tracce di calcio normalizzate in diversi colori (Figura 3A) chiamando le funzioni di processo e plottaggio nella finestra di comando.
- Se il codice non rileva la maggior parte degli eventi di calcio nelle singole tracce, modificare i parametri incorporati all'interno della casella di ottimizzazione della configurazione chiamando la funzione opt_config e regolando i valori, ad esempio diminuendo la soglia per i dati filtrati passa-corto a tre volte la deviazione standard del periodo di base, ovvero i primi 30 fotogrammi della serie T.
- Selezionare il pulsante Processo nella casella di ottimizzazione per applicare i nuovi parametri.
NOTA: Al fine di rilevare e classificare i segnali, la traccia di calcio normalizzata è filtrata a passaggio lungo e passaggio di banda, il che aiuta a smussare i dati per le stime di ampiezza e larghezza, ma anche a determinare se i segnali sono picchi singoli, multi-picchi o plateau (Figura 3B). - Restituisci i dati come file .csv che contiene informazioni spaziali sulle regioni di interesse e sui picchi identificati chiamando la funzione output_data nella finestra di comando. Assegnare al file un nome univoco per ulteriori analisi in un programma di statistiche.
NOTA: I passaggi 4.1.24-4.1.31 descrivono il metodo per analizzare gli eventi di calcio dei periciti che riscaldano utilizzando l'analisi dei ROI basati sull'attività. - Ripetere i passaggi 4.1.8.-4.1.16 per importare il filmato di calcio, deselezionare i canali e chiamare la funzione CellScan nella piattaforma di programmazione.
- Il codice chiederà "Quale metodo di rilevamento del ROI si desidera utilizzare?". Digitare il numero 6 per selezionare l'identificazione automatica della regione di interesse in base all'attività e al cambiamento di fluorescenza in 3 dimensioni (x, y e tempo; "Algoritmo 3D FLIKA").
- Tracciate i risultati elaborati per visualizzare le aree di interesse identificate come colori diversi chiamando le funzioni di processo e plottaggio nella finestra di comando. Ogni ROI si distingue nel tempo e nello spazio e viene rappresentato come una maschera sovrapposta (Figura 4).
- Se l'algoritmo non rileva ROI chiaramente visibili a occhio, modificare i parametri incorporati all'interno della casella di ottimizzazione chiamando la funzione opt_config e regolando i valori, ad esempio aumentando il filtro gaussiano che attenua i dati nel tempo (di 2 s) e diminuendo la soglia per trovare i ROI a 3 volte la deviazione standard della linea di base.
- Selezionare il pulsante di processo nella casella di ottimizzazione per applicare i nuovi parametri. Con il processo di ottimizzazione dovrebbero essere identificati più ROI (Figura 4B).
- Traccia il ROI come un filmato per identificare chiaramente le aree di attività (delineate nei colori dell'arcobaleno) modificando la modalità della casella predefinita in filmato all'interno della finestra di ottimizzazione per un'ulteriore visualizzazione.
- Generare i dati come file csv chiamando la funzione output_data nella finestra di comando. Questo file può essere analizzato ulteriormente in un programma di statistiche.
NOTA: I parametri di analisi possono essere regolati per adattarsi a qualsiasi tipo di segnale cellulare dinamico (calcio, rapporti FRET, ecc.). Tutti i passaggi precedenti possono essere automatizzati con un semplice codice di programmazione al fine di elaborare in batch molti film di calcio con le stesse impostazioni.
- Analisi del flusso sanguigno a scansione di linea.
- Importare il file di dati del kymograph di scansione della linea acquisito nella sezione 3.5 nel software di programmazione.
- Il codice chiederà "Cosa viene mostrato sui canali 1 e 2?". Definisci cosa c'è su ogni canale quando richiesto. In questo esempio, il canale 1 è vuoto (tipo 0) e il canale 2 è blood_plasma (tipo 1).
- Eseguire la funzione di analisi del diametro sulla scansione della linea chiamando la funzione LineScanDiam, che apre una casella per selezionare l'area corrispondente al diametro nel kymograph (Figura 5B, a sinistra).
- Disegnare una scatola al di fuori dei confini di fluorescenza del kymografo che corrisponde al diametro del vaso.
- Elaborare questa classe di dati chiamando la funzione di processo per misurare l'intera larghezza a mezzo massimo per il diametro del recipiente e generare un grafico (Figura 5C) con la funzione plot.
- Generare i dati come file csv chiamando la funzione output_data nella finestra di comando. Questo file può essere analizzato ulteriormente in un programma di statistiche.
- Eseguire l'analisi della trasformazione del radon della velocità chiamando la funzione LineScanVel, che apre una casella per selezionare l'area che corrisponde alla velocità RBC nel kymograph (Figura 5B, a destra).
- Disegna una casella all'interno del bordo della fluorescenza del kymografo che corrisponde alla velocità del vaso.
- Elaborare questa classe di dati chiamando la funzione di processo per calcolare la velocità, il flusso e la densità lineare dei globuli rossi dall'angolo delle strisce nella fluorescenza. Generare un plottaggio (Figura 5D) con la funzione plot.
- Generare i dati come file csv chiamando la funzione output_data nella finestra di comando. Questo file può essere analizzato ulteriormente in un programma di statistiche.
NOTA: I kymografi devono avere una fluorescenza chiara con bordi ben definiti tra gli spazi neri affinché l'analisi del diametro e della velocità sia accurata (Figura 5A, B). È molto importante disegnare le linee ortogonali e parallele in modo preciso, altrimenti non sarà possibile un'analisi affidabile dei chimografi. Analogamente all'analisi del calcio con gli algoritmi di elaborazione delle immagini, i parametri per i calcoli di diametro e velocità possono essere ottimizzati.
Risultati
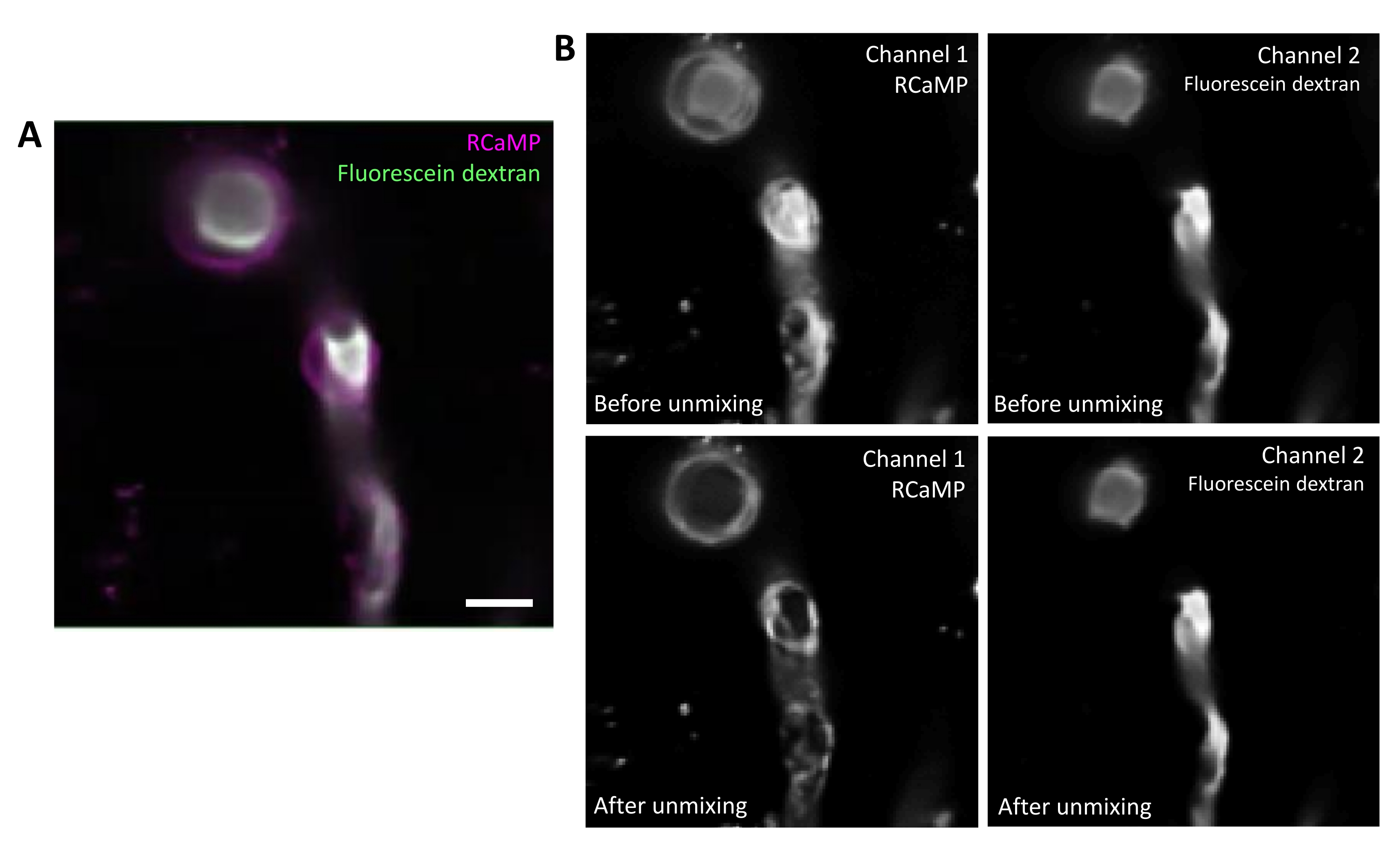
La fluoresceina-destrano ha un ampio spettro di emissione che può sanguinare attraverso il canale rosso, influenzando il rilevamento RCaMP nei periciti che rivestono (Figura 2A). Lo smixing spettrale dopo l'acquisizione dei dati nel programma software riduce il sanguinamento della fluoresceina attraverso (Figura 2B, inferiore), migliorando il rilevamento del segnale di calcio nelle successive fasi di analisi.
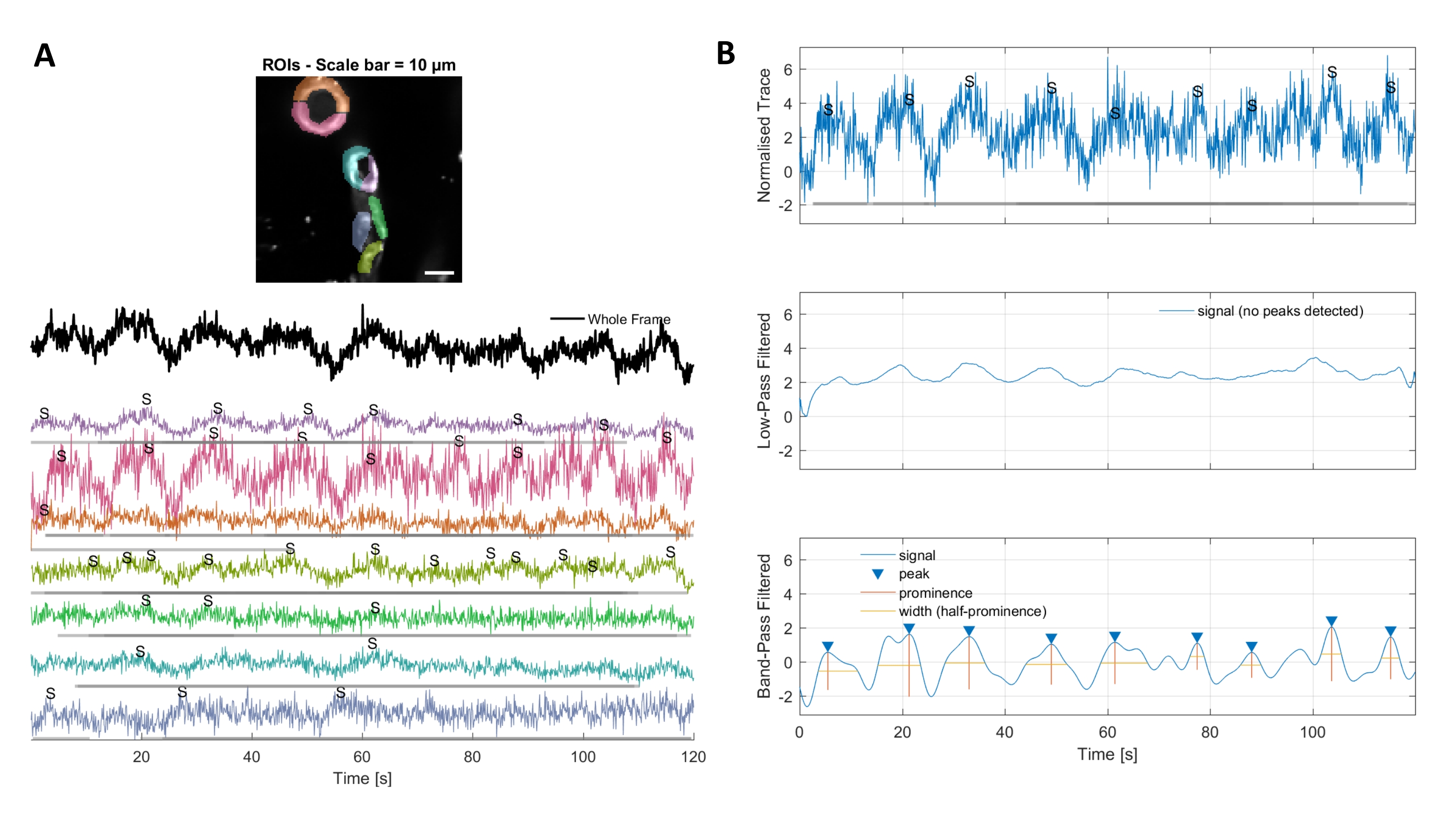
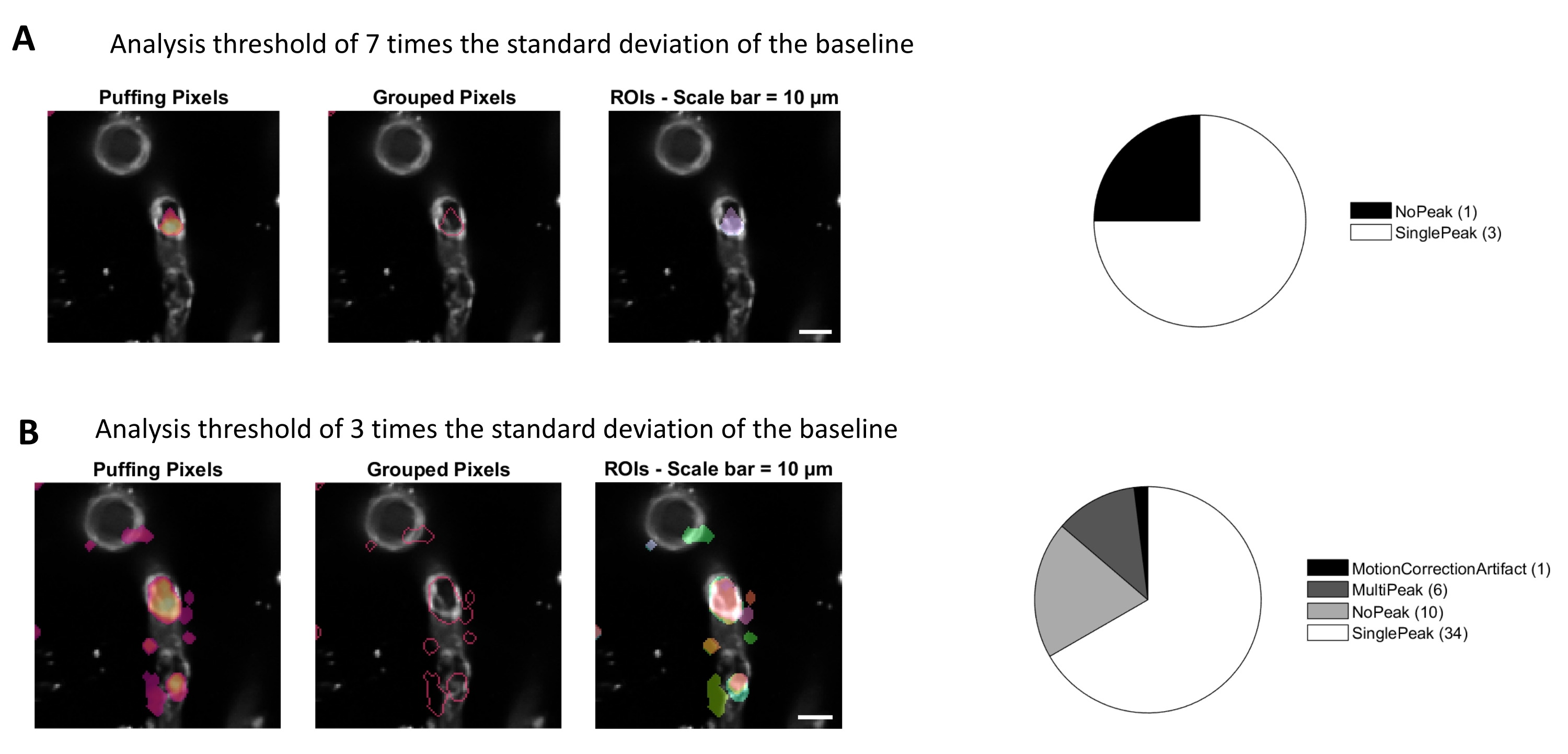
L'analisi del calcio con gli algoritmi di elaborazione delle immagini utilizzati in questo protocollo consente diversi approcci per identificare i ROI e le fluttuazioni intracellulari del calcio (cioè i segnali di calcio). La selezione manuale delle strutture cellulari consente di rilevare le fluttuazioni di calcio all'interno di queste regioni (Figura 3A), compresi diversi tipi di picchi di segnale, come picchi singoli e multi-picchi, dopo che le tracce di calcio normalizzate sono filtrate passa-basso e passa-banda (Figura 3B). Inoltre, i ROI vengono identificati raggruppando insieme i pixel attivi in cui l'intensità della fluorescenza cambia nel tempo utilizzando algoritmi di elaborazione delle immagini sviluppati da Ellefsen et al. 201416 e Barrett et al. 201817 (Figura 4). Questo può essere applicato a qualsiasi segnale cellulare dinamico regolando il tempo, la soglia e i parametri spaziali per comprendere la dimensione e la forma previste del segnale. Diminuendo la soglia per l'identificazione del segnale si trovano più regioni di interesse (Figura 4B).
Kymografi emodinamici luminosi e chiari possono essere analizzati per misurare il diametro e la velocità RBC nei vasi sanguigni vicino ai periciti che si avvolgivano (Figura 5A, B). Il diametro è calcolato a partire dall'intera larghezza a metà massimo della fluorescenza (Figura 5C). La velocità RBC è approssimata dalle strisce fatte da globuli rossi non etichettati, dove l'angolo viene immesso in una trasformazione di Radon per calcolare la velocità, il flusso (celle / s) e la densità lineare (celle / mm; Figura 5D). I kymografi di scarsa qualità in cui vi è saturazione di fluorescenza, scarso rapporto segnale/rumore o movimento del campo di imaging (Figura 6A) creano grafici inaffidabili con punti di errore (croci rosse) in cui i dati non possono essere determinati (Figura 6B, C). La qualità dei dati acquisiti è fondamentale per un buon esito e seguire i passaggi descritti in questo protocollo garantisce buoni risultati.

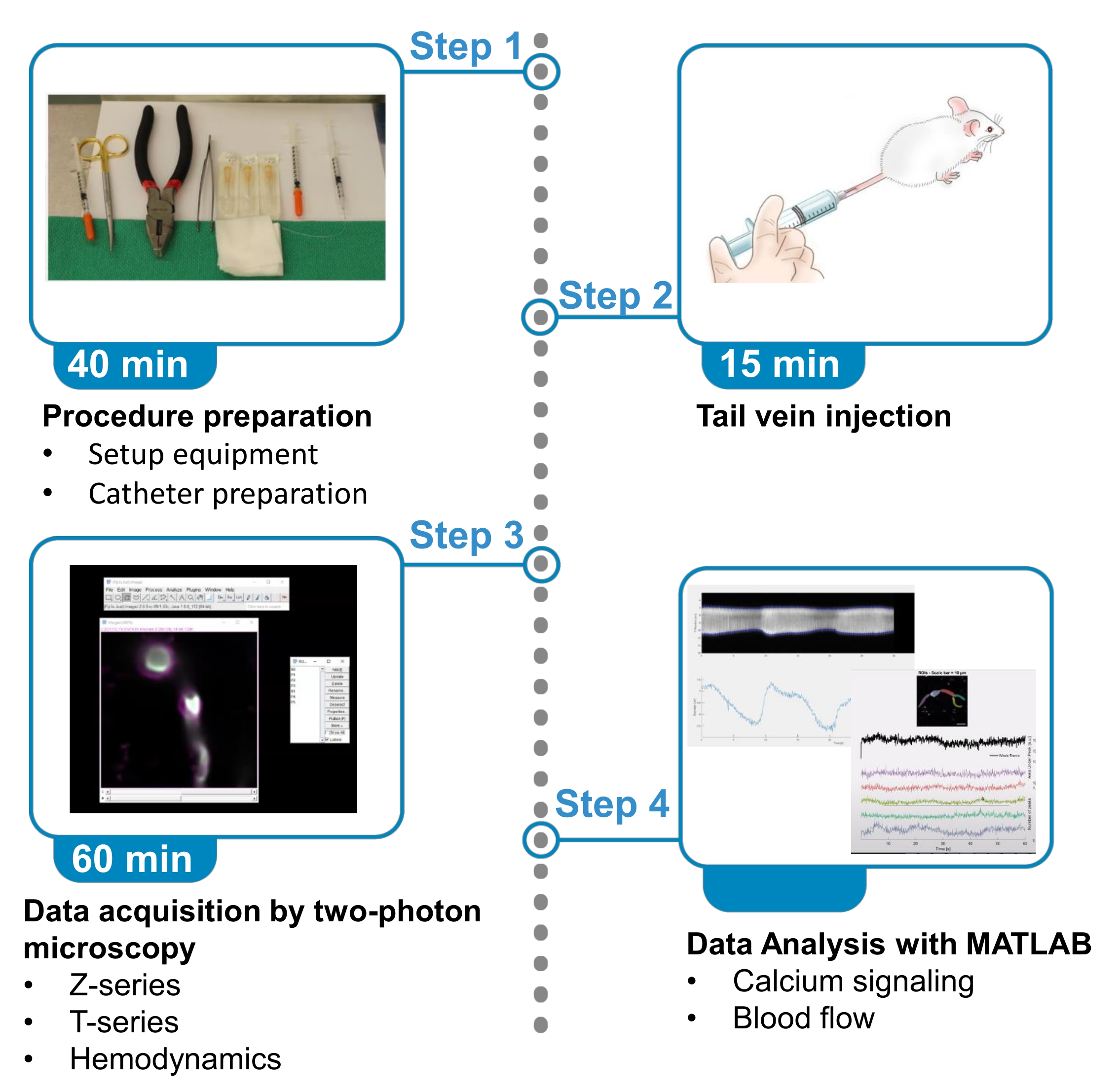
Figura 1. Riepilogo del protocollo. Il protocollo presenta i passaggi per acquisire e analizzare immagini fluorescenti di calcio da periciti che riscaldano il cervello e dati sul flusso sanguigno dai vasi sanguigni vicini in topi anestetizzati. Il protocollo è diviso in 4 passaggi. 1) Preparazione della procedura: messa a posto dell'attrezzatura e preparazione del catetere; 2) Iniezione della vena della coda; 3) Acquisizione dati mediante microscopia a due fotoni; 4) Analisi dei dati con algoritmi di elaborazione delle immagini. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2. Unmixing spettrale di fluorofori. A) Immagine media rappresentativa del vaso sanguigno marcato con periciti e fluoresceina-destrano di RCaMP da un'acquisizione della serie T. Barra di scala = 10 μm.B) Superiore: quando si considerano i singoli canali, il bleed-through dal canale 2 è evidente nel canale 1 (a sinistra). Inferiore: dopo l'unmixing spettrale, il sanguinamento viene ridotto e il segnale proveniente da RCaMP è più prominente nella struttura dei periciti. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3. ROI selezionati a mano e tracce di calcio ottimizzate. A) Le regioni di interesse selezionate nel software di elaborazione delle immagini utilizzato (forme arcobaleno) possono essere utilizzate per identificare le tracce del segnale di calcio. B) I picchi di segnale provenienti da tracce normalizzate sono identificati dal passa-basso e dal passaggio di banda filtrando i dati. Abbiamo definito la soglia del segnale come 3 volte la deviazione standard del periodo di base (primi 30 fotogrammi) e qualsiasi picco oltre questa soglia è stato considerato un segnale (traccia inferiore). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4. ROI automatizzati e basati sull'attività per l'analisi del calcio. Gli stessi dati sono stati analizzati con una soglia di 7 volte la deviazione standard della linea di base (A) e 3 volte la deviazione standard della linea di base (B). Diminuendo la soglia per l'identificazione dei pixel attivi si trovano più ROI (B) e picchi di segnale (grafico a torta) all'interno dei periciti. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5. Misure emodinamiche del kymografo. A) Esempio di scansione della linea attraverso la nave. B) Esempio di chimografi ben definiti per diametro (a sinistra) e velocità (a destra). Le striature nere all'interno della banda destra di fluorescenza corrispondono ai globuli rossi.C) Analisi del diametro con chiare fluttuazioni vasomotion. D) Analisi della velocità con grafici per asse Y = flusso RBC (celle /s), densità di linea (celle / mm), velocità (mm / s) e angolo di striscia (grado), rapporto segnale / rumore (unità arbitrarie, a.u.), asse X = tempo (sec). Fare clic qui per visualizzare una versione più grande di questa figura.
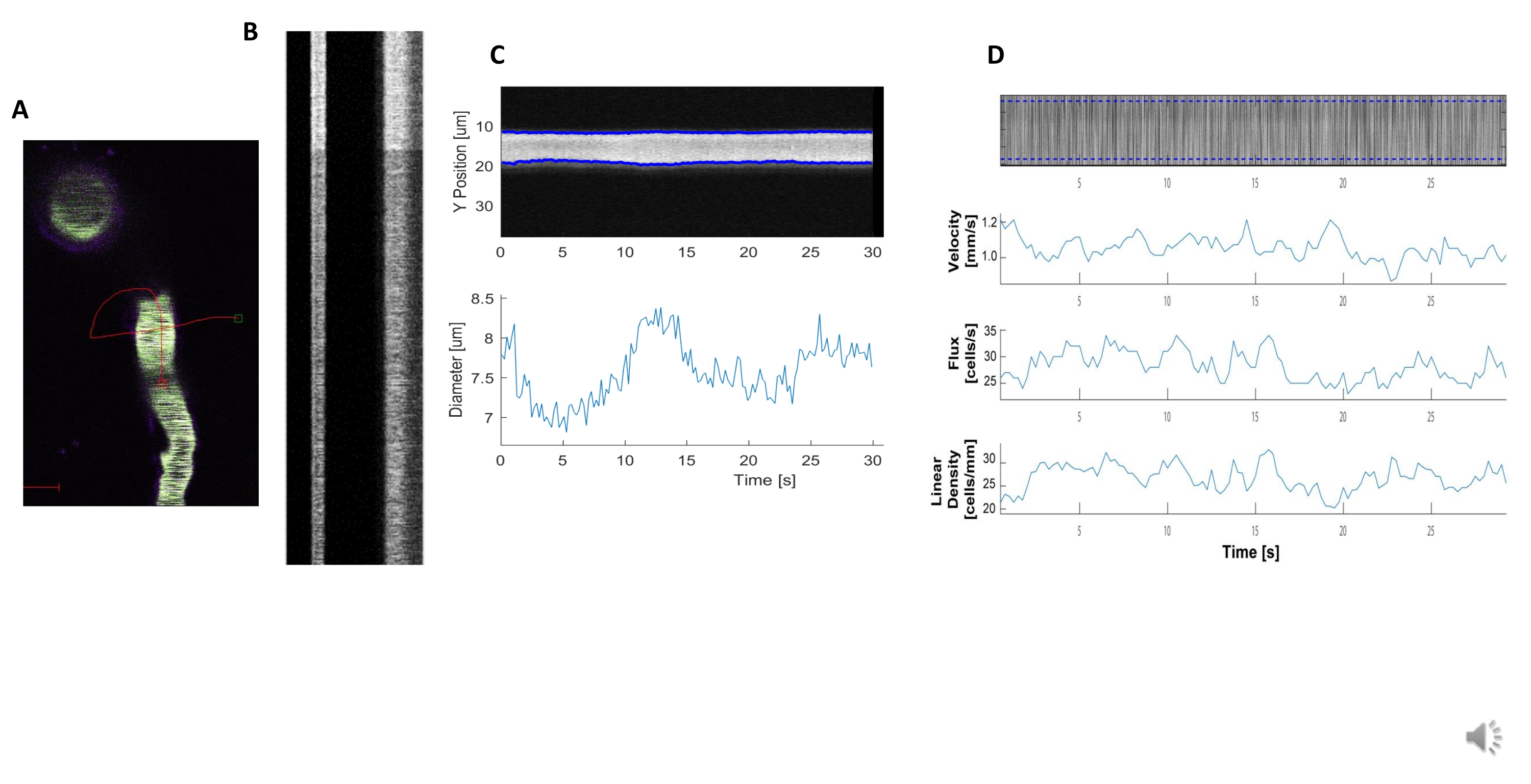{kind=link}

Figura 6. Rappresentazione di misure emodinamiche di scarsa qualità. A) Esempi di chimografi di scarsa qualità con saturazione di fluorescenza, scarso rapporto segnale/rumore o movimento del campo di imaging durante l'acquisizione. B e C) Grafici simili alla Figura 7 dei dati di diametro e velocità che hanno punti di errore (punti rossi) a causa della scarsa qualità dei kymografi. (immagine E, asse Y=Diametro (μm), asse X=tempo (sec); immagine F, asse Y= flusso RBC (celle/s), densità di linea (celle/mm), velocità (mm/s) e angolo di striscia (grado), rapporto segnale/rumore (u.a.), asse X=tempo (sec). Fare clic qui per visualizzare una versione più grande di questa figura.
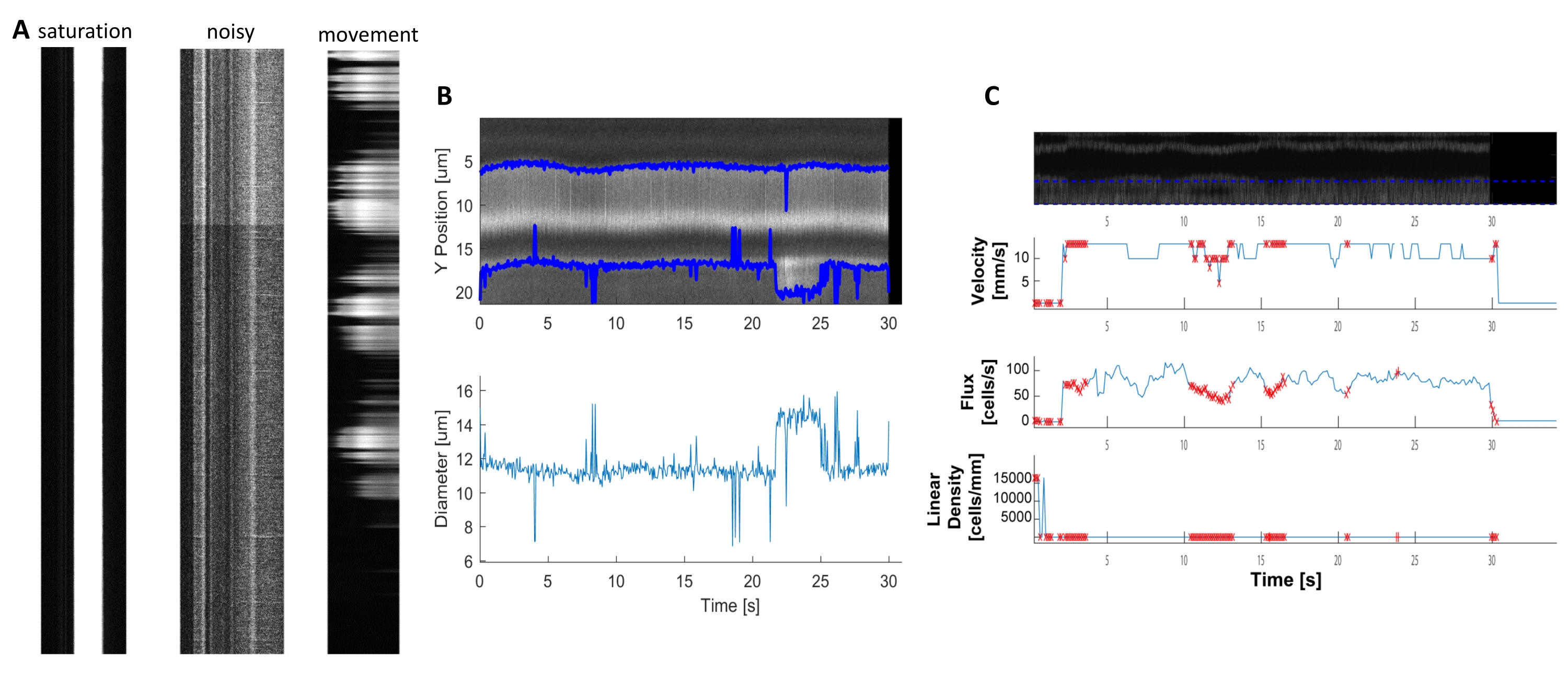{kind=link}
Discussione
Il presente metodo fornisce dettagli sull'iniezione della vena della coda del topo con un catetere, l'acquisizione di immagini al microscopio a due fotoni per pile di profondità, i filmati di segnalazione del calcio cellulare, la creazione di kymografi emodinamici e l'analisi del calcio e dell'emodinamica con i nostri algoritmi di elaborazione delle immagini17 (Figura 1). Ci sono diversi vantaggi di queste tecniche che migliorano il risultato dell'imaging in vivo e riducono il tempo, le risorse e lo stress animale durante la sessione. In primo luogo, l'uso di un catetere per l'iniezione della vena della coda fornisce un maggiore controllo sull'ago, sulla siringa e sulla quantità di sostanza iniettata nella circolazione del topo. Inoltre, impedisce l'iniezione di colorante nel tessuto della coda, risparmiando costosi reagenti. In secondo luogo, utilizziamo topi transgenici che esprimono sensori di calcio geneticamente codificati nei periciti che si avvolsero e dimostrano come localizzarli all'interno della rete vascolare cerebrale con uno z-stack di profondità, che facilita l'identificazione e il trasferimento delle cellule nelle successive sessioni di imaging a lungo termine. Questo è un fattore importante negli studi sui periciti e garantisce una corretta classificazione cellulare6,7. In terzo luogo, forniamo i nostri parametri per la raccolta di filmati di calcio e dati di scansione emodinamica della linea che sono un buon punto di partenza per misurare i segnali cellulari dinamici. Infine, presentiamo i nostri algoritmi di elaborazione delle immagini17, una cassetta degli attrezzi completa per l'elaborazione delle immagini che contiene più approcci per la pre-elaborazione delle immagini (come lo smixing spettrale), l'analisi delle immagini di calcio e l'analisi emodinamica (diametro, velocità, ecc.). Questi algoritmi possono generare grafici per una visualizzazione rapida e semplice dei dati, riducendo al minimo il livello di competenza dell'utente richiesto per analizzare i risultati. Inoltre, può essere automatizzato con poche righe di codice per elaborare rapidamente in batch più set di dati con gli stessi parametri. Ciò può potenzialmente migliorare la visualizzazione dei dati e l'investimento di tempo del ricercatore.
La chiave per raccogliere buoni dati di imaging del calcio è regolare la potenza del laser e le impostazioni PMT per una chiara acquisizione del segnale di fluorescenza, ma anche raccogliere dati a un frame rate sufficiente per catturare l'intero evento di calcio. I dati in questo protocollo sono stati acquisiti a 10-11 fotogrammi al secondo, che cattura le oscillazioni di calcio più lente nei periciti che si riscaldano. Ci sono anche diversi passaggi durante l'analisi che possono migliorare il risultato dell'analisi. In primo luogo, l'unmixing spettrale è utile se vi è una sovrapposizione significativa tra gli spettri di emissione dei fluorofori (Figura 2). La fluoresceina-destrano è stata utilizzata in questo protocollo perché è un coniugato destrano economico e disponibile in commercio che viene comunemente usato per le misurazioni emodinamiche5. L'unmixing spettrale aiuta a ripulire i dati per una migliore rilevazione dei segnali di calcio, ma potrebbero essere utilizzati anche fluorofori alternativi con spettri di emissione più stretti. In secondo luogo, la selezione manuale delle strutture cellulari come ROI (Figura 3) è utile per classificare gli eventi di calcio in diverse regioni subcellulari come il soma o i rami di processo. La selezione del ROI basata sull'attività (Figura 4)16 fornisce maggiori informazioni spaziali e temporali sui singoli eventi di calcio. Questo può essere utile quando si determina la frequenza degli eventi di calcio in una determinata area o la propagazione degli eventi ad altre aree cellulari. L'uso di software di programmazione per analizzare i dati di imaging può far risparmiare ai ricercatori ore di tempo quando i dati vengono elaborati in batch, ma richiede un investimento iniziale di tempo per regolare i parametri per risultati ottimali. I fattori più importanti sono la dimensione prevista (in μm2) della regione attiva e la durata del segnale (devono essere definiti il tempo minimo del segnale e il tempo massimo del segnale). I ricercatori devono prima esaminare alcuni film di esempio della serie T per determinare al meglio quali parametri si adattano ai loro dati. Infine, i dati di scarsa qualità acquisiti al microscopio possono ostacolare notevolmente l'analisi del calcio e dell'emodinamica (Figura 6). Pertanto, è necessario prestare attenzione per ottimizzare le impostazioni di acquisizione del microscopio all'inizio. Con questi fattori in mente, questo protocollo che può essere adattato per adattarsi all'imaging del calcio o all'analisi di altri segnali cellulari dinamici (ad esempio, sodio fluorescente, potassio, metabolita o fluttuazioni di tensione) in altri tessuti o tipi di cellule.
Esistono diverse limitazioni a questo protocollo. In primo luogo, i dati vengono raccolti in anestesia, che influisce sull'attività cerebrale e potrebbe influire sul flusso sanguigno. Immagini simili possono essere eseguite in topi svegli che sono addestrati ad accettare la fissazione della testa per risultati più fisiologici. Inoltre, è importante ricordare che raccogliamo immagini 2-dimensionali di una cellula tridimensionale e di un vaso sanguigno in vivo. Pertanto, possiamo catturare solo una fazione degli eventi di calcio all'interno di queste cellule o il flusso sanguigno in una singola sezione del vaso sanguigno alla volta.
Un'altra limitazione da notare è che l'imaging del calcio a due fotone è sensibile agli artefatti di movimento, in cui il movimento dentro e fuori dal piano focale può essere scambiato per fluttuazioni di calcio. Questo protocollo è stato eseguito in anestesia, che limita il movimento dell'animale; tuttavia, gli artefatti di movimento possono essere introdotti dalla frequenza respiratoria del topo, dalla frequenza cardiaca, dal possibile gonfiore dei tessuti e, nel caso di periciti incatenanti, contrazione dei vasi o vasomozione 4,6,18,19. Gli artefatti di movimento possono essere mitigati da diverse strategie. I pacchetti di elaborazione delle immagini utilizzati in questo protocollo includono una fase di correzione del movimento opzionale, che utilizza un motore di convoluzione 2D per allineare le immagini all'interno della serie T in base allavascolarizzazione visibile 13,17. I fotogrammi con cambiamenti significativi nel piano focale sono identificati da questo algoritmo e possono essere esclusi dall'analisi. Inoltre, è possibile utilizzare strategie statistiche all'interno dei pacchetti di elaborazione dell'imaging, come un punteggio Z quando si generano le tracce di fluorescenza per normalizzare le fluttuazioni di calcio indotte dal movimento20. L'approccio più robusto per tenere conto degli artefatti di movimento nell'imaging a due fotoli è quello di combinare l'espressione di due indicatori fluorescenti all'interno della stessa cellula, come un indicatore di calcio (ad esempio, GCaMP) e un reporter fluorescente (ad esempio, mCherry) che è indipendente dal calcio. Le fluttuazioni nel reporter fluorescente possono quindi essere attribuite al movimento e vengono sottratte dal segnale dell'indicatore del calcio per normalizzare gli artefatti di movimento.
Lo scopo di questo protocollo è quello di fornire una chiara comprensione di come raccogliere dati ottimali di imaging del calcio e del flusso sanguigno in vivo e di presentare nuovi metodi e strumenti di analisi che i ricercatori possono implementare al fine di migliorare i loro risultati. Queste tecniche possono essere applicate per studiare il ruolo di diverse popolazioni di periciti nel controllo del flusso sanguigno o in diversi stati di malattia cerebrale. Questi parametri di imaging possono anche essere utilizzati per studiare il calcio e il flusso sanguigno in altri tipi di cellule e sistemi di organi e principi simili si applicano ad altre tecniche di imaging dinamico rese possibili da altri sensori geneticamente codificati, oltre al calcio.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
J. Meza è supportato da borse di studio di Mitacs e Research Manitoba. Il finanziamento per questo lavoro è stato fornito da Canadian Institutes for Health Research, Research Manitoba, Manitoba Medical Service Foundation, finanziamenti di start-up dall'Università di Manitoba e Brain Canada attraverso il Canada Brain Research Fund, con il sostegno finanziario di Health Canada e Azrieli Foundation. Le opinioni qui espresse non rappresentano necessariamente le opinioni del Ministro della Salute o del Governo del Canada.
Materiali
| Name | Company | Catalog Number | Comments |
| Acta2-RCaMP1.07 | The Jackson Laboratory | 28345 | In the video protocol the animal model used is a female mouse of 10 months, 1 day old. |
| Applicators (Regular) | Bisco | X-80250P | |
| BioFormats package for MATLAB | NA | NA | Denominated in this protocol as "image processing packages". Available in: https://docs.openmicroscopy.org/bio-formats/ |
| CHIPS MATLAB toolbox | NA | NA | Denomitaded in this protocol as "image processing algorithms". Barrett MJP, Ferrari KD, Stobart JL, Holub M, Weber B. CHIPS: an Extensible Toolbox for Cellular and Hemodynamic Two-Photon Image Analysis. Neuroinformatics. 2018;16(1):145-147. doi:10.1007/s12021-017-9344-y. Available in: https://github.com/EIN-lab/CHIPS |
| Clear Ultrasound Gel, Medium viscosity | HealthCare Plus | UGC250 | |
| Dextran, fluorescein, 70,000 MW, anionic | Thermo Fisher Scientific | D1823 | |
| Dextran, Texas Red, 70,000 MW, neutral | Thermo Fisher Scientific | D1830 | |
| Eye Lube Plus | Optixcare | NA | |
| FIJI | Image J | NA | Denominated in this protocol as "image processor software". Available in: https://imagej.net/Fiji/Downloads |
| GCaMP6sfl/fl | The Jackson Laboratory | ||
| Head Post fixing platform | University of Zurich | NA | |
| Ketamine (Narketan 100 mg/mL) | Vetoquinol | 440893 | |
| MATLAB R2020b | NA | Denominated in this protocol as "programming platform ". Available in: https://www.mathworks.com/downloads/ | |
| Needle 0.3mmx25mm | BD PrecisionGlide | 305128 | |
| Objective XLUMPLFLN20XW | Olympus | NA | https://www.olympus-lifescience.com/en/objectives/lumplfln-w/ |
| PDGFRβ-CreERT2 | The Jackson Laboratory | 30201 | |
| Polyethylene Tubing, PE10 I.D. 28mm (0.11”) O.D. 61mm (.024”) | BD Intramedic | 427401 | |
| Prairie View | Bruker Fluorescence Microscopy | NA | https://www.bruker.com/en/products-and-solutions/fluorescence-microscopy/multiphoton-microscopes/ultima-in-vitro.html |
| Ultima In Vitro Multiphoton Microscope | Bruker Fluorescence Microscopy | NA | https://www.bruker.com/en/products-and-solutions/fluorescence-microscopy/multiphoton-microscopes/ultima-in-vitro.html |
| Under Tank Heater | Reptitherm U.T.H | E169064 | |
| Xylazine (Rompun 20 mg/mL) | Bayer HealthCare | 2169592 |
Riferimenti
- Armulik, A., Genové, G., Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Developmental Cell. 21 (2), 193-215 (2011).
- Armulik, A., et al. Pericytes regulate the blood-brain barrier. Nature. 468 (7323), 557-561 (2010).
- Berthiaume, A. -. A., et al. Dynamic remodeling of pericytes in vivo maintains capillary coverage in the adult mouse brain. Cell Reports. 22 (1), 8-16 (2018).
- Rungta, R. L., Chaigneau, E., Osmanski, B. -. F. F., Charpak, S. Vascular compartmentalization of functional hyperemia from the synapse to the pia. Neuron. 99 (2), 362-375 (2018).
- Shen, Z., Lu, Z., Chhatbar, P. Y., O'Herron, P., Kara, P. An artery-specific fluorescent dye for studying neurovascular coupling. Nature Methods. 9 (3), 273-276 (2012).
- Gonzales, A. L., et al. Contractile pericytes determine the direction of blood flow at capillary junctions. Proceedings of the National Academy of Sciences of the United States of America. 117 (43), 27022-27033 (2020).
- Hartmann, D. a., et al. Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics. 2 (4), 041402 (2015).
- Grant, R. I., et al. Organizational hierarchy and structural diversity of microvascular pericytes in adult mouse cortex. Journal of Cerebral Blood Flow & Metabolism. 39 (3), 411-425 (2017).
- Hill, R. A., Tong, L., Yuan, P., Murikinati, S., Gupta, S., Grutzendler, J. Regional blood flow in the normal and ischemic brain is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron. 87 (1), 95-110 (2015).
- 28345 - STOCK Tg(RP23-370F21-RCaMP1.07)B3-3Mik/J. Jackson Laboratory Available from: https://www.jax.org/strain/028345 (2021)
- Ohkura, M., Sasaki, T., Kobayashi, C., Ikegaya, Y., Nakai, J. An improved genetically encoded red fluorescent Ca2+ indicator for detecting optically evoked action potentials. PLoS ONE. 7 (7), (2012).
- Stobart, J. L., et al. Long-term in vivo calcium imaging of astrocytes reveals distinct cellular compartment responses to sensory stimulation. Cerebral Cortex. 28 (1), 184-198 (2018).
- Stobart, J. L., et al. Cortical circuit activity evokes rapid astrocyte calcium signals on a similar timescale to neurons. Neuron. 98 (4), 726-735 (2018).
- Mostany, R., Portera-Cailliau, C. A craniotomy surgery procedure for chronic brain imaging. JoVE. (12), e680 (2008).
- Lin, X., et al. Imaging neural activity in the primary somatosensory cortex using Thy1-GCaMP6s transgenic mice. JoVE. (143), e56297 (2019).
- Ellefsen, K. L., Settle, B., Parker, I., Smith, I. F. An algorithm for automated detection, localization and measurement of local calcium signals from camera-based imaging. Cell Calcium. 56 (3), 147-156 (2014).
- Barrett, M. J. P., Ferrari, K. D., Stobart, J. L., Holub, M., Weber, B. CHIPS: an extensible toolbox for cellular and hemodynamic two-photon image analysis. Neuroinformatics. 16, 145-147 (2018).
- Hall, C. N., et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 508 (1), 55-60 (2014).
- Nilsson, H., Aalkjaer, C. Vasomotion: mechanisms and physiological importance. Molecular interventions. 3 (2), 79-89 (2003).
- Rungta, R. L., et al. Vascular arbors in layer II / III somatosensory cortex. Communications Biology. , (2021).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati