
Method Article
微生物分子シグネチャを用いた河川における水圧破砕の影響評価
要約
ここでは、水と堆積物の微生物群集を分析することによって、近くの河川に対する水圧破砕の影響を調査するプロトコルを提示します。
要約
一般に「フラッキング」と呼ばれる油圧破砕(HF)は、高圧水、砂、化学物質の混合物を使用して岩石を破壊し、石油とガスを放出します。このプロセスは、以前は入手できなかった資源へのアクセスを提供し、現在は米国の天然ガス全体の3分の2を生産しているので、米国のエネルギー産業に革命をもたらしました。フラッキングは米国経済にプラスの影響を与えているが、いくつかの研究は、その有害な環境への影響を強調している。特に懸念されるのは、流域全体の健康に不釣り合いな大きな影響を与えるため、特に重要なヘッドウォーターストリームに対するフラッキングの影響です。これらのストリーム内の細菌は、存在する細菌とその乱れた流れの豊富さは、それ以外の比較可能だが乱れない流れのものとは異なると予想されるので、ストリームの健康の指標として使用することができます。したがって、このプロトコルは、ストリームがフラッキングによって影響を受けたかどうかを判断するために細菌コミュニティを使用することを目的としています。このためには、フラッキング(影響を受ける可能性がある)や上流の近くの小川や、フラッキング活動の異なる流域(影響を受けていない)からの堆積物と水のサンプルを収集する必要があります。これらのサンプルは、次いで核酸抽出、ライブラリー調製、およびシーケンシングを行い、微生物群集組を調査する。相関分析と機械学習モデルは、その後、コミュニティの変動を説明する特徴を特定し、フラッキングの影響に対する予測バイオマーカーを特定するために使用することができます。これらの方法は、フラッキングに近いヘッドウォーターストリーム間の微生物群集の様々な違いを明らかにし、フラッキング活動の環境への影響に関する将来の調査の基礎となる。
概要
水圧破砕(HF)、または「フラッキング」は、化石燃料の需要が増加し続けるにつれてますます普及している天然ガス抽出の方法です。この技術は、高出力の掘削装置を使用して、メタンが豊富なシェール鉱床に水、砂、および化学物質のブレンドを注入し、通常は閉じ込められたガスを放出する。
これらの非伝統的な収穫技術は比較的新しいため、近くの水路に対するそのような慣行の影響を調査することが重要です。フラッキング活動は、機器輸送と井戸パッド建設のための広大な土地のクリアを義務付けています。井戸パッド2ごとに約1.2~1.7ヘクタールの土地をクリアする必要があり、システム3の流出と水質に影響を与える可能性があります。どのような殺生物剤が使用されているかを含め、フラッキング流体の正確な化学組成を取り巻く透明性の欠如があります。さらに、フラッキング排水は非常に生理学的になる傾向があります。さらに、廃水には、金属や天然放射性物質2が含まれていてもよい。そのため、人的ミスや機器の故障によるフラッキング液の漏出や流出の可能性が懸念される。
河川生態系は、周囲の景観4の変化に非常に敏感であることが知られており、流域全体の生物多様性5と適切な栄養サイクリング6を維持するために重要です。微生物は淡水の流れで最も豊富な生物であり、栄養循環、生分解、一次生産に不可欠です。微生物群集の組成および機能は、perturbanceに対する感受性のために生態系に関する情報を得るための素晴らしいツールとして機能し、最近の研究では、フラッキング活動7,8に近い状態に基づいて観察された細菌集合体の明確な変化が示されている。例えば、ベージェリンキア、バークホルデリア、メサノバクテリウムはフラッキング近くの小川で濃縮されたと同定され、シュードノカルディア、ニトロスピラ、ロドバクターはフラッキング7の近くにない小川で濃縮された。
16SリボソームRNA(rRNA)遺伝子の次世代シーケンシングは、全ゲノムシーケンシングアプローチ9よりも速くかつ安価である細菌群組成を決定する手頃な方法である。分子生態学の分野における一般的な実践は、16S rRNA遺伝子の高可変V4領域をシーケンシング分解能に使用する方法であり、多くの場合、同定の広い範囲を有する属レベルまで下がることが、予測不可能な環境試料に理想的である。この技術は、公開された研究で広く実装されており、水生環境に対するフラッキング操作の影響を特定するために成功裏に利用されています7,8.しかし、細菌は16S rRNA遺伝子のコピー数が変化し、検出された存在量10に影響を与えるのは注目に値する。これを説明するためのいくつかのツールがありますが、その有効性は疑わしい10です。有病率が急速に増加し、この弱点を欠いているもう一つの方法は、すべてのRNAが配列決定され、研究者が活性細菌とその遺伝子発現の両方を同定することを可能にするメタトランスクリプトームシーケンシングです。
したがって、以前に発表された研究7、8、11、12の方法とは対照的に、このプロトコルは、微生物群集機能(メタトランスクリプトミクス)を調査するためのサンプル収集、保存、処理、および分析もカバーしています。ここに記載されているステップは、研究者が抗菌性遺伝子を含む彼らの流れの微生物によって発現される遺伝子と経路にどのような影響を与えたかを見ることができます。さらに、サンプル収集に関する詳細レベルが向上しました。いくつかのステップとメモは経験豊富な研究者には明らかに見えるかもしれませんが、研究を始めたばかりの人にとっては非常に貴重な場合があります。
ここでは、我々の研究室の数年間の経験に基づいて、近くの河川にフラッキングが及ぼす影響を調査する手段として、細菌遺伝データを生成するためのサンプル収集および処理の方法を説明する。これらのデータは、下流アプリケーションで使用して、フラッキング状態に対応する差異を識別できます。
プロトコル
核酸抽出のための堆積物サンプルの収集
- 滅菌50 mL円錐管を流水に浸します。不要な人体汚染を避けるために、サンプル採取時に手袋を着用してください。このステップは、海岸から、または水の中にある場合は上流に向いているかのどちらかを実行します。
- 円錐管が水没している間、キャップを取り外し、約3mLの堆積物を深さ1〜3cmから円錐形チューブにすくい取ります。
- 水から円錐形のチューブを取り出し、堆積物サンプル(約1 mL)を覆う薄い層を除くすべての水を捨てる。
- 1000 μL ピペットと適切なピペットチップを使用して、3 mL の DNA/RNA 保存剤 (保存剤の仕様については 、材料表 を参照) を、収集したサンプルに加えます。ピペットの先端を滅菌ピペットチップボックスに入れておき、使用前に直ちに取り付け、使用後に廃棄してください。キャップされた円錐形チューブを10回反転し、防腐剤とサンプルが完全に混合されていることを確認します。
注 ステップ 1.4 は必要ありませんが、後で堆積物から RNA を抽出する場合は強くお勧めします。 - サンプルコレクションの残りの部分については、氷の上にサンプルを置きます。回収から戻ったら、サンプルを16S分析(DNA)、-70°C(メタトランスクリプトミクス分析(RNA)に使用する場合は-20°Cで冷凍庫に保管します。
2. 核酸抽出用のフィルターコレクション
- 滅菌1 Lボトルのキャップを取り外します。上流または海岸から向いている間、ボトルを上に流水で満たし、それを捨てなさい。ボトルの条件を整えるために、このプロセスをさらに2回繰り返します。ボトル全体を4回目に充填し、キャップします。
注:1 Lボトルを再利用する場合は、2分間10%漂白剤で洗い流し、続いて脱イオン水で3回、エタノール70%で1回洗い流し、最終的に設定でオートクレーブする:121.1°Cで30分の暴露時間と15分乾燥時間を使用して滅菌することができます。オートクレーブの間に、ボトルがプロセスで圧縮されるのを避けるために、ボトルのキャップは非常に緩くする必要があります。 - 安定した表面に着いたら、滅菌ルアーロックシリンジを使用し、フルボリュームを描画します。その後、シリンジを無菌およびDNA/RNAフリーの直径1.7cmのポリエーテルスルホンフィルターに0.22 μmの細孔サイズで接続し、プランジャーを押してフィルターを通してボリューム全体を押し込みます。ボトルに集められた総容積(1L)がフィルターに押し込まれるまで、このプロセスを繰り返します。
注: シリンジの体積は、フィルタを通して押される水の総量が追跡される場合、可変することができます。しかしながら、一般的には、60mLが好ましい。1 Lは理想的ですが、逸話的には、少なくとも200 mLの体積は、DNAおよびRNAの抽出に十分なバイオマス(mL当たり約20,000細胞を想定)を依然として収集する可能性が高い。 - 約20mL相当の空気をシリンジに引き上げ、フィルターを通して押し込んで、フィルターから余分な水を取り除きます。
注: ステップ 2.4 が実行された場合、防腐剤の損失を防ぐことができます。 - P1000マイクロピペットを使用して、フィルターを水平に保持したまま、フィルターの大きな開口部(シリンジに取り付けられた場所)を通して排出して、DNA/RNA 保存剤を2 mL加えます。ピペットの先端は、保存剤がフィルタに入るようにピペットが押し下げられたとき、フィルターのバレル内にあるべきです。使用後にチップを変更します。
注:堆積物の収集と同様に、このステップは必要ではありませんが、特にRNAの場合は、後で核酸収量を増加させ、強く推奨されます。 - パラフィンフィルムの1つの正方形を剥がし、シールするフィルターの各開口部/端の周りにしっかりと包みます。パラフィンフィルムを包んだフィルターを滅菌サンプルバッグに入れ、収集中に袋全体を氷の上に置きます。
注: フィルターのラップに使用される側が無菌、すなわち、以前は環境にさらされていないことを確認してください。 - サンプリングから戻ると、16Sの場合は-20°C、メタ転写学の場合は-70°Cでフィルターを保管してください。
3. 核酸の抽出と定量
- サンプルの移管を開始する前に、10%の漂白剤と70%エタノールで作業領域を清掃してください。
- 堆積物(ステップ1.5から)の場合、一般的に、サンプルの〜0.25 gを使用してください。70%エタノールのビーカーに浸し、サンプル間でエタノールを焼き払って金属工具を殺菌します。
- フィルター(ステップ2.6から)の場合は、抽出のためにフィルターペーパーを無菌チューブに移動します。これを行うには、次の手順に従います。
- アルミ箔を折り畳んで、折り目の内側が外部環境にさらされないようにして、121.1 °Cと5分の乾燥時間で折り曲げた部分をオートクレーブします。
- 70%エタノールと開炎でバイスグリップを殺菌します。その後、バイスグリップを使用して、滅菌表面のフィルターケーシングを開き、ケーシングからコアを取り除きます。
- 滅菌メスを使用して、上部と下部をスライスしてから縫い目に沿ってスライスすることで、フィルターペーパーをコアから切り離します。滅菌ピンセットを使用して濾紙を折り、メスを使用してフィルターを小さくカットします。
- 抽出のためにマイクロ遠心チューブにフィルター片を入れる。サンプルの不要な汚染につながるため、フィルターペーパーが滅菌されていない表面や核酸が存在する可能性のある表面に接触していないことを確認してください。
- 前述のとおりにDNA分離を行うか、市販のカラムベースキットを使用して行います(「材料表」を参照)。以下に、一覧に記載されている商用キットの手順を簡単に説明します。
- サンプル内の細胞をビーズチューブに移し、少なくとも5分間高速で細胞破壊器に施します。遠心分離機と滅菌マイクロ遠心チューブに上清を転送します。
- 上清(1:1ボリューム)にリシスバッファーを追加し、提供されたフィルタ(黄色)に転送します。フィルターを遠心分離します。
- フィルターを新しい滅菌マイクロ遠心チューブに移します。準備バッファー(400 μL)を加え、遠心分離機を加え、流れを通して廃棄します。
- 洗浄バッファー(700 μL)を加え、遠心分離機を加え、流れを通して廃棄します。次に、洗浄バッファー(400 μL)、遠心分離機を加え、再度流れを廃棄します。
- フィルターを新しい滅菌マイクロ遠心チューブに移します。50 μLのDNase/RNaseフリーウォーターでエルテを使用し、遠心分離機の前に室温で5分間座らせます。
- その間、キューバケーション期間中に、III-HRCフィルターをコレクションチューブに入れ、HRC調製溶液(600μL)を添加し、続いて8,000xgで3分の遠心分離ステップを行います。
- 準備されたフィルターを無菌マイクロ遠心チューブに移動します。溶出したDNAをステップ3.4.5からこのフィルターに移し、遠心分離機を16,000 x g で3分間転送します。流れには抽出されたDNAが含まれています。
- 堆積物とフィルターの両方のDNA抽出物を-20 °Cに保存します。
注:DNA抽出物は、安定した温度、限られた光暴露、有害な汚染物質14を仮定して-20°Cで約8年間保存することができます。 - メーカーのプロトコルに従ってRNAの分離を行います。RNA抽出物を-80°Cに保存します。
- サンプル内の細胞をビーズチューブに移し、少なくとも5分間高速で細胞破壊器に施します。遠心分離機と滅菌マイクロ遠心チューブに上清を転送します。
- 上清(1:1ボリューム)にリシスバッファーを追加し、提供された列(黄色)に転送します。柱を遠心分離します。
- 流れに95-100%エタノールの等しい量を加え、上下に5回ピペットで混ぜます。
- IICGカラム(緑色)を無菌マイクロ遠心チューブに置きます。混合溶液をカラムと遠心分離機に移します。
- 洗浄バッファー(400 μL)を加え、遠心分離機を加え、流れを通して廃棄します。
- DNase Iの5 μLおよび75 μLのDNA消化バッファーをカラムに加え、室温で15分間インキュベートします。
- 準備バッファー(400 μL)、遠心分離機を追加し、流れを通して廃棄します。
- 洗浄バッファー(700 μL)を加え、遠心分離機を加え、流れを通して廃棄します。次に、洗浄バッファー(400 μL)、遠心分離機を加え、再度流れを廃棄します。
- カラムを新しい滅菌マイクロ遠心チューブに移します。50 μLのDNase/RNaseフリーウォーターでエルテを使用し、遠心分離する前に5分間座らせます。
- そのインキュベーション期間中、III-HRCフィルターを回収チューブに入れ、HRC調製溶液(600μL)を加えて、8,000xgで3分の遠心分離ステップを行います。
- 準備されたフィルターを無菌マイクロ遠心チューブに移動します。溶出したRNAをステップ3.6.9からこのフィルターに移し、遠心分離機を16,000 x g で3分間移動します。流れには抽出されたRNAが含まれています。
注:RNA抽出物は、15を劣化し始める前に1年間しか保存できません。DNAとRNAの両方の抽出物は、凍結融解を繰り返すことによって分解されます。一部のプロトコルは、同じサンプル16,17からDNAとRNAの両方を抽出することを可能にする。
- 抽出したDNAおよびRNAサンプルを、フッ素計または分光光度計で定量化します。例えば、フッ素計DNA濃度値については 表1 を参照してください。分光光度計定量プロトコルの例については、参照18を参照してください。 材料表 に記載されているキットを使用した沈み込みDNA濃度値は一般的に1〜40 ng/μLの範囲ですが、フィルターDNA濃度値は0.5~10 ng/μLの 範囲である傾向 があります。 通常は 0.5 から 5 ng/μL の範囲です。
4. 16S rRNAライブラリの作成
- 10%漂白剤と70%エタノールで作業領域を清掃してください。作業領域は、層流条件(層流フード)を生成することができる密閉空間でなければなりません。
- DNA抽出物(ステップ3.5から)を使用し、層流条件下で16S rRNA19 のV4超可変領域を増幅する地球マイクロバイオームのウェブサイトに記載されているような標準的なPCRプロトコルで16S rRNAアンプリコンシーケンシングのサンプルを準備します。
- 先に説明した2%アガロースゲルを調製し、17を固化させる。7 μLのPCR産物と13 μLのDNaseフリーウォーターを混合します。ゲルローディング染料を最終濃度の1倍に加えます。アガロースが固まったら、このPCR製品を2%アガロースゲルに混ぜてロードします。
注:代わりに、プレキャストゲルを使用して、これらのゲルは、より速く実行し、事前に作られています。 - 地球マイクロバイオームのプロトコルを使用して、16S rRNA V4アンプリコンの増幅に成功した386のバンドサイズを確認するために、60〜90分間90 Vでゲルを実行します。
5. DNA 16S rRNA ライブラリの精製
- 明るいバンドを生み出したサンプル用のPCR製品を10 μL、適切なサイズの無菌マイクロ遠心チューブでかすかなバンドを生み出したサンプルに13 μLプールします。
- 得られたプールの濃度を蛍光計または分光光度計を使用して確認し、以前と同様に2%のアガロースゲルを調製する。理想的には、プールは少なくとも10 ng /μLの濃度を有し、ほとんどのサンプルは約25ng/μLの濃度を有する必要があります。
- 濃度と体積が許し、2%アガロースゲルのウェルで150〜200 ngの周りに負荷を与える。
- ゲルを90ボルトで60~90分間動かします。
- 2%アガロースゲルを実行して、プールされたライブラリを浄化します。
- ゲルから386bpのDNAバンドを物品化し、先に説明した市販のキットを用いてプールされたライブラリを精製する20.精製したDNAを10 mMトリスCl(pH 8.5)の30 μLで溶出します。このステップは、DNAまたはRNA抽出とは異なる領域で実行して、将来の汚染を防ぎ、ゲルを切断するとPCRアンプリコンが実験者と周辺領域の両方に広がります。
- フッ素計または分光光度計を使用して、精製プールの濃度を確認します。精製がうまくいけば、その濃度は未精製プールの少なくとも半分でなければなりません。一般的に、最終濃度は5〜20 ng/μLの範囲でなければなりません。
- 次世代シーケンシング用に精製ライブラリを送信します。輸送容器にドライアイスを入れて、輸送中に冷たく保たれるようにしてください。
6. RNAライブラリの作成と精製
- いくつかの市販キットを使用してRNAライブラリを作成できます。どちらの使用でも、滅菌層流動環境で作業している間に書かれたように、製造業者のプロトコルに従ってください。 資料一覧 のキットのプロトコルの非常に要約されたバージョンは、以下に示されています 21.
- 最初のストランドcDNA合成マスターミックス(ヌクレアーゼフリー水8μL、ファーストストランド合成エニスムミックス2μL)を作り、サンプルに加えます。プロトコルで指定された条件でサンプルをサーモサイクラーに入れます。
- 2番目の鎖cDNA合成マスターミックス(2番目の鎖合成反応バッファーの8 μL、4 μL第2鎖合成酵素ミックス、および48μLのヌクレアーゼフリー水)を氷上に作り、サンプルに加えます。サーモサイクラーに16°Cに1時間置きます。
- 提供されたビーズ(144 μL)を加え、2つの80%エタノールの和を行って反応を浄化します(200 μL)。
- 提供されたTEバッファー(53 μL)でエルテを、清浄なPCRチューブに50μLの上清を移します。PCRチューブを氷の上に置きます。
- 氷の上にエンドプレップマスターミックス(エンドプレップ反応バッファーの7 μLとエンドプレップ酵素ミックスの3 μL)を作り、PCRチューブに加えます。プロトコルで指定された条件を持つサーモサイクラーに PCR チューブを配置します。
- 希釈アダプター(2.5 μL)、ライゲーションマスターミックス(30 μL)、ライゲーションエンハンサー(1 μL)溶液を氷の上に混ぜます。混合溶液をサンプルに加え、20°Cで15分間サーモサイクラーに入れます。
- 提供されたビーズ(87 μL)を添加し、TEを17 μLだけ添加する以外は、以前と同様にエタノールの溶解と溶出を行って反応を浄化します。
- インデックス(10 μL)とQ5マスターミックス(25 μL)のソリューションを追加し、プロトコルに記載されている条件を持つサーモサイクラーに配置します。
- 提供されたビーズ(45 μL)を添加し、2個のエタノールのスミッシュ(200 μL)を加え、23 μLのTEで溶出して反応を浄化します。20 μL をクリーン PCR チューブに移します。
- バイオアナライザー、蛍光計、または分光光度計を使用して、RNAの検出可能な濃度をライブラリで確認します。
- メタトランスクリプトライブラリをほぼ等量比でプールします。
- 250と400 bpの間の物品化断片を除いて、16Sライブラリ精製のための同じプロトコルに従ってライブラリを精製する。16Sライブラリは増幅領域を表す明確なバンドを持っていたのに対し、ここでの結果はスミアである。
- 以前と同様に、精製されたライブラリーの濃度を確認してください。
- 精製したライブラリをドライアイスでシーケンシング施設に送ります。
注:あるいは、RNA抽出物は図書館の準備とシーケンシングのために大学や民間企業に送ることができます。
7. 微生物群集解析
- シーケンス処理が完了したら、サンプル データにアクセスします。使用できるコンピュータにダウンロードします。
注: 理想的には、デバイスには 16 ギガバイト以上の RAM が必要です。コンピューティング要件の説明 (Qiime2 の場合) については、https://forum.qiime2.org/t/recommended-specifications-to-run-qiime2/9808 を参照してください。 - 16S rRNAデータを分析するには、モトゥール、QIIME2、Rなどのソフトウェアを使用します。QIIME2 16S 解析チュートリアルの例については、ここ https://docs.qiime2.org/2020.11/tutorials/moving-pictures/ を参照してください。
- メタトランスクリプトミクス(RNA)データの場合、HUMAnN2およびATLASを使用して、サンプルに存在する遺伝子と経路を決定します。
注: 多様性とランダムフォレスト分析で最高潮に達するメタトランスクリプトミクスパイプラインの例は 、補足情報ファイルに記載されています。すべてのコマンドは、例えば、Macユーザーのためのターミナルなどのコマンドラインを介して実行されます。
結果
DNAおよびRNA抽出の成功は、様々な装置やプロトコルを用いて評価することができます。一般的に、いずれかの検出可能な濃度は、抽出が成功したと結論付けるのに十分であると考えられる。 その後、表 1 を 調べると、1 つを除くすべての抽出が成功と呼ばれる。このステップでの失敗は、多くの場合、低い初期バイオマス、不良サンプル保存、または抽出中の人為的ミスによるものです。フィルターの場合、濃度が検出以下であっても抽出が成功している可能性がある。これらの抽出物がPCR(16Sを行う場合)またはライブラリ調製後の検出可能な濃度(メタトランスクリプトミクス)のバンドを得ない場合、それらは本当に失敗した可能性が高いです。
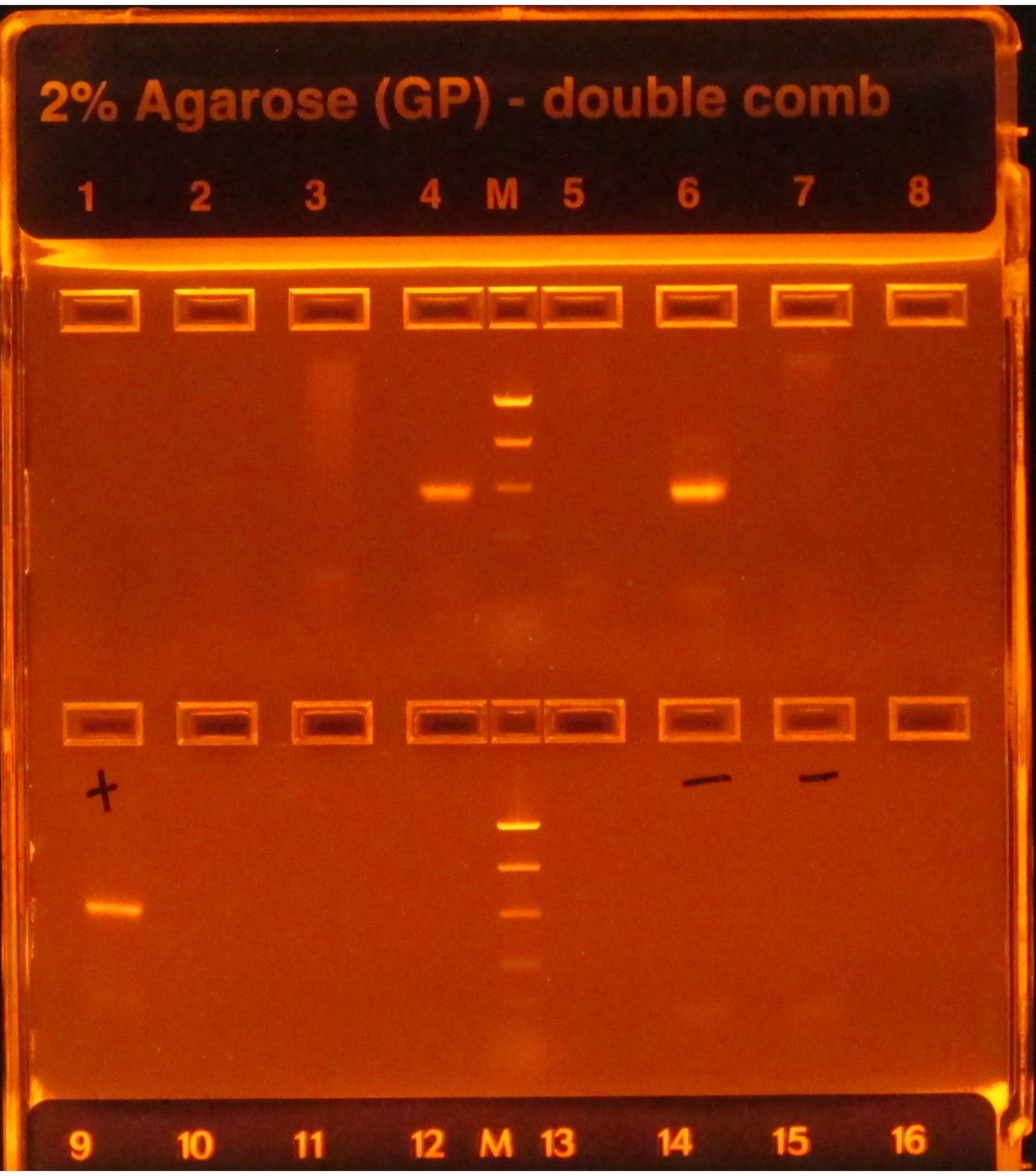
16Sプロトコルに従う場合、 図1のウェル4と6に見られるようにPCR増幅後の明るいバンドは成功を示し、一番上の行の他のウェルに見られるようにバンドの欠如は失敗を示します。さらに、ゲルレーンの明るいバンドに負のPCRコントロールを含む場合も、負のコントロールに影響を与える汚染がサンプルに影響を与えないと考える危険があるため、障害を示します。
16Sとメタトランスクリプトミクスの両方について、配列決定の成功は、得られた配列数を見ることによって評価することができる(図2)。16S サンプルには、最低 1,000 個のシーケンスを含める必要があり、少なくとも 5,000 個が理想的です (図 2A)。同様に、メタトランスクリプト法サンプルは、最低500,000個の配列を持ち、少なくとも2,000,000個が理想的である必要があります(図2B)。これらの最小値よりも少ないシーケンスを持つサンプルは、細菌群を正確に表さない可能性があるため、分析には使用しないでください。ただし、最小と理想の間に収まっているサンプルは使用できますが、その範囲に多数のサンプルが含まれる場合は、より慎重に解釈する必要があります。
その後のダウンストリーム分析の成功は、期待される出力ファイルが取得されたかどうかに基づいて単純に判断できます。いずれにせよ、QIIME2やR(図3)などのプログラムは、フラッキングに基づいて細菌群集間の潜在的な有意差の評価を可能にするべきである。 図3 のデータは、16Sおよびメタトランスクリプトミクス分析のために13の異なるストリームで21の異なる部位から堆積物サンプルを収集することによって得られた。これらの21のサイトのうち、そのうちの12はフラッキング活動の下流にあり、HF+に分類され、そのうちの9つはフラッキング活動の上流か、フラッキングが起こっていない流域のいずれかでした。これらのストリームは HF-に分類されました。フラッキング活動の存在に加えて、ストリームはそれ以外の場合は同等でした。
これらの違いは、フラッキングステータスに基づいて一貫した構成シフトの形を取る可能性があります。その場合、図 3A および図 3Bのように、HF+ サンプルと HF- サンプルは PCoA プロットで互いに離れてクラスター化することが期待されます。これらの明らかな変化が単なる聖任法のアーティファクトではないことを確認するには、さらなる統計分析が必要です。例えば、図3Aおよび図3Bが基づく距離行列上のPERMANOVA22検定は、フラッキング状態に基づいて明らかにされた有意なクラスタリングに基づいているため、プロットで観察された分離は、聖任のアーティファクトではなく、サンプルの細菌群集間の違いと一致することを意味する。重要なPERMANOVAまたはANOSIMの結果は、HF+サンプルとHF-サンプルの間の一貫した違いを強く示すもので、HF+サンプルがフラッキングの影響を受けたことを示し、p値が高い場合はサンプルが影響を受けないことを示します。メタトランスクリプトデータも同様に、同じ方法を用いて視覚化および評価することができる。
微分特性(微生物または機能)を調べると、サンプルも影響を受けているという証拠が明らかになります。差分フィーチャを決定する方法の 1 つは、ランダム フォレスト モデルを作成することです。ランダムフォレストモデルを使用して、サンプルのフラッキング状態をどの程度適切に分類できるかを確認できます。モデルが偶然予想以上に優れている場合、それはフラッキングステータスに依存する違いの追加の証拠になります。さらに、最も重要な予測変数は、サンプルを正しく区別するために最も重要な機能を明らかにするでしょう(図3C)。これらの機能はまた、フラッキングステータスに基づいて一貫して異なる値を持っていたでしょう。これらの差動機能が決定されると、文献をレビューして、以前にフラッキングに関連付けられているかどうかを確認できます。しかし、ほとんどの場合、16S rRNA組成データしか使用していないため、差動関数を決定する研究を見つけるのは難しいかもしれません。したがって、差動機能の影響を評価するために、考えられる方法の1つは、それらが以前にフラッキング液で一般的に使用される殺生物剤に対する潜在的な耐性と関連しているかどうか、または非常に生理的な条件を容認するのに役立つかどうかを確認することです。さらに、対象の分類の機能プロファイルを調べると、フラッキングの影響の証拠が明らかになる可能性があります(図 3D)。例えば、タクソンがランダムフォレストモデルによって差と識別された場合、HF+サンプル中の抗菌性プロファイルをHF-サンプルのプロファイルと比較することができ、それらが大きく異なる場合は、殺生物剤を含むフラッキング流体がストリームに入ることを示唆する可能性があります。
| サンプル ID | 濃度(ng/μL) |
| 1 | 1.5 |
| 2 | 1.55 |
| 3 | 0.745 |
| 4 | 0.805 |
| 5 | 7.82 |
| 6 | 0.053 |
| 7 | 0.248 |
| 8 | 0.945 |
| 9 | 1.82 |
| 10 | 0.804 |
| 11 | 0.551 |
| 12 | 1.69 |
| 13 | 4.08 |
| 14 | Below_Detection |
| 15 | 7.87 |
| 16 | 0.346 |
| 17 | 2.64 |
| 18 | 1.15 |
| 19 | 0.951 |
表1:フッ素計1xDSDNA高感度アッセイに基づくDNA濃度の例。 14を除くすべてのこれらのサンプルの抽出は、検出可能な量のDNAを有するため成功すると考えられる。

図1:PCR産物を含むe-gelの例 ゲルはUV光の下で事前染色され、視覚化され、その上に存在するDNAが輝く原因となる。PCRは、(はしごに基づいて)予想されるサイズの単一の明るいバンドを持っていたので、最初の行の井戸4と6のサンプルに対して働いた。他の6つのウェルのサンプルのPCRは、バンドを作り出さなかったため失敗しました。陽性対照(第1ウェル、2行目)は明るいバンドを有し、PCRが適切に行われたことを示し、負の対照(ウェル6および7、2列目)はバンドを持っていなかったので、サンプルが汚染されなかったことを示す。陰性のバンドがサンプルと同じくらい明るい場合、PCRはサンプルが汚染の結果だけではないアンプリコンを持っていると仮定するのは危険であるため、障害と見なされていました。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図2:シーケンスカウントの例 (A)16S のシーケンスカウント例。これらの16Sサンプルのほとんどすべてが1,000以上の配列を持っていました。1,000未満の配列を持つ非常に少数は、細菌群集を正確に表現するのに十分な配列を持っていなかったため、下流の分析から除外する必要があります。いくつかのシーケンスは1,000から5,000のシーケンスの間を持っていました。理想的ではありませんが、それらは最低限を超えるのでまだ使用できるでしょう、そしてサンプルの大半は理想的な最低5,000を超えています。(B) メタトランスクリプトミクスの例カウント。すべてのサンプルは、最小(500,000)と理想的な最小(2,000,000)のシーケンス数の両方を超えました。したがって、シーケンスは、それらのすべてのために成功し、すべてのダウンストリーム分析で使用することができました。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図3: 分析例 (A) 重み付き Unifrac 距離行列を使用して計算された座標に基づく PCoA プロットが、QIIME2 を通じて作成され、視覚化された。(B) QIIME2 からエクスポートされた重み付け Unifrac 距離行列で計算された座標に基づく PCoA プロット。座標は、R.メタデータベクトルのPhyloseqとggplot2パッケージを使用して視覚化され、ビーガンパッケージを使用してプロットに適合しました。各ポイントはサンプルの細菌群を表し、より類似したコミュニティ組成物を示す近い点を有する。これらの16Sの沈積サンプルのフラッキング状態に基づくクラスタリングが観察された(PERMANOVA、p=0.001)。さらに、このベクターは、HF+サンプルがバリウム、臭化物、ニッケル、亜鉛のより高いレベルを有する傾向があることを明らかにし、HF-サンプルと比較して異なる細菌群集組に対応した。(C) サンプル間のフラッキング状態を予測するために細菌の存在量をどこで使用できるかをテストしたランダムフォレストモデルの最良の予測因子のプロット。ランダム フォレスト モデルは、ランダム フォレスト パッケージを使用して R を通じて作成されました。上位20個の予測変数は、不純物の減少(HF+およびHF-サンプルをグループ化した数の尺度)と同様に、ジニ指数の平均減少の形で、サンプルを分離するために利用される。(D)メタトランスクリプトデータに基づくバークホルデニアル性プロファイルの抗菌性プロファイルを示す円グラフ。シーケンスは、彼らが属している分類を決定するためにKraken2で最初にアノケン付きにしました。BLASTは、これらのアコード化された配列とMEGARes 2.0データベースと共に使用され、どの抗菌性遺伝子(「MEG_#」の形で)が積極的に発現されているかを決定しました。その後、バークホルデリアのメンバーによって発現された抗菌性遺伝子を抽出し、そのタクサの中で最も流行したものを確認した。メタトランスクリプトオミクスは、コストと時間がかかりますが、16Sデータではできない機能分析を可能にします。特に、Kraken2はHUMAnN2の代わりにこの例の分析に使用されました。クラーケン2はHUMAnN2よりも速いです。しかし、組成、寄与、機能(遺伝子)やHUMAnN2のような経路の代わりに、組成情報のみを出力します。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
補足ファイル: メタトランスクリプトミクスパイプラインの例。 このファイルをダウンロードするには、ここをクリックしてください。
ディスカッション
この論文に記載されている方法は、2014年から2018年の間に私たちのグループが発表したいくつかの研究の過程で開発され、洗練されており、まもなく出版のための論文を提出する3年間のプロジェクトで水生コミュニティにフラッキングが及ぼす影響を調査する共同プロジェクトに成功しました。これらの方法は、プロジェクトの残りの部分で引き続き使用されます。さらに、河川や生態系に対するフラッキングの影響を調査している他の現在の文献では、サンプルの収集、処理、および分析に関する同様の方法が7、8、10、11に関する記述されています。しかし、これらの論文はいずれもメタトランスクリプト分析を利用し、この論文は、これらの分析が近くの河川に対するフラッキングの影響を解明するためにどのように使用できるかを最初に説明した。さらに、サンプル収集のためにここに示す方法は、汚染を避けるために取られたステップと同様に、より詳細です。
プロトコルの最も重要なステップの 1 つは、初期サンプルの収集と保存です。フィールドサンプリングとコレクションには、収集中に無菌または無菌環境を維持することが困難な場合があります。このステップの間、サンプルを汚染することを避けることは重要である。これを行うには、手袋を着用し、滅菌容器と工具のみがサンプルに接触するようにする必要があります。サンプルは、核酸の分解を緩和するために、採取後すぐに氷の上に置く必要があります。また、収集時に市販の核酸保存剤を添加すると、核酸収率を増加させ、採取後のサンプルの保存期間を長くすることができます。核酸抽出が行われるたびに、適切な量のサンプルを使用することが重要であり、抽出に使用されるスピンフィルターを詰まらせ過ぎる可能性がありますが(それらを使用するプロトコルの場合)、あまりにも少なすぎると低収率になる可能性があります。使用するキットの説明に従ってください。
フィールドコレクションと同様に、特に最適でない沈着サンプル(大量の砂利や岩石を含むサンプル)や水サンプルなどの低核酸収率サンプルを使用する場合は、核酸抽出やサンプル調製中に汚染を回避または最小限に抑えることも重要です。したがって、サンプル収集と同様に、汚染を減らすために、これらすべてのステップで手袋を着用する必要があります。さらに、ラボの手順で使用されるすべての作業面は、10%の漂白剤溶液で拭くことによって事前に滅菌し、その後に70%のエタノール溶液を使用する必要があります。ピペット化ステップ(3-6)では、ピペット自体による汚染を避けるためにフィルターチップを使用し、チップは非滅菌表面に触れるたびに変更される必要があります。ピペットを含むラボ作業に使用されるすべてのツールは、漂白剤とエタノール溶液で前後に拭き取る必要があります。汚染を評価するには、核酸抽出およびPCR反応の各セットに抽出ブランクおよびネガ(滅菌液)を含める必要があります。抽出後の定量化により陰性のDNA/RNAの検出可能量が明らかになった場合、十分なサンプルが残っていれば抽出を繰り返すことができます。PCR の陰性サンプルが増幅を示す場合は、ソースを特定するためにトラブルシューティングを実行し、サンプルを再実行する必要があります。低レベルの汚染を考慮して、抽出ブランクとPCRネガを配列決定して、必要に応じて計算解析中に汚染物質を特定して除去することをお勧めします。逆に、PCR 増幅もさまざまな原因で失敗する可能性があります。環境試料の場合、PCR反応の阻害が原因であることが多く、これはTaqポリメラーゼ23に干渉する様々な物質によるものとなり得る。阻害が疑われる場合、PCR等グレードの水( 材料表を参照)を使用してDNA抽出物を希釈することができます。
このプロトコルには、いくつかの顕著な制限と潜在的な困難があります。サンプル収集は、水と堆積物の両方のサンプルに挑戦することができます。十分なバイオマスを得るためには、理想的には1Lの流水をフィルターに押し込む必要があります。フィルターの細孔は、微生物を捕獲するために小さくする必要がありますが、堆積物をトラップすることもできます。最近の降雨により多くの土砂が水中にある場合、フィルターが詰まってフィルターを通してボリューム全体を押し込むのが困難になる可能性があります。堆積物の収集では、収集中の堆積物の深さを推定するのは難しい場合があります。さらに、小石や岩石は核酸収量の低下につながり、微生物群集の正確な表現ではない可能性があるため、収集された堆積物が主に土壌であることを確認することが重要です。最後に、特に防腐剤を使用しない場合は、採取後にサンプルを氷の上に保管することが重要です。
このプロトコルはメタトランスクリプトと 16S の両方のラボ プロトコルを対象としていますが、これらの 2 つの方法は、プロセスとデータの種類の両方で非常に異なっていることを強調する必要があります。16S rRNA遺伝子は、一般的に標的領域であり、細菌および古細菌で高度に保存されており、サンプル中の細菌群集を特徴付けるための有用なものである。標的化された具体的なアプローチではあるが、種レベルの分解能は達成不可能であることが多く、新たに発散した種や株の特徴付けは困難である。それどころか、メタトランスクリプトミクスは、サンプル内に存在するすべての活性遺伝子および微生物を捕捉する、より広範なアプローチである。16Sは同定のためのデータのみを提供するのに対し、メタトランスクリプトミクスは発現した遺伝子および代謝経路のような機能的なデータを提供することができる。どちらも貴重であり、組み合わせると、どの細菌が存在し、どの遺伝子が発現しているかが明らかにできます。
本論文では、フラッキングの研究における16S rRNAおよびメタトランスクリプト分析の両方に対するフィールド収集およびサンプル処理の方法について説明する。さらに、低バイオマスサンプルからの高品質DNA/RNAの収集方法や長期保存方法を詳しく説明しています。ここで説明する方法は、微生物群集の構造と機能を調べることによって、フラッキングが近くの小川にどのような影響を与えるかを学ぶ取り組みの中で、サンプルの収集と処理に関する私たちの経験の集大成です。微生物は障害に迅速に対応し、その結果、どの微生物が存在し、それらが発現する遺伝子は、生態系に対するフラッキングの影響に関する情報を提供することができます。全体として、これらの方法は、フラッキングがこれらの重要な生態系にどのような影響を与えるかについての理解において非常に貴重である可能性があります。
開示事項
著者らは開示するものは何もない。
謝辞
著者らは、これらの方法の開発につながったプロジェクトの資金源を認めたいと考えています: ハワード・ヒューズ医学研究所 (http://www.hhmi.org) 先進学と学部科学教育プログラムを通じて, ならびに国立科学財団 (http://www.nsf.gov) による NSF賞 DBI-1248096 および CBET-1805549.
資料
| Name | Company | Catalog Number | Comments |
| 200 Proof Ethanol | Thermo Fisher Scientific | A4094 | 400 mL need to be added to Buffer PE (see Qiagen QIAQuck Gel Extraction kit protocol) and 96 mL needs to be added to the DNA/RNA Wash Buffer (see ZymoBIOMICS DNA/RNA Miniprep kit protocol). Additional ethanol is needed for the ZymoBIOMICS DNA/RNA Miniprep and NEBNext® Ultra™ II RNA Library Prep with Sample Purification Beads kits. |
| Agarose | Thermo Fisher Scientific | BP1356-100 | 100 g per bottle. 0.6 g of agarose would be needed to make one 2% 30 mL gel. |
| Disinfecting Bleach | Walmart (Clorox) | No catalog number | Use a 10% bleach solution for cleaning the work area before and after lab procedures |
| DNA gel loading dye | Thermo Fisher Scientific | R0611 | Each user-made (i.e. non-e-gel) should include loading dye with all of the samples in the ratio of 1 µL dye to 5 µL sample |
| DNA ladder | MilliporeSigma | D3937-1VL | A ladder should be run on every gel/e-gel |
| DNA/RNA Shield (2x) | Zymo Research | R1200-125 | 3 mL per sediment sample (50 mL conical) and 2 mL per water sample (filter) |
| Ethidium bromide | Thermo Fisher Scientific | BP1302-10 | Used for staining user-made e-gels |
| Forward Primer | Integrated DNA Technologies (IDT) | 51-01-19-06 | 0.5 µL per PCR reaction |
| Isopropanol | MilliporeSigma | 563935-1L | Generally less than 2 mL per library. Volume needed varies by mass of excised gel fragment (see Qiagen QIAQuick Gel Extraction kit protocol). |
| PCR-grade water | MilliporeSigma | 3315932001 | 13 µL per PCR reaction (assuming 1 µL of sample DNA template is used) |
| Platinum Hot Start PCR Master Mix (2x) | Thermo Fisher Scientific | 13000012 | 10 µL per PCR reaction |
| Reverse Primer | Integrated DNA Technologies (IDT) | 51-01-19-07 | 0.5 µL per PCR reaction |
| TBE Buffer (Tris-borate-EDTA) | Thermo Fisher Scientific | B52 | 1 L of 10x TBE buffer (30 mL of 1x TBE buffer would be needed to make one 30 mL gel) |
| 1 L bottle | Thermo Fisher Scientific | 02-893-4E | One needed per stream (the same bottle can be used for multiple streams if it is sterilized between uses) |
| 1.5 mL Microcentrifuge tubes | MilliporeSigma | BR780400-450EA | 5 microcentrifuge tubes are needed per DNA extraction and an additional 3 are needed to purify RNA (see ZymoBIOMICS DNA/RNA Miniprep kit protocol) |
| 2% Agarose e-gel | Thermo Fisher Scientific | G401002 | Each gel can run 10 samples (so 9 with a PCR negative and 8 if the extraction negative is run on the same gel) |
| 50 mL Conicals | CellTreat | 229421 | 1 50 mL conical needed per sediment samples |
| 500 mL Beaker | MilliporeSigma | Z740580 | Only 1 needed (for flame sterilization) |
| Aluminum foil | Walmart (Reynolds KITCHEN) | No number | Aluminum foil can be folded and autoclaved. The part not exposed to the environment can then be used as a sterile, DNA and RNA free surface for processing filters (one folded piece per filter to avoid cross-contamination) |
| Autoclave | Gettinge | LSS 130 | Only one needed |
| Centrifuge | MilliporeSigma | EP5404000138-1EA | Only 1 needed |
| Cooler | ULINE | S-22567 | Just about any cooler can be used. This one is listed due to being made of foam, making it lighter and thus easier to take along for field sampling. |
| Disruptor Genie | Bio-Rad | 3591456 | Only one needed |
| Electrophoresis chamber | Bio-Rad | 1664000EDU | Only 1 needed |
| Electrophoresis power supply | Bio-Rad | 1645050 | Only 1 needed |
| Freezer (-20 C) | K2 SCIENTIFIC | K204SDF | One needed to store DNA extracts |
| Freezer (-80 C) | K2 SCIENTIFIC | K205ULT | One needed to store RNA extracts |
| Gloves | Thermo Fisher Scientific | 19-020-352 | The catalog number is for Medium gloves. |
| Heat block | MilliporeSigma | Z741333-1EA | Only one needed |
| Lab burner | Sterlitech | 177200-00 | Only one needed |
| Laminar Flow Hood | AirClean Systems | AC624LFUV | Only 1 needed |
| Library purification kit | Qiagen | 28704 | One kit has enough for 50 reactions |
| Magnet Plate | Alpaqua | A001219 | Only one needed |
| Microcentrifuge | Thermo Fisher Scientific | 75004061 | Only one needed |
| Micropipette (1000 µL volume) | Pipette.com | L-1000 | Only 1 needed |
| Micropipette (2 µL volume) | Pipette.com | L-2 | Only 1 needed |
| Micropipette (20 µL volume) | Pipette.com | L-20 | Only 1 needed |
| Micropipette (200 µL volume) | Pipette.com | L-200R | Only 1 needed |
| NEBNext Ultra II RNA Library Prep with Sample Purification Beads | New England BioLabs Inc. | E7775S | One kit has enough reagents for 24 samples. |
| Parafilm | MilliporeSigma | P7793-1EA | 2 1" x 1" squares are needed per filter |
| PCR Tubes | Thermo Fisher Scientific | AM12230 | One tube needed per reaction |
| Pipette tips (for 1000 µL volume) | Pipette.com | LF-1000 | Pack of 576 tips |
| Pipette tips (for 20 µL volume) | Pipette.com | LF-20 | Pack of 960 tips |
| Pipette tips (for 200 µL volume) | Pipette.com | LF-250 | Pack of 960 tips |
| PowerWulf ZXR1+ computer cluster | PSSC Labs | No number | This is just an example of a supercomputer powerful enough to perform metatranscriptomics analysis in a timely manner. Only one needed. |
| Qubit fluorometer starter kit | Thermo Fisher Scientific | Q33239 | Comes with a Qubit 4 fluorometer, enough reagent for 100 DNA assays, and 500 Qubit tubes |
| Scoopula | Thermo Fisher Scientific | 14-357Q | Only one needed |
| Sterile blades | AD Surgical | A600-P10-0 | One needed per filter |
| Sterivex-GP Pressure Filter Unit | MilliporeSigma | SVGP01050 | 1 filter needed per water sample |
| Thermocycler | Bio-Rad | 1861096 | Only one needed |
| Vise-grip | Irwin | 2078500 | Only one needed (for cracking open the filters) |
| Vortex-Genie 2 | MilliporeSigma | Z258415-1EA | Only 1 needed |
| WHIRL-PAK bags | ULINE | S-22729 | 1 needed per filter |
| ZymoBIOMICS DNA/RNA Miniprep kit | Zymo Research | R2002 | One kit has enough reagents for 50 samples. |
参考文献
- The process of unconventional natural gas production. US EPA Available from: https://www.epa.gov/uog/process-unconventional-natural-gas-production (2013)
- Brittingham, M. C., Maloney, K. O., Farag, A. M., Harper, D. D., Bowen, Z. H. Ecological risks of shale oil and gas development to wildlife, aquatic resources, and their habitats. Environmental Science & Technology. 48 (19), 11034-11047 (2014).
- McBroom, M., Thomas, T., Zhang, Y. Soil erosion and surface water quality impacts of natural gas development in East Texas, USA. Water. 4 (4), 944-958 (2012).
- Maloney, K. O., Weller, D. E. Anthropogenic disturbance, and streams: land use and land-use change affect stream ecosystems via multiple pathways. Freshwater Biology. 56 (3), 611-626 (2011).
- Meyer, J. L., et al. The contribution of headwater streams to biodiversity in river networks1. JAWRA Journal of the American Water Resources Association. 43 (1), 86-103 (2007).
- Alexander, R. B., Boyer, E. W., Smith, R. A., Schwarz, G. E., Moore, R. B. The role of headwater streams in downstream water quality. Journal of the American Water Resources Association. 43 (1), 41-59 (2007).
- Ulrich, N., et al. Response of aquatic bacterial communities to hydraulic fracturing in Northwestern Pennsylvania: A five-year study. Scientific Reports. 8 (1), 5683 (2018).
- Chen See, J. R., et al. Bacterial biomarkers of Marcellus shale activity in Pennsylvania. Frontiers in Microbiology. 9, 1697 (2018).
- Rausch, P., et al. Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome. 7 (1), 133 (2019).
- Louca, S., Doebeli, M., Parfrey, L. W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome. 6 (1), 41 (2018).
- Trexler, R., et al. Assessing impacts of unconventional natural gas extraction on microbial communities in headwater stream ecosystems in Northwestern Pennsylvania. Frontiers in Microbiology. 5, 522 (2014).
- Mumford, A. C., et al. Shale gas development has limited effects on stream biology and geochemistry in a gradient-based, multiparameter study in Pennsylvania. Proceedings of the National Academy of Sciences. 117 (7), 3670-3677 (2020).
- JoVE Core Biology DNA Isolation. Journal of Visualized Experiments Available from: https://www.jove.com/cn/science-education/10814/dna-isolation (2020)
- Oxford Gene Technology DNA Storage and Quality. OGT Available from: https://www.ogt.com/resources/literature/403_dna_storage_and_quality (2011)
- ThermoFisher SCIENTIFIC Technical Bulletin #159: Working with RNA. Thermoscientific Available from: https://www.thermofisher.com/us/en/home/references/ambion-tech-support/nuclease-enzymes/general-articles/working-with-rna.html (2020)
- QIAGEN AllPrep DNA/RNA Mini Kit. Qiagen Available from: https://www.qiagen.com/us/products/discovery-and-translational-research/dna-rna-purification/multianalyte-and-virus/allprep-dnarna-mini-kit/#orderinginformation (2020)
- ZymoBIOMICS DNA/RNA Miniprep Kit. Zymo Research Available from: https://www.zymoresearch.com/products/zymobiomics-dna-rna-miniprep-kit (2020)
- Desjardins, P., Conklin, D. NanoDrop microvolume quantitation of nucleic acids. Journal of Visualized Experiments. (45), e2565 (2010).
- 16S Illumina amplicon protocol: Earth microbiome project. Earth microbiome project Available from: https://earthmicrobiome.org/protocols-and-standards/16s/ (2018)
- Gel Purification: Binding, washing and eluting a sample | Protocol. Journal of Visualized Experiments Available from: https://www.jove.com/v/5063/gel-purification (2020)
- New England Biolabs protocol for the use with NEBNext Poly(A) mRNA magnetic isolation module (E7490) and NEBNext Ultra II RNA library prep kit for Illumina (E7770, E7775). New England Biolabs Available from: https://www.neb.com/protocols/2017/03/04/protocol-for-use-with-purified-mrna-or-rrna-depleted-rna-and-nebnext-ultra-ii-rna-library-prep-ki (2020)
- Anderson, M. J. Permutational multivariate analysis of variance (PERMANOVA). Wiley StatsRef: Statistics Reference Online. , 1-15 (2017).
- Schrader, C., Schielke, A., Ellerbroek, L., Johne, R. PCR inhibitors - occurrence, properties and removal. Journal of Applied Microbiology. 113 (5), 1014-1026 (2012).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved