
Method Article
Bewertung der Auswirkungen von Hydraulic Fracturing auf Bäche mit mikrobiellen molekularen Signaturen
In diesem Artikel
Zusammenfassung
Hier präsentieren wir ein Protokoll, um die Auswirkungen des Hydraulic Fracturing auf nahe gelegene Bäche zu untersuchen, indem wir ihre mikrobiellen Wasser- und Sedimentgemeinschaften analysieren.
Zusammenfassung
Hydraulic Fracturing (HF), allgemein als "Fracking" bezeichnet, verwendet eine Mischung aus Hochdruckwasser, Sand und Chemikalien, um Gesteine zu brechen und Öl und Gas freizusetzen. Dieser Prozess revolutionierte die US-Energieindustrie, da er Zugang zu Ressourcen bietet, die zuvor nicht verfügbar waren und jetzt zwei Drittel des gesamten Erdgases in den Vereinigten Staaten produzieren. Obwohl Fracking einen positiven Einfluss auf die US-Wirtschaft hatte, haben mehrere Studien seine schädlichen Umweltauswirkungen hervorgehoben. Besonders besorgniserregend ist die Wirkung von Fracking auf Quellbäche, die aufgrund ihrer unverhältnismäßig großen Auswirkungen auf die Gesundheit des gesamten Wassereinzugsgebiets besonders wichtig sind. Die Bakterien innerhalb dieser Ströme können als Indikatoren für die Gesundheit des Baches verwendet werden, da die vorhandenen Bakterien und ihre Häufigkeit in einem gestörten Strom voraussichtlich von denen in einem ansonsten vergleichbaren, aber ungestörten Strom abweichen. Daher zielt dieses Protokoll darauf ab, die Bakteriengemeinschaft zu nutzen, um festzustellen, ob Bäche durch Fracking beeinträchtigt wurden. Zu diesem Zweck müssen Sediment- und Wasserproben aus Bächen in der Nähe von Fracking (potenziell betroffen) und stromaufwärts oder in einem anderen Wassereinzugsgebiet der Fracking-Aktivität (unbeeinträchtigt) gesammelt werden. Diese Proben werden dann der Nukleinsäureextraktion, Bibliotheksvorbereitung und Sequenzierung unterzogen, um die Zusammensetzung der mikrobiellen Gemeinschaft zu untersuchen. Korrelationsanalysen und maschinelle Lernmodelle können anschließend verwendet werden, um zu identifizieren, welche Merkmale die Variation in der Gemeinschaft explanativ sind, sowie die Identifizierung prädiktiver Biomarker für die Auswirkungen von Fracking. Diese Methoden können eine Vielzahl von Unterschieden in den mikrobiellen Gemeinschaften zwischen Quellströmen aufdecken, basierend auf der Nähe zum Fracking, und dienen als Grundlage für zukünftige Untersuchungen zu den Umweltauswirkungen von Fracking-Aktivitäten.
Einleitung
Hydraulic Fracturing (HF) oder "Fracking" ist eine Methode der Erdgasgewinnung, die sich mit der weiter steigenden Nachfrage nach fossilen Brennstoffen immer mehr durchgesetzt hat. Diese Technik besteht darin, hochleistungsfähige Bohrgeräte zu verwenden, um eine Mischung aus Wasser, Sand und Chemikalien in methanreiche Schieferlagerstätten zu injizieren, in der Regel um eingeschlossene Gase freizusetzen1.
Da diese unkonventionellen Erntetechniken relativ neu sind, ist es wichtig, die Auswirkungen solcher Praktiken auf nahe gelegene Wasserstraßen zu untersuchen. Fracking-Aktivitäten erfordern die Rodung großer Landstriche für den Transport von Ausrüstung und den Bau von Bohrplätzen. Für jedes Bohrfeld2müssen etwa 1,2-1,7 Hektar Land gerodet werden, was sich möglicherweise auf den Abfluss und die Wasserqualität des Systemsauswirkt 3. Es gibt einen Mangel an Transparenz über die genaue chemische Zusammensetzung von Fracking-Flüssigkeit, einschließlich der verwendeten Biozide. Darüber hinaus neigt Fracking-Abwasser dazu, stark salzhaltig zu sein2. Weiterhin kann das Abwasser Metalle und natürlich vorkommende radioaktive Stoffe enthalten2. Daher ist die Möglichkeit von Leckagen und Verschüttungen von Fracking-Flüssigkeit aufgrund von menschlichem Versagen oder Fehlfunktionen der Ausrüstung besorgniserregend.
Es ist bekannt, dass Bachökosysteme sehr empfindlich auf Veränderungen in den umliegenden Landschaftenreagieren 4 und wichtig für die Erhaltung der Biodiversität5 und den richtigen Nährstoffkreislauf6 innerhalb des gesamten Wassereinzugsgebiets sind. Mikroben sind die am häufigsten vorkommenden Organismen in Süßwasserströmen und daher für den Nährstoffkreislauf, den biologischen Abbau und die Primärproduktion unerlässlich. Die Zusammensetzung und Funktion der mikrobiellen Gemeinschaft dient aufgrund ihrer Empfindlichkeit gegenüber Störungen als großartige Werkzeuge, um Informationen über das Ökosystem zu gewinnen, und neuere Forschungen haben deutliche Verschiebungen in den beobachteten Bakterienansammlungen aufgrund der Nähe zur Fracking-Aktivität gezeigt7,8. Zum Beispiel wurden Beijerinckia, Burkholderiaund Methanobacterium als angereichert in Bächen in der Nähe von Fracking identifiziert, während Pseudonocardia, Nitrospiraund Rhodobacter in den Bächen nicht in der Nähe von Fracking angereichert wurden7.
Die Next-Generation-Sequenzierung des ribosomalen RNA-Gens (rRNA) 16S ist eine erschwingliche Methode zur Bestimmung der Zusammensetzung der Bakteriengemeinschaft, die schneller und billiger ist als die Sequenzierung des gesamten Genoms9. Eine gängige Praxis im Bereich der molekularen Ökologie ist es, die hochvariable V4-Region des 16S rRNA-Gens für die Sequenzierungsauflösung zu verwenden, oft bis auf die Gattungsebene mit einem breiten Identifikationsbereich9, da sie ideal für unvorhersehbare Umweltproben ist. Diese Technik wurde in veröffentlichten Studien weit verbreitet und wurde erfolgreich eingesetzt, um die Auswirkungen von Fracking-Operationen auf die aquatische Umwelt zu identifizieren7,8. Es ist jedoch erwähnenswert, dass Bakterien unterschiedliche Kopienzahlen des 16S rRNA-Gens haben, was ihre nachgewiesenen Häufigkeiten beeinflusst10. Es gibt ein paar Werkzeuge, um dies zu erklären, aber ihre Wirksamkeit istfraglich 10. Eine weitere Praxis, die schnell an Prävalenz zunimmt und der diese Schwäche fehlt, ist die metatranskriptomische Sequenzierung, bei der die gesamte RNA sequenziert wird, so dass Forscher sowohl aktive Bakterien als auch ihre Genexpression identifizieren können.
Daher umfasst dieses Protokoll im Gegensatz zu Methoden in den zuvorveröffentlichtenStudien7,8,11,12auch die Probenentnahme, Konservierung, Verarbeitung und Analyse zur Untersuchung der mikrobiellen Gemeinschaftsfunktion (Metatranskriptomik). Die hier beschriebenen Schritte ermöglichen es den Forschern zu sehen, welche Auswirkungen Fracking auf die Gene und Signalwege hatte, die von Mikroben in ihren Strömen exprimiert werden, einschließlich antimikrobieller Resistenzgene. Darüber hinaus wird der Detaillierungsgrad für die Probenentnahme verbessert. Obwohl einige der Schritte und Notizen für erfahrene Forscher offensichtlich erscheinen mögen, könnten sie für diejenigen, die gerade erst mit der Forschung beginnen, von unschätzbarem Wert sein.
Hier beschreiben wir Methoden zur Probenentnahme und -verarbeitung zur Generierung bakterieller genetischer Daten als Mittel, um die Auswirkungen von Fracking auf nahe gelegene Bäche auf der Grundlage der mehrjährigen Erfahrung unserer Labore zu untersuchen. Diese Daten können in nachgelagerten Anwendungen verwendet werden, um Unterschiede zu identifizieren, die dem Fracking-Status entsprechen.
Protokoll
1. Entnahme von Sedimentproben zur Nukleinsäureextraktion
- Tauchen Sie ein steriles 50 mL konisches Rohr in das Bachwasser. Tragen Sie während der Probenentnahme Handschuhe, um unerwünschte menschliche Kontaminationen zu vermeiden. Führen Sie diesen Schritt entweder vom Ufer aus oder stromaufwärts, wenn Sie im Wasser sind.
- Während das konische Rohr untergetaucht ist, entfernen Sie die Kappe und schaufeln Sie damit etwa 3 mL Sediment aus einer Tiefe von 1 bis 3 cm in das konische Rohr.
- Entfernen Sie das konische Rohr aus dem Wasser und entsorgen Sie das gesamte Wasser, mit Ausnahme einer dünnen Schicht, die die Sedimentprobe bedeckt (ca. 1 ml).
- Mit einer 1000 μL Pipette und geeigneten Pipettenspitzen 3 mL DNA/RNA-Konservierungsmittel (siehe Materialtabelle für die Konservierungsmittelspezifikationen) in die gesammelte Probe geben. Bewahren Sie die Pipettenspitzen in einer sterilen Pipettenspitzenbox auf und befestigen Sie sie erst unmittelbar vor Gebrauch und entsorgen Sie sie nach Gebrauch. 10-mal das verschlossene konische Röhrchen umkehren, um sicherzustellen, dass konservierungsmittel und Probe gründlich gemischt werden.
HINWEIS Schritt 1.4 ist nicht notwendig, wird aber dringend empfohlen, wenn später RNA aus den Sedimenten extrahiert werden soll. - Legen Sie die Proben für den Rest der Probenentnahme auf Eis. Nach der Rückkehr aus der Entnahme in einem Gefrierschrank bei -20 °C lagern, wenn die Proben für die 16S-Analyse (DNA) verwendet werden sollen, oder -70 °C, wenn sie für die Metatranskriptomik-Analyse (RNA) verwendet werden sollen.
2. Filtersammlung zur Nukleinsäureextraktion
- Entfernen Sie den Verschluss einer sterilen 1 L Flasche. Wenn Sie stromaufwärts oder vom Ufer aus blicken, füllen Sie die Flasche mit Bachwasser nach oben und werfen Sie sie dann aus. Wiederholen Sie diesen Vorgang noch zwei weitere Male, um die Flasche zu konditionieren. Füllen Sie die gesamte Flasche ein viertes Mal und verschließen Sie sie.
HINWEIS: Wenn Sie eine 1-L-Flasche wiederverwenden, kann sie sterilisiert werden, indem Sie 2 Minuten lang mit 10% Bleichmittel spülen, gefolgt von dreimaligem Spülen mit entionisiertem Wasser und dann einmal mit 70% Ethanol und schließlich Autoklavieren mit Einstellungen: 30 min Einwirkzeit bei 121,1 ° C und 15 Minuten Trocknungszeit. Während des Autoklavierens sollte der Verschluss der Flasche sehr locker sein, um zu vermeiden, dass die Flasche dabei komprimiert wird. - Sobald Sie sich auf einer stabilen Oberfläche befinden, verwenden Sie eine sterile Luer-Lock-Spritze und ziehen Sie ein volles Volumen auf. Verbinden Sie dann die Spritze mit einem sterilen und DNA/RNA-freien Polyethersulfonfilter mit 1,7 cm Durchmesser und einer Porengröße von 0,22 μm und drücken Sie das gesamte Volumen durch den Filter, indem Sie den Kolben ganz nach unten drücken. Wiederholen Sie diesen Vorgang, bis das in der Flasche gesammelte Gesamtvolumen (1 L) durch den Filter gedrückt wird.
HINWEIS: Das Volumen der Spritze kann variabel sein, wenn die Gesamtmenge des durch den Filter gedrückten Wassers verfolgt wird. Im Allgemeinen werden jedoch 60 ml bevorzugt. Während 1 L anekdotisch ideal ist, würde ein Volumen von mindestens 200 ml wahrscheinlich immer noch genug Biomasse (vorausgesetzt ~ 20.000 Zellen pro ml) für die Extraktion von DNA und RNA sammeln. - Entfernen Sie überschüssiges Wasser aus dem Filter, indem Sie etwa 20 ml Luft in die Spritze ziehen und durch den Filter drücken.
HINWEIS: Dies hilft, den Verlust des Konservierungsmittels zu verhindern, wenn Schritt 2.4 durchgeführt wird. - Fügen Sie mit einer P1000-Mikropipette 2 ml eines DNA/RNA-Konservierungsmittels hinzu, indem Sie es durch die größere Öffnung des Filters (wo es an der Spritze befestigt war) abführen, während Sie den Filter horizontal halten. Die Spitze der Pipette sollte sich im Zylinder des Filters befindet, wenn die Pipette gedrückt wird, um sicherzustellen, dass das Konservierungsmittel in den Filter gelangt. Ändern Sie die Spitze nach jedem Gebrauch.
HINWEIS: Wie bei der Sedimentsammlung ist dieser Schritt nicht notwendig, wird aber für eine spätere erhöhte Nukleinsäureausbeute, insbesondere für RNA, dringend empfohlen. - Ziehen Sie ein Quadrat Paraffinfolie ab und wickeln Sie es fest um jede Öffnung / jedes Ende des Filters, um es zu versiegeln. Legen Sie den mit Paraffinfolie umwickelten Filter in einen sterilen Probenbeutel und legen Sie den gesamten Beutel während der Entnahme auf Eis.
HINWEIS: Stellen Sie sicher, dass die Seite, die zum Umwickeln des Filters verwendet wird, steril ist, d. h. nicht zuvor der Umgebung ausgesetzt war. - Lagern Sie nach der Rückkehr von der Probenahme filtern bei -20 °C für 16S oder -70 °C für die Meta-Transkriptomik.
3. Nukleinsäureextraktion und Quantifizierung
- Reinigen Sie den Arbeitsbereich mit 10% Bleichmittel und 70% Ethanol, bevor Sie mit dem Probentransfer beginnen.
- Verwenden Sie für Sedimente (ab Schritt 1.5) im Allgemeinen ~ 0,25 g Probe. Flammensterilisieren Sie ein Metallwerkzeug, indem Sie es in ein Becherglas mit 70% Ethanol tauchen und das Ethanol zwischen den Proben verbrennen.
- Bei Filtern (ab Schritt 2.6) das Filterpapier zur Extraktion in ein steriles Röhrchen geben. Führen Sie dazu die folgenden Schritte aus.
- Erzeugen Sie eine sterile, DNA- und RNA-freie Oberfläche, indem Sie Aluminiumfolie so falten, dass der innere Teil der Falte nicht der äußeren Umgebung ausgesetzt ist, und das gefaltete Stück mit den Einstellungen 121,1 ° C und 5 min Trocknungszeit autoklavieren.
- Sterilisieren Sie einen Schraubstockgriff mit 70% Ethanol und einer offenen Flamme. Dann verwenden Sie den Schraubstockgriff, um das Filtergehäuse auf der sterilen Oberfläche aufzubrechen und den Kern aus dem Gehäuse zu entfernen.
- Verwenden Sie ein steriles Skalpell, um das Filterpapier vom Kern wegzuschneiden, indem Sie oben und unten und dann entlang der Naht schneiden. Falten Sie das Filterpapier mit einer sterilen Pinzette und schneiden Sie den Filter dann mit dem Skalpell in kleine Stücke.
- Legen Sie die Filterstücke zur Extraktion in ein Mikrozentrifugenrohr. Achten Sie darauf, dass das Filterpapier nicht mit Oberflächen in Berührung kommt, die nicht sterilisiert sind oder bei denen Nukleinsäure vorhanden sein könnte, da dies zu einer unerwünschten Kontamination der Probe führen würde.
- Führen Sie die DNA-Isolierung wie zuvor beschrieben13 oder mit einem handelsüblichen säulenbasierten Kit durch (siehe Materialtabelle). Die Schritte für das aufgeführte kommerzielle Kit werden im Folgenden kurz beschrieben.
- Lyse die Zellen in der Probe, indem Sie sie in ein Perlenröhrchen übertragen und sie mindestens 5 Minuten lang mit hoher Geschwindigkeit einem Zelldisruptor zuweisen. Zentrifugieren und den Überstand in ein steriles Mikrozentrifugenröhrchen überführen.
- Fügen Sie dem Überstand einen Lysepuffer hinzu (1:1 Volumen) und übertragen Sie ihn in den bereitgestellten Filter (gelb). Zentrifuge den Filter.
- Geben Sie den Filter in ein neues steriles Mikrozentrifugenröhrchen. Fügen Sie den Präparationspuffer (400 μL) hinzu, zentrifugieren Sie ihn und verwerfen Sie den Durchfluss.
- Waschpuffer (700 μL) hinzufügen, Zentrifuge zentrifugieren und den Durchfluss verwerfen. Dann Waschpuffer (400 μL), Zentrifuge hinzufügen und den Durchfluss wieder verwerfen.
- Geben Sie den Filter in ein neues steriles Mikrozentrifugenröhrchen. Mit 50 μL DNase/RNase-freiem Wasser eluiert und vor dem Zentrifugen 5 min bei Raumtemperatur einlegen lassen.
- Bereiten Sie während dieser Zeit in der Cubation den III-HRC-Filter vor, indem Sie ihn in ein Auffangrohr legen und die HRC-Vorbereitungslösung (600 μL) hinzufügen, gefolgt von einem Zentrifugationsschritt von 3 min bei 8.000 x g.
- Bewegen Sie den vorbereiteten Filter auf ein steriles Mikrozentrifugenröhrchen. Übertragen Sie die eluierte DNA aus Schritt 3.4.5 auf diesen Filter und zentrifugieren Sie bei 16.000 x g für 3 min. Der Durchfluss enthält die extrahierte DNA.
- Lagern Sie DNA-Extrakte für Sedimente und Filter bei -20 °C.
HINWEIS: DNA-Extrakte können etwa 8 Jahre bei -20 °C gelagert werden, vorausgesetzt, dass eine stabile Temperatur, begrenzte Lichteinwirkung und keine schädlichen Verunreinigungenbestehen 14. - Führen Sie eine RNA-Isolierung gemäß dem Protokoll des Herstellers durch. Lagern Sie RNA-Extrakte bei -80 °C.
- Lyse die Zellen in der Probe, indem sie in ein Perlenröhrchen überführt und mindestens fünf Minuten lang mit hoher Geschwindigkeit einem Zelldisruptor ausgesetzt werden. Zentrifugieren und den Überstand in ein steriles Mikrozentrifugenröhrchen überführen.
- Fügen Sie dem Überstand (1:1 Volumen) einen Lysepuffer hinzu und übertragen Sie ihn in die bereitgestellte Spalte (gelb). Zentrifugieren Sie die Säule.
- Fügen Sie ein gleiches Volumen von 95-100% Ethanol zum Durchfluss hinzu und mischen Sie durch fünfmales Pipettieren nach oben und unten.
- Legen Sie die IICG-Säule (grün) auf ein steriles Mikrozentrifugenröhrchen. Die Mischlösung auf die Säule und Zentrifuge übertragen.
- Waschpuffer (400 μL), Zentrifuge hinzufügen und den Durchfluss verwerfen.
- 5 μL DNase I und 75 μL DNA-Verdauungspuffer in die Säule geben und 15 Minuten bei Raumtemperatur inkubieren.
- Fügen Sie einen Vorbereitungspuffer (400 μL) hinzu, zentrifugieren Sie ihn und verwerfen Sie den Durchfluss.
- Waschpuffer (700 μL) hinzufügen, Zentrifuge zentrifugieren und den Durchfluss verwerfen. Dann Waschpuffer (400 μL), Zentrifuge hinzufügen und den Durchfluss wieder verwerfen.
- Die Säule wird in ein neues steriles Mikrozentrifugenröhrchen überführen. Mit 50 μL DNase/RNase freiem Wasser eluiert und vor dem Zentrifugalen 5 min einlegen lassen.
- Bereiten Sie während dieser Inkubationszeit den III-HRC-Filter vor, indem Sie ihn in ein Sammelrohr geben und die HRC-Vorbereitungslösung (600 μL) hinzufügen, gefolgt von einem Zentrifugationsschritt von 3 min bei 8.000 x g.
- Bewegen Sie den vorbereiteten Filter auf ein steriles Mikrozentrifugenröhrchen. Übertragen Sie die eluierte RNA aus Schritt 3.6.9 auf diesen Filter und zentrifugieren Sie bei 16.000 x g für 3 min. Der Durchfluss enthält die extrahierte RNA.
HINWEIS: RNA-Extrakte können nur ein Jahr lang gelagert werden, bevor sie beginnen,15abzubauen. Sowohl DNA- als auch RNA-Extrakte werden durch wiederholtes Auftauen abgebaut. Einige Protokolle erlauben die Extraktion von DNA und RNA aus derselben Probe16,17.
- Quantifizieren Sie die extrahierten DNA- und RNA-Proben mit einem Fluorometer oder einem Spektralphotometer. Siehe Tabelle 1 zum Beispiel Fluorometer-DNA-Konzentrationswerte. Ein Beispiel für ein Spektralphotometer-Quantifizierungsprotokoll finden Sie unter Referenz18. Sediment-DNA-Konzentrationswerte mit dem in der Materialtabelle aufgeführten Kit liegen im Allgemeinen zwischen 1 und 40 ng / μL, während die Filter-DNA-Konzentrationswerte tendenziell zwischen 0,5 und 10 ng / μL liegen. Sediment-RNA-Konzentrationswerte mit dem in der Materialtabelle aufgeführten Kit liegen im Allgemeinen zwischen 1 und 20 ng / μL, während die Filter-RNA-Konzentrationswerte tendenziell niedriger sind. typischerweise im Bereich von 0,5 bis 5 ng/μL.
4. 16S rRNA-Bibliothekserstellung
- Reinigen Sie den Arbeitsbereich mit 10% Bleichmittel und 70% Ethanol. Der Arbeitsbereich sollte ein geschlossener Raum sein, der laminare Strömungsbedingungen erzeugen kann (laminare Strömungshaube).
- Verwenden Sie die DNA-Extrakte (aus Schritt 3.5) und bereiten Sie Proben für die 16S rRNA-Amplicon-Sequenzierung mit einem Standard-PCR-Protokoll vor, wie es auf der Website des Earth Microbiome beschrieben ist, das die hypervariable V4-Region von 16S rRNA19 unter laminaren Strömungsbedingungen amplifiziert.
- Bereiten Sie ein 2% ige Agarosegel wie zuvor beschrieben vor und lassen Sie es17erstarren. Mischen Sie 7 μL PCR-Produkt und 13 μL DNase-freies Wasser. Fügen Sie einen Gel-Ladefarbstoff zu einer Endkonzentration von 1x hinzu. Sobald Agarose erstarrt ist, laden Sie diese PCR-Produktmischung auf ein 2% ige agarosegel.
HINWEIS: Alternativ kann stattdessen ein vorgefertigtes Gel verwendet werden, da diese Gele schneller laufen und vorgefertigt werden. - Führen Sie das Gel bei 90 V für 60-90 Minuten aus, um die Bandgröße von 386 als erfolgreiche Amplifikation für 16S rRNA V4-Amplicons unter Verwendung des Protokolls des Erdmikrobioms zu überprüfen.
5. DNA 16S rRNA Bibliotheksreinigung
- Pool 10 μL PCR-Produkte für die Proben, die helle Bänder ergaben, und 13 μL für die Proben, die schwache Bänder in einem entsprechend großen sterilen Mikrozentrifugenröhrchen ergaben.
- Überprüfen Sie die Konzentration des resultierenden Pools mit einem Fluorometer oder Spektralphotometer und bereiten Sie wie zuvor ein 2% iges Agarosegel vor. Idealerweise sollte der Pool eine Konzentration von mindestens 10 ng/μL aufweisen, und die meisten Proben sollten eine Konzentration von etwa 25 ng/μL aufweisen.
- Wenn Konzentration und Volumen zulässig sind, laden Sie etwa 150-200 ng in eine Vertiefung von 2% Agarosegel.
- Lassen Sie das Gel für 60-90 min bei 90 Volt laufen.
- Reinigen Sie die gepoolte Bibliothek, indem Sie ein 2% iges Agarosegel ausführen.
- Entfernen Sie das 386 bp DNA-Band aus dem Gel und reinigen Sie die gepoolte Bibliothek mit einem kommerziell erhältlichen Kit, wie zuvor beschrieben20. Eluiert die gereinigte DNA mit 30 μL 10 mM Tris-Cl (pH 8,5). Führen Sie diesen Schritt in einem anderen Bereich als der DNA- oder RNA-Extraktion durch, um eine zukünftige Kontamination zu verhindern, da das Schneiden des Gels PCR-Amplicons sowohl auf den Experimentator als auch auf die Umgebung verteilt.
- Überprüfen Sie die Konzentration des gereinigten Pools mit einem Fluorometer oder Spektralphotometer. Wenn die Reinigung gut verlief, sollte die Konzentration mindestens die Hälfte der ungereinigten Pools sein. Im Allgemeinen sollte die Endkonzentration zwischen 5 und 20 ng/μL liegen.
- Senden Sie die gereinigten Bibliotheken für die Sequenzierung der nächsten Generation. Stellen Sie sicher, dass sie während des Transports kalt gehalten werden, indem Sie Trockeneis in den Versandbehälter einschließen.
6. Erstellung und Reinigung von RNA-Bibliotheken
- Mehrere kommerzielle Kits können verwendet werden, um RNA-Bibliotheken zu erstellen. Befolgen Sie für welches Verwendetes auch immer, befolgen Sie das Protokoll des Herstellers, wie es geschrieben wurde, während Sie in einer sterilen Laminar-Flow-Umgebung arbeiten. Eine sehr zusammengefasste Version des Protokolls für Kit in der Materialtabelle ist unten21dargestellt.
- Machen Sie den ersten Strang cDNA-Synthese-Master-Mix (8 μL nukleasefreies Wasser und 2 μL First Strand Synthesis Enyzme Mix) und fügen Sie ihn der Probe hinzu. Legen Sie die Probe unter den im Protokoll angegebenen Bedingungen in den Thermocycler.
- Machen Sie den zweiten Strang cDNA-Synthese-Master-Mix (8 μL Second Strand Synthesis Reaction Buffer, 4 μL Second Strand Synthesis Enzyme Mix und 48 μL nukleasefreies Wasser) auf Eis und fügen Sie ihn der Probe hinzu. Eine Stunde lang in einen thermocycler eingestellten Thermocycler mit 16 °C geben.
- Reinigen Sie die Reaktion, indem Sie die bereitgestellten Perlen (144 μL) hinzufügen und zwei 80% Ethanolwaschungen (200 μL) durchführen.
- Eluieren Sie mit dem bereitgestellten TE-Puffer (53 μL) und übertragen Sie 50 μL des Überstands in ein sauberes PCR-Röhrchen. Legen Sie die PCR-Röhre auf Eis.
- Machen Sie den End Prep Master Mix (7 μL End Prep Reaction Buffer und 3 μL End Prep Enzyme Mix) auf Eis und geben Sie ihn in das PCR-Röhrchen. Legen Sie das PCR-Röhrchen in einen Thermocycler mit den im Protokoll angegebenen Bedingungen.
- Mischen Sie die Lösungen des verdünnten Adapters (2,5 μL), des Ligation Master Mix (30 μL) und des Ligation Enhancer (1 μL) auf Eis. Die Mischlösungen in die Probe geben und für 15 min bei 20 °C in einen Thermocycler geben.
- Reinigen Sie die Reaktion, indem Sie die bereitgestellten Perlen (87 μL) hinzufügen und Ethanolwäschen (200 μL) und Elution wie zuvor durchführen, außer nur 17 μL TE hinzufügen.
- Fügen Sie Indizes (10 μL) und die Q5 Master Mix (25 μL) Lösung hinzu und legen Sie sie in einen Thermocycler mit den im Protokoll beschriebenen Bedingungen.
- Reinigen Sie die Reaktion, indem Sie die bereitgestellten Perlen (45 μL) hinzufügen und zwei Ethanolwäschen (200 μL) hinzufügen und mit 23 μL TE eluieren. 20 μL in eine saubere PCR-Röhre überführen.
- Überprüfen Sie die Bibliotheken mit einem Bioanalyzer, Fluorometer oder Spektralphotometer auf nachweisbare Konzentrationen von RNA.
- Bündeln Sie die metatranskriptomischen Bibliotheken in einem ungefähr äquimolaren Verhältnis.
- Reinigen Sie die Bibliothek nach dem gleichen Protokoll für die Reinigung der 16S-Bibliothek, mit Ausnahme von Verbrauchsfragmenten zwischen 250 und 400 bp. Während die 16S-Bibliothek ein deutliches Band hatte, das den verstärkten Bereich darstellte, ist das Ergebnis hier ein Abstrich.
- Überprüfen Sie die Konzentration der gereinigten Bibliothek wie zuvor.
- Versenden Sie die gereinigte Bibliothek mit Trockeneis an eine Sequenziereinrichtung.
HINWEIS: Alternativ können RNA-Extrakte zur Bibliotheksvorbereitung und -sequenzierung an eine Universität oder ein privates Unternehmen gesendet werden.
7. Mikrobielle Gemeinschaftsanalyse
- Sobald die Sequenzierung abgeschlossen ist, greifen Sie auf die Beispieldaten zu. Laden Sie es auf einen brauchbaren Computer herunter.
HINWEIS: Idealerweise sollte das Gerät über mindestens 16 Gigabyte RAM verfügen. Eine Erläuterung der Computeranforderungen (für Qiime2) finden Sie unter https://forum.qiime2.org/t/recommended-specifications-to-run-qiime2/9808. - Verwenden Sie Software wie mothur, QIIME2 und R, um 16S rRNA-Daten zu analysieren. Hier finden Sie https://docs.qiime2.org/2020.11/tutorials/moving-pictures/ ein Beispiel für ein QIIME2 16S Analyse-Tutorial.
- Für Metatranskriptomik-Daten (RNA) verwenden Sie HUMAnN2 und ATLAS, um zu bestimmen, welche Gene und Wege in den Proben vorhanden sind.
HINWEIS: Ein Beispiel für eine Metatranskriptomik-Pipeline, die in einer Diversitäts- und Zufallsgesamtstrukturanalyse gipfelt, wird in der Datei "Ergänzende Informationen" vorgestellt. Alle Befehle werden über die Befehlszeile ausgeführt, z. B. Terminal für Mac-Benutzer.
Ergebnisse
Der Erfolg von DNA- und RNA-Extraktionen kann mit einer Vielzahl von Geräten und Protokollen bewertet werden. Im Allgemeinen wird jede nachweisbare Konzentration von beiden als ausreichend angesehen, um zu dem Schluss zu kommen, dass die Extraktion erfolgreich war. Bei der Untersuchung von Tabelle 1 würden dann alle Extraktionen, bis auf eine, als erfolgreich bezeichnet. Das Versagen bei diesem Schritt ist oft auf geringe anfängliche Biomasse, schlechte Probenkonservierung oder menschliches Versagen während der Extraktion zurückzuführen. Bei Filtern kann die Extraktion auch dann erfolgreich gewesen sein, wenn die Konzentration unter dem Nachweis liegt. Wenn diese Extrakte keine Bänder für PCR (wenn 16S) oder eine nachweisbare Konzentration nach der Bibliotheksvorbereitung (Metatranskriptomik) ergeben, dann sind sie wahrscheinlich wirklich gescheitert.
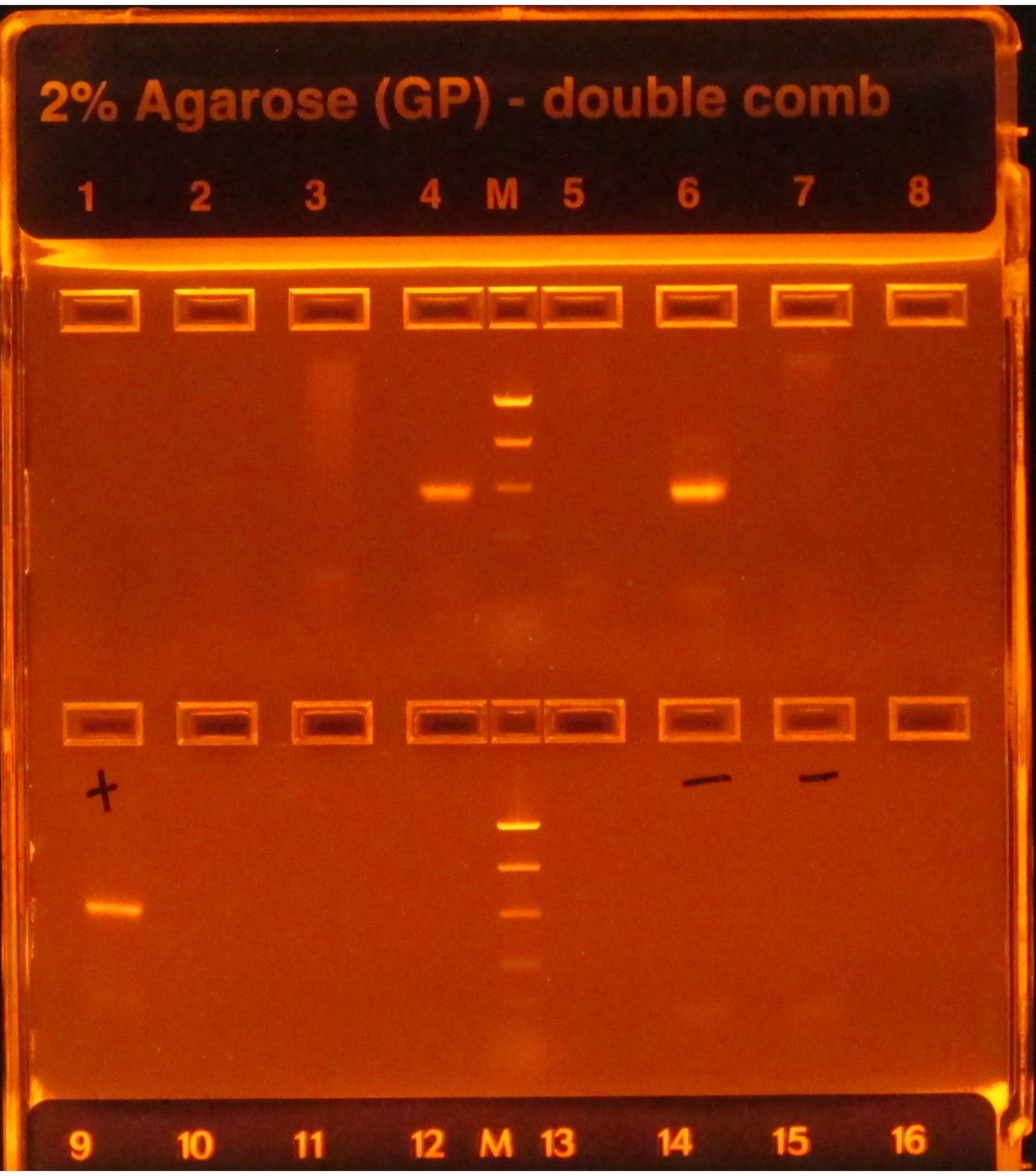
Wenn das 16S-Protokoll befolgt wird, zeigen helle Bänder nach PCR-Amplifikation, wie in den Bohrungen 4 und 6 in Abbildung 1zu sehen, erfolgend an, während ein Mangel an Bändern, wie in den anderen Bohrungen in der oberen Reihe, auf ein Versagen hinweist. Darüber hinaus würde ein helles Band in der Gelbahn, das eine negative PCR-Kontrolle enthält, auch auf einen Fehler hinweisen, da es riskant wäre anzunehmen, dass die Kontamination, die sich auf die Negativkontrolle(en) auswirkt, die Proben nicht beeinflusst.
Sowohl für die 16S- als auch für die Metatranskriptomik kann der Erfolg der Sequenzierung anhand der Anzahl der erhaltenen Sequenzen bewertet werden (Abbildung 2). 16S-Proben sollten mindestens 1.000 Sequenzen aufweisen, wobei mindestens 5.000 ideal sind(Abbildung 2A). Ebenso sollten Metatranskriptomik-Proben mindestens 500.000 Sequenzen aufweisen, wobei mindestens 2.000.000 ideal sind(Abbildung 2B). Proben mit weniger Sequenzen als diesen Mindestsequenzen sollten nicht für Analysen verwendet werden, da sie ihre Bakteriengemeinschaft möglicherweise nicht genau darstellen. Proben, die zwischen dem Minimum und dem Ideal liegen, können jedoch weiterhin verwendet werden, obwohl die Ergebnisse vorsichtiger interpretiert werden sollten, wenn viele Proben in diesen Bereich fallen.
Der Erfolg der nachfolgenden nachgelagerten Analyse kann einfach anhand der Frage bestimmt werden, ob die erwarteten Ausgabedateien erhalten wurden oder nicht. Programme wie QIIME2 und R (Abbildung 3) sollten es jedenfalls ermöglichen, mögliche signifikante Unterschiede zwischen den Bakteriengemeinschaften auf basis von Fracking zu bewerten. Die Daten für Abbildung 3 wurden durch das Sammeln von Sedimentproben von einundzwanzig verschiedenen Standorten an dreizehn verschiedenen Strömen für die 16S- und Metatranskriptomik-Analyse erhalten. Von diesen einundzwanzig Standorten befanden sich zwölf von ihnen stromabwärts der Fracking-Aktivität und klassifiziert als HF +, und neun von ihnen befanden sich entweder stromaufwärts der Fracking-Aktivität oder in einem Wassereinzugsgebiet, in dem Fracking nicht stattfand; diese Ströme wurden als HF- klassifiziert. Neben dem Vorhandensein von Fracking-Aktivitäten waren die Bäche ansonsten vergleichbar.
Diese Unterschiede könnten die Form konsistenter Kompositionsverschiebungen auf der Grundlage des Fracking-Status annehmen. Wenn dies der Fall wäre, würde erwartet, dass HF+- und HF-Proben in einem PCoA-Diagramm voneinander getrennt gruppiert werden, wie dies in Abbildung 3A und Abbildung 3B der Fall ist. Um zu bestätigen, dass diese scheinbaren Verschiebungen nicht nur ein Artefakt der Ordinationsmethode sind, sind weitere statistische Analysen erforderlich. Zum Beispiel ergab ein PERMANOVA22-Test auf der Entfernungsmatrix, auf der Abbildung 3A und Abbildung 3B basieren, eine signifikante Clusterung basierend auf dem Fracking-Status, was bedeutet, dass die im Diagramm beobachtete Trennung mit Unterschieden zwischen den Bakteriengemeinschaften der Proben übereinstimmt, anstatt ein Artefakt der Ordination. Ein signifikantes PERMANOVA- oder ANOSIM-Ergebnis ist ein starker Hinweis auf konsistente Unterschiede zwischen HF+- und HF-Proben, was darauf hindeuten würde, dass die HF+-Proben durch Fracking beeinträchtigt wurden, während ein hoher p-Wert darauf hindeuten würde, dass die Proben nicht betroffen waren. Metatranskriptomische Daten können ebenfalls mit den gleichen Methoden visualisiert und ausgewertet werden.
Die Untersuchung differentieller Merkmale (Mikroben oder Funktionen) kann Hinweise darauf ergeben, dass auch Proben betroffen sind. Eine Methode zum Bestimmen differentieller Features besteht darin, ein zufälliges Gesamtstrukturmodell zu erstellen. Das Zufallswaldmodell kann verwendet werden, um zu sehen, wie gut der Fracking-Status der Proben korrekt klassifiziert werden kann. Wenn das Modell besser abschneidet als zufällig erwartet, wäre dies ein zusätzlicher Beweis für Unterschiede, die vom Fracking-Status abhängen. Darüber hinaus würden die wichtigsten Prädiktoren zeigen, welche Merkmale für die korrekte Differenzierung von Stichproben am wichtigsten sind (Abbildung 3C). Auch diese Merkmale hätten dann aufgrund des Fracking-Status durchweg unterschiedliche Werte gehabt. Sobald diese differenziellen Merkmale bestimmt sind, kann die Literatur überprüft werden, um zu sehen, ob sie zuvor mit Fracking in Verbindung gebracht wurden. Es kann jedoch schwierig sein, Studien zu finden, die differentielle Funktionen bestimmen, da die meisten nur 16S rRNA-Zusammensetzungsdaten verwendet haben. Um die Auswirkungen von Differentialfunktionen zu bewerten, wäre es daher eine mögliche Methode, zu sehen, ob sie zuvor mit einer potenziellen Resistenz gegen Biozide in Verbindung gebracht wurden, die üblicherweise in Fracking-Flüssigkeiten verwendet werden, oder ob sie bei der Tolerierung von stark salzhaltigen Bedingungen helfen könnten. Darüber hinaus könnte die Untersuchung des Funktionsprofils eines Taxons von Interesse Hinweise auf die Auswirkungen von Fracking aufdecken (Abbildung 3D). Wenn beispielsweise ein Taxon durch das Zufallswaldmodell als differentiell identifiziert wird, könnte sein antimikrobielles Resistenzprofil in HF+-Proben mit seinem Profil in HF-Proben verglichen werden, und wenn sie sich stark unterscheiden, könnte dies darauf hindeuten, dass Fracking-Flüssigkeit, die Biozide enthält, in den Strom gelangt ist.
| Beispiel-ID | Konzentration (ng/μL) |
| 1 | 1.5 |
| 2 | 1.55 |
| 3 | 0.745 |
| 4 | 0.805 |
| 5 | 7.82 |
| 6 | 0.053 |
| 7 | 0.248 |
| 8 | 0.945 |
| 9 | 1.82 |
| 10 | 0.804 |
| 11 | 0.551 |
| 12 | 1.69 |
| 13 | 4.08 |
| 14 | Below_Detection |
| 15 | 7.87 |
| 16 | 0.346 |
| 17 | 2.64 |
| 18 | 1.15 |
| 19 | 0.951 |
Tabelle 1: Beispiel DNA-Konzentrationen basierend auf Fluorometer 1x DS DNA Hochempfindlichkeitsassay. Extraktionen für alle diese Proben, mit Ausnahme von 14, würden aufgrund nachweisbarer DNA-Mengen als erfolgreich angesehen werden.

Abbildung 1: Beispiel E-Gel mit PCR-Produkten. Das Gel wurde vorgefärbt und unter UV-Licht visualisiert, wodurch die darauf vorhandene DNA zum Leuchten kam. Die PCR funktionierte für die Proben in den Bohrungen 4 und 6 in der ersten Reihe, da beide ein einziges helles Band der erwarteten Größe (basierend auf der Leiter) hatten. Die PCR für die Proben in den anderen sechs Bohrungen schlug fehl, da sie keine Bänder produzierten. Die Positivkontrolle (erste Bohrung, zweite Reihe) hatte ein helles Band, was darauf hindeutet, dass die PCR ordnungsgemäß durchgeführt wurde, und die Negativkontrollen (Vertiefungen 6 und 7, zweite Reihe) hatten keine Bänder, was darauf hindeutet, dass die Proben nicht kontaminiert waren. Wenn ein Negativ ein Band hätte, das so hell ist wie die Proben, wäre die PCR als Fehler angesehen worden, da es riskant wäre anzunehmen, dass die Proben Amplicons enthielten, die nicht nur das Ergebnis einer Kontamination waren. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Anzahl der Beispielsequenzen. (A) 16S Beispielsequenzzählungen. Fast alle diese 16S-Proben hatten über 1.000 Sequenzen. Die wenigen, die weniger als 1.000 Sequenzen hatten, sollten von nachgelagerten Analysen ausgeschlossen werden, da sie nicht genügend Sequenzen hatten, um ihre Bakteriengemeinschaften genau darzustellen. Mehrere Sequenzen hatten zwischen 1.000 und 5.000 Sequenzen; Obwohl sie nicht ideal sind, wären sie immer noch verwendbar, da sie das absolute Minimum überschreiten, und die Mehrheit der Proben überschreitet auch das ideale Minimum von 5.000. (B) Metatranscriptomics Beispiel zählt. Alle Proben überschritten sowohl die minimale (500.000) als auch die ideale minimale (2.000.000) Anzahl von Sequenzen. Daher war die Sequenzierung für alle von ihnen erfolgreich, und sie konnten alle in der nachgelagerten Analyse verwendet werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Beispielanalyse. (A) PCoA-Diagramm basierend auf Koordinaten, die mit einer gewichteten Unifrac-Entfernungsmatrix berechnet wurden, die durch QIIME2 erstellt und visualisiert wurde. (B) PCoA-Diagramm basierend auf Koordinaten, die mit der aus QIIME2 exportierten Gewichteten Unifrac-Entfernungsmatrix berechnet wurden. Die Koordinaten wurden mit den Paketen Phyloseq und ggplot2 in R visualisiert. Jeder Punkt repräsentiert die Bakteriengemeinschaft einer Probe, wobei nähere Punkte auf ähnlichere Gemeinschaftszusammensetzungen hinweisen. Für diese 16S-Sedimentproben wurde eine Clusterung basierend auf dem Fracking-Status beobachtet (PERMANOVA, p=0,001). Darüber hinaus zeigen die Vektoren, dass die HF+-Proben tendenziell höhere Konzentrationen von Barium, Bromid, Nickel und Zink aufwiesen, was einer anderen Zusammensetzung der Bakteriengemeinschaft im Vergleich zu den HF-Proben entsprach. (C) Diagramm der besten Prädiktoren für ein zufälliges Waldmodell, das testete, wo bakterielle Häufigkeiten verwendet werden könnten, um den Fracking-Status unter den Proben vorherzusagen. Das Zufallsgesamtstrukturmodell wurde über R mit dem randomForest-Paket erstellt. Die Top-20-Prädiktoren werden ebenso gezeigt wie die daraus resultierende Abnahme der Verunreinigung (Maß für die Anzahl der HF+ und HF-Proben, die zusammen gruppiert sind) in Form der mittleren Abnahme des Gini-Index, wenn sie zur Trennung von Proben verwendet werden. (D) Kreisdiagramm, das das antimikrobielle Resistenzprofil des Burkholderiales-Profils auf der Grundlage metatranskriptomischer Daten zeigt. Sequenzen wurden zunächst mit Kraken2 kommentiert, um zu bestimmen, zu welchen Taxa sie gehörten. BLAST wurde dann mit diesen annotierten Sequenzen und der MEGARes 2.0-Datenbank verwendet, um festzustellen, welche antimikrobiellen Resistenzgene (in Form von "MEG_#") aktiv exprimiert wurden. Antimikrobielle Resistenzgene, die von Mitgliedern von Burkholderiales exprimiert wurden, wurden dann extrahiert, um zu sehen, welche unter diesen Taxa am häufigsten waren. Die Metatranskriptomik ist zwar kostspieliger und zeitaufwendiger, ermöglicht jedoch funktionelle Analysen wie diese, die mit 16S-Daten nicht durchgeführt werden können. Bemerkenswerterweise wurde kraken2 für diese Beispielanalyse anstelle von HUMAnN2 verwendet. Kraken2 ist schneller als HUMAnN2; Es gibt jedoch nur kompositorische Informationen aus, anstatt Zusammensetzung, Beitrag und Funktionen (Gene) und Signalwege wie HUMAnN2. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Ergänzungsdatei: Ein Beispiel für eine Metatranscriptomics-Pipeline. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Die in diesem Artikel beschriebenen Methoden wurden im Laufe mehrerer Studien entwickelt und verfeinert, die von unserer Gruppe zwischen 2014 und 20187,8,10 veröffentlicht wurden, und wurden erfolgreich in einem Kooperationsprojekt eingesetzt, um die Auswirkungen von Fracking auf aquatische Gemeinschaften in einem dreijährigen Projekt zu untersuchen, das in Kürze ein Papier zur Veröffentlichung einreichen wird. Diese Methoden werden im weiteren Verlauf des Projekts weiter eingesetzt. Darüber hinaus beschreiben andere aktuelle Literatur, die die Auswirkungen von Fracking auf Bäche und Ökosysteme untersucht, ähnliche Methoden für die Probenentnahme, -verarbeitung und -analyse7,8,10,11. Keine dieser Arbeiten verwendete jedoch metatranskriptomische Analysen, so dass dieses Papier das erste ist, das beschreibt, wie diese Analysen verwendet werden können, um die Auswirkungen von Fracking auf nahe gelegene Bäche aufzuklären. Darüber hinaus sind die hier vorgestellten Methoden zur Probenentnahme detaillierter, ebenso wie die Maßnahmen zur Vermeidung von Kontaminationen.
Einer der wichtigsten Schritte unseres Protokolls ist die Erstentnahme und Konservierung von Proben. Die Probenahme und Entnahme vor Ort bringt bestimmte Herausforderungen mit sich, da die Aufrechterhaltung einer aseptischen oder sterilen Umgebung während der Entnahme schwierig sein kann. Während dieses Schritts ist es wichtig, eine Kontamination der Proben zu vermeiden. Dazu sollten Handschuhe getragen werden, und nur sterile Behälter und Werkzeuge sollten mit Proben in Berührung kommen dürfen. Die Proben sollten auch sofort nach der Entnahme auf Eis gelegt werden, um den Abbau der Nukleinsäure zu mildern. Die Zugabe eines handelsüblichen Nukleinsäurekonservierungsmittels bei der Entnahme kann auch die Nukleinsäureausbeute erhöhen und es ermöglichen, Proben nach der Entnahme für längere Zeit zu lagern. Wann immer eine Nukleinsäureextraktion durchgeführt wird, ist es wichtig, die entsprechende Menge an Probe zu verwenden, zu viel kann Spinfilter verstopfen, die für die Extraktion verwendet werden (für die Protokolle, die sie verwenden), aber zu wenig kann zu niedrigen Ausbeuten führen. Befolgen Sie unbedingt die Anweisungen für das verwendete Kit.
Ähnlich wie bei der Feldentnahme ist die Vermeidung oder Minimierung von Kontaminationen auch bei der Nukleinsäureextraktion und Probenvorbereitung wichtig, insbesondere bei der Arbeit mit Proben mit geringer Nukleinsäureausbeute, wie suboptimalen Sedimentproben (Proben, die eine große Menge Kies oder Gestein enthalten) oder Wasserproben. Daher sollten wie bei der Probenentnahme bei all diesen Schritten Handschuhe getragen werden, um die Kontamination zu reduzieren. Darüber hinaus sollten alle Arbeitsflächen, die während der Laborprozeduren verwendet werden, zuvor durch Abwischen mit einer 10% igen Bleichlösung sterilisiert werden, gefolgt von einer 70% igen Ethanollösung. Für Pipettierschritte (3-6) sollten Filterspitzen verwendet werden, um Verunreinigungen durch die Pipette selbst zu vermeiden, wobei die Spitzen jedes Mal gewechselt werden, wenn sie eine nicht sterile Oberfläche berühren. Alle werkzeuge, die für laborarbeiten verwendet werden, einschließlich Pipetten, sollten vorher und nachher mit den Bleichmittel- und Ethanollösungen abgewischt werden. Zur Beurteilung der Kontamination sollten Extraktionsrohlinge und -negative (sterile Flüssigkeit) bei jeder Nukleinsäureextraktion und PCR-Reaktion einbezogen werden. Wenn die Quantifizierung nach Extraktionen eine nachweisbare Menge an DNA/RNA in den Negativen ergibt, können Extraktionen wiederholt werden, wenn noch genügend Probe übrig ist. Wenn negative Proben für PCR eine Verstärkung zeigen, sollte eine Fehlerbehebung durchgeführt werden, um die Quelle zu bestimmen, und dann sollten die Proben erneut ausgeführt werden. Um geringen Kontaminationsgraden Rechnung zu tragen, wird empfohlen, Extraktionsrohlinge und PCR-Negative so zu sequenzieren, dass die Verunreinigungen identifiziert und gegebenenfalls während der computergestützten Analyse entfernt werden können. Umgekehrt könnte die PCR-Amplifikation auch aufgrund einer Vielzahl von Ursachen versagen. Bei Umweltproben ist oft die Hemmung der PCR-Reaktion der Schuldige, was auf eine Vielzahl von Substanzen zurückzuführen sein kann, die die Taq-Polymerase23stören. Bei Verdacht auf Hemmung kann PCR-Wasser (siehe Materialtabelle)zur Verdünnung der DNA-Extrakte verwendet werden.
Dieses Protokoll hat einige bemerkenswerte Einschränkungen und potenzielle Schwierigkeiten. Die Probenentnahme kann sowohl für Wasser- als auch für Sedimentproben eine Herausforderung darstellen. Um genügend Biomasse zu erhalten, muss idealerweise 1 L Bachwasser durch einen Filter gedrückt werden. Die Poren des Filters müssen klein sein, um Mikroben einzufangen, können aber auch Sedimente einfangen. Wenn sich aufgrund der jüngsten Regenfälle viel Sediment im Wasser befindet, kann der Filter verstopfen, was es schwierig macht, das gesamte Volumen durch den Filter zu drücken. Für die Sedimentsammlung kann es schwierig sein, die Tiefe des Sediments während der Sammlung abzuschätzen. Darüber hinaus ist es wichtig sicherzustellen, dass das gesammelte Sediment überwiegend Aus Erde besteht, da Kieselsteine und Gesteine zu einer geringeren Nukleinsäureausbeute führen und möglicherweise keine genaue Darstellung der mikrobiellen Gemeinschaft sind. Schließlich ist es auch wichtig, dass die Proben nach der Entnahme auf Eis gehalten werden, insbesondere wenn kein Konservierungsmittel verwendet wird.
Obwohl dieses Protokoll sowohl Metatranskriptomik als auch 16S-Laborprotokolle abdeckt, sollte betont werden, dass sich diese beiden Methoden sowohl im Prozess als auch in der Art der daten, die sie liefern, sehr unterscheiden. Das 16S rRNA-Gen ist eine häufig anvisierte Region, die in Bakterien und Archaeen stark konserviert ist und zur Charakterisierung der Bakteriengemeinschaft in einer Probe nützlich ist. Obwohl es sich um einen gezielten und spezifischen Ansatz handelt, ist die Auflösung auf Artenebene oft unerreichbar, und die Charakterisierung neu divergierender Arten oder Stämme ist schwierig. Im Gegensatz dazu ist die Metatranskriptomik ein breiterer Ansatz, der alle aktiven Gene und Mikroben in einer Probe erfasst. Während 16S nur Daten zur Identifizierung liefert, kann die Metatranskriptomik funktionelle Daten wie exprimierten Gene und Stoffwechselwege liefern. Beide sind wertvoll und in Kombination können sie aufdecken, welche Bakterien vorhanden sind und welche Gene sie exprimieren.
Dieser Artikel beschreibt Methoden zur Feldentnahme und Probenverarbeitung sowohl für 16S rRNA- als auch für metatranskriptomische Analysen im Rahmen der Untersuchung von Fracking. Darüber hinaus werden Sammlungsmethoden für hochwertige DNA/RNA aus Proben mit geringer Biomasse und für die Langzeitlagerung beschrieben. Die hier beschriebenen Methoden sind der Höhepunkt unserer Erfahrungen mit der Probenentnahme und -verarbeitung in unseren Bemühungen, zu erfahren, wie sich Fracking auf nahe gelegene Bäche auswirkt, indem wir die Struktur und Funktion ihrer mikrobiellen Gemeinschaften untersuchen. Mikroben reagieren schnell auf Störungen, und folglich können die vorhandenen Mikroben und die Gene, die sie exprimieren, Aufschluss über die Auswirkungen von Fracking auf Ökosysteme geben. Insgesamt könnten diese Methoden für unser Verständnis der Auswirkungen von Fracking auf diese wichtigen Ökosysteme von unschätzbarem Wert sein.
Offenlegungen
Die Autoren haben nichts preiszugeben.
Danksagungen
Die Autoren möchten die Finanzierungsquellen für die Projekte anerkennen, die zur Entwicklung dieser Methoden geführt haben, wobei diese Quellen sind: das Howard Hughes Medical Institute (http://www.hhmi.org) durch das Precollege and Undergraduate Science Education Program sowie durch die National Science Foundation (http://www.nsf.gov) durch NSF-Auszeichnungen DBI-1248096 und CBET-1805549.
Materialien
| Name | Company | Catalog Number | Comments |
| 200 Proof Ethanol | Thermo Fisher Scientific | A4094 | 400 mL need to be added to Buffer PE (see Qiagen QIAQuck Gel Extraction kit protocol) and 96 mL needs to be added to the DNA/RNA Wash Buffer (see ZymoBIOMICS DNA/RNA Miniprep kit protocol). Additional ethanol is needed for the ZymoBIOMICS DNA/RNA Miniprep and NEBNext® Ultra™ II RNA Library Prep with Sample Purification Beads kits. |
| Agarose | Thermo Fisher Scientific | BP1356-100 | 100 g per bottle. 0.6 g of agarose would be needed to make one 2% 30 mL gel. |
| Disinfecting Bleach | Walmart (Clorox) | No catalog number | Use a 10% bleach solution for cleaning the work area before and after lab procedures |
| DNA gel loading dye | Thermo Fisher Scientific | R0611 | Each user-made (i.e. non-e-gel) should include loading dye with all of the samples in the ratio of 1 µL dye to 5 µL sample |
| DNA ladder | MilliporeSigma | D3937-1VL | A ladder should be run on every gel/e-gel |
| DNA/RNA Shield (2x) | Zymo Research | R1200-125 | 3 mL per sediment sample (50 mL conical) and 2 mL per water sample (filter) |
| Ethidium bromide | Thermo Fisher Scientific | BP1302-10 | Used for staining user-made e-gels |
| Forward Primer | Integrated DNA Technologies (IDT) | 51-01-19-06 | 0.5 µL per PCR reaction |
| Isopropanol | MilliporeSigma | 563935-1L | Generally less than 2 mL per library. Volume needed varies by mass of excised gel fragment (see Qiagen QIAQuick Gel Extraction kit protocol). |
| PCR-grade water | MilliporeSigma | 3315932001 | 13 µL per PCR reaction (assuming 1 µL of sample DNA template is used) |
| Platinum Hot Start PCR Master Mix (2x) | Thermo Fisher Scientific | 13000012 | 10 µL per PCR reaction |
| Reverse Primer | Integrated DNA Technologies (IDT) | 51-01-19-07 | 0.5 µL per PCR reaction |
| TBE Buffer (Tris-borate-EDTA) | Thermo Fisher Scientific | B52 | 1 L of 10x TBE buffer (30 mL of 1x TBE buffer would be needed to make one 30 mL gel) |
| 1 L bottle | Thermo Fisher Scientific | 02-893-4E | One needed per stream (the same bottle can be used for multiple streams if it is sterilized between uses) |
| 1.5 mL Microcentrifuge tubes | MilliporeSigma | BR780400-450EA | 5 microcentrifuge tubes are needed per DNA extraction and an additional 3 are needed to purify RNA (see ZymoBIOMICS DNA/RNA Miniprep kit protocol) |
| 2% Agarose e-gel | Thermo Fisher Scientific | G401002 | Each gel can run 10 samples (so 9 with a PCR negative and 8 if the extraction negative is run on the same gel) |
| 50 mL Conicals | CellTreat | 229421 | 1 50 mL conical needed per sediment samples |
| 500 mL Beaker | MilliporeSigma | Z740580 | Only 1 needed (for flame sterilization) |
| Aluminum foil | Walmart (Reynolds KITCHEN) | No number | Aluminum foil can be folded and autoclaved. The part not exposed to the environment can then be used as a sterile, DNA and RNA free surface for processing filters (one folded piece per filter to avoid cross-contamination) |
| Autoclave | Gettinge | LSS 130 | Only one needed |
| Centrifuge | MilliporeSigma | EP5404000138-1EA | Only 1 needed |
| Cooler | ULINE | S-22567 | Just about any cooler can be used. This one is listed due to being made of foam, making it lighter and thus easier to take along for field sampling. |
| Disruptor Genie | Bio-Rad | 3591456 | Only one needed |
| Electrophoresis chamber | Bio-Rad | 1664000EDU | Only 1 needed |
| Electrophoresis power supply | Bio-Rad | 1645050 | Only 1 needed |
| Freezer (-20 C) | K2 SCIENTIFIC | K204SDF | One needed to store DNA extracts |
| Freezer (-80 C) | K2 SCIENTIFIC | K205ULT | One needed to store RNA extracts |
| Gloves | Thermo Fisher Scientific | 19-020-352 | The catalog number is for Medium gloves. |
| Heat block | MilliporeSigma | Z741333-1EA | Only one needed |
| Lab burner | Sterlitech | 177200-00 | Only one needed |
| Laminar Flow Hood | AirClean Systems | AC624LFUV | Only 1 needed |
| Library purification kit | Qiagen | 28704 | One kit has enough for 50 reactions |
| Magnet Plate | Alpaqua | A001219 | Only one needed |
| Microcentrifuge | Thermo Fisher Scientific | 75004061 | Only one needed |
| Micropipette (1000 µL volume) | Pipette.com | L-1000 | Only 1 needed |
| Micropipette (2 µL volume) | Pipette.com | L-2 | Only 1 needed |
| Micropipette (20 µL volume) | Pipette.com | L-20 | Only 1 needed |
| Micropipette (200 µL volume) | Pipette.com | L-200R | Only 1 needed |
| NEBNext Ultra II RNA Library Prep with Sample Purification Beads | New England BioLabs Inc. | E7775S | One kit has enough reagents for 24 samples. |
| Parafilm | MilliporeSigma | P7793-1EA | 2 1" x 1" squares are needed per filter |
| PCR Tubes | Thermo Fisher Scientific | AM12230 | One tube needed per reaction |
| Pipette tips (for 1000 µL volume) | Pipette.com | LF-1000 | Pack of 576 tips |
| Pipette tips (for 20 µL volume) | Pipette.com | LF-20 | Pack of 960 tips |
| Pipette tips (for 200 µL volume) | Pipette.com | LF-250 | Pack of 960 tips |
| PowerWulf ZXR1+ computer cluster | PSSC Labs | No number | This is just an example of a supercomputer powerful enough to perform metatranscriptomics analysis in a timely manner. Only one needed. |
| Qubit fluorometer starter kit | Thermo Fisher Scientific | Q33239 | Comes with a Qubit 4 fluorometer, enough reagent for 100 DNA assays, and 500 Qubit tubes |
| Scoopula | Thermo Fisher Scientific | 14-357Q | Only one needed |
| Sterile blades | AD Surgical | A600-P10-0 | One needed per filter |
| Sterivex-GP Pressure Filter Unit | MilliporeSigma | SVGP01050 | 1 filter needed per water sample |
| Thermocycler | Bio-Rad | 1861096 | Only one needed |
| Vise-grip | Irwin | 2078500 | Only one needed (for cracking open the filters) |
| Vortex-Genie 2 | MilliporeSigma | Z258415-1EA | Only 1 needed |
| WHIRL-PAK bags | ULINE | S-22729 | 1 needed per filter |
| ZymoBIOMICS DNA/RNA Miniprep kit | Zymo Research | R2002 | One kit has enough reagents for 50 samples. |
Referenzen
- The process of unconventional natural gas production. US EPA Available from: https://www.epa.gov/uog/process-unconventional-natural-gas-production (2013)
- Brittingham, M. C., Maloney, K. O., Farag, A. M., Harper, D. D., Bowen, Z. H. Ecological risks of shale oil and gas development to wildlife, aquatic resources, and their habitats. Environmental Science & Technology. 48 (19), 11034-11047 (2014).
- McBroom, M., Thomas, T., Zhang, Y. Soil erosion and surface water quality impacts of natural gas development in East Texas, USA. Water. 4 (4), 944-958 (2012).
- Maloney, K. O., Weller, D. E. Anthropogenic disturbance, and streams: land use and land-use change affect stream ecosystems via multiple pathways. Freshwater Biology. 56 (3), 611-626 (2011).
- Meyer, J. L., et al. The contribution of headwater streams to biodiversity in river networks1. JAWRA Journal of the American Water Resources Association. 43 (1), 86-103 (2007).
- Alexander, R. B., Boyer, E. W., Smith, R. A., Schwarz, G. E., Moore, R. B. The role of headwater streams in downstream water quality. Journal of the American Water Resources Association. 43 (1), 41-59 (2007).
- Ulrich, N., et al. Response of aquatic bacterial communities to hydraulic fracturing in Northwestern Pennsylvania: A five-year study. Scientific Reports. 8 (1), 5683 (2018).
- Chen See, J. R., et al. Bacterial biomarkers of Marcellus shale activity in Pennsylvania. Frontiers in Microbiology. 9, 1697 (2018).
- Rausch, P., et al. Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome. 7 (1), 133 (2019).
- Louca, S., Doebeli, M., Parfrey, L. W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome. 6 (1), 41 (2018).
- Trexler, R., et al. Assessing impacts of unconventional natural gas extraction on microbial communities in headwater stream ecosystems in Northwestern Pennsylvania. Frontiers in Microbiology. 5, 522 (2014).
- Mumford, A. C., et al. Shale gas development has limited effects on stream biology and geochemistry in a gradient-based, multiparameter study in Pennsylvania. Proceedings of the National Academy of Sciences. 117 (7), 3670-3677 (2020).
- JoVE Core Biology DNA Isolation. Journal of Visualized Experiments Available from: https://www.jove.com/cn/science-education/10814/dna-isolation (2020)
- Oxford Gene Technology DNA Storage and Quality. OGT Available from: https://www.ogt.com/resources/literature/403_dna_storage_and_quality (2011)
- ThermoFisher SCIENTIFIC Technical Bulletin #159: Working with RNA. Thermoscientific Available from: https://www.thermofisher.com/us/en/home/references/ambion-tech-support/nuclease-enzymes/general-articles/working-with-rna.html (2020)
- QIAGEN AllPrep DNA/RNA Mini Kit. Qiagen Available from: https://www.qiagen.com/us/products/discovery-and-translational-research/dna-rna-purification/multianalyte-and-virus/allprep-dnarna-mini-kit/#orderinginformation (2020)
- ZymoBIOMICS DNA/RNA Miniprep Kit. Zymo Research Available from: https://www.zymoresearch.com/products/zymobiomics-dna-rna-miniprep-kit (2020)
- Desjardins, P., Conklin, D. NanoDrop microvolume quantitation of nucleic acids. Journal of Visualized Experiments. (45), e2565 (2010).
- 16S Illumina amplicon protocol: Earth microbiome project. Earth microbiome project Available from: https://earthmicrobiome.org/protocols-and-standards/16s/ (2018)
- Gel Purification: Binding, washing and eluting a sample | Protocol. Journal of Visualized Experiments Available from: https://www.jove.com/v/5063/gel-purification (2020)
- New England Biolabs protocol for the use with NEBNext Poly(A) mRNA magnetic isolation module (E7490) and NEBNext Ultra II RNA library prep kit for Illumina (E7770, E7775). New England Biolabs Available from: https://www.neb.com/protocols/2017/03/04/protocol-for-use-with-purified-mrna-or-rrna-depleted-rna-and-nebnext-ultra-ii-rna-library-prep-ki (2020)
- Anderson, M. J. Permutational multivariate analysis of variance (PERMANOVA). Wiley StatsRef: Statistics Reference Online. , 1-15 (2017).
- Schrader, C., Schielke, A., Ellerbroek, L., Johne, R. PCR inhibitors - occurrence, properties and removal. Journal of Applied Microbiology. 113 (5), 1014-1026 (2012).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten