
Method Article
TCR-のpMHCが結合を測定します
要約
This manuscript describes how to conduct (single molecule) Förster Resonance Energy Transfer (FRET)- based assays to measure the binding dynamics between T-cell antigen receptor (TCR) and antigenic peptide-loaded MHC molecules as they occur within the immunological synapse of a T-cell in contact with a functionalized planar supported lipid bilayer.
要約
T-cells are remarkably specific and effective when recognizing antigens in the form of peptides embedded in MHC molecules (pMHC) on the surface of Antigen Presenting Cells (APCs). This is despite T-cell antigen receptors (TCRs) exerting usually a moderate affinity (µM range) to antigen when binding is measured in vitro1. In view of the molecular and cellular parameters contributing to T-cell antigen sensitivity, a microscopy-based methodology has been developed as a means to monitor TCR-pMHC binding in situ, as it occurs within the synapse of a live T-cell and an artificial and functionalized glass-supported planar lipid bilayer (SLB), which mimics the cell membrane of an Antigen presenting Cell (APC) 2. Measurements are based on Förster Resonance Energy Transfer (FRET) between a blue- and red-shifted fluorescent dye attached to the TCR and the pMHC. Because the efficiency of FRET is inversely proportional to the sixth power of the inter-dye distance, one can employ FRET signals to visualize synaptic TCR-pMHC binding. The sensitive of the microscopy approach supports detection of single molecule FRET events. This allows to determine the affinity and off-rate of synaptic TCR-pMHC interactions and in turn to interpolate the on-rate of binding. Analogous assays could be applied to measure other receptor-ligand interactions in their native environment.
概要
T細胞が抗原を認識する方法のより基本的な理解は、T細胞とAPCとの間に形成された免疫学的シナプスの中に、すなわち、適切な場所で見ることが必要です。ここでは、分子の結合反応速度のみセルラー力、膜構造と膜タンパク質との間の横方向の相互作用だけでなく、シナプス特有の幾何学的制約を含む細胞パラメーター、に大きく依存するが、関連する相互作用パートナーの固有の生化学的特性によって決定されていません3。生化学的アプローチは、それらが関連するシナプス膜の少なくとも一つの破壊を必要とするような分解能が制限されます。この理由のためにFRETベースのイメージング法は、抗原pMHCs 2 TCRの結合をモニターするために開発されました。ここでは、T細胞は、組換えとサイト特異的に標識されたTCRβ反応性単鎖抗体フラグメント(SCFのV)で装飾され、計画に直面しています蛍光標識された抗原ペプチド、共刺激分子および接着タンパク質を搭載したMHCクラスII分子を保有ARガラスサポート脂質二重層(のSLB)。シナプスは、色素で標識したTCRと全反射蛍光(TIRF)顕微鏡によってバルクおよび単一分子レベルで監視することができFRETの色素標識のpMHC結果、間の結合。
この記事では、T細胞のシナプスをアッセイするためのSLBを利用する機能的T細胞のカルシウムフラックスアッセイを介してそれらの完全性を検証する、バルクでFRETの測定を行い、単一分子の感度で、取得したデータを分析する方法を詳細に説明します。勧告は、二層の官能化のために必要な適切に準拠したタンパク質を生産することに提供されています。適切なTIRF顕微鏡の二層形成とセットアップに関するより具体的な情報については4を背中合わせに発表され、追加のパブリックアクセスJoveの出版物を参照してください。
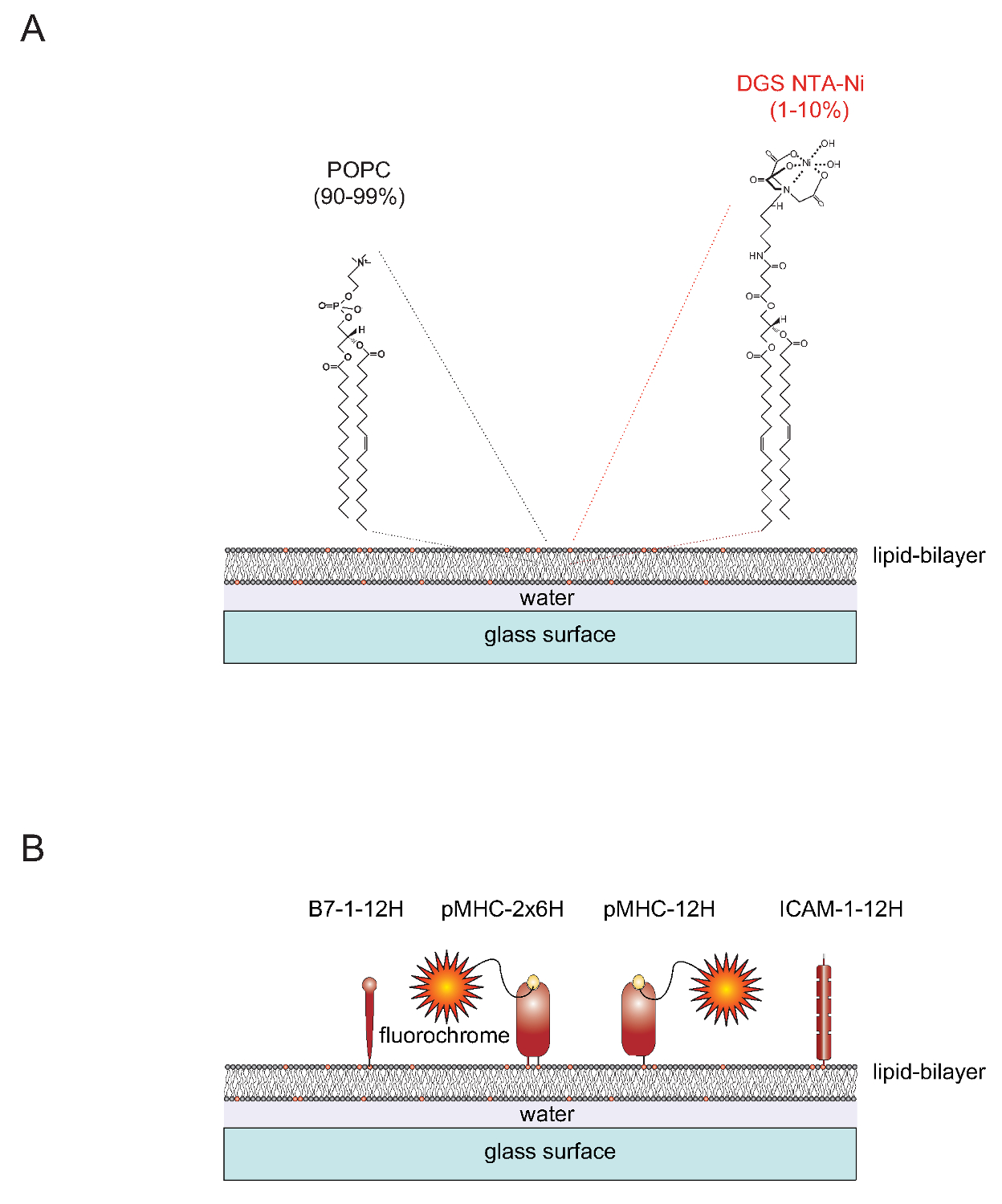
10トン ">のSLBの自然官能化のSLBは、容易に(短い:POPC、90から99%)2脂質1-パルミトイル-2-オレオイル-SN-gylcero -3-ホスホコリンを含む単層小胞(SUV車)から生成することができ、1,2- dioleoyl- SNを -グリセロ-3 - {[N(5-アミノ-1-カルボキシペンチル)イミノ二酢酸]スクシニル}(ショート:DGS NTA-Ni系、1〜10%)。 SUVが隣接する平面二重層4を形成するために、きれいなガラススライド上に広げました。 DGS-NTA-Niは、合成NTA-Niの含有頭部基( 図1A)とポリヒスチジン媒介複合体形成を介してポリヒスチジンタグ融合タンパク質を固定するのに役立ちます。安定な会合のために1は、一般的に12個のヒスチジン(ICAM-1-12H、B7-1 -12H)( 図1B)を含むつのタグでネイティブの膜貫通ドメインおよび接着タンパク質のICAM-1および共刺激分子B7-1の細胞質尾部を置き換えます。ペプチド負荷クラスII分子IE kは2つの膜埋め込 み(α及びβ)のポリペプチド鎖を含みます秒。両鎖の膜貫通/細胞質ドメインは、6つのヒスチジンそれぞれ(α6Hβ6HまたはIE K -2x6H IEのk)を含むタグと交換する必要があります。代替、12個のヒスチジンとα鎖を拡張し、タグなし(12Hの βの0HまたはIEのk個の -12HαIE kに上昇を与える)β鎖の細胞外ドメインを残すように、満足のいく結果( 図1B)を生じさせます。
pMHCsの部位特異的標識
それは意味の平衡結合定数にFRET測定利回りを変換できるようにするために、化学量論的および部位特異的のpMHCにラベルを付けることが重要です。これは、組換えヒスチジンタグ付きMHCクラスII分子2,5のペプチド結合溝にロードされた合成ペプチドの化学的標識化によって達成することができます。ペプチドは、WとしてT細胞エピトープの全ての残基を含みますエルシステインに続く短いC末端リンカー(GGS)として(例えば蛾シトクロムc(MCC)ペプチドANERADLIAYLKQATK- GGSCに、リンカーは太字で示されています)。このシステインは、マレイミド色素誘導体を用いた化学量論的にペプチドを標識するために使用されます。この時点で、特別な注意は、システイン含有ペプチドを定量的色素結合の検証に専念する必要があります。ペプチド - 色素付加物のHPLC精製は、推奨およびエレクトロスプレーイオン化質量分析によって従うべき有します。 (染料なし)ペプチド遊離体に対応する任意の記録質量は不完全なラベリングを反映しています。これが本当であれば、標識が定量的とみなされるまで、HPLC精製ペプチドは、色素標識の連続したラウンドに供されるべきです。この方法は、試料のイオン化のためにレーザー照射することを含むようにMALDI-TOF質量分析を避けるべきであることに留意されたいです。ペプチド質量を読み出すため、underrepreされる前に、この処理は、添付の敏感な蛍光色素分子を分解する色素結合のsents度。
価単鎖F V断片を用いた細胞結合TCRの間接まだサイト特異的標識
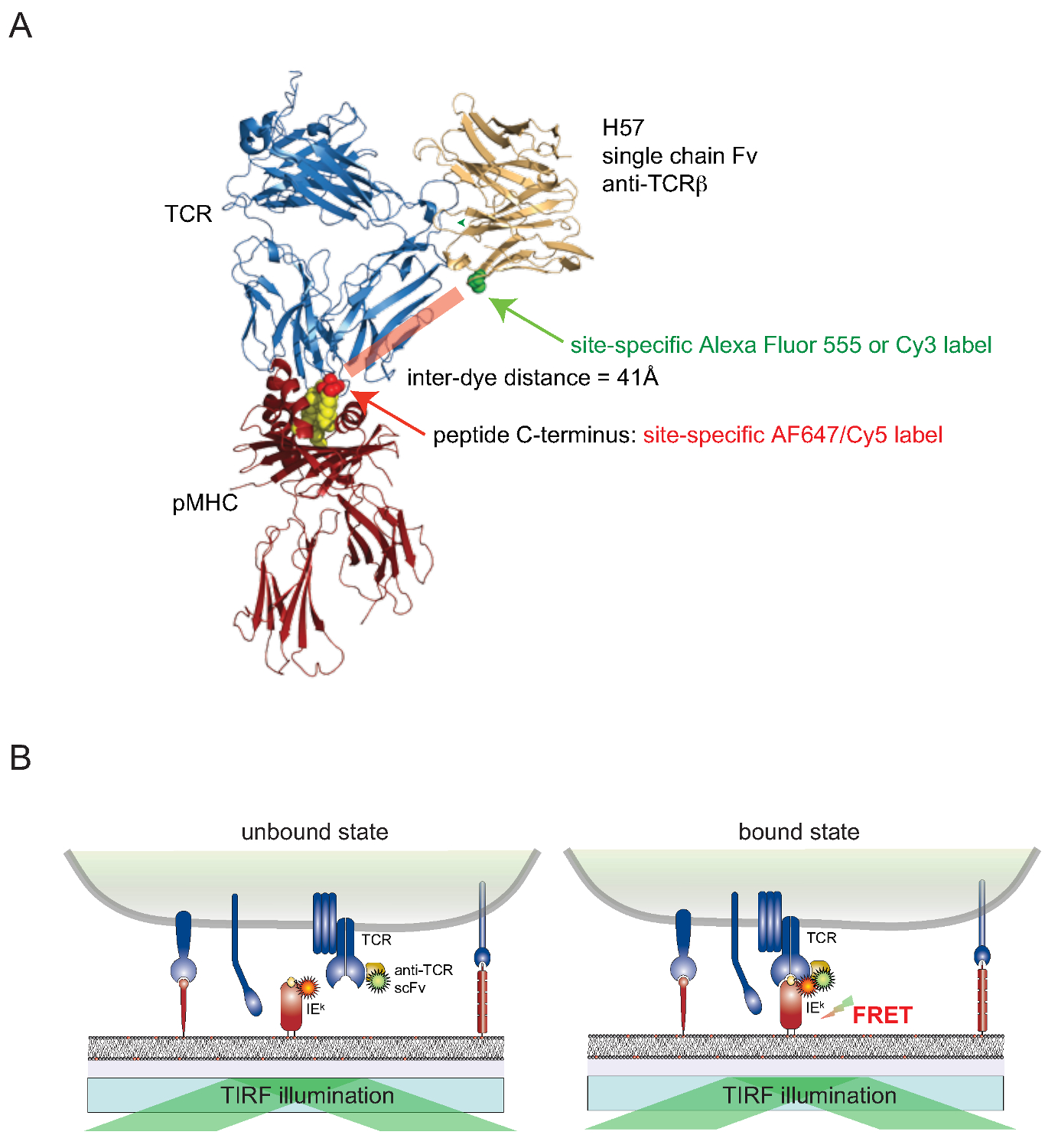
それはまだ部位特異的に生細胞の表面関連タンパク質をセルに色素を結合するために挑戦しています。表面露出のTCRは、このハードルを克服するために、TCRβ -反応性モノクローナル抗体H57-197 2の遺伝子からの一価の単鎖バージョン(SCF V)が構築されています。 TCRとの複合体中のこの抗体の結晶構造は、合理的に(対応するFRETパートナーの染料が結合している)MHC関連ペプチドのC末端に近接したセリン残基をするために置換されたバージョンを設計することができシステイン残基。この変異体システインはその後、色素コンジュゲート( 図2)のための受容体として機能します。
FRETを記録するための方法論
NT ">バルクFRET値は、選択されたインター色素距離との関係を検証することが最も適しているとこのTCR-のpMHC結合システム 2で測定効率をFRET。また、バルクのFRET測定は、シナプスTCR-のpMHCの親和性(で定性的および定量的な差を明らかに下記参照)とプロトコルセクション3.2。FRET効率を定量化するための様々なアプローチは、文献6で導入されました。この記事では、FRETを介して記録されています(a)は、ドナーアクセプター漂白後の回復、及びビアを
(b)は、FRETアクセプター放出を感作。
最初の方法は、(a)は、容易に光退色することができるFRETアクセプター、むしろ光安定であり、ドナーの使用を必要とします。それに加えて、光退色アクセプターはもはやドナー蛍光を消光することが可能であることを保証しないことが重要です。同じ検出チャネル(ドナー)を定量化するために使用されるように、補正は係数ありませんD全く色収差がこの方法論は、シンプルで信頼性の高いレンダリングする、考慮しなければなりません。しかし、定量的な測定は、同一試料の場で繰り返すことはできませんし、FRETの変化が経時的に記録することはできません。 (アクセプター漂白の前に)最初のFRETドナーおよび(FRETアクセプター漂白後の)第二FRETドナー画像取得の間を通過する時間を最小限に分子拡散または高速漂白工程をを目的としなければならない細胞の運動性によって引き起こされる影響を回避します。これは、照明及び漂白時間を最小限にするために、FRETアクセプターの励起波長の強力なレーザ光源を使用することを推奨しています。
これに対し、(b)は、感作されたFRET発光測定のアプローチにFRETドナーが励起され、FRETアクセプターの発光は、FRETアクセプターチャネル中で観察されます。 FRETアクセプターシグナルの変化は、赤方偏移アクセプターチャネル(トンにFRETドナーの時間が、放射にわたって記録することができますermed bleedthrough)と正確に決定し、記録FRETアクセプターチャネルから減算する必要がドナーの励起を経由して、アクセプター相互励起をFRET。このためのFRETドナーに対応するFRETアクセプタ画像が空間的に整列されなければなりません。
単一分子の検出(SM)のイベントをFRET
励起源、感度カメラとノイズ減衰全反射顕微鏡としてレーザーを使用すると、単一のフルオロフォアの蛍光は簡単に時間をかけて追跡することができます。同様に分子間smFRET事象の検出のために真です。しかし、合併症は、FRETドナーbleedthroughとFRETアクセプターの相互励起によって引き起こされる可能性があり、したがって、細心の注意がsmFRET実験における蛍光体濃度を調整する際に注意しなければなりません。
以下に提供されたプロトコル(プロトコル部4)でTCRは少量でFRETアクセプタとして高い豊富とのpMHCにおけるFRETドナーとして選ばれました。 FRを減衰させます十分に、蛍光のSCF Vと非蛍光のSCF VとTCRの90から70パーセントとTCRの10〜30%を飾るbleedthrough ET供与。それは単一分子FRETチャネルと共焦点であるため、ここではFRETアクセプターチャネルは単一分子チャネルとして選択されました。これはsmFRET検証の基礎であり、FRETアクセプターの単一分子とsmFRETイベントを揃えることができます。
smFRETの測定によってシナプスオフ速度の抽出
両方のFRETドナーの光退色とアクセプターをFRETは、単一分子からの相互作用の半減期を抽出する際、考慮される必要はありトレースをFRET。単一のドナー-アクセプター対Nのような外見(0)の開始時に観察可能なFRET-信号の数は、受容体-リガンド複合体のアンバインドと光退色の両方で時間をかけて削減されます。次のように与えられた時間N(T)での複合体の生存数は数学的に表現することができます。

光退色項exp(-t /τ 漂白剤 )で、時間t はすなわち 、漂白のみ照射中に発生する観測の数 Nとの積(なぜなら、非連続的な、個別の観察モードの病気照明時間tで記述されています)。運動項のEXP-(T /τ オフ )内の時間tは、観測値の数の積であり、nおよび単一のFRET観察のための時間t 遅れ ( すなわち 、動力学的ア ンバインドが継続的に行われます)。式1は、のように表すことができます。

用語τ 漂白剤/τ 病気デ漂白は、その指数関数のために発生し、期待値漂白剤 >のように定義されるまで、観測数をcribes。次のように方程式2を簡略化することができます。

時間 tの後に観察FRET事象のフレーム数N(t)の期待値は、遅れ )>直接実験から決定されます。これは、実験で選択された観察(T 遅れ )、τ オフ(k offをオフ速度の逆数)のための未知の値の間に設定可能な時間に依存し、漂白が発生する前に、観測数の期待値を漂白します > 。
したがって、Tのうちの少なくとも二つの値のための期待値遅れ )>の計算ラグは漂白 >とオフ τの実験的な決意を可能にします。
FRETベースの測定によってシナプスの2D-K D値を抽出します
TCR占有すなわち A、バウンドのTCRと合計のTCRとの比率を測定し、シナプスの2D-K D値を決定する中心的です。式(4)によると、この用語は、限り、FRETアクセプターとしてのTCRは、FRETドナーおよびpMHCsとして機能するようにして測定FRET収率に正比例します。

= TCR占有で、C =変換係数
Cは、FRETシステムに依存し、フルオロフォアを用いた、定数です。以下に示すように、実験的に決定することができます。式5ときによれば、2D-K Dに変換することができます。前の二重層のT細胞を添加するTCRリガンドの初期密度が知られています。これは、SLB接続タンパク質の高移動度であり、またのSLBは、リガンド2のほぼ無尽蔵の貯蔵器を提供するからです。

[のpMHCの初期 ]と=のpMHCの初期密度の前のT細胞の添加に
式では4と5一つは簡単に、TCRのpMHCとの間のシナプス2D-K D を決定することができます 。これは、最も確実に、アクセプター漂白(プロトコルセクション3.1を参照)の後、ドナーの回復に基づいて、FRET測定で行われます。
しかし、C私はFRET FRET強度との関係を測定し、TCRの占有率Aを決定しなければならない(バックグラウンドについて補正し、FRETドナーおよびアクセプタbleedthroughクロス励起FRET)。このため、あります単一のTCR関連FRETドナーフルオロフォア(例えば、Cy3またはAF555)SMの平均蛍光強度との比Rを知る必要があります私は、ドナーと単一分子の平均強度をFRETのイベントは、私がFRET SM FRET。 Rは、蛍光検出のために使用される質問、発光フィルターとカメラ内FRET系に依存します。
TCRは、直接式(6)に従って決定することができる人数

R = SMと私はFRETドナー / SMをFRET
RはにつながるH57 scFv-たCy3 / Cy5ののpMHC-システムのための1.45のように決定しました。
私は1.45•/バルクI TCR-のCy3をFRET =バルク
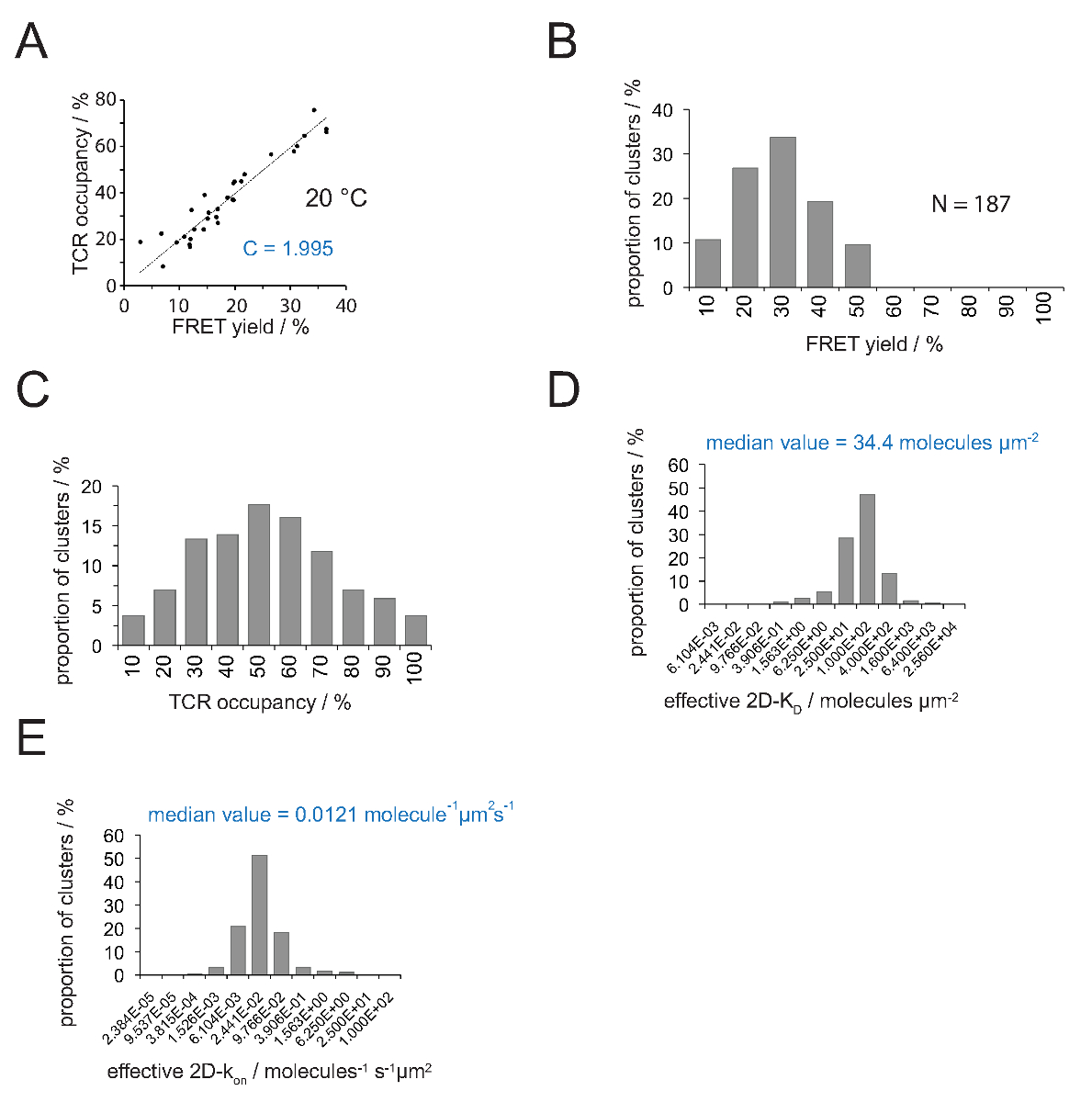
TCR占有a及びFRET歩留まりとの関係は、FRETドナーによって決定することができる復旧アクセプター漂白後のY。線形近似の図4A【選択勾配に示すように、この両方のパラメータの数TCRマ イクロクラスターのために互いに対してプロットされているため(式4)からの変換係数Cを示しています 。
1.995までのCy3 / Cy5のFRETのpMHC・システムと、(b)適用顕微鏡システムの構成- 図4(a)に示すように、Cは、(A)H57のSCF Vの金額。 TCRは、以下のように容易に推定することができる人数:
TCR占有A = 1.995•得FRET
プロトコル
1.タンパク質産生
1.1。二層に常駐するタンパク質:B7-1、ICAM-1、のpMHC(例えば、IEのK /ペプチド)
- B7-1 -12H、ICAM-1-12H
- エクスプレスB7-1-12Hおよびバキュロウイルス発現系を用いて、分泌天然のタンパク質として大量にあるICAM-1-12H構築します。
- モノQ陰イオン交換クロマトグラフィーおよびS200サイズ排除クロマトグラフィー、続いてNi-NTAアフィニティークロマトグラフィーを介して培養上清からタンパク質を精製します。
- そのようなアレクサフルオロ488(またはFITC)のNHSスクシンイミジルエステル製剤としてアミン反応性染料、アレクサフルオロ555(またはCy3の)、アレクサフルオロ647(またはCy5)で精製したタンパク質の1つのアリコートにラベルを付けます。
- S200サイズ排除クロマトグラフィーの後、(アレクサフルオロ488、FITC)488nmで555または552 nmでの280nmでのタンパク質吸収色素吸収を比較することにより、染料製造業者の仕様書に従って単量体タンパク質のラベリングの程度を決定します(アレクサフルオロ555またはCy3の)または647 nmの(アレクサフルオロ647またはCy5)。
注:この比率は、後に、色素標識されたタンパク質のバルク蛍光シグナルからSLB上のタンパク質濃度を決定するために必要とされます。 - PBS中で-20℃プラス50%グリセロールで標識されていないと、フルオロフォアで標識されたタンパク質を保管してください。
- (ここではIEのk個の -2x6HまたはIEのk個の -12H)のpMHC
- 後で定量的選択肢5,7の蛍光団コンジュゲートペプチドと交換することができるはるかに安い紫外線切断可能なペプチド代替の存在下で大腸菌で発現した封入体からMHCクラスIIをリフォールディング。
- 標準的な技術(のNi-NTAアフィニティークロマトグラフィー、モノQ陰イオン交換クロマトグラフィー、S200ゲル濾過)によるリフォールディングのpMHC複合体を精製します。
- 蛍光性ペプチド5、7とUV切断可能なペプチドを交換します。
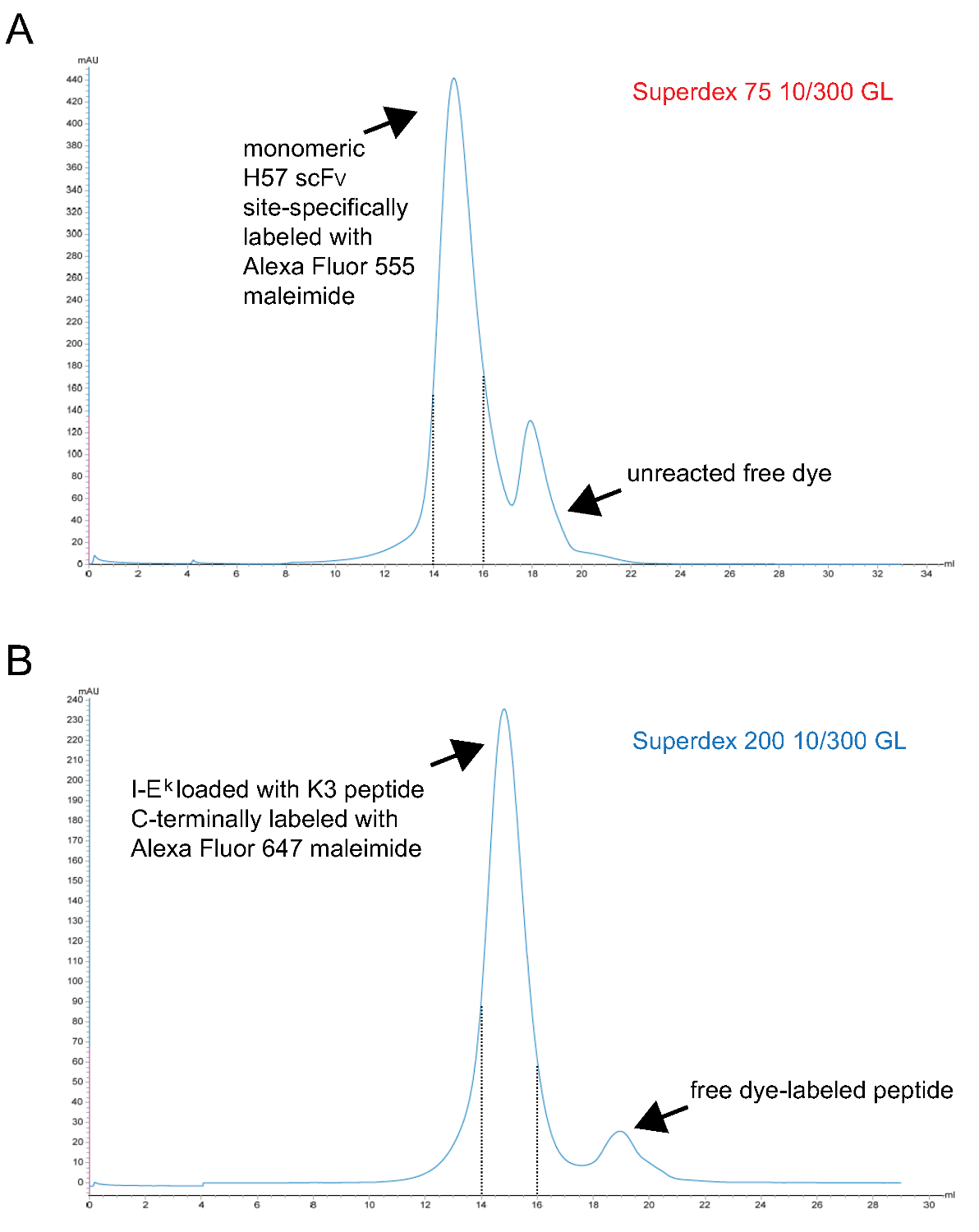
- 単量体蛍光のpMHC複合体Fを精製inally S200ゲル濾過による。代表的なクロマトグラムを図3に示されています。
- 分光光度法により定量的ペプチド負荷を確認してください。
- PBS / 50%グリセロール中で-20℃で保存タンパク質。
1.2。単鎖抗体断片(SCF V S)の生成、部位特異的標識のためのシステインの導入
- 封入体からのscF Vのリフォールディング大腸菌で発現させることができる。ここで津本や同僚8によって考案されたプロトコルが封入体が完全に還元され、その後徐々ににより、週の途中でリフォールディング、6 M塩化グアニジニウムで最初に展開されている続いていますタンパク質アンフォールディング塩化グアニジニウムの濃度を低下させます。
- 10キロダルトンの分子カットオフを有する分子フィルターユニットを使用して、適切に折りたたまれたSCF Vを濃縮します。
- S200ゲルろ過によって濃縮物を精製します。
- 単量体のscFにラベルを付けます(TCEP)の存在下で、B> V。タンパク質のモル比で最高の標識反応を実行します。もはや2時間以上のために、室温で2:わずか1の染料を。
注意:TCEPはphosphorous-ベースの還元剤であり、これらの濃度9でマレイミドと反応しません。それ故に、それは染料マレイミド誘導体と反応するまで、減少、不対スルフヒドリル基を維持するために、標識反応中に存在することができます。 - 最後にS75ゲル濾過により単量体のscF Vラベル精製します。代表的なクロマトグラムを図3に示されています。
- タンパク質比の分光光度法:色素を決定します。
- PBS / 50%グリセロール中で-20℃で保存タンパク質。
2.カルシウムフラックスの測定
- 50μgのFURA-2-AMを含む新鮮なバイアルを取り出し、50μlの水を含まないDMSO中でそれを溶解します。
- 250〜400グラムで2分間5ミリリットルポリプロピレン丸底チューブに10 6 T細胞をスピンダウン。
- 再懸濁したT細胞を室温で200ハンクス液を含むμlの画像形成媒体に加えて、カルシウム/マグネシウム及び1%オボアルブミンで、FURA-2-AMストック溶液1μlを追加(1:200希釈)を、細胞懸濁液を混合してインキュベート30分間室温で。
- 画像バッファに一度T細胞を洗浄します。このために、上清を除去し、5×10 6個の最終細胞密度で200μlの画像バッファ( すなわち、で細胞ペレットを再懸濁し、室温でイメージング緩衝液で細胞を含有するチューブを充填し、手順2で説明したように細胞をペレット化細胞ml -1の)。細胞は、カルシウム測定のために直ちに使用するか、または3時間まで氷上で保存することができます。
- 画像バッファ(37℃)で官能化されたSLBの場所細胞。すぐにT細胞は、SLBに接触し始めると、30分ごとに15〜30秒の画像の次のセットを取得します:
excitat340±5 nmのイオン、510 +/- 40 nmの発光検出
380±5 nmで励起、510 +/- 40 nmの発光検出
DIC(オプション)
注意:それぞれの露光時間は、励起光源の強度に依存します。満足な結果が得られた場合に最大可能強度値の約1/4(半分)に340nmで(380nmの)チャンネル量の画素強度。 340 nmで励起フラ-2の値が増加し、380 nmで励起されたものは、T細胞活性化の際に減少させることに留意してください。 - 実行に20〜25分は、20〜50μgのミリリットル-1の最終濃度で予熱した抗のpMHC阻止抗体の50〜100μlを添加します。抗体はすべてpMHCsを飽和し、結果としてT細胞は、抗原を認識し、カルシウムをフラックス処理を停止しなくなります。別の5用の画像を記録し続ける - T細胞の非活性状態に対応する細胞内カルシウム濃度のベースラインを取得するために10分< / LI>
- 340 nmおよび何の細胞を含まない少なくとも1000画素の関心領域内の380 nm励起、での平均バックグラウンド蛍光シグナルを決定します。 340 nmおよび380 nmで励起し、すべての測定されたFURA-2画像を背景減算し、340 nmの/ 380nmの強度個々の細胞の比率または細胞群を計算します。 (のpMHCの完全な抗体遮断薬で、 すなわち )5前回フレームの割合を通じてすべての比率を分割することにより強度比を正規化。
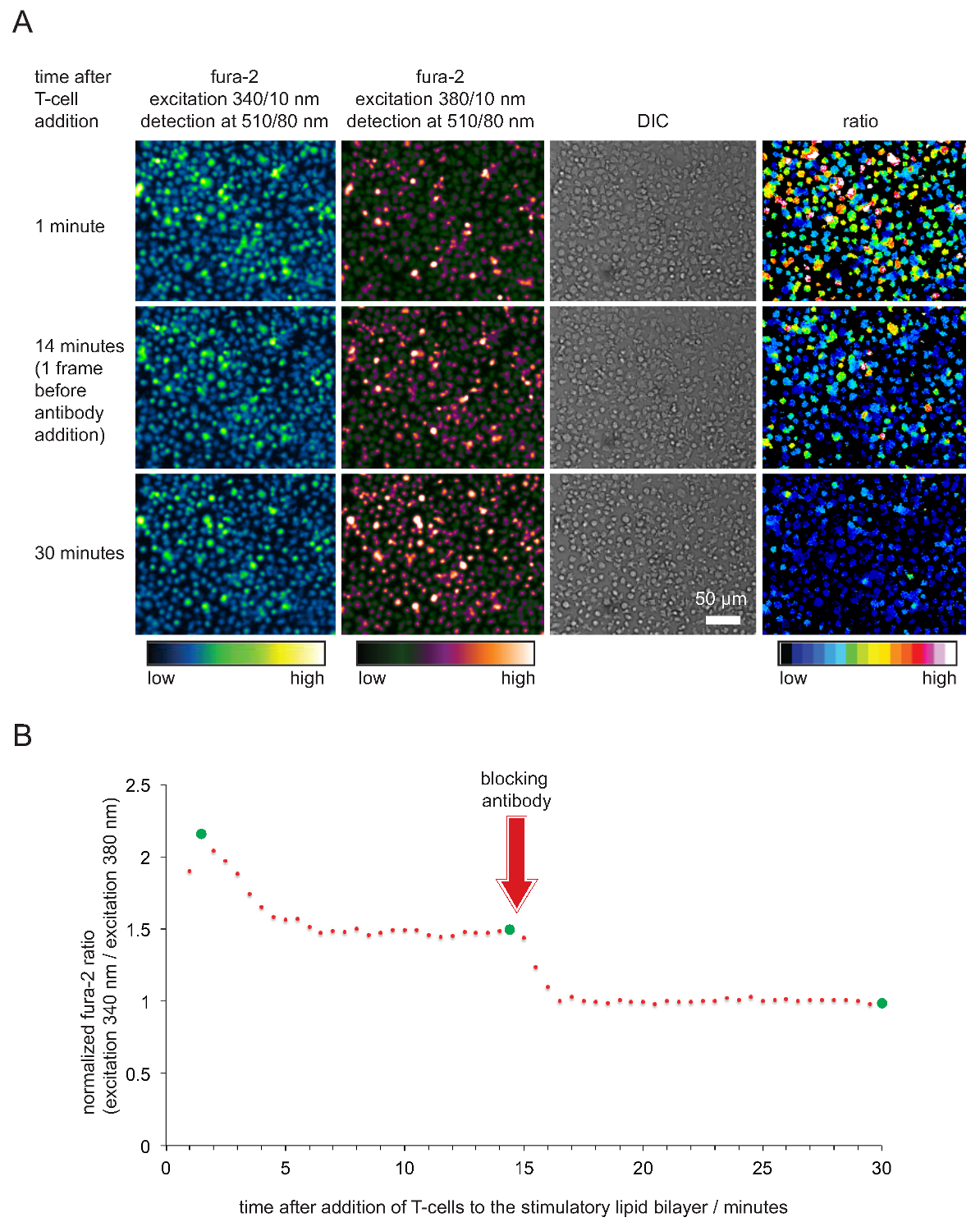
- 時間に対する正規化されたフラ-2比をプロットします。注:セルおよび二重層が良好な状態にある場合、FURA-2の340nm / 380nmの強度比は、一般的に、細胞が二層に接触してきた15〜45秒後、2と5の間の値を採用。比率は、それらは、少なくとも20分以上のために一定のまま1.6と2の間の値にドロップします。値は、唯一の抗のpMHC抗体の添加後1に低下する必要があります。典型的な例は、 図5に示されています。
3.1。 H57scF Vで 、TCRの装飾
- 250〜400グラムで2分間5ミリリットルポリプロピレン丸底チューブに10 6 T細胞をスピンダウン。
- メディア、フリック細胞ペレットを静かにデカントし、細胞懸濁液にのscF V(濃度〜1 mg / mlの)0.3μlを添加します。バルクFRETのための測定は、色素標識のscFvを採用しています。単一分子のための測定は、非標識のscFv(5〜9重量部、不対システインを含まない)と、色素標識のscFv(1部、1対になっていない色素結合されたシステインが含まれている)のミックスを使用しFRET。
- 15分間氷上で細胞をインキュベートし、氷冷画像バッファを使用して、連続遠心分離を介して細胞を2回洗浄します。
注:細胞は、結合のscF V(0°C〜4時間2でのSCF V解離のT 1/2)の有意な損失なしに、氷上で保存することができます。 TCRトランスジェニック(および必要に応じて、また由来の初代T細胞の純度Rag-T細胞は、T細胞の培養物に刺激するために追加されたペプチドに応答して()7細胞分裂まで増殖する唯一の細胞であるので1/2欠損マウス)及びインビトロでの刺激は、98%以上です。 B細胞はアポトーシスを起こしていないと栽培の7〜10日後にはもはや生きています。いくつかの樹状細胞はまだ生き残るために容易に彼らの独特な形態のものではないだけで差別することができますが、また、彼らは、H57抗TCRベータscFvフラグメントに結合しないので。
3.2。アクセプター漂白後のドナーの回復を経由して測定をFRET
注意:22.5℃で50分に、TCR-H57のSCF V複合体の半減期は、氷上で4時間にのぼることを覚えておいてください、37℃で6.8分に(と氷上で約4時間に)2。限りH57のscF Vは FRETドナーとして機能するように、測定されたFRETの収量は、しかし、対雑音比が増大する信号H57のSCF V解離に敏感ではありません増加H57解離と。
- SLB含むAF647 / Cy5標識pMHCsを準備するだけでなく、非標識ICAM-1とB7 Axmann ら 4に係ります 。
- ハンクス液を含むイメージング媒体の撮像室のPBSプラスカルシウム/マグネシウム及び1%卵白アルブミンを交換します。ウェルに画像バッファのこのピペット400μlのために、慎重に混合してウェルから400μlのを削除します。この手順を3〜4回繰り返します。いつでも空気にSLBを公開しないでください。
- 顕微鏡ステージ上にイメージング室を配置し、蛍光(AF647、Cy5の)SLBはクリアな視界に入ってくるまで、フォーカスを調整します。
- 潜望鏡の並進ステージの移動を経由して目的の焦点面の周囲に光軸に集束ビームを平行に変換することによって、全反射照明を設定します。
- TIRFモードの励起レーザビームを微調整するには、AF555 / Cy3で飾ら追加T細胞はのscF Vをラベル、それらをSLBに落ち着くしましょう。注:基底細胞膜は、SLBと上向きに着目すると見えてくるセルの無い他の部分に加えて、フォーカスがある場合の全反射照明の条件が満たされています。 TIRFは、適切に調整されていない場合は、SLBと接触していない蛍光T細胞の原形質膜の部分は、リングとして現れます。この場合TIRF照明が達成されるまで、潜望鏡の並進ステージと、レーザビームを調整します。
- 指示された順にと矢継ぎ早に次の6つの画像( 図6)を取得します :
セルの画像を撮影する(I1、オプション)白色光、
前漂白剤パルスにFRETアクセプタ(のpMHC)の画像を撮影する(I2、オプション)647 nmの励起(低消費電力)、
前漂白剤パルスにFRETドナー(TCR)の画像を撮影する(I3)514 nmの励起(低消費電力)、
647 nmの励起(高出力)フォトブリーチFRETアクセプタへ(I4)、 />(I5)514 nmの励起(低消費電力)漂白剤パルスに続くFRETドナー(TCR)の画像を取るために、
(I6)647 nmの励起(低消費電力)は、完全なFRETアクセプター漂白を確認します。 - 後で分析するための画像を相関させることができるようにするために画像(I3)とできるだけ短い(I5)の間の経過時間をおいてください。まだT細胞の適切なイメージングを可能にし、可能な限り低い電力レベル、での励起を使用することによって最小限にFRETドナー漂白をしてください。
- 関心領域(ROI)を選んで、 例えば 、全体のシナプスまたは個々のTCRマ イクロクラスター、(I3)での平均強度を決定する(= I(3))と、(I5)で(= I(5))。バックグラウンド減算のため、照明スポットの外側に、同じ大きさのROIを選択(3)または(5)とその平均強度を決定する(I(背景))。 FRETの収量を決定するには、次の操作を実行します。
G "/>(式7)
注:これは、絶対的なFRETレベルに関連し、それはドナー画像(I5)との間の漂白及び(I3)を補正する必要があることに留意すべきです。
3.3。感作された放射を経由して測定をFRET
- すべての発光チャンネルで蛍光を発する多色ビーズを用いた空間的なドナーとアクセプターチャネルのアライメントを行います。色収差による両チャンネル間の空間的なシフトは、超位置決め個々のビーズによって決定することができ、すべての次の2色の画像ペア10のための2つのチャネルの一方の補正のために適用されなければなりません。
- 単独のFRETドナー蛍光団を含むSLBを用いたドナーbleedthroughの程度を決定します。これは、非標識のpMHCを有する脂質二重層にFRETドナーで標識されたT細胞を使用することも可能です。背景には、最初のビューの照明されたフィールドの外側を決定した後、両方のチャネルから減算されます。このように平均BA ckground補正された2対応するROIの強度(I ドナーチャンネルと私はチャンネルを受容体)に決定されます。次のようにbleedthrough係数(BTC)を計算します。
 (式8)
(式8)
注:このBTCは、特定の染料とフィルタセットの組み合わせのための定数です。 - 次のようにFRETドナーbleedthrough画像を計算します。
 (式9)
(式9) - エキサイティングSLBによってアクセプター相互励起がFRETドナー励起光( 例えば 、514ナノメートル)を有する第1単独のFRETアクセプターフルオロフォア(例えば、IE K / MCC-アレクサフルオロ647)を含む、その後、アクセプターの励起光(例えば、647で決定NM)。次のようにクロス励起係数(CEC)を計算するためにFRETアクセプターチャネル内でバックグラウンドを差し引いた画像を使用してください:
ftp_upload / 53157 / 53157eq10.jpg "/>(式10) - CECは、ドナーの励起に用いるレーザ強度に依存するように、各測定日に、それを決定します。単にクロス励起されて生成された画像を計算するために、得られたCECを使用してください。
 (式11)
(式11) - 次のようにbleedthrough、クロス励起のために補正されたFRET画像を計算します。
 (式12)
(式12)
注:絶対FRETシグナル(ただし、相対的なFRET収率)は、T細胞の膜に結合したTCRからH57のscF Vの解離に敏感です。 TCR-FRETプローブの過度の損失を回避するために、二重層へのT細胞の添加後の最初の2分以内に37℃で測定を実行することを目指しています。定量的(≥95%)、TCRのラベルを持つ測定は、最初の3分後内で、または22.5℃以下でのみ可能です二重層のT細胞の添加。
4.単一分子FRET測定
- 試料で1-5キロワット/μm2での強度を生じさせるために、両方のレーザーのパワーを調整します。詳細については、Axmann らを参照してください。4。
- 非標識のscF V(5-9部品)およびCy3 / AF555標識のscF V(1部)の混合物と上記のようにラベルT細胞。注:この方法では、TCRの一部のみをFRETドナーでマークされます。これは、検出可能な相互作用の数は減少しますが、ドナーbleedthroughから発生するノイズは、個々の単一の分子がイベントをFRET解決するときに重要である、(約5 1/2 1/2 10にして)有意に減少されます。
注:それははるかに大きな時間の範囲内でSLB結合のpMHC(サブ秒からのTCRの解離よりも(数分〜数時間)に発生したとして、TCRからH57のscFVプローブの解離が、測定には影響しません第二範囲)。 - 顕微鏡ステージ上のAF647標識pMHCsだけでなく、ICAMおよびB7を搭載し、SLBを置き、二重層を明確に見えてくるようにフォーカスを調整します。
- オプション:シ ナプスを除く照明の分野の大部分をマスクする(。Axmannら 4に示すように)励起経路にスリット開口を挿入します。無漂白IEのK / MCC(C)-Alexaフルーア647 FRETアクセプター分子は、照明の領域に移動することができますこの方法です。
- (画像バッファ付き)二重層にH57のscF V飾られたT細胞を追加し、シナプスが視野に表示されるまで待ちます。
- 立て続けに2色検出を用いて10〜20の画像セットのシーケンスを取ります。
I1)励起514 nmの
I2)励起647 nmの - 1〜5ミリ秒のための画像を公開し、画像のセットとして取得します。
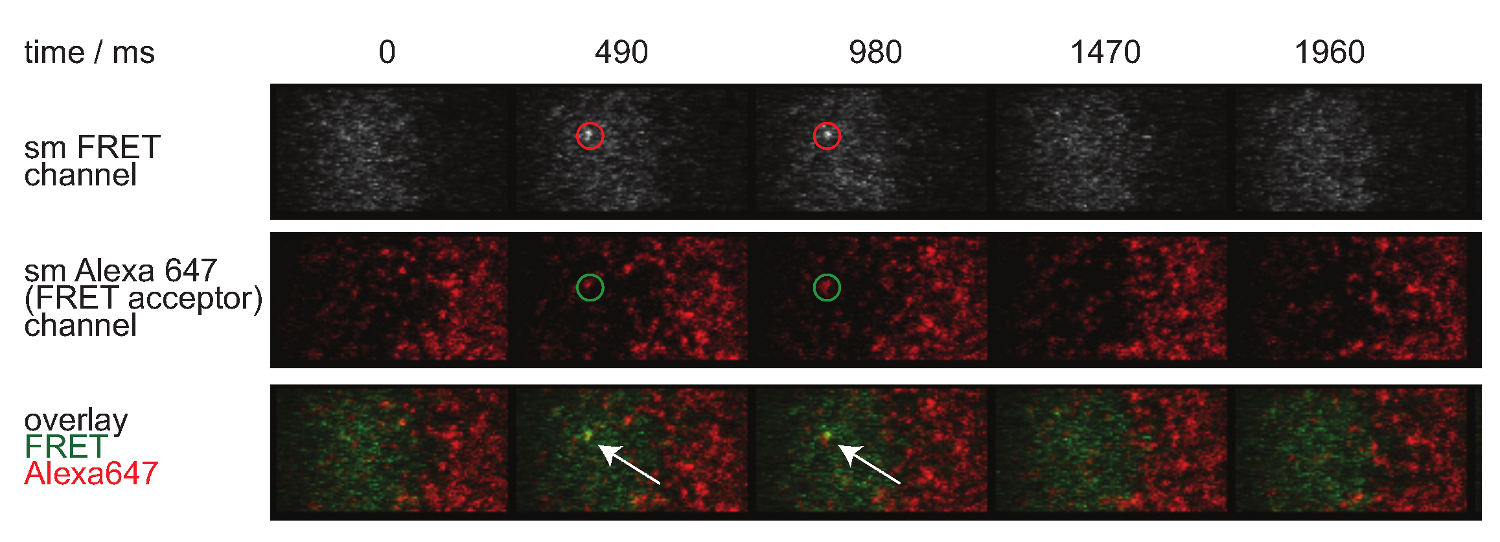
- イベントをFRET単一分子のアイデンティティを評価するために、ドナーとアクセプタbleedthroughクロスexcitaに関する同様の補正を適用しまする。また、単一分子は、イベントが単一の受容体分子を整列させるために持って一歩(また図7を参照)に表示され、消えますFRET。
- オフ率決意
- 単一分子のレコードトレースは、いくつかの取得時間フレームのイベントをFRET。
注:この例では(斜体)オフ速度5c.c7間TCRとIE K / K3は、4つの異なる遅延時間で25℃で測定された(42ミリ秒、490ミリ秒、1007ミリ、1989ミリ秒)。 - 一覧表1に示すように、それらのトレース長に応じてトレースをFRET。
- 示すように、(着色番号は、表1から取得されます)逆累積減衰関数(表2)に、表1に変換します。
- 結果として減衰関数を正規化するには、その特定のグループ内のすべてのトレースの和で表2のトレース数を割ります。時間フレームの数に対して正規化した値をプロットします。ゼロが含まれている最後の時間枠を省略する場合には、減衰を読み取ることができますILY単一指数関数( 図8A)を装備。
- X = T 遅れ = 0.042: 図8(b)に示すように 、期待値遅れ )>、すなわち 、 この例で用いられる遅延時間t の遅れに対して上記で決定減衰関数の指数の負の逆数を(プロットSとy = 遅れ )> = 1 / 0.662 = 1.511、X = 0.49秒とy = 1 / 0.902 = 1.101、X = 1.007秒とy = 1 / 1.131 = 0.884、X = 1.989秒とy = 1 / 1.591 = 0.629)。
- 式3に基づいて、これは、原点と科学的データ分析プログラムの非線形フィッティング関数を用いてフィットτ オフと漂白剤 >を行うことができます。
注:この例に示されている最高のフィットは2.12 +/- 0.23秒のオフ τが得られ、1.53 +/- 0.06秒の漂白します >。 - HALを計算•LN オフ =τ オフT 1/2とオフ1/2トン相互作用のFライフ(2)(この例では1.47秒)。
- 単一分子のレコードトレースは、いくつかの取得時間フレームのイベントをFRET。
- 2D-K Dの決意
- 式6を用いて使用さFRET色素ペアの変換係数Cを決定した (図4A)上記のように。
- 個々のTCRマ イクロクラスターまたは全体のシナプス( 図3Bおよび5) のためのFRET収量を決定します。
- 式4変換を使用して、TCRの占有にすべての個々のFRET利回り( 図4C)。
- 2D-K D S内にすべてのTCR占有率を変換するための式11を適用します。以下に示すように、シナプス結合が不均一です。シナプスのK Dに意味のある尺度は、すべての測定されたミクロクラスター( 図4D) の(赤で表示)の中央値です。
- S上の2D-kの計算
- K D とオフ実験的に決定さKとK D値をオフ / K = 上の kの質量作用の法則との kを計算します。
注: 図4(IE K / MCCは25℃で5c.c7 TCRとの相互作用)に示した実験のためのオフシナプスkは0.41秒-1です。 図4E に示すように、したがってK Dプロット ( 図4D)は、 プロット上のkに変換することができます。
- K D とオフ実験的に決定さKとK D値をオフ / K = 上の kの質量作用の法則との kを計算します。
結果
FURA-2カルシウムを介した細胞内カルシウムの記録染料、ならびに刺激効力を確認するために、その後の細胞解析、ひいてはのSLBの機能は、 図4に示されている。明らかになるように、カルシウム濃度は、T細胞の上昇(として発現ベースラインはすぐに、すぐに彼らは刺激するSLBに落ち着くように)1である正規化FURA-2 340nm / 380nmの比。カルシウムレベルがまもなくTCR結合とするT細胞活性化を終了からブロックpMHCs抗体を添加した後にベースラインレベルに戻ります。
図6は、(図4に示されている)FRET収率を測定するために使用され、2D-K D値を計算するのに役立つ受容漂白、後のFRETドナー回復を伴う典型的な実験を示しています。 FRET Aの迅速かつ完全な切除後、AF555を介しラベルTCRを表すFRETドナー強度の増加を、注意してくださいcceptor種(ここではpMHCsに関連したAF647)。また、明らかにFRETチャンネルに強い減少、 すなわち 、FRETドナー励起下でのFRETアクセプターチャネルは、FRETアクセプター漂白した後、あります。ほとんど見えない残りの信号は、ドナーbleedthroughをFRETに相当します。個々のTCRマ イクロクラスターまたは全体のシナプス内の収率は示さ測定された強度値( 図6B)に基づいて計算されるFRET。
図7には、2つの時間枠に表示単一分子FRETイベントの軌跡と時間経過を示しています。導入に上記で概説したように、このような挙動は、両方の結合シナプスTCR-のpMHCの崩壊と光退色が原因で発生します。これらの2つの寄与とを区別するために、実験的な取得時間フレームは、期間中に変化させることがあります:光退色は一定のまま、FRETイベント軌道長さの変化が唯一の結合速度によって引き起こされます。 W tracelengthsの定量、 HICH は 、図7に示されるオフ速度及び漂白の計算のための基礎を形成する、3の表1に提供されています。
2D-K D Sの決意は、FRETドナーラベルのTCRのためのバルクのFRET利回りの記録が必要です。実験的に推定された定数 C( 図4)を使用すると、TCRのマイクロクラスターまたは全体のシナプスについて測定FRETの収量は、TCRに占有変換することができ、 すなわち 、のpMHC-従事し、総TCRの割合( 図4C) 。前のT細胞の添加にSLBに存在する既知のpMHC密度で、値は、シナプスの2D-K D値( 図4D)を決定するために適用することができます。オン率は質量作用の法則を用いて計算することができる( 上の 2D-K = 2D-K / 2D-K D をオフ )を決定し、シナプスからオフ速度および2D-KD値。
1 "SRC =" /ファイル/ ftp_upload / 53157 / 53157fig1.jpg "/>
図1.平面ガラスサポート脂質二重層(SLB)システムの概要図。 (A)のSLBは、対応する脂質からなるPOPC(90から99までパーセント)と合成脂質DGSのNi-NTA(1-10%)で構成され、きれいなガラス表面は単層小胞(SUV車)で充電しているときに自発的に形成されています。 (B)は、一旦形成されると、そのようなのSLBは、T細胞のためのAPCとして機能するように、B7-1タンパク質およびICAM-1接着タンパク質共刺激、pMHCs由来の可溶性ポリヒスチジンタグ化細胞外部分で官能化することができます。 SLBの準備の詳細についてはAxmann ら 4を参照してください。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

Figu その場で結合TCR-のpMHCを定量する2.再フェルスター共鳴エネルギー移動に基づくアッセイ。 (A)H57単鎖フラグメントと複合体TCRの複合構造のpMHC係合は、本明細書に記載のFRETベースのアプローチを示しています。約41Aは、FRETを受けて対応する二つの蛍光団を分離する短い距離に注意してください。フルオロフォア-マレイミドのためのアクセプター部位は、緑と赤で示されている。(B) インサイチュでのTCRのpMHC相互作用を検出する原理が示されています。のみのscF Vは特定の複合体を形成するTCRとpMHCs(ここではIEのk)を 、-decorated、測定可能なFRETシグナルを生じさせる。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
"/>
図のscF Vの単量体およびペプチド負荷IE kの-2x6H分子に最終ゲル濾過工程生じさせる3.クロマトグラム。X軸はミリリットルでの保持容量を表し、Y軸は任意の単位で280 nmで(AU)の吸光度を示します。(A)H57のscF Vの部位特異的にアレクサフルオロ555マレイミドで標識したタンパク質は、(ステップ1.2.5)からの未反応染料を分離するために、S75クロマトグラフィーに付しました。間隔14〜15ミリリットル(破線)を保持するために対応する画分を標識した単量体H57のscF Vを表しています。UV-切断可能なANP空間ホルダーペプチドと複合体を形成する分子は、UV照射、部位特異的にアレックスと一緒にインキュベートされていた-2x6H(B)IEのK 647マレイミド標識ペプチド、最終的に遊離ペプチド(ステップ1.1.2.4)からタンパク質を分離するためにS200クロマトグラフィーに供しました。破線の間隔が適切に折り畳まれ、単量体pMHCsが含まれています。(A、B) は0.7ミリリットルのサンプルは、ラム酒(0ミリリットル点)の開始時にカラムに適用しました。集めた画分を濃縮しました。タンパク質対色素比は(-20°で保存)、PBS / 50%グリセロールに対して透析前にphotospectrometryによって決定した。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

図4の決定2D-K D SとS の 2D-K。(A)、アクセプター漂白及びTCR占有が実験的に測定したドナーの回復によって決定されるように、FRETの収量との相関関係。セクション4.2で説明したように、TCRの占有率は、個々のTCRマイクロクラスターについて決定することができます。データの線形適合が線で表示されている、の傾きは、TCRとの比Cに等しいです。占有とFRET収率。Cが採用FRET系のための一定の特定およびフルオロフォア(ここでのCy3およびCy5)です。この例では、(B)1.988を得た収率データは、個々のTCRマ イクロクラスターについて決定したFRET(N = 187、温度= 24℃)アクセプター漂白の供与体を介して回収。ヒストグラムの棒の下の数字は、区間内の上限値を示す。(A)で決定された定数 Cと測定されたFRETの利回りを乗じて(B)に示したデータの(C)変換 。バーの下の数字は、区間内の上限値を示している。(D)ヒストグラム(半対数、ベース= 4)個々のTCRマ イクロクラスターについて測定した2D-K D Sの分布を示しました。中央値は、2D-K D が青色で表示されます。バーの下の数字は、(E)(D)に示すヒストグラム。区間内の上限を示すint型に変換されました-histogram 上のOA 2D-K(半対数、ベース= 4)24℃(0.41秒-1)のためのオフシナプスKを採用。値の決定の中央値の2D-Kが青色で表示されます。データはもともとHuppa らに出版された。2、ここに新しい形式で可視化されている。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

T細胞刺激およびイメージングのために使用のSLBの図5の機能検証。 FURA-2を搭載した(A)TCRトランスジェニックT細胞ブラストは、抗原pMHCs、ICAM-1およびB-7を保有刺激SLBに直面しました。携帯電話340 nmおよび380 nmで励起されたFURA-2排出量だけでなく、DIC画像を記録しました。示されるように、発光強度の比の値は34で励起0と380 nmのは、右側のパネルに表示されます。実験に抗体14分を遮断するのpMHCの添加は、休止T細胞のそれに匹敵する細胞内カルシウムレベルが減少した。(B)刺激のSLBを接触させ、T細胞の平均FURA-2比の典型的な時間的プロファイルを特徴としています抗体媒介遮断後の抗原を奪われた非活性化T細胞またはT細胞に比べ2~4倍高い細胞内カルシウムの立ち上がりによる。グリーンの円は、(A)に示した時点を示している。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

図6は、バルクFRETアクセプター漂白後のFRETドナー回収を通して測定される収率をFRET。 (A)図は、典型的なシナプスのFRET測定の一例です。左側に、トップに表示されるように一連の画像は、FRETドナーとFRETアクセプターチャネル(ビームスプリッタの詳細については、Axmann ら 4を参照)を生じさせる放射ビームスプリッタを使用して取得されました。左DICの画像に示す線は、T細胞のシナプスの境界を示します。 (B)ならびにFRETアクセプター漂白(ステップ4)の後にFRETドナーチャネル中の強度の増加はFRETアクセプターチャネル内の強度の損失を注意個々のシナプス領域または全体シナプスために示されているように効率を定量化することができるFRET。検査のために、FRETアクセプター漂白前と後の画像は2つのルックアップテーブル(LUT、緑と物理学)を使用して示されている。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

図7.単一分子は、イベントが表示され、insingleステップを消え、完全に単一のFRETアクセプターフルオロフォアで整列されている。単一分子FRETイベントの時間経過が示されているFRET。画像は、裏面照射型EMCCDカメラを用いて得た。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

H57のscF V -AF555由来観察可能なFRETシグナルのオフ = 1 / K測定smFRET軌道からオフ τの決定図8(A)の正規化累積和は、(IEの<認識5c.c7 TCRトランスジェニックT細胞ブラストを飾っSUP> / K3-AF647は、4つの異なるタイムラグ(42ミリ秒、490ミリ秒、1007ミリ、1989ミリ秒)のため24℃)での観測の合計数の関数としてプロットしたK。単一指数フィット関数が。期待値ラグ )>の負の逆数に対応を生じさせる(B) の期待値は遅延tの遅れに対してプロットし、遅れ )>式を用いて適合=τ オフ / {(τ オフ / 漂白 >)+ T 遅れ }τをオフに生成すると、漂白剤 >。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
ディスカッション
in situでのタンパク質-タンパク質相互作用を測定することは、TCRのpMHC-11に結合するような低親和性相互作用を扱う場合は特に、非常に望ましいです。オン速度、ならびにこのような相互作用の安定性を大幅に結合が起こるような状況下で、特定の影響を受けるためです。低侵襲FRETベースのイメージング手法は、原理的にはこのように、このようなタスクに最適です、まだ最初に克服しなければならないハードルの数を含みます。携帯自家蛍光によって発生するノイズは、測定の感度を制限するため、最小限に保たれるべきです。 TIRF顕微鏡は、理想的な選択13-15のタンパク質で飾ら平面ガラスサポート脂質二重層の形で、非常によく12本必要性を提供していますが、ガラススライドの官能化を必要とします。一部reconstitutiveアプローチのもう一つの利点は、組換え二重層常駐FRETパートナーがずっとmとすることができるということです鉱石は容易に細胞表面に発現するタンパク質で可能であるよりも小さく、明るい蛍光体で定量的な部位特異的かつ合理的な方法でラベルされました。以前に2テストされたようなTCRは、組換えのscF V S、T細胞認識に影響を与えないものでタグ付けされています。また、SLBのタンパク質組成物は、例えばpMHCsの密度およびアクセサリー因子の選択は、1つの特定のニーズに調整することができます。我々は以前、刺激pMHCsの様々な密度で実験を行っているが、 オフ 2D-Kおよび2D-K D 2で有意差を検出していません。
これまでのところ、ここでのMHCクラスII分子の認識は、主に両端が開いているので、蛍光体の結合のためのリンカーを含む、より大きなペプチドを収容し、それらのペプチド結合溝、の性質上、のみを扱ってきました。いくつかのケースでは、このようなアプローチは、MHC CLAを標識するために働く可能性がありますSS私は16分子が、大きな注意が実験での使用を確認するために取られるべきです。表面プラズモン共鳴によってインビトロで測定されるT細胞増殖アッセイ、ならびに動力学結合のpMHC-TCRを介して測定することができる抗原に対するT細胞の感受性を、リンカーおよびフルオロフォアの添加によって影響されてはなりませんペプチド。代わりに、私は自分自身を分子MHCクラスは、重鎖(未発表の観察)の配列内の不対システインの導入と部位特異的に標識することができます。
適切な分子プローブを使用して任意のシナプスのタンパク質 - タンパク質相互作用は、原理的には、本明細書中に記載の方法で研究することができます。このようなプローブは、 例えば、のscF V Sまたは設計されたアンキリンリピートタンパク質(ダルピン)17、一価であるべきであり、関心の相互作用に影響を与えることなく、安定的に彼らの標的に結合する必要があります。もちろん、構造informationが合理的なプローブ設計のための非常に望ましいですが、絶対に必要ではありません。 FRETパートナーの新しいペアを確立するときは、最初に一括でFRETを記録し、分析することをお勧めします。標識結合の部位は、FRETシグナルを最大化するために、また、測定されたFRET収率がインター染料距離に基づいて異なることを確認するために、大幅に変化させることができます。システムが最適化されると、単一分子は、信号が10〜30%に多量のFRETパートナーのラベルを制限し、単一分子は照明の分野で解決可能になるまで少量のFRETパートナーを漂白することによって記録することができるFRET。
少なくとも最後にではなく、それはのSLBは、生理的形質膜のすべての側面一部に近似するがないことに留意すべきです。このような膜の曲率と柔軟性、ドメイン区画、細胞骨格の再編成および細胞運動だけでなく、表面発現膜タンパク質の高いさまざまな資質としては、SLBので表されていないが、Tに影響を与える可能性があります彼は調査中で処理します。多くの努力は、TIRFイメージングにアクセスできない生理的シナプスにおける単一分子分解能でのタンパク質 - タンパク質相互作用をモニタリングすることを可能にするの画像診断法を確立するために投資する必要があります。
開示事項
The authors declare that they have no competing financial interest.
謝辞
MAは行財政支援のためのオーストリア科学基金(FWF、J3086-B11)と感謝のシュレーディンガーの交わりマックスプランク協会によってサポートされていました。 GSとJHは、ウィーン科学技術基金(WWTF、LS13-030)によってサポートされていました。
資料
| Name | Company | Catalog Number | Comments |
| LB-media | Fisher Scientific | 10000713 | bacterial expression |
| Sf900 II | Life Technologies | 10227402 | insect cell media for baculo virus production |
| Insect-XPRESS with L-glutamine (Lonza) | Fisher Scientific | 10564038 | insect cell media for baculo virus expression |
| Sf9 cells | Life Technologies | 11496-015 | cells for virus production and expansion |
| High Five Cells | Life Technologies | B855-02 | cells for potein expression |
| LB-media | Fisher Scientific | 10000713 | bacterial expression |
| Centramate System | Pall | protein concentartion from large volumes | |
| Centramate cassette 10kDa cutoff | Pall | OS010T12 | protein concentartion from large volumes |
| Amicon Ultra-15 Centrifugal Filter Units | EMD Millipore | UFC900308 | protein concentartion |
| Amicon Ultra-4 Centrifugal Filter Units | EMD Millipore | UFC800308 | protein concentartion |
| Amicon Stirred Ultrafiltration Cell Model 200 mL | EMD Millipore | 5123 | protein concentartion |
| Äkta pure 25L | GE Healthcare | 29-0182-24 | protein purification |
| Superdex 200 10/300 GL | GE Healthcare | 17-5175-01 | protein purification |
| Superdex 75 10/300 GL | GE Healthcare | 17-5174-01 | protein purification |
| Mono Q 5/50GL | GE Healthcare | 17-5166-01 | protein purification |
| Ni Sepharose 6 Fast Flow | GE Healthcare | 17-5318-01 | protein purification |
| Tricorn 10/20 column | GE Healthcare | 28-4064-13 | protein purification |
| Gilson HPLC system | Gilson | purificationof fluorochrome-coupled peptides | |
| Pursuit XRs C18, 5 µm particle size, 21.2*250mm column size | Agilent | A6000250X212 | purificationof fluorochrome-coupled peptides |
| Pursuit XRs C18, 5 µm particle size, 21.2*50 mm column size | Agilent | A6000050G212 | purificationof fluorochrome-coupled peptides |
| Tricorn 10/20 column | GE Healthcare | 28-4064-13 | protein purification |
| Gilson HPLC system | Gilson | purificationof fluorochrome-coupled peptides | |
| Pursuit XRs C18, 5 µm particle size, 21.2*250mm column size | Agilent | A6000250X212 | purificationof fluorochrome-coupled peptides |
| Pursuit XRs C18, 5 µm particle size, 21.2*50 mm column size | Agilent | A6000050G212 | purificationof fluorochrome-coupled peptides |
| Cy3 maleimide | GE Healthcare | PA23031 | site-specific protein labeling via mutant unpaired cysteines |
| Cy5 maleimide | GE Healthcare | PA25031 | site-specific protein labeling via mutant unpaired cysteines |
| Alexa Fluor 555 C2 Maleimide | Life Technologies | A-20346 | site-specific protein labeling via mutant unpaired cysteines |
| Alexa Fluor 647 C2 Maleimide | Life Technologies | A-20347 | site-specific protein labeling via mutant unpaired cysteines |
| Fura-2, AM, cell permeant | Life Technologies | F-1221 | calcium-sensitive dye for cell labeling |
| dimethyl sulfoxide | Sigma Aldrich | 151874 | for dissolving fura-2 am |
| Hank's Balanced Salt Solution plus calcium/magnesium | Fisher Scientific | 10225362 | imaging buffer |
| PBS | Life Technologies | 14190-136 | |
| Bovine Serum Albumin lyophilized powder | Sigma Aldrich | A2153 | supplement for imaging buffer |
| 14-4-4S antibody | affimetrix eBioscience | 14-5980-81 | blocking antibody for H2-I-Ek (recognized by the 5c.c7, 2B4 and AND TCR) |
| 5 ml polypropylene round-bottom tube | Becton Dickinson | FALCON 352063 | |
| 0.22 μm Ultrafree-MC centrifugal filter unit | EMD Millipore | UFC30GV0S | |
| Syringe filter 0.2µm | Millipore | GVWP04700 | |
| TetraSpeck Microspheres, 0.1 µm, fluorescent blue/green/orange/dark red | Life technologies | T-7279 | |
| Microscope for fura-2-based calcium measurements | LEICA | DMI4000B | |
| Microscope for (single molecule) FRET measurements | LEICA/ZEISS/NIKON/OLYMPUS | for details please refer to parallel JoVE contribution by Axmann et al. | |
| planar supported lipid bilayers | for details please refer to parallel JoVE contribution by Axmann et al. | ||
| RPMI 1640, with L-Glutamine | Life Technologies | 11554416 | T-cell media |
| non-essential amino acid 100X | Hyclone | SH30238.01 | T-cell media supplement |
| penicillin/streptomycin/L-glutamine 100x | Life Technologies | 12000226 | T-cell media supplement |
| 2-mercaptoethanol | Sigma Aldrich | M6250 | T-cell media supplement |
| mouse interleukin-2 recombinant protein | BPS Bioscience | 90185-B | T-cell media supplement |
| Research Grade Fetal Bovine Serum | Hyclone | SV30160.03 | T-cell media supplement |
| Origin (analysis program) | OrigenLab | http://www.originlab.com/ | non-linear fitting of two parameters (tauoff, [ntlag]) |
参考文献
- Garcia, K. C., Adams, J. J., Feng, D., Ely, L. K. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat Immunol.. 10, 143-147 (2009).
- Huppa, J. B., et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature.. 463, 963-967 (2010).
- Huppa, J. B., Davis, M. M. The interdisciplinary science of T-cell recognition. Advances in immunology.. 119, 1-50 (2013).
- Axmann, M., Schuetz, G. J., Huppa, J. B. Single Molecule Microscopy on Planar Supported Bilayers. Journal of Vizualized Experiments J. Vis. Exp.. 101, e53158(2015).
- Xie, J., et al. Photocrosslinkable pMHC monomers stain T cells specifically and cause ligand-bound TCRs to be preferentially transported to the cSMAC. Nat Immunol. 13, 674-680 (2012).
- Jares-Erijman, E. A., Jovin, T. M. FRET imaging. Nat Biotechnol. 21, 1387-1395 (2003).
- Toebes, M., et al. Design and use of conditional MHC class I ligands. Nat Med. 12, 246-251 (2006).
- Tsumoto, K., et al. Highly efficient recovery of functional single-chain Fv fragments from inclusion bodies overexpressed in Escherichia coli by controlled introduction of oxidizing reagent--application to a human single-chain Fv fragment. J Immunol Methods. 219, 119-129 (1998).
- Ruegg, U. T., Rudinger, J. Reductive cleavage of cystine disulfides with tributylphosphine. Methods Enzymol. 47, 111-116 (1977).
- Ruprecht, V., Brameshuber, M., Schütz, G. J. Two-color single molecule tracking combined with photobleaching for the detection of rare molecular interactions in fluid biomembranes. Soft Matter. 6, 568-581 (2010).
- Dustin, M. L., Bromley, S. K., Davis, M. M., Zhu, C. Identification of self through two-dimensional chemistry and synapses. Annu Rev Cell Dev Biol. 17, 133-157 (2001).
- Axelrod, D. Cell-substrate contacts illuminated by total internal reflection fluorescence. The Journal of cell biology. 89, 141-145 (1981).
- Grakoui, A., et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 285, 221-227 (1999).
- Kaizuka, Y., Douglass, A. D., Varma, R., Dustin, M. L., Vale, R. D. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc Natl Acad Sci USA. 104, 20296-20301 (2007).
- Varma, R., Campi, G., Yokosuka, T., Saito, T., Dustin, M. L. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 25, 117-127 (2006).
- Purbhoo, M. A., Irvine, D. J., Huppa, J. B., Davis, M. M. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol. 5, 524-530 (2004).
- Binz, H. K., Stumpp, M. T., Forrer, P., Amstutz, P., Pluckthun, A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol. 332, 489-503 (2003).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved