
Method Article
Évaluation de l’impact de la fracturation hydraulique sur les cours d’eau à l’aide de signatures moléculaires microbiennes
Dans cet article
Résumé
Nous présentons ici un protocole pour étudier les impacts de la fracturation hydraulique sur les cours d’eau voisins en analysant leurs communautés microbiennes d’eau et de sédiments.
Résumé
La fracturation hydraulique (HF), communément appelée « fracturation hydraulique », utilise un mélange d’eau à haute pression, de sable et de produits chimiques pour fracturer les roches, libérant du pétrole et du gaz. Ce processus a révolutionné l’industrie énergétique américaine, car il donne accès à des ressources qui étaient auparavant inaccessibles et produit maintenant les deux tiers du gaz naturel total aux États-Unis. Bien que la fracturation hydraulique ait eu un impact positif sur l’économie américaine, plusieurs études ont mis en évidence ses effets néfastes sur l’environnement. L’effet de la fracturation hydraulique sur les cours d’eau d’amont, qui est particulièrement important en raison de leur impact disproportionné sur la santé de l’ensemble du bassin versant, est particulièrement préoccupant. Les bactéries à l’intérieur de ces cours d’eau peuvent être utilisées comme indicateurs de la santé des cours d’eau, car on s’attendrait à ce que les bactéries présentes et leur abondance dans un cours d’eau perturbé diffèrent de celles d’un cours d’eau autrement comparable mais non perturbé. Par conséquent, ce protocole vise à utiliser la communauté bactérienne pour déterminer si les cours d’eau ont été touchés par la fracturation hydraulique. À cette fin, des échantillons de sédiments et d’eau provenant de cours d’eau situés à proximité de la fracturation hydraulique (potentiellement touchés) et en amont ou dans un autre bassin versant d’activité de fracturation (non affecté) doivent être prélevés. Ces échantillons sont ensuite soumis à l’extraction d’acides nucléiques, à la préparation de la bibliothèque et au séquençage pour étudier la composition de la communauté microbienne. L’analyse corrélationnelle et les modèles d’apprentissage automatique peuvent ensuite être utilisés pour identifier les caractéristiques qui expliquent la variation dans la communauté, ainsi que pour identifier les biomarqueurs prédictifs de l’impact de la fracturation hydraulique. Ces méthodes peuvent révéler une variété de différences dans les communautés microbiennes entre les cours d’eau d’amont, en fonction de la proximité de la fracturation hydraulique, et servir de base à de futures études sur l’impact environnemental des activités de fracturation.
Introduction
La fracturation hydraulique (HF), ou « fracturation », est une méthode d’extraction de gaz naturel, qui est devenue de plus en plus répandue à mesure que la demande de combustibles fossiles continue d’augmenter. Cette technique consiste à utiliser des équipements de forage de grande puissance pour injecter un mélange d’eau, de sable et de produits chimiques dans des gisements de schiste riches en méthane, généralement pour libérer des gaz piégés1.
Étant donné que ces techniques de récolte non conventionnelles sont relativement nouvelles, il est important d’étudier les effets de telles pratiques sur les cours d’eau avoisinants. Les activités de fracturation hydraulique exigent le défrichement de vastes étendues de terrain pour le transport de l’équipement et la construction de plates-formes de puits. Environ 1,2 à 1,7 hectare de terrain doivent être défrichés pour chaque plate-forme de puits2,ce qui pourrait avoir une incidence sur le ruissellement et la qualité de l’eau du système3. Il y a un manque de transparence concernant la composition chimique exacte du fluide de fracturation, y compris les biocides utilisés. De plus, les eaux usées de fracturation ont tendance à être trèssalines 2. En outre, les eaux usées peuvent contenir des métaux et des substances radioactives naturelles2. Par conséquent, la possibilité de fuites et de déversements de liquide de fracturation dus à une erreur humaine ou à un dysfonctionnement de l’équipement est préoccupante.
Les écosystèmes fluviaux sont connus pour être très sensibles aux changements dans les paysages environnants4 et sont importants pour le maintien de la biodiversité5 et le cycle approprié des nutriments6 dans l’ensemble du bassin versant. Les microbes sont les organismes les plus abondants dans les cours d’eau douce et sont donc essentiels au cycle des nutriments, à la biodégradation et à la production primaire. La composition et la fonction de la communauté microbienne sont d’excellents outils pour obtenir des informations sur l’écosystème en raison de leur sensibilité à la perturbance, et des recherches récentes ont montré des changements distincts dans les assemblages bactériens observés en fonction de la proximité de l’activité de fracturation7,8. Par exemple, Beijerinckia, Burkholderiaet Methanobacterium ont été identifiés comme enrichis dans des cours d’eau proches de la fracturation hydraulique tandis que Pseudonocardia, Nitrospiraet Rhodobacter ont été enrichis dans les cours d’eau non proches de la fracturation7.
Le séquençage de nouvelle génération du gène de l’ARN ribosomique (ARNr) 16S est une méthode abordable de détermination de la composition de la communauté bactérienne qui est plus rapide et moins chère que le séquençage du génome entierapproche 9. Une pratique courante dans le domaine de l’écologie moléculaire consiste à utiliser la région V4 très variable du gène de l’ARNr 16S pour la résolution du séquençage, souvent jusqu’au niveau du genre avec un large champ d’identification9, car elle est idéale pour les échantillons environnementaux imprévisibles. Cette technique a été largement mise en œuvre dans des études publiées et a été utilisée avec succès pour identifier l’impact des opérations de fracturation sur les milieux aquatiques7,8. Cependant, il convient de noter que les bactéries ont des nombres de copies variables du gène de l’ARNr 16S, ce qui affecte leur abondance détectée10. Il existe quelques outils pour expliquer cela, mais leur efficacité est discutable10. Une autre pratique dont la prévalence augmente rapidement et qui n’a pas cette faiblesse est le séquençage métatranscriptomique, dans lequel tout l’ARN est séquencé, ce qui permet aux chercheurs d’identifier à la fois les bactéries actives et l’expression de leurs gènes.
Par conséquent, contrairement aux méthodes utilisées dans les études7,8 ,11,12publiées précédemment, ce protocole couvre également la collecte, la conservation, le traitement et l’analyse d’échantillons pour étudier la fonction de la communauté microbienne (métatranscriptomique). Les étapes détaillées ici permettent aux chercheurs de voir quel impact, le cas échéant, la fracturation hydraulique a eu sur les gènes et les voies exprimés par les microbes dans leurs flux, y compris les gènes de résistance aux antimicrobiens. De plus, le niveau de détail présenté pour la collecte d’échantillons est amélioré. Bien que plusieurs des étapes et des notes puissent sembler évidentes pour les chercheurs expérimentés, elles pourraient être inestimables pour ceux qui commencent tout juste la recherche.
Nous décrivons ici les méthodes de collecte et de traitement des échantillons afin de générer des données génétiques bactériennes afin d’étudier l’impact de la fracturation hydraulique sur les cours d’eau voisins sur la base de plusieurs années d’expérience de nos laboratoires. Ces données peuvent être utilisées dans les applications en aval pour identifier les différences correspondant à l’état de la fracturation.
Protocole
1. Prélèvement d’échantillons de sédiments pour l’extraction des acides nucléiques
- Immergez un tube conique stérile de 50 mL dans l’eau du ruisseau. Portez des gants lors du prélèvement des échantillons pour éviter d’introduire une contamination humaine indésirable. Effectuez cette étape soit à partir du rivage, soit face à l’amont si vous êtes dans l’eau.
- Pendant que le tube conique est immergé, retirez le bouchon et utilisez-le pour ramasser environ 3 mL de sédiments d’une profondeur de 1 à 3 cm dans le tube conique.
- Retirez le tube conique de l’eau et déversez toute l’eau, à l’exception d’une fine couche recouvrant l’échantillon de sédiments (environ 1 mL).
- À l’aide d’une pipette de 1000 μL et d’embouts de pipette appropriés, ajouter 3 mL d’agent de conservation ADN/ARN (voir le tableau des matériaux pour les spécifications de l’agent de conservation) à l’échantillon recueilli. Conservez les embouts de pipette dans une boîte à embouts de pipette stérile et ne les attachez que immédiatement avant utilisation et jetés après utilisation. Inverser le tube conique bouché 10 fois pour s’assurer que l’agent de conservation et l’échantillon sont bien mélangés.
NOTE L’étape 1.4 n’est pas nécessaire, mais elle est fortement recommandée si l’ARN doit être extrait des sédiments plus tard. - Placez les échantillons sur de la glace pour le reste du prélèvement d’échantillons. Au retour de la collecte, conserver au congélateur à -20 °C si les échantillons doivent être utilisés pour l’analyse 16S (ADN), ou à -70 °C, s’ils doivent être utilisés pour l’analyse métatranscriptomique (ARN).
2. Collecte de filtres pour l’extraction des acides nucléiques
- Retirez le bouchon d’une bouteille stérile de 1 L. En faisant face à l’amont ou à partir du rivage, remplissez la bouteille d’eau de ruisseau jusqu’au sommet, puis jetez-la. Répétez ce processus deux fois de plus pour conditionner la bouteille. Remplissez la bouteille entière une quatrième fois et bouchontez-la.
REMARQUE: Si vous réutilisez une bouteille de 1 L, elle peut être stérilisée en rinçant avec 10% d’eau de Javel pendant 2 min, suivie d’un rinçage trois fois à l’eau désionisée puis une fois avec 70% d’éthanol, et enfin en autoclavant avec réglages: 30 min de temps d’exposition à 121,1 ° C et 15 min de temps de séchage. Pendant l’autoclavage, le bouchon de la bouteille doit être très lâche pour éviter que la bouteille ne soit comprimée dans le processus. - Une fois sur une surface stable, utilisez une seringue stérile Luer lock et tirez un volume complet. Ensuite, connectez la seringue à un filtre en polyéthersulfone stérile et sans ADN / ARN de 1,7 cm de diamètre avec une taille de pore de 0,22 μm et poussez tout le volume à travers le filtre en appuyant sur le piston vers le bas. Répétez ce processus jusqu’à ce que le volume total recueilli dans la bouteille (1 L) soit poussé à travers le filtre.
REMARQUE: Le volume de la seringue peut être variable si, la quantité totale d’eau poussée à travers le filtre est suivie. Cependant, généralement, 60 mL est préféré. Bien que 1 L soit idéal, de manière anecdotique, un volume d’au moins 200 mL recueillerait probablement encore suffisamment de biomasse (en supposant environ 20 000 cellules par mL) pour l’extraction de l’ADN et de l’ARN. - Retirez l’excès d’eau du filtre en puisant environ 20 mL d’air dans la seringue et en la poussant à travers le filtre.
REMARQUE: Cela aidera à prévenir la perte de l’agent de conservation si l’étape 2.4 est effectuée. - À l’aide d’une micropipette P1000, ajoutez 2 mL d’un agent de conservation DE l’ADN/ARN en le déchargeant à travers la plus grande ouverture du filtre (où il a été attaché à la seringue) tout en maintenant le filtre horizontalement. L’extrémité de la pipette doit se trouver dans le barillet du filtre lorsque la pipette est enfoncée pour s’assurer que l’agent de conservation pénètre dans le filtre. Changez l’embout après chaque utilisation.
REMARQUE: Comme pour la collecte des sédiments, cette étape n’est pas nécessaire, mais elle est fortement recommandée pour augmenter le rendement en acides nucléiques plus tard, en particulier pour l’ARN. - Décollez un carré de film de paraffine et enroulez-le hermétiquement autour de chaque ouverture/ extrémité du filtre pour le sceller. Placez le filtre enveloppé de film de paraffine dans un sac d’échantillon stérile, puis placez le sac entier sur de la glace pendant le prélèvement.
REMARQUE: Assurez-vous que le côté utilisé pour envelopper le filtre est stérile, c’est-à-dire qu’il n’a pas été exposé à l’environnement auparavant. - Au retour de l’échantillonnage, conserver les filtres à -20 °C pour 16S ou -70 °C pour la méta-transcriptomique.
3. Extraction et quantification des acides nucléiques
- Nettoyez la zone de travail avec 10% d’eau de Javel et 70% d’éthanol avant de commencer le transfert de l’échantillon.
- Pour les sédiments (à partir de l’étape 1.5), utilisez généralement ~0,25 g d’échantillon. Stérilisez à la flamme un outil métallique en le trempant dans un bécher de 70% d’éthanol et en brûlant l’éthanol entre les échantillons.
- Pour les filtres (à partir de l’étape 2.6), déplacez le papier filtre dans un tube stérile pour l’extraction. Pour ce faire, suivez les étapes ci-dessous.
- Créez une surface stérile, sans ADN et ARN en pliant une feuille d’aluminium de sorte que la partie interne du pli ne soit pas exposée à l’environnement extérieur et en autoclavant la pièce pliée avec les réglages: 121,1 ° C et 5 min de temps de séchage.
- Stériliser une visière avec 70% d’éthanol et une flamme nue. Utilisez ensuite la poignée pour ouvrir le boîtier du filtre sur la surface stérile et retirer le noyau du boîtier.
- Utilisez un scalpel stérile pour couper le papier filtre du noyau en tranchant en haut et en bas, puis le long de la couture. Pliez le papier filtre à l’aide d’une pince à épilester stérile, puis coupez le filtre en petits morceaux à l’aide du scalpel.
- Placez les pièces filtrantes dans un tube de microcentrifugation pour l’extraction. Assurez-vous que le papier filtre n’entre pas en contact avec des surfaces qui ne sont pas stérilisées ou qui pourraient contenir de l’acide nucléique, car cela entraînerait une contamination indésirable de l’échantillon.
- Effectuer l’isolement de l’ADN comme décrit précédemment13 ou en utilisant un kit à base de colonnes disponible dans le commerce (voir tableau des matériaux). Les étapes du kit commercial répertoriées sont brièvement décrites ci-dessous.
- Lyser les cellules de l’échantillon en le transférant dans un tube à billes et en le soumettant à un perturbateur cellulaire à grande vitesse pendant au moins 5 min. Centrifuger et transférer le surnageant dans un tube de microcentrifugation stérile.
- Ajouter un tampon de lyse au surnageant (volume 1:1) et transférer au filtre fourni (jaune). Centrifugez le filtre.
- Transférer le filtre dans un nouveau tube de microcentrifugation stérile. Ajouter le tampon de préparation (400 μL), centrifuger et jeter le flux à travers.
- Ajouter le tampon de lavage (700 μL), centrifuger et jeter le flux à travers. Ajoutez ensuite un tampon de lavage (400 μL), une centrifugeuse et jetez à nouveau le flux.
- Transférer le filtre dans un nouveau tube de microcentrifugation stérile. Éluer avec 50 μL d’eau sans DNase/RNase et laisser reposer pendant 5 min à température ambiante avant de centrifuger.
- Pendant cette période de cubation, préparer le filtre III-HRC en le plaçant dans un tube de collecte et en y ajoutant la solution de préparation HRC (600 μL), suivie d’une étape de centrifugation de 3 min à 8 000 x g.
- Déplacez le filtre préparé sur un tube de microcentrifugation stérile. Transférer l’ADN élué de l’étape 3.4.5 à ce filtre et centrifuger à 16 000 x g pendant 3 min. Le flux à travers contient l’ADN extrait.
- Stocker les extraits d’ADN pour les sédiments et les filtres à -20 °C.
REMARQUE: Les extraits d’ADN peuvent être conservés pendant environ 8 ans à -20 ° C en supposant une température stable, une exposition limitée à la lumière et aucun contaminant nocif14. - Effectuez l’isolement de l’ARN conformément au protocole du fabricant. Conserver les extraits d’ARN à -80 °C.
- Lyser les cellules de l’échantillon en le transférant dans un tube à billes et en le soumettant à un perturbateur cellulaire à grande vitesse pendant au moins cinq minutes. Centrifuger et transférer le surnageant dans un tube de microcentrifugation stérile.
- Ajouter un tampon de lyse au surnageant (volume 1:1) et transférer dans la colonne fournie (jaune). Centrifugez la colonne.
- Ajouter un volume égal d’éthanol à 95-100% au flux et mélanger en pipetant cinq fois vers le haut et vers le bas.
- Placez la colonne IICG (verte) sur un tube de microcentrifugation stérile. Transférer la solution mélangée dans la colonne et la centrifugeuse.
- Ajouter le tampon de lavage (400 μL), centrifuger et jeter le flux à travers.
- Ajouter 5 μL de DNase I et 75 μL de tampon de digestion de l’ADN à la colonne et incuber à température ambiante pendant 15 minutes.
- Ajouter le tampon de préparation (400 μL), centrifuger et éliminer le flux.
- Ajouter le tampon de lavage (700 μL), centrifuger et jeter le flux à travers. Ajoutez ensuite un tampon de lavage (400 μL), une centrifugeuse et jetez à nouveau le flux.
- Transférer la colonne dans un nouveau tube de microcentrifugation stérile. Éluer avec 50 μL d’eau libre de DNase/RNase et laisser reposer pendant 5 min avant de centrifuger.
- Pendant cette période d’incubation, préparer le filtre III-HRC en le plaçant dans un tube de collecte et en y ajoutant la solution de préparation HRC (600 μL), suivie d’une étape de centrifugation de 3 min à 8 000 x g.
- Déplacez le filtre préparé sur un tube de microcentrifugation stérile. Transférer l’ARN élué de l’étape 3.6.9 vers ce filtre et centrifuger à 16 000 x g pendant 3 min. Le flux à travers contient l’ARN extrait.
REMARQUE: Les extraits d’ARN ne peuvent être conservés que pendant un an avant de commencer à se dégrader15. Les extraits d’ADN et d’ARN sont dégradés par congélation-décongélation répétée. Certains protocoles permettent l’extraction de l’ADN et de l’ARN du même échantillon16,17.
- Quantifier les échantillons d’ADN et d’ARN extraits à l’aide d’un fluoromètre ou d’un spectrophotomètre. Voir le tableau 1 par exemple les valeurs de concentration de l’ADN du fluoromètre. Pour un exemple de protocole de quantification du spectrophotomètre, voir la référence18. Les valeurs de concentration d’ADN des sédiments avec le kit répertorié dans la Table des matériaux varient généralement de 1 à 40 ng / μL, tandis que les valeurs de concentration de l’ADN du filtre ont tendance à varier de 0,5 à 10 ng / μL. Les valeurs de concentration de l’ARN des sédiments avec le kit répertorié dans la table des matériaux varient généralement d’environ 1 à 20 ng / μL, tandis que les valeurs de concentration de l’ARN du filtre ont tendance à être plus faibles, allant généralement de 0,5 à 5 ng/μL.
4. Création d’une bibliothèque d’ARNr 16S
- Nettoyez la zone de travail avec 10% d’eau de Javel et 70% d’éthanol. La zone de travail doit être un espace clos capable de produire des conditions d’écoulement laminaire (hotte à écoulement laminaire).
- Utilisez les extraits d’ADN (à partir de l’étape 3.5) et préparez des échantillons pour le séquençage de l’amplicon de l’ARNr 16S avec un protocole PCR standard, tel que celui décrit sur le site Web du microbiome terrestre qui amplifie la région hypervariable V4 de l’ARNr19 16S dans des conditions d’écoulement laminaire.
- Préparez un gel d’agarose à 2% comme décrit précédemment et laissez-le se solidifier17. Mélanger 7 μL de produit PCR et 13 μL d’eau sans DNase. Ajouter un colorant de chargement de gel à une concentration finale de 1x. Une fois l’agarose solidifiée, chargez ce mélange de produits PCR sur un gel d’agarose à 2%.
REMARQUE: Alternativement, un gel préfabriqué peut être utilisé à la place, car ces gels fonctionnent plus rapidement et sont préfabriqués. - Exécutez le gel à 90 V pendant 60 à 90 minutes pour vérifier la taille de la bande de 386 comme amplification réussie pour les amplicons V4 à ARNr 16S, en utilisant le protocole du microbiome terrestre.
5. Purification de la bibliothèque d’ARNr ADN 16S
- Regrouper 10 μL de produits de PCR pour les échantillons qui ont produit des bandes brillantes et 13 μL pour les échantillons qui ont donné des bandes faibles dans un tube de microcentrifugation stérile de taille appropriée.
- Vérifiez la concentration de la piscine résultante à l’aide d’un fluoromètre ou d’un spectrophotomètre et préparez un gel d’agarose à 2% comme auparavant. Idéalement, la piscine devrait avoir une concentration d’au moins 10 ng / μL, et la plupart des échantillons devraient avoir une concentration d’environ 25 ng / μL.
- Si la concentration et le volume le permettent, chargez environ 150-200 ng dans un puits de gel d’agarose à 2%.
- Faire fonctionner le gel pendant 60-90 min à 90 volts.
- Purifiez la bibliothèque regroupée en exécutant un gel d’agarose à 2%.
- Excisez la bande d’ADN de 386 bp du gel et purifiez la bibliothèque regroupée à l’aide d’un kit disponible dans le commerce comme décrit précédemment20. Éluez l’ADN purifié avec 30 μL de 10 mM de Tris-Cl (pH 8,5). Effectuez cette étape dans une zone différente de l’extraction de l’ADN ou de l’ARN pour éviter toute contamination future, car couper le gel propagera des amplicons PCR sur l’expérimentateur et la zone environnante.
- Vérifiez la concentration de la piscine purifiée à l’aide d’un fluoromètre ou d’un spectrophotomètre. Si la purification s’est bien passée, sa concentration devrait être d’au moins la moitié de la piscine non purifiée. En général, la concentration finale doit varier de 5 à 20 ng/μL.
- Envoyez les bibliothèques purifiées pour le séquençage de nouvelle génération. Assurez-vous qu’ils sont conservés au froid pendant le transport en incluant de la glace carbonique dans le conteneur d’expédition.
6. Création et purification d’une bibliothèque d’ARN
- Plusieurs kits commerciaux peuvent être utilisés pour créer des bibliothèques d’ARN. Quel que soit celui utilisé, suivez le protocole du fabricant tel qu’il est écrit lorsque vous travaillez dans un environnement à flux laminaire stérile. Une version très résumée du protocole pour le kit dans la table des matériaux est présentée ci-dessous21.
- Préparez le mélange maître de synthèse de l’ADNc du premier brin (8 μL d’eau sans nucléase et 2 μL de mélange d’enyzme de synthèse du premier brin) et ajoutez-le à l’échantillon. Placez l’échantillon dans le thermocycleur dans les conditions spécifiées dans le protocole.
- Préparez le mélange maître de synthèse de l’ADNc du deuxième brin (8 μL de tampon de réaction de synthèse du deuxième brin, 4 μL de mélange d’enzymes de synthèse du deuxième brin et 48 μL d’eau sans nucléase) sur de la glace et ajoutez-le à l’échantillon. Placer dans un thermocycleur réglé à 16 °C pendant une heure.
- Purifier la réaction en ajoutant les billes fournies (144 μL) et en effectuant deux lavages à l’éthanol à 80 % (200 μL).
- Éluez avec le tampon TE fourni (53 μL) et transférez 50 μL du surnageant dans un tube PCR propre. Placez le tube PCR sur de la glace.
- Préparez le mélange maître de préparation finale (7 μL de tampon de réaction de préparation finale et 3 μL de mélange d’enzymes de préparation finale) sur de la glace et ajoutez-le au tube de PCR. Placez le tube PCR dans un thermocycleur avec les conditions spécifiées dans le protocole.
- Mélanger les solutions d’adaptateur dilué (2,5 μL), de mélange maître de ligature (30 μL) et de ligature-amplificateur (1 μL) sur de la glace. Ajouter les solutions mélangées à l’échantillon et placer dans un thermocycleur pendant 15 min à 20 °C.
- Purifier la réaction en ajoutant les billes fournies (87 μL) et en effectuant des lavages à l’éthanol (200 μL) et une élution comme auparavant, sauf ajouter seulement 17 μL de TE.
- Ajouter les indices (10 μL) et la solution Q5 Master Mix (25 μL) et placer dans un thermocycleur dans les conditions décrites dans le protocole.
- Purifier la réaction en ajoutant les billes fournies (45 μL) et en effectuant un ajout de deux lavages à l’éthanol (200 μL) et élué avec 23 μL de TE. Transférer 20 μL dans un tube PCR propre.
- Vérifiez les bibliothèques pour les concentrations détectables d’ARN à l’aide d’un bioanalyseur, d’un fluoromètre ou d’un spectrophotomètre.
- Regrouper les bibliothèques métatranscriptomiques dans un rapport à peu près équinomolaire.
- Purifier la bibliothèque en suivant le même protocole pour la purification de la bibliothèque 16S, à l’exception des fragments d’accise entre 250 et 400 bp. Alors que la bibliothèque 16S avait une bande distincte représentant la région amplifiée, le résultat ici est un frottis.
- Vérifiez la concentration de la bibliothèque purifiée comme précédemment.
- Expédiez la bibliothèque purifiée avec de la glace carbonique à une installation de séquençage.
REMARQUE: Alternativement, les extraits d’ARN peuvent être envoyés à une université ou à une entreprise privée pour la préparation et le séquençage de la bibliothèque.
7. Analyse de la communauté microbienne
- Une fois le séquençage terminé, accédez aux données de l’échantillon. Téléchargez-le sur un ordinateur utilisable.
REMARQUE: Idéalement, l’appareil devrait avoir au moins 16 gigaoctets de RAM. Pour une discussion sur les exigences informatiques (pour Qiime2), voir https://forum.qiime2.org/t/recommended-specifications-to-run-qiime2/9808. - Utilisez des logiciels, tels que mothur, QIIME2 et R, pour analyser les données d’ARNr 16S. Voir ici https://docs.qiime2.org/2020.11/tutorials/moving-pictures/ pour un exemple de didacticiel d’analyse QIIME2 16S.
- Pour les données métatranscriptomiques (ARN), utilisez HUMAnN2 et ATLAS pour déterminer quels gènes et voies sont présents dans les échantillons.
REMARQUE : Un exemple de pipeline métatranscriptomique aboutissant à une analyse de la diversité et des forêts aléatoires est présenté dans le fichier d’informations supplémentaires. Toutes les commandes sont exécutées via la ligne de commande, par exemple, Terminal pour les utilisateurs Mac.
Résultats
Le succès des extractions d’ADN et d’ARN peut être évalué à l’aide d’une variété d’équipements et de protocoles. En général, toute concentration détectable de l’un ou l’autre est considérée comme suffisante pour conclure que l’extraction a réussi. En examinant le tableau 1, toutes les extractions, à l’exception d’une seule, seraient qualifiées de réussies. L’échec à cette étape est souvent dû à une faible biomasse initiale, à une mauvaise conservation de l’échantillon ou à une erreur humaine lors de l’extraction. Dans le cas des filtres, l’extraction peut avoir réussi même si la concentration est inférieure à la détection. Si ces extraits ne donnent pas de bandes pour la PCR (si vous faites 16S) ou une concentration détectable après la préparation de la bibliothèque (métatranscriptomique), alors ils ont probablement vraiment échoué.
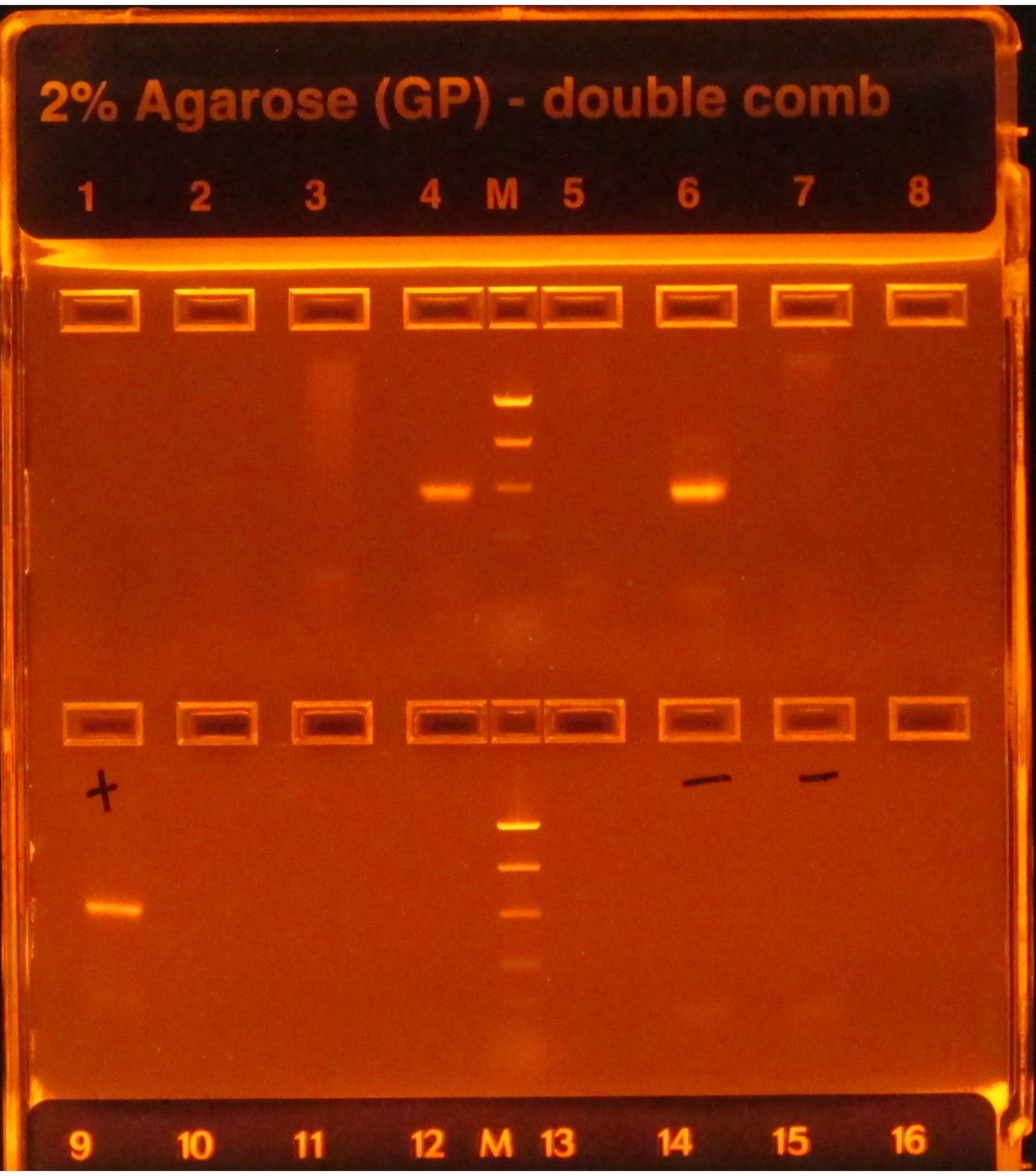
Si le protocole 16S est suivi, les bandes lumineuses suivant l’amplification par PCR, comme on le voit dans les puits 4 et 6 de la figure 1,indiquent un succès, tandis qu’un manque de bandes, comme on le voit dans les autres puits de la rangée supérieure, indique une défaillance. De plus, une bande lumineuse dans la voie de gel qui contient un contrôle PCR négatif indiquerait également une défaillance, car il serait risqué de supposer que la contamination ayant une incidence sur le ou les témoins négatifs n’a pas affecté les échantillons.
Pour le 16S et la métatranscriptomique, le succès du séquençage peut être évalué en regardant le nombre de séquences obtenues (Figure 2). Les échantillons 16S doivent avoir un minimum de 1 000 séquences, avec au moins 5 000 étant idéales(Figure 2A). De même, les échantillons métatranscriptomiques doivent avoir un minimum de 500 000 séquences, avec au moins 2 000 000 d’idéal(Figure 2B). Les échantillons avec moins de séquences que ces minimums ne doivent pas être utilisés pour les analyses, car ils peuvent ne pas représenter avec précision leur communauté bactérienne. Cependant, les échantillons qui se situent entre le minimum et l’idéal peuvent toujours être utilisés, bien que les résultats doivent être interprétés avec plus de prudence si de nombreux échantillons se situent dans cette fourchette.
Le succès de l’analyse ultérieure en aval peut être déterminé simplement sur la base de l’obtention ou non des fichiers de sortie attendus. Quoi qu’il en soit, les programmes, tels que QIIME2 et R(figure 3),devraient permettre d’évaluer les différences significatives potentielles entre les communautés bactériennes en fonction de la fracturation hydraulique. Les données de la figure 3 ont été obtenues en recueillant des échantillons de sédiments de vingt et un sites différents dans treize cours d’eau différents pour l’analyse 16S et métatranscriptomique. Sur ces vingt et un sites, douze d’entre eux étaient en aval de l’activité de fracturation et classés comme HF+, et neuf d’entre eux étaient soit en amont de l’activité de fracturation, soit dans un bassin versant où la fracturation hydraulique n’avait pas lieu; ces cours d’eau ont été classés comme HF-. Outre la présence d’activités de fracturation, les cours d’eau étaient par ailleurs comparables.
Ces différences pourraient prendre la forme de changements de composition cohérents basés sur l’état de la fracturation hydraulique. Si tel était le cas, on s’attendrait à ce que les échantillons HF+ et HF- se regroupent les uns des autres dans un diagramme PCoA, comme c’est le cas dans les figures 3A et 3B. Pour confirmer que ces changements apparents ne sont pas seulement un artefact de la méthode d’ordination, une analyse statistique plus approfondie est nécessaire. Par exemple, un test PERMANOVA22 sur la matrice de distance sur laquelle sont basées les figures 3A et 3B a révélé un regroupement significatif basé sur l’état de fracturation, ce qui signifie que la séparation observée dans le graphique est cohérente avec les différences entre les communautés bactériennes des échantillons, au lieu d’un artefact d’ordination. Un résultat significatif de PERMANOVA ou d’ANOSIM est une forte indication de différences constantes entre les échantillons HF+ et HF-, ce qui indiquerait que les échantillons HF+ ont été touchés par la fracturation hydraulique, tandis qu’une valeur p élevée indiquerait que les échantillons n’ont pas été touchés. Les données métatranscriptomiques peuvent également être visualisées et évaluées à l’aide des mêmes méthodes.
L’examen des caractéristiques différentielles (microbes ou fonctions) peut révéler des preuves que des échantillons ont également été touchés. Une méthode pour déterminer les entités différentielles consiste à créer un modèle de forêt aléatoire. Le modèle de forêt aléatoire peut être utilisé pour voir dans quelle mesure l’état de fracturation des échantillons peut être correctement classé. Si le modèle fonctionne mieux que prévu par hasard, ce serait une preuve supplémentaire des différences en fonction de l’état de la fracturation. De plus, les prédicteurs les plus importants révéleraient quelles caractéristiques étaient les plus importantes pour différencier correctement les échantillons(figure 3C). Ces caractéristiques auraient également eu des valeurs constamment différentes en fonction de l’état de la fracturation hydraulique. Une fois ces caractéristiques différentielles déterminées, la littérature peut être examinée pour voir si elles ont déjà été associées à la fracturation hydraulique. Cependant, il peut être difficile de trouver des études qui ont déterminé les fonctions différentielles, car la plupart n’ont utilisé que des données de composition de l’ARNr 16S. Par conséquent, pour évaluer les implications des fonctions différentielles, une méthode possible serait de voir si elles ont déjà été associées à une résistance potentielle aux biocides couramment utilisés dans les fluides de fracturation ou si elles pourraient aider à tolérer des conditions hautement salines. De plus, l’examen du profil fonctionnel d’un taxon d’intérêt pourrait révéler des preuves de l’impact de la fracturation hydraulique(figure 3D). Par exemple, si un taxon est identifié comme différentiel par le modèle forestier aléatoire, son profil de résistance aux antimicrobiens dans les échantillons HF+ pourrait être comparé à son profil dans les échantillons HF- et s’ils diffèrent considérablement, cela pourrait suggérer que le liquide de fracturation contenant des biocides est entré dans le flux.
| SampleID (ID de l’échantillon | Concentration (ng/μL) |
| 1 | 1.5 |
| 2 | 1.55 |
| 3 | 0.745 |
| 4 | 0.805 |
| 5 | 7.82 |
| 6 | 0.053 |
| 7 | 0.248 |
| 8 | 0.945 |
| 9 | 1.82 |
| 10 | 0.804 |
| 11 | 0.551 |
| 12 | 1.69 |
| 13 | 4.08 |
| 14 | Below_Detection |
| 15 | 7.87 |
| 16 | 0.346 |
| 17 | 2.64 |
| 18 | 1.15 |
| 19 | 0.951 |
Tableau 1 : Exemples de concentrations d’ADN basées sur le test de haute sensibilité de l’ADN Fluoromètre 1x DS. Les extractions pour tous ces échantillons, à l’exception de 14, seraient considérées comme réussies en raison de quantités détectables d’ADN.

Figure 1: Exemple d’e-gel avec des produits PCR. Le gel a été pré-coloré et visualisé sous une lumière UV, faisant briller tout ADN présent sur lui. La PCR a fonctionné pour les échantillons des puits 4 et 6 de la première rangée, car ils avaient tous deux une seule bande lumineuse de la taille attendue (basée sur l’échelle). La PCR pour les échantillons des six autres puits a échoué, car ils n’ont produit aucune bande. Le témoin positif (premier puits, deuxième rangée) avait une bande brillante, ce qui indique que la PCR a été effectuée correctement, et les témoins négatifs (puits 6 et 7, deuxième rangée) n’avaient pas de bandes, ce qui indique que les échantillons n’étaient pas contaminés. Si un négatif avait une bande aussi brillante que les échantillons, la PCR aurait été considérée comme un échec car il serait risqué de supposer que les échantillons avaient des amplicons qui n’étaient pas seulement le résultat d’une contamination. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2: Exemples de comptages de séquences. (A) 16S exemples de séquences. Presque tous ces échantillons 16S avaient plus de 1 000 séquences. Les rares qui avaient moins de 1 000 séquences devraient être exclus des analyses en aval, car ils avaient des séquences insuffisantes pour représenter avec précision leurs communautés bactériennes. Plusieurs séquences avaient entre 1 000 et 5 000 séquences; Bien qu’ils ne soient pas idéaux, ils seraient toujours utilisables puisqu’ils dépassent le strict minimum, et la majorité des échantillons dépassent également le minimum idéal de 5 000. (B) L’exemple de métatranscriptomique compte. Tous les échantillons ont dépassé à la fois le nombre minimum (500 000) et le nombre minimum idéal (2 000 000) de séquences. Par conséquent, le séquençage a été un succès pour chacun d’entre eux, et ils ont tous pu être utilisés dans l’analyse en aval. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3: Exemple d’analyse. (A) Diagramme PCoA basé sur des coordonnées calculées avec une matrice de distance unifrac pondérée créée et visualisée via QIIME2. (B) Diagramme PCoA basé sur les coordonnées calculées avec la matrice de distance unifrac pondérée exportée à partir de QIIME2. Les coordonnées ont été visualisées à l’aide des paquets Phyloseq et ggplot2 dans R. Les vecteurs de métadonnées ont été ajustés au tracé à l’aide du paquet Vegan. Chaque point représente la communauté bactérienne d’un échantillon, avec des points plus proches indiquant des compositions de communauté plus similaires. Un regroupement basé sur l’état de fracturation de ces échantillons de sédiments 16S a été observé (PERMANOVA, p = 0,001). En outre, les vecteurs révèlent que les échantillons HF + avaient tendance à avoir des niveaux plus élevés de baryum, de bromure, de nickel et de zinc, ce qui correspondait à une composition de communauté bactérienne différente par rapport aux échantillons HF-. (C) Graphique des meilleurs prédicteurs pour un modèle forestier aléatoire qui a testé où les abondances bactériennes pouvaient être utilisées pour prédire l’état de fracturation parmi les échantillons. Le modèle de forêt aléatoire a été créé via R à l’aide du package randomForest. Les 20 principaux prédicteurs sont présentés ainsi que les diminutions d’impuretés qui en résultent (mesure du nombre d’échantillons HF+ et HF- regroupés) sous la forme d’une diminution moyenne de l’indice de Gini lorsqu’ils sont utilisés pour séparer les échantillons. (D) Diagramme circulaire montrant le profil de résistance aux antimicrobiens du profil Burkholderiales basé sur des données métatranscriptomiques. Les séquences ont d’abord été annotées avec Kraken2 pour déterminer à quels taxons elles appartenaient. BLAST a ensuite été utilisé avec ces séquences annotées et la base de données MEGARes 2.0 pour déterminer quels gènes de résistance aux antimicrobiens (sous la forme de « MEG_# ») étaient activement exprimés. Les gènes de résistance aux antimicrobiens exprimés par les membres des Burkholderiales ont ensuite été extraits pour voir lesquels étaient les plus répandus parmi ces taxons. Bien que plus coûteuse et plus longue, la métatranscriptomique permet des analyses fonctionnelles, comme celle-ci, qui ne peut pas être effectuée avec des données 16S. Notamment, Kraken2 a été utilisé pour cet exemple d’analyse, au lieu de HUMAnN2. Kraken2 est plus rapide que HUMAnN2; cependant, il ne produit que des informations de composition, au lieu de la composition, de la contribution, des fonctions (gènes) et des voies comme le fait HUMAnN2. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Fichier supplémentaire : Exemple de pipeline métatranscriptomique. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Les méthodes décrites dans cet article ont été développées et affinées au cours de plusieurs études publiées par notre groupe entre 2014 et 2018 7,8,10 et ont été utilisées avec succès dans un projet collaboratif visant à étudier les impacts de la fracturation hydraulique sur les communautés aquatiques dans un projet de trois ans qui soumettra bientôt un article pour publication. Ces méthodes continueront d’être utilisées pendant le reste du projet. En outre, d’autres publications actuelles sur l’impact de la fracturation hydraulique sur les cours d’eau et les écosystèmes décrivent des méthodes similaires de collecte, de traitement etd’analysedes échantillons7,8,10,11. Cependant, aucun de ces articles n’a utilisé l’analyse métatranscriptomique, ce qui fait de cet article le premier à décrire comment ces analyses peuvent être utilisées pour élucider l’impact de la fracturation hydraulique sur les cours d’eau voisins. De plus, les méthodes présentées ici pour le prélèvement d’échantillons sont plus détaillées, tout comme les mesures prises pour éviter la contamination.
L’une des étapes les plus importantes de notre protocole est la collecte et la conservation initiales des échantillons. L’échantillonnage et la collecte sur le terrain comportent certains défis, car il peut être difficile de maintenir un environnement aseptique ou stérile pendant la collecte. Au cours de cette étape, il est essentiel d’éviter de contaminer les échantillons. Pour ce faire, des gants doivent être portés et seuls les contenants et les outils stériles doivent être autorisés à entrer en contact avec les échantillons. Les échantillons doivent également être immédiatement placés sur la glace après le prélèvement afin d’atténuer la dégradation des acides nucléiques. L’ajout d’un agent de conservation commercial des acides nucléiques lors de la collecte peut également augmenter le rendement en acides nucléiques et permettre aux échantillons d’être conservés pendant de plus longues périodes après la collecte. Chaque fois que l’extraction d’acide nucléique est effectuée, il est important d’utiliser la quantité appropriée d’échantillon, trop peut obstruer les filtres de spin utilisés pour l’extraction (pour les protocoles qui les utilisent), mais trop peu peut entraîner de faibles rendements. Assurez-vous de suivre les instructions pour le kit utilisé.
À l’instar de la collecte sur le terrain, il est également important d’éviter ou de minimiser la contamination lors de l’extraction et de la préparation des échantillons d’acides nucléiques, en particulier lorsque vous travaillez avec des échantillons à faible rendement en acides nucléiques, tels que des échantillons de sédiments sous-optimaux (échantillons contenant une grande quantité de gravier ou de roches) ou des échantillons d’eau. Par conséquent, comme pour le prélèvement d’échantillons, des gants doivent être portés pendant toutes ces étapes afin de réduire la contamination. De plus, toutes les surfaces de travail utilisées pendant les procédures de laboratoire doivent être stérilisées au préalable en essuyant avec une solution d’eau de Javel à 10%, suivie d’une solution d’éthanol à 70%. Pour les étapes de pipetage (3-6), les embouts de filtre doivent être utilisés pour éviter la contamination due à la pipette elle-même, les embouts étant changés chaque fois qu’ils touchent une surface non stérile. Tous les outils utilisés pour les travaux de laboratoire, y compris les pipettes, doivent être essuyés avant et après avec les solutions d’eau de Javel et d’éthanol. Pour évaluer la contamination, les blancs et les négatifs d’extraction (liquide stérile) doivent être inclus lors de chaque série d’extractions d’acides nucléiques et de réactions de PCR. Si la quantification après les extractions révèle une quantité détectable d’ADN/ARN dans les négatifs, les extractions peuvent être répétées s’il reste suffisamment d’échantillon. Si des échantillons négatifs pour la PCR montrent une amplification, un dépannage doit être effectué pour déterminer la source, puis les échantillons doivent être réexécutés. Pour tenir compte des faibles niveaux de contamination, il est recommandé de séquencer les blancs d’extraction et les négatifs pcR afin que les contaminants puissent être identifiés et éliminés, si nécessaire, lors de l’analyse informatique. Inversement, l’amplification par PCR pourrait également échouer en raison de diverses causes. Pour les échantillons environnementaux, l’inhibition de la réaction PCR est souvent le coupable, ce qui peut être dû à une variété de substances interférant avec la Taq polymérase23. Si une inhibition est suspectée, de l’eau de qualité PCR (voir Tableau des matériaux)peut être utilisée pour diluer les extraits d’ADN.
Ce protocole présente quelques limites notables et difficultés potentielles. La collecte d’échantillons peut être difficile pour les échantillons d’eau et de sédiments. Afin d’obtenir suffisamment de biomasse, idéalement, 1 L d’eau de ruisseau doit être poussé à travers un filtre. Les pores du filtre doivent être petits pour capturer les microbes, mais peuvent également piéger les sédiments. Si beaucoup de sédiments sont dans l’eau en raison des précipitations récentes, le filtre peut se boucher, ce qui rend difficile de pousser tout le volume à travers le filtre. Pour la collecte des sédiments, il peut être difficile d’estimer la profondeur des sédiments pendant la collecte. De plus, il est important de s’assurer que les sédiments recueillis sont principalement du sol, car les cailloux et les roches entraîneront une baisse du rendement en acides nucléiques et peuvent ne pas être une représentation précise de la communauté microbienne. Enfin, il est également essentiel que les échantillons soient conservés sur la glace après la collecte, surtout si un agent de conservation n’est pas utilisé.
Bien que ce protocole couvre à la fois la métatranscriptomique et les protocoles de laboratoire 16S, il convient de souligner que ces deux méthodes sont très différentes à la fois dans le processus et dans le type de données qu’elles fournissent. Le gène de l’ARNr 16S est une région couramment ciblée, hautement conservée dans les bactéries et les archées, et utile pour caractériser la communauté bactérienne dans un échantillon. Bien qu’il s’agit d’une approche ciblée et spécifique, la résolution au niveau de l’espèce est souvent inaccessible, et il est difficile de caractériser les espèces ou les souches nouvellement divergentes. Au contraire, la métatranscriptomique est une approche plus large qui capture tous les gènes actifs et les microbes présents dans un échantillon. Alors que 16S ne fournit que des données pour l’identification, la métatranscriptomique peut fournir des données fonctionnelles telles que les gènes exprimés et les voies métaboliques. Les deux sont précieux et lorsqu’ils sont combinés, ils peuvent révéler quelles bactéries sont présentes et quels gènes ils expriment.
Cet article décrit les méthodes de collecte sur le terrain et de traitement des échantillons pour les analyses d’ARNr 16S et métatranscriptomiques dans le contexte de l’étude de la fracturation hydraulique. En outre, il détaille les méthodes de collecte d’ADN / ARN de haute qualité à partir d’échantillons à faible biomasse et pour le stockage à long terme. Les méthodes décrites ici sont l’aboutissement de nos expériences en matière de collecte et de traitement d’échantillons dans le cadre de nos efforts visant à apprendre l’impact de la fracturation hydraulique sur les cours d’eau voisins en examinant la structure et la fonction de leurs communautés microbiennes. Les microbes réagissent rapidement aux perturbations et, par conséquent, quels microbes sont présents et les gènes qu’ils expriment peuvent fournir des informations sur les effets de la fracturation hydraulique sur les écosystèmes. Dans l’ensemble, ces méthodes pourraient être inestimables pour comprendre l’impact de la fracturation hydraulique sur ces écosystèmes importants.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Les auteurs aimeraient reconnaître les sources de financement pour les projets qui ont conduit au développement de ces méthodes, ces sources étant: le Howard Hughes Medical Institute (http://www.hhmi.org) par le biais du programme d’enseignement des sciences préuniversitaire et de premier cycle, ainsi que par la National Science Foundation (http://www.nsf.gov) par le biais des prix NSF DBI-1248096 et CBET-1805549.
matériels
| Name | Company | Catalog Number | Comments |
| 200 Proof Ethanol | Thermo Fisher Scientific | A4094 | 400 mL need to be added to Buffer PE (see Qiagen QIAQuck Gel Extraction kit protocol) and 96 mL needs to be added to the DNA/RNA Wash Buffer (see ZymoBIOMICS DNA/RNA Miniprep kit protocol). Additional ethanol is needed for the ZymoBIOMICS DNA/RNA Miniprep and NEBNext® Ultra™ II RNA Library Prep with Sample Purification Beads kits. |
| Agarose | Thermo Fisher Scientific | BP1356-100 | 100 g per bottle. 0.6 g of agarose would be needed to make one 2% 30 mL gel. |
| Disinfecting Bleach | Walmart (Clorox) | No catalog number | Use a 10% bleach solution for cleaning the work area before and after lab procedures |
| DNA gel loading dye | Thermo Fisher Scientific | R0611 | Each user-made (i.e. non-e-gel) should include loading dye with all of the samples in the ratio of 1 µL dye to 5 µL sample |
| DNA ladder | MilliporeSigma | D3937-1VL | A ladder should be run on every gel/e-gel |
| DNA/RNA Shield (2x) | Zymo Research | R1200-125 | 3 mL per sediment sample (50 mL conical) and 2 mL per water sample (filter) |
| Ethidium bromide | Thermo Fisher Scientific | BP1302-10 | Used for staining user-made e-gels |
| Forward Primer | Integrated DNA Technologies (IDT) | 51-01-19-06 | 0.5 µL per PCR reaction |
| Isopropanol | MilliporeSigma | 563935-1L | Generally less than 2 mL per library. Volume needed varies by mass of excised gel fragment (see Qiagen QIAQuick Gel Extraction kit protocol). |
| PCR-grade water | MilliporeSigma | 3315932001 | 13 µL per PCR reaction (assuming 1 µL of sample DNA template is used) |
| Platinum Hot Start PCR Master Mix (2x) | Thermo Fisher Scientific | 13000012 | 10 µL per PCR reaction |
| Reverse Primer | Integrated DNA Technologies (IDT) | 51-01-19-07 | 0.5 µL per PCR reaction |
| TBE Buffer (Tris-borate-EDTA) | Thermo Fisher Scientific | B52 | 1 L of 10x TBE buffer (30 mL of 1x TBE buffer would be needed to make one 30 mL gel) |
| 1 L bottle | Thermo Fisher Scientific | 02-893-4E | One needed per stream (the same bottle can be used for multiple streams if it is sterilized between uses) |
| 1.5 mL Microcentrifuge tubes | MilliporeSigma | BR780400-450EA | 5 microcentrifuge tubes are needed per DNA extraction and an additional 3 are needed to purify RNA (see ZymoBIOMICS DNA/RNA Miniprep kit protocol) |
| 2% Agarose e-gel | Thermo Fisher Scientific | G401002 | Each gel can run 10 samples (so 9 with a PCR negative and 8 if the extraction negative is run on the same gel) |
| 50 mL Conicals | CellTreat | 229421 | 1 50 mL conical needed per sediment samples |
| 500 mL Beaker | MilliporeSigma | Z740580 | Only 1 needed (for flame sterilization) |
| Aluminum foil | Walmart (Reynolds KITCHEN) | No number | Aluminum foil can be folded and autoclaved. The part not exposed to the environment can then be used as a sterile, DNA and RNA free surface for processing filters (one folded piece per filter to avoid cross-contamination) |
| Autoclave | Gettinge | LSS 130 | Only one needed |
| Centrifuge | MilliporeSigma | EP5404000138-1EA | Only 1 needed |
| Cooler | ULINE | S-22567 | Just about any cooler can be used. This one is listed due to being made of foam, making it lighter and thus easier to take along for field sampling. |
| Disruptor Genie | Bio-Rad | 3591456 | Only one needed |
| Electrophoresis chamber | Bio-Rad | 1664000EDU | Only 1 needed |
| Electrophoresis power supply | Bio-Rad | 1645050 | Only 1 needed |
| Freezer (-20 C) | K2 SCIENTIFIC | K204SDF | One needed to store DNA extracts |
| Freezer (-80 C) | K2 SCIENTIFIC | K205ULT | One needed to store RNA extracts |
| Gloves | Thermo Fisher Scientific | 19-020-352 | The catalog number is for Medium gloves. |
| Heat block | MilliporeSigma | Z741333-1EA | Only one needed |
| Lab burner | Sterlitech | 177200-00 | Only one needed |
| Laminar Flow Hood | AirClean Systems | AC624LFUV | Only 1 needed |
| Library purification kit | Qiagen | 28704 | One kit has enough for 50 reactions |
| Magnet Plate | Alpaqua | A001219 | Only one needed |
| Microcentrifuge | Thermo Fisher Scientific | 75004061 | Only one needed |
| Micropipette (1000 µL volume) | Pipette.com | L-1000 | Only 1 needed |
| Micropipette (2 µL volume) | Pipette.com | L-2 | Only 1 needed |
| Micropipette (20 µL volume) | Pipette.com | L-20 | Only 1 needed |
| Micropipette (200 µL volume) | Pipette.com | L-200R | Only 1 needed |
| NEBNext Ultra II RNA Library Prep with Sample Purification Beads | New England BioLabs Inc. | E7775S | One kit has enough reagents for 24 samples. |
| Parafilm | MilliporeSigma | P7793-1EA | 2 1" x 1" squares are needed per filter |
| PCR Tubes | Thermo Fisher Scientific | AM12230 | One tube needed per reaction |
| Pipette tips (for 1000 µL volume) | Pipette.com | LF-1000 | Pack of 576 tips |
| Pipette tips (for 20 µL volume) | Pipette.com | LF-20 | Pack of 960 tips |
| Pipette tips (for 200 µL volume) | Pipette.com | LF-250 | Pack of 960 tips |
| PowerWulf ZXR1+ computer cluster | PSSC Labs | No number | This is just an example of a supercomputer powerful enough to perform metatranscriptomics analysis in a timely manner. Only one needed. |
| Qubit fluorometer starter kit | Thermo Fisher Scientific | Q33239 | Comes with a Qubit 4 fluorometer, enough reagent for 100 DNA assays, and 500 Qubit tubes |
| Scoopula | Thermo Fisher Scientific | 14-357Q | Only one needed |
| Sterile blades | AD Surgical | A600-P10-0 | One needed per filter |
| Sterivex-GP Pressure Filter Unit | MilliporeSigma | SVGP01050 | 1 filter needed per water sample |
| Thermocycler | Bio-Rad | 1861096 | Only one needed |
| Vise-grip | Irwin | 2078500 | Only one needed (for cracking open the filters) |
| Vortex-Genie 2 | MilliporeSigma | Z258415-1EA | Only 1 needed |
| WHIRL-PAK bags | ULINE | S-22729 | 1 needed per filter |
| ZymoBIOMICS DNA/RNA Miniprep kit | Zymo Research | R2002 | One kit has enough reagents for 50 samples. |
Références
- The process of unconventional natural gas production. US EPA Available from: https://www.epa.gov/uog/process-unconventional-natural-gas-production (2013)
- Brittingham, M. C., Maloney, K. O., Farag, A. M., Harper, D. D., Bowen, Z. H. Ecological risks of shale oil and gas development to wildlife, aquatic resources, and their habitats. Environmental Science & Technology. 48 (19), 11034-11047 (2014).
- McBroom, M., Thomas, T., Zhang, Y. Soil erosion and surface water quality impacts of natural gas development in East Texas, USA. Water. 4 (4), 944-958 (2012).
- Maloney, K. O., Weller, D. E. Anthropogenic disturbance, and streams: land use and land-use change affect stream ecosystems via multiple pathways. Freshwater Biology. 56 (3), 611-626 (2011).
- Meyer, J. L., et al. The contribution of headwater streams to biodiversity in river networks1. JAWRA Journal of the American Water Resources Association. 43 (1), 86-103 (2007).
- Alexander, R. B., Boyer, E. W., Smith, R. A., Schwarz, G. E., Moore, R. B. The role of headwater streams in downstream water quality. Journal of the American Water Resources Association. 43 (1), 41-59 (2007).
- Ulrich, N., et al. Response of aquatic bacterial communities to hydraulic fracturing in Northwestern Pennsylvania: A five-year study. Scientific Reports. 8 (1), 5683 (2018).
- Chen See, J. R., et al. Bacterial biomarkers of Marcellus shale activity in Pennsylvania. Frontiers in Microbiology. 9, 1697 (2018).
- Rausch, P., et al. Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome. 7 (1), 133 (2019).
- Louca, S., Doebeli, M., Parfrey, L. W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome. 6 (1), 41 (2018).
- Trexler, R., et al. Assessing impacts of unconventional natural gas extraction on microbial communities in headwater stream ecosystems in Northwestern Pennsylvania. Frontiers in Microbiology. 5, 522 (2014).
- Mumford, A. C., et al. Shale gas development has limited effects on stream biology and geochemistry in a gradient-based, multiparameter study in Pennsylvania. Proceedings of the National Academy of Sciences. 117 (7), 3670-3677 (2020).
- JoVE Core Biology DNA Isolation. Journal of Visualized Experiments Available from: https://www.jove.com/cn/science-education/10814/dna-isolation (2020)
- Oxford Gene Technology DNA Storage and Quality. OGT Available from: https://www.ogt.com/resources/literature/403_dna_storage_and_quality (2011)
- ThermoFisher SCIENTIFIC Technical Bulletin #159: Working with RNA. Thermoscientific Available from: https://www.thermofisher.com/us/en/home/references/ambion-tech-support/nuclease-enzymes/general-articles/working-with-rna.html (2020)
- QIAGEN AllPrep DNA/RNA Mini Kit. Qiagen Available from: https://www.qiagen.com/us/products/discovery-and-translational-research/dna-rna-purification/multianalyte-and-virus/allprep-dnarna-mini-kit/#orderinginformation (2020)
- ZymoBIOMICS DNA/RNA Miniprep Kit. Zymo Research Available from: https://www.zymoresearch.com/products/zymobiomics-dna-rna-miniprep-kit (2020)
- Desjardins, P., Conklin, D. NanoDrop microvolume quantitation of nucleic acids. Journal of Visualized Experiments. (45), e2565 (2010).
- 16S Illumina amplicon protocol: Earth microbiome project. Earth microbiome project Available from: https://earthmicrobiome.org/protocols-and-standards/16s/ (2018)
- Gel Purification: Binding, washing and eluting a sample | Protocol. Journal of Visualized Experiments Available from: https://www.jove.com/v/5063/gel-purification (2020)
- New England Biolabs protocol for the use with NEBNext Poly(A) mRNA magnetic isolation module (E7490) and NEBNext Ultra II RNA library prep kit for Illumina (E7770, E7775). New England Biolabs Available from: https://www.neb.com/protocols/2017/03/04/protocol-for-use-with-purified-mrna-or-rrna-depleted-rna-and-nebnext-ultra-ii-rna-library-prep-ki (2020)
- Anderson, M. J. Permutational multivariate analysis of variance (PERMANOVA). Wiley StatsRef: Statistics Reference Online. , 1-15 (2017).
- Schrader, C., Schielke, A., Ellerbroek, L., Johne, R. PCR inhibitors - occurrence, properties and removal. Journal of Applied Microbiology. 113 (5), 1014-1026 (2012).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.