
Method Article
Evaluating the Impact of Hydraulic Fracturing on Streams using Microbial Molecular Signatures
In This Article
Summary
Here, we present a protocol to investigate the impacts of hydraulic fracturing on nearby streams by analyzing their water and sediment microbial communities.
Abstract
Hydraulic fracturing (HF), commonly called "fracking", uses a mixture of high-pressure water, sand, and chemicals to fracture rocks, releasing oil and gas. This process revolutionized the U.S. energy industry, as it gives access to resources that were previously unobtainable and now produces two-thirds of the total natural gas in the United States. Although fracking has had a positive impact on the U.S. economy, several studies have highlighted its detrimental environmental effects. Of particular concern is the effect of fracking on headwater streams, which are especially important due to their disproportionately large impact on the health of the entire watershed. The bacteria within those streams can be used as indicators of stream health, as the bacteria present and their abundance in a disturbed stream would be expected to differ from those in an otherwise comparable but undisturbed stream. Therefore, this protocol aims to use the bacterial community to determine if streams have been impacted by fracking. To this end, sediment, and water samples, from streams near fracking (potentially impacted) and upstream or in a different watershed of fracking activity (unimpacted) must be collected. Those samples are then subjected to nucleic acid extraction, library preparation, and sequencing to investigate microbial community composition. Correlational analysis and machine learning models can subsequently be employed to identify which features are explanative of variation in the community, as well as identification of predictive biomarkers for fracking's impact. These methods can reveal a variety of differences in the microbial communities among headwater streams, based on the proximity to fracking, and serve as a foundation for future investigations on the environmental impact of fracking activities.
Introduction
Hydraulic fracturing (HF), or "fracking", is a method of natural gas extraction, which has become increasingly prevalent as the demand for fossil fuels continues to rise. This technique consists of using high-powered drilling equipment to inject a blend of water, sand, and chemicals into methane-rich shale deposits, usually to release trapped gasses1.
Because these unconventional harvesting techniques are relatively new, it is important to investigate the effects of such practices on nearby waterways. Fracking activities mandate the clearing of large swaths of land for equipment transportation and well pad construction. Approximately 1.2-1.7 hectares of land must be cleared for each well pad2, potentially impacting runoff and water quality of the system3. There is a lack of transparency surrounding the exact chemical composition of fracking fluid, including what biocides are used. Additionally, fracking wastewater tends to be highly saline2. Furthermore, the wastewater may contain metals and naturally occurring radioactive substances2. Therefore, the possibility of leaks and spills of fracking fluid due to human error or equipment malfunction is concerning.
Stream ecosystems are known to be very sensitive to changes in surrounding landscapes4 and are important for maintaining biodiversity5 and proper nutrient cycling6 within the entire watershed. Microbes are the most abundant organisms in freshwater streams and thus, are essential to nutrient cycling, biodegradation, and primary production. Microbial community composition and function serve as great tools to gain information on the ecosystem due to their sensitivity to perturbance, and recent research has shown distinct shifts in observed bacterial assemblages based on proximity to fracking activity7,8. For example, Beijerinckia, Burkholderia, and Methanobacterium were identified as enriched in streams near fracking while Pseudonocardia, Nitrospira, and Rhodobacter were enriched in the streams not near fracking7.
Next generation sequencing of the 16S ribosomal RNA (rRNA) gene is an affordable method of determining bacterial community composition that is faster and cheaper than whole genome sequencing approaches9. A common practice within the field of molecular ecology is to use the highly variable V4 region of the 16S rRNA gene for sequencing resolution, often down to the genus level with a wide scope of identification9, as it is ideal for unpredictable environmental samples. This technique has been implemented widely in published studies and has been successfully utilized to identify the impact of fracking operations on aquatic environments7,8. However, it is worth noting that bacteria have varying copy numbers of the 16S rRNA gene, which affects their detected abundances10. There are a few tools to account for this, but their efficacy is questionable10. Another practice that is quickly growing in prevalence and lacks this weakness is metatranscriptomic sequencing, in which all RNA is sequenced, allowing researchers to identify both active bacteria and their genes expression.
Therefore, in contrast to methods in previously published studies7,8,11,12, this protocol also covers sample collection, preservation, processing, and analysis for investigating microbial community function (metatranscriptomics). The steps detailed herein allow researchers to see what impact, if any, fracking has had on the genes and pathways expressed by microbes in their streams, including antimicrobial resistance genes. Moreover, the level of detail presented for sample collection is improved. Although several of the steps and notes may seem obvious to experienced researchers, they could be invaluable to those just starting research.
Herein, we describe methods for sample collection and processing to generate bacterial genetic data as a means to investigate the impact of fracking on nearby streams based on our labs' several years of experience. These data can be used in downstream applications to identify differences corresponding to fracking status.
Protocol
1. Collection of sediment samples for nucleic acid extraction
- Submerge a sterile 50 mL conical tube into the stream water. Wear gloves during sample collection to avoid introducing unwanted human contamination. Perform this step either from the shore or facing upstream if in the water.
- While the conical tube is submerged, remove the cap, and use it to scoop approximately 3 mL of sediment from a depth of 1 to 3 cm into the conical tube.
- Remove the conical tube from the water and dump out all water, except for a thin layer covering the sediment sample (approximately 1 mL).
- Using a 1000 µL pipette and appropriate pipette tips, add 3 mL of DNA/RNA preservative (see Table of Materials for the preservative specifications) to the collected sample. Keep the pipette tips in a sterile pipette tip box and only attach them immediately before use and discarded after use. Invert the capped conical tube 10 times to ensure the preservative and sample are thoroughly mixed.
NOTE Step 1.4 is not necessary, but it is strongly recommended if RNA is to be extracted from the sediments later. - Place the samples on ice for the rest of sample collection. Upon returning from collection, store in a freezer at -20 °C if the samples are to be used for 16S analysis (DNA), or -70 °C, if they are to be used for metatranscriptomics analysis (RNA).
2. Filter collection for nucleic acid extraction
- Remove the cap of a sterile 1 L bottle. While facing upstream or from the shore, fill the bottle with stream water to the top and then dump it out. Repeat this process two more times to condition the bottle. Fill the entire bottle a fourth time and cap it.
NOTE: If reusing a 1 L bottle, it can be sterilized by rinsing with 10% bleach for 2 min, followed by rinsing three times with deionized water and then once with 70% ethanol, and finally autoclaving with settings: 30 min exposure time at 121.1 °C and 15 min drying time. During autoclaving, the cap on the bottle should be very loose to avoid the bottle being compressed in the process. - Once on a stable surface, use a sterile Luer lock syringe and draw up a full volume. Then connect the syringe to a sterile and DNA/RNA-free 1.7 cm diameter polyethersulfone filter with a pore size of 0.22 µm and push the entire volume through the filter by pressing the plunger all the way down. Repeat this process until the total volume collected in the bottle (1 L) is pushed through the filter.
NOTE: The volume of the syringe can be variable, if, the total amount of water pushed through the filter is tracked. However, generally, 60 mL is preferred. While 1 L is ideal, anecdotally, a volume of at least 200 mL would likely still collect enough biomass (assuming ~20,000 cells per mL) for the extraction of DNA and RNA. - Remove excess water from the filter by drawing up roughly 20 mL worth of air into the syringe and pushing it through the filter.
NOTE: This will help prevent loss of the preservative if step 2.4 is performed. - Using a P1000 micropipette, add 2 mL of a DNA/RNA preservative by discharging it through the filter's larger opening (where it was attached to the syringe) while holding the filter horizontally. The tip of the pipette should be within the barrel of the filter when the pipette is depressed to ensure the preservative enters the filter. Change the tip after each use.
NOTE: As with the sediment collection, this step is not necessary, but it is strongly recommended for increased nucleic acid yield later, especially for RNA. - Peel off one square of paraffin film and wrap it tightly around each opening/end of the filter to seal. Place the paraffin film wrapped filter into a sterile sample bag and then place the entire bag on ice during collection.
NOTE: Ensure that the side used to wrap the filter is sterile, i.e., not previously exposed to the environment. - Upon return from sampling, store filters at -20 °C for 16S or -70 °C for meta-transcriptomics.
3. Nucleic acid extraction and quantification
- Clean the work area with 10% Bleach and 70% Ethanol before beginning sample transfer.
- For sediment (from step 1.5), generally, use ~0.25 g of sample. Flame sterilize a metal tool by dipping it in a beaker of 70% ethanol and burning the ethanol off between samples.
- For filters (from step 2.6), move the filter paper into a sterile tube for extraction. To do so follow the steps below.
- Create a sterile, DNA and RNA free-surface by folding aluminum foil so that the inner part of the fold is not exposed to the outside environment and autoclaving the folded piece with the settings: 121.1 °C and 5 min drying time.
- Sterilize a vise-grip with 70% ethanol and an open flame. Then use the vise-grip to break open the filter casing on the sterile surface and remove the core from the casing.
- Use a sterile scalpel to cut the filter paper away from the core by slicing at the top and bottom and then along the seam. Fold the filter paper using sterile tweezers and then cut the filter into small pieces using the scalpel.
- Place the filter pieces in a microcentrifuge tube for extraction. Make sure that the filter paper does not come into contact with any surfaces which are not sterilized or that could have nucleic acid present, as this would lead to unwanted contamination of the sample.
- Perform DNA isolation as described previously13 or by using a commercially available column-based kit (see Table of Materials). The steps for the commercial kit listed are briefly described below.
- Lyse the cells within the sample by transferring it to a bead tube and subjecting it to a cell disruptor at high speed for at least 5 min. Centrifuge and transfer the supernatant to a sterile microcentrifuge tube.
- Add lysis buffer to the supernatant (1:1 volume) and transfer to the provided filter (yellow). Centrifuge the filter.
- Transfer the filter to a new sterile microcentrifuge tube. Add the preparation buffer (400 µL), centrifuge, and discard the flow through.
- Add wash buffer (700 µL), centrifuge, and discard the flow through. Then add wash buffer (400 µL), centrifuge, and discard the flow through again.
- Transfer the filter to a new sterile microcentrifuge tube. Elute with 50 µL of DNase/RNase free water and let sit for 5 min at room temperature before centrifuging.
- During that in cubation period, prepare the III-HRC filter by placing it in a collection tube and adding the HRC prep solution (600 µL) to it, followed by a centrifugation step of 3 min at 8,000 x g.
- Move the prepared filter onto a sterile microcentrifuge tube. Transfer the eluted DNA from step 3.4.5 to this filter and centrifuge at 16,000 x g for 3 min. The flow through contains the extracted DNA.
- Store DNA extracts for both sediments and filters at -20 °C.
NOTE: DNA extracts can be stored for around 8 years at -20 °C assuming stable temperature, limited light exposure, and no harmful contaminants14. - Perform RNA isolation as per the manufacturer's protocol. Store RNA extracts at -80 °C.
- Lyse the cells within the sample by transferring it to a bead tube and subjecting it to a cell disruptor at high speed for at least five minutes. Centrifuge and transfer the supernatant to a sterile microcentrifuge tube.
- Add lysis buffer to the supernatant (1:1 volume) and transfer to the provided column (yellow). Centrifuge the column.
- Add an equal volume of 95-100% ethanol to the flow through and mix by pipetting up and down five times.
- Place the IICG Column (green) on a sterile microcentrifuge tube. Transfer the mixed solution to the column and centrifuge.
- Add wash buffer (400 µL), centrifuge, and discard the flow through.
- Add 5 µL of DNase I and 75 µL of DNA digestion buffer to the column and incubate at room temperature for 15 minutes.
- Add prep buffer (400 µL), centrifuge, and discard the flow through.
- Add wash buffer (700 µL), centrifuge, and discard the flow through. Then add wash buffer (400 µL), centrifuge, and discard the flow through again.
- Transfer the column to a new sterile microcentrifuge tube. Elute with 50 µL of DNase/RNase free water and let sit for 5 min before centrifuging.
- During that incubation period, prepare the III-HRC filter by placing it in a collection tube and adding the HRC prep solution (600 µL) to it, followed by a centrifugation step of 3 min at 8,000 x g.
- Move the prepared filter onto a sterile microcentrifuge tube. Transfer the eluted RNA from step 3.6.9 to this filter and centrifuge at 16,000 x g for 3 min. The flow through contains the extracted RNA.
NOTE: RNA extracts can only be stored for one year before they start to degrade15. Both DNA and RNA extracts are degraded by repeated freeze-thawing. Some protocols allow for the extraction of both DNA and RNA from the same sample16,17.
- Quantify the extracted DNA and RNA samples using a fluorometer or a spectrophotometer. See Table 1 for example fluorometer DNA concentration values. For an example spectrophotometer quantification protocol, see reference18. Sediment DNA concentration values with the kit listed in Table of Materials generally range from 1 to 40 ng/µL, while filter DNA concentration values tend to range from 0.5 to 10 ng/µL. Sediment RNA concentration values with the kit listed in Table of Materials generally range from around 1 to 20 ng/µL, while filter RNA concentration values tend to be lower, typically ranging from 0.5 to 5 ng/µL.
4. 16S rRNA library creation
- Clean the work area with 10% Bleach and 70% Ethanol. The work area should be an enclosed space capable of producing laminar flow conditions (laminar flow hood).
- Use the DNA extracts (from step 3.5) and prepare samples for 16S rRNA amplicon sequencing with a standard PCR protocol, such as the one described on the Earth Microbiome's website that amplifies the V4 hypervariable region of 16S rRNA19 under laminar flow conditions.
- Prepare a 2% agarose gel as described previously and let it solidify17. Mix 7 µL of PCR product and 13 µL of DNase free water. Add a gel loading dye to a final concentration of 1x. Once agarose is solidified, load this PCR products mix on a 2% agarose gel.
NOTE: Alternatively, a pre-cast gel can be used instead, as these gels run faster and come pre-made. - Run the gel at 90 V for 60-90 min to check for the band size of 386 as successful amplification for 16S rRNA V4 amplicons, using the Earth Microbiome's protocol.
5. DNA 16S rRNA library purification
- Pool 10 µL of PCR products for the samples that yielded bright bands and 13 µL for the samples that yielded faint bands in an appropriately sized sterile microcentrifuge tube.
- Check the concentration of the resulting pool using a fluorometer or spectrophotometer and prepare a 2% agarose gel as before. Ideally, the pool should have a concentration of at least 10 ng/µL, and most samples should have had a concentration of around 25 ng/µL.
- Concentration and volume permitting, load around 150-200 ng in a well of 2% agarose gel.
- Run the gel for 60-90 min at 90 volts.
- Purify the pooled library by running a 2% agarose gel.
- Excise the 386 bp DNA band from the gel and purify the pooled library using a commercially available kit as described previously20. Elute the purified DNA with 30 µL of 10 mM Tris-Cl (pH 8.5). Perform this step in a different area than DNA or RNA extraction to prevent future contamination, as cutting the gel will spread PCR amplicons onto both the experimenter and the surrounding area.
- Check the concentration of the purified pool using a fluorometer or spectrophotometer. If purification went well, its concentration should be at least half of the unpurified pool's. Generally, the final concentration should range from 5 to 20 ng/µL.
- Send the purified libraries for next generation sequencing. Ensure that they are kept cold during transport by including dry ice in the shipping container.
6. RNA library creation and purification
- Several commercial kits can be utilized to create RNA libraries. For whichever one is used, follow the manufacturer's protocol as written while working in a sterile laminar flow environment. A very summarized version of the protocol for kit in the Table of Materials is presented below21.
- Make the first strand cDNA synthesis master mix (8 µL of nuclease-free water and 2 µL of First Strand Synthesis Enyzme Mix) and add it to the sample. Place the sample in the thermocycler with the conditions specified in the protocol.
- Make the second strand cDNA synthesis master mix (8 µL of Second Strand Synthesis Reaction Buffer, 4 µL Second Strand Synthesis Enzyme Mix, and 48 µL of nuclease-free water) on ice and add it to the sample. Place in a thermocycler set to 16 °C for one hour.
- Purify the reaction by adding the provided beads (144 µL) and performing two 80% ethanol washes (200 µL).
- Elute with the provided TE buffer (53 µL) and transfer 50 µL of the supernatant to a clean PCR tube. Place the PCR tube on ice.
- Make the end prep master mix (7 µL of End Prep Reaction Buffer and 3 µL of End Prep Enzyme Mix) on ice and add it to the PCR tube. Place the PCR tube in a thermocycler with the conditions specified in the protocol.
- Mix the Diluted Adaptor (2.5 µL), Ligation Master Mix (30 µL) and Ligation Enhancer (1 µL) solutions on ice. Add the mixed solutions to the sample and place in a thermocycler for 15 min at 20 °C.
- Purify the reaction by adding the provided beads (87 µL) and performing ethanol washes (200 µL) and elution as before, except only add 17 µL of TE.
- Add indices (10 µL) and the Q5 Master Mix (25 µL) solution and place in a thermocycler with the conditions described in the protocol.
- Purify the reaction by adding the provided beads (45 µL) and performing an addition two ethanol washes (200 µL) and elute with 23 µL of TE. Transfer 20 µL to a clean PCR tube.
- Check the libraries for detectable concentrations of RNA using a Bioanalyzer, fluorometer, or spectrophotometer.
- Pool the metatranscriptomic libraries in a roughly equimolar ratio.
- Purify the library following the same protocol for the 16S library purification, except excise fragments between 250 and 400 bp. Whereas the 16S library had a distinct band representing the amplified region, the result here is a smear.
- Check the concentration of the purified library as before.
- Ship the purified library with dry ice to a sequencing facility.
NOTE: Alternatively, RNA extracts can be sent to a university or private company for library preparation and sequencing.
7. Microbial community analysis
- Once sequencing is complete, access the sample data. Download it to a usable computer.
NOTE: Ideally, the device should have at least 16 gigabytes of RAM. For a discussion of computing requirements (for Qiime2), see https://forum.qiime2.org/t/recommended-specifications-to-run-qiime2/9808. - Use software, such as mothur, QIIME2, and R, to analyze 16S rRNA data. See here https://docs.qiime2.org/2020.11/tutorials/moving-pictures/ for an example QIIME2 16S analysis tutorial.
- For metatranscriptomics (RNA) data, use HUMAnN2 and ATLAS to determine which genes and pathways are present in the samples.
NOTE: An example metatranscriptomics pipeline culminating in diversity and random forest analysis is presented in the Supplemental Information file. All commands are run through command line, e.g., Terminal for Mac users.
Results
The success of DNA and RNA extractions can be evaluated using a variety of equipment and protocols. Generally, any detectable concentration of either is considered sufficient to conclude that the extraction was successful. Examining Table 1 then, all extractions, except for one, would be dubbed successful. Failure at this step is often due to low initial biomass, poor sample preservation, or human error during extraction. In the case of filters, extraction may have been successful even if the concentration is below detection. If those extracts do not yield bands for PCR (if doing 16S) or a detectable concentration after library preparation (metatranscriptomics), then they likely did truly fail.
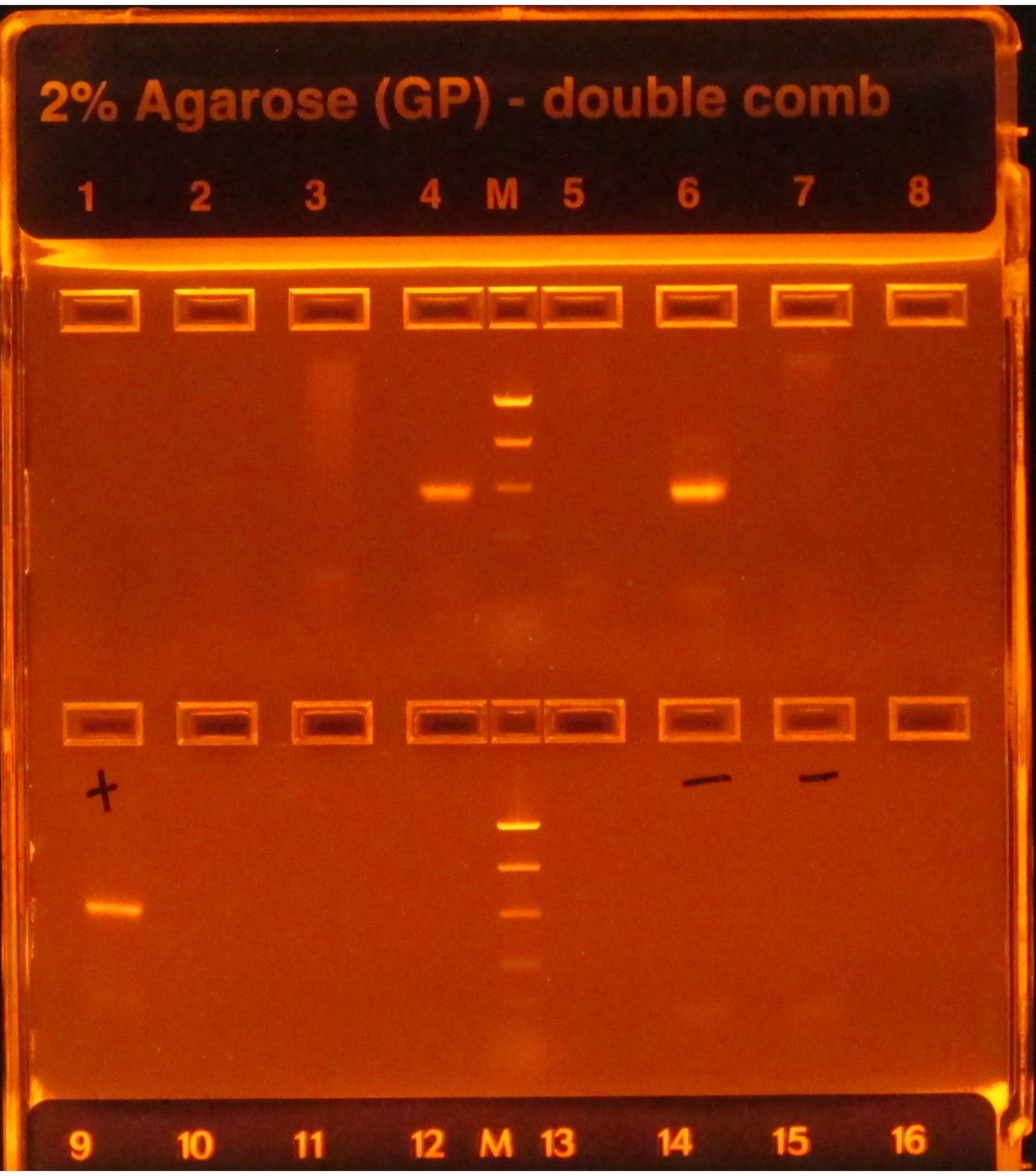
If the 16S protocol is followed, bright bands following PCR amplification, as seen in wells 4 and 6 in Figure 1, indicate success, while a lack of bands, as seen in the other wells in the top row, indicates failure. Moreover, a bright band in the gel lane that contains a negative PCR control would also indicate a failure since it would be risky to assume that the contamination impacting the negative control(s) did not affect the samples.
For both 16S and metatranscriptomics, the success of sequencing can be evaluated by looking at the number of sequences obtained (Figure 2). 16S samples should have a minimum of 1,000 sequences, with at least 5,000 being ideal (Figure 2A). Likewise, metatranscriptomics samples should have a minimum of 500,000 sequences, with at least 2,000,000 being ideal (Figure 2B). Samples with fewer sequences than those minimums should not be used for analyses, as they may not accurately represent their bacterial community. However, samples that fall between the minimum and ideal can still be used though results should be interpreted more cautiously if many samples fall in that range.
The success of subsequent downstream analysis can be determined simply on the basis of whether the expected output files were obtained or not. At any rate, programs, such as QIIME2 and R (Figure 3), should allow for the evaluation of potential significant differences among the bacterial communities based on fracking. The data for Figure 3 was obtained by collecting sediment samples from twenty-one different sites at thirteen different streams for 16S and metatranscriptomics analysis. Of those twenty-one sites, twelve of them were downstream of fracking activity and classified as HF+, and nine of them were either upstream of fracking activity or in a watershed where fracking was not occurring; these streams were classified as HF-. Besides the presence of fracking activity, the streams were otherwise comparable.
Those differences could take the form of consistent compositional shifts based on fracking status. If that were the case, HF+ and HF- samples would be expected to cluster apart from each other in a PCoA plot, as is the case in Figure 3A and Figure 3B. To confirm that those apparent shifts are not just an artifact of the ordination method, further statistical analysis is needed. For example, a PERMANOVA22 test on the distance matrix that Figure 3A and Figure 3B are based on revealed significant clustering based on fracking status, meaning that the separation observed in the plot is consistent with differences among the samples' bacterial communities, instead of an artifact of ordination. A significant PERMANOVA or ANOSIM result is a strong indication of consistent differences between HF+ and HF- samples, which would indicate that the HF+ samples were impacted by fracking, while a high p-value would indicate that the samples were not impacted. Metatranscriptomic data can likewise be visualized and evaluated using the same methods.
Examining differential features (microbes or functions) can reveal evidence that samples have been impacted too. One method of determining differential features is to create a random forest model. The random forest model can be used to see how well the samples' fracking status can be correctly classified. If the model performs better than expected by chance, that would be additional evidence of differences dependent on fracking status. Moreover, the most important predictors would reveal which features were most important for correctly differentiating samples (Figure 3C). Those features also then would have had consistently different values based on fracking status. Once those differential features are determined, the literature can be reviewed to see if they have been previously associated with fracking. However, it may be challenging to find studies that determined differential functions, as most have only used 16S rRNA compositional data. Therefore, for evaluating the implications of differential functions, one possible method would be to see if they have been previously associated with potential resistance to biocides commonly used in fracking fluid or if they could aid in tolerating highly saline conditions. Furthermore, examining the functional profile of a taxon of interest could reveal evidence of fracking's impact (Figure 3D). For example, if a taxon is identified as differential by the random forest model, its antimicrobial resistance profile in HF+ samples could be compared to its profile in HF- samples and if they differ greatly, that could suggest that fracking fluid containing biocides entered the stream.
| SampleID | Concentration (ng/µL) |
| 1 | 1.5 |
| 2 | 1.55 |
| 3 | 0.745 |
| 4 | 0.805 |
| 5 | 7.82 |
| 6 | 0.053 |
| 7 | 0.248 |
| 8 | 0.945 |
| 9 | 1.82 |
| 10 | 0.804 |
| 11 | 0.551 |
| 12 | 1.69 |
| 13 | 4.08 |
| 14 | Below_Detection |
| 15 | 7.87 |
| 16 | 0.346 |
| 17 | 2.64 |
| 18 | 1.15 |
| 19 | 0.951 |
Table 1: Example DNA concentrations based on Fluorometer 1x DS DNA high sensitivity assay. Extractions for all these samples, except for 14, would be considered successful due to having detectable amounts of DNA.

Figure 1: Example e-gel with PCR products. The gel was pre-stained and visualized under a UV light, causing any DNA present on it to glow. PCR worked for the samples in wells 4 and 6 in the first row, as they both had one single bright band of the expected size (based on the ladder). PCR for the samples in the other six wells failed, as they did not produce any bands. The positive control (first well, second row) had a bright band, indicating that PCR was performed properly, and the negative controls (wells 6 and 7, second row) did not have any bands, indicating that samples were not contaminated. If a negative had a band as bright as the samples, PCR would have been considered a failure since it would be risky to assume that the samples had amplicons that were not just the result of contamination. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Example sequence counts. (A) 16S example sequence counts. Nearly all these 16S samples had over 1,000 sequences. The very few that had less than 1,000 sequences should be excluded from downstream analyses, as they had insufficient sequences to accurately represent their bacterial communities. Several sequences had between 1,000 and 5,000 sequences; while not ideal, they would still be usable since they exceed the bare minimum, and the majority of samples exceed the ideal minimum of 5,000 as well. (B) Metatranscriptomics example counts. All samples exceeded both the minimum (500,000) and ideal minimum (2,000,000) number of sequences. Therefore, sequencing was successful for all of them, and they could all be used in downstream analysis. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Example analysis. (A) PCoA plot based on coordinates calculated with a Weighted Unifrac distance matrix created and visualized through QIIME2. (B) PCoA plot based on coordinates calculated with the Weighted Unifrac distance matrix exported from QIIME2. The coordinates were visualized using the Phyloseq and ggplot2 packages in R. Metadata vectors were fitted to the plot using the Vegan package. Each point represents a sample's bacterial community, with closer points indicating more similar community compositions. Clustering based on fracking status for these 16S sediment samples was observed (PERMANOVA, p=0.001). Furthermore, the vectors reveal that the HF+ samples tended to have higher levels of Barium, Bromide, Nickel, and Zinc, which corresponded to different bacterial community composition compared to the HF- samples. (C) Plot of best predictors for a random forest model that tested where bacterial abundances could be used to predict fracking status among the samples. The random forest model was created through R using the randomForest package. The top 20 predictors are shown as well as the resulting decreases in impurity (measure of the number of HF+ and HF- samples grouped together) in the form of Mean Decrease in Gini Index when they are utilized to separate samples. (D) Pie chart showing the antimicrobial resistance profile of the Burkholderiales profile based on metatranscriptomic data. Sequences were first annotated with Kraken2 to determine which taxa they belonged to. BLAST was then used with those annotated sequences and the MEGARes 2.0 database to determine which antimicrobial resistance genes (in the form of "MEG_#") were being actively expressed. Antimicrobial resistance genes expressed by members of Burkholderiales were then extracted to see which ones were most prevalent among that taxa. While more costly and time-consuming, metatranscriptomics does allow for functional analyses, such as this which cannot be done with 16S data. Notably, Kraken2 was used for this example analysis, instead of HUMAnN2. Kraken2 is faster than HUMAnN2; however, it only outputs compositional information, instead of composition, contribution, and functions (genes) and pathways like HUMAnN2 does. Please click here to view a larger version of this figure.
{kind=link}
Supplementary File: An example metatranscriptomics pipeline. Please click here to download this file.
Discussion
The methods described in this paper have been developed and refined over the course of several studies published by our group between 2014 and 20187,8,10 and have been employed successfully in a collaborative project to investigate the impacts of fracking on aquatic communities in a three year project that will soon submit a paper for publication. These methods will continue to be utilized over the course of the remainder of the project. Additionally, other current literature investigating the impact of fracking on streams and ecosystems describe similar methods for sample collection, processing, and analysis7,8,10,11. However, none of those papers utilized metatranscriptomic analysis, making this paper the first to describe how those analyses can be used to elucidate fracking's impact on nearby streams. Furthermore, the methods presented here for sample collection are more detailed, as are the steps taken to avoid contamination.
One of the most important steps of our protocol is initial sample collection and preservation. Field sampling and collection comes with certain challenges, as maintaining an aseptic or sterile environment during collection can be difficult. During this step, it is vital to avoid contaminating samples. To do this, gloves should be worn, and only sterile containers and tools should be allowed to come into contact with samples. Samples should also be immediately placed on ice after collection to mitigate nucleic acid degradation. Adding a commercial nucleic acid preservative upon collection can also increase nucleic acid yield and allow samples to be stored for longer periods of time after collection. Whenever nucleic acid extraction is performed, it is important to use the appropriate amount of sample, too much can clog spin filters used for extraction (for those protocols that make use of them) but too little can result in low yields. Be sure to follow the instructions for whichever kit is used.
Similar to field collection, avoiding or minimizing contamination is also important during nucleic acid extraction and sample preparation, especially when working with low nucleic acid yield samples, such as suboptimal sediment samples (samples containing a large amount of gravel or rocks) or water samples. Therefore, as with sample collection, gloves should be worn during all these steps to reduce contamination. Additionally, all work surfaces used during lab procedures should be sterilized beforehand by wiping with a 10% bleach solution, followed by a 70% ethanol solution. For pipetting steps (3-6), filter tips should be used to avoid contamination due to the pipette itself, with tips being changed every time they touch a non-sterile surface. All tools used for lab work, including pipettes, should be wiped down before and after with the bleach and ethanol solutions. To evaluate contamination, extraction blanks and negatives (sterile liquid) should be included during every set of nucleic acid extractions and PCR reactions. If quantification after extractions reveals a detectable amount of DNA/RNA in the negatives, extractions can be repeated if there is sufficient sample left. If negative samples for PCR show amplification, troubleshooting should be performed to determine the source and then the samples should be rerun. To account for low levels of contamination, it is recommended that extraction blanks and PCR negatives be sequenced so that the contaminants can be identified and removed, if necessary, during computational analysis. Conversely, PCR amplification could also fail due to a variety of causes. For environmental samples, inhibition of the PCR reaction is often the culprit, which can be due to a variety of substances interfering with Taq polymerase23. If inhibition is suspected, PCR grade water (see Table of Materials) can be used to dilute the DNA extracts.
This protocol has a few notable limitations and potential difficulties. Sample collection can be challenging for both water and sediment samples. In order to get enough biomass, ideally 1 L of stream water needs to be pushed through a filter. The pores of the filter need to be small to capture microbes but can also trap sediment. If a lot of sediment is in the water due to recent rainfall, the filter can clog making it difficult to push the entire volume through the filter. For sediment collection, it can be challenging to estimate the depth of sediment during collection. Furthermore, it is important to ensure that the sediment collected is predominantly soil, as pebbles and rocks will lead to lower nucleic acid yield and may not be an accurate representation of the microbial community. Lastly, it is vital as well that samples are kept on ice after collection, especially if a preservative is not used.
Though this protocol covers both metatranscriptomics and 16S lab protocols, it should be emphasized that these two methods are very different in both process and in the type of data they provide. The 16S rRNA gene is a commonly targeted region, highly conserved in bacteria and archaea, and useful for characterizing the bacterial community in a sample. Although a targeted and specific approach, species level resolution is often unattainable, and characterizing newly diverged species or strains is difficult. Contrarily, metatranscriptomics is a broader approach that captures all the active genes and microbes present within a sample. Whereas 16S provides only data for identification, metatranscriptomics can provide functional data such as expressed genes and metabolic pathways. Both are valuable and when combined, they can reveal which bacteria are present and which genes they are expressing.
This paper describes methods for field collection and sample processing for both 16S rRNA and metatranscriptomic analyses in the context of studying fracking. Additionally, it details collection methods for high quality DNA/RNA from low biomass samples and for long-term storage. The methods described here are the culmination of our experiences with sample collection and processing in our efforts to learn how fracking impacts nearby streams through examining the structure and function of their microbial communities. Microbes respond quickly to disturbances, and consequently, which microbes are present and the genes they express can provide information about the effects of fracking on ecosystems. Overall, these methods could be invaluable in our understanding of how fracking impacts these important ecosystems.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors would like to acknowledge the funding sources for the projects that led to the development of these methods, with those sources being: the Howard Hughes Medical Institute (http://www.hhmi.org) through the Precollege and Undergraduate Science Education Program, as well as by the National Science Foundation (http://www.nsf.gov) through NSF awards DBI-1248096 and CBET-1805549.
Materials
| Name | Company | Catalog Number | Comments |
| 200 Proof Ethanol | Thermo Fisher Scientific | A4094 | 400 mL need to be added to Buffer PE (see Qiagen QIAQuck Gel Extraction kit protocol) and 96 mL needs to be added to the DNA/RNA Wash Buffer (see ZymoBIOMICS DNA/RNA Miniprep kit protocol). Additional ethanol is needed for the ZymoBIOMICS DNA/RNA Miniprep and NEBNext® Ultra™ II RNA Library Prep with Sample Purification Beads kits. |
| Agarose | Thermo Fisher Scientific | BP1356-100 | 100 g per bottle. 0.6 g of agarose would be needed to make one 2% 30 mL gel. |
| Disinfecting Bleach | Walmart (Clorox) | No catalog number | Use a 10% bleach solution for cleaning the work area before and after lab procedures |
| DNA gel loading dye | Thermo Fisher Scientific | R0611 | Each user-made (i.e. non-e-gel) should include loading dye with all of the samples in the ratio of 1 µL dye to 5 µL sample |
| DNA ladder | MilliporeSigma | D3937-1VL | A ladder should be run on every gel/e-gel |
| DNA/RNA Shield (2x) | Zymo Research | R1200-125 | 3 mL per sediment sample (50 mL conical) and 2 mL per water sample (filter) |
| Ethidium bromide | Thermo Fisher Scientific | BP1302-10 | Used for staining user-made e-gels |
| Forward Primer | Integrated DNA Technologies (IDT) | 51-01-19-06 | 0.5 µL per PCR reaction |
| Isopropanol | MilliporeSigma | 563935-1L | Generally less than 2 mL per library. Volume needed varies by mass of excised gel fragment (see Qiagen QIAQuick Gel Extraction kit protocol). |
| PCR-grade water | MilliporeSigma | 3315932001 | 13 µL per PCR reaction (assuming 1 µL of sample DNA template is used) |
| Platinum Hot Start PCR Master Mix (2x) | Thermo Fisher Scientific | 13000012 | 10 µL per PCR reaction |
| Reverse Primer | Integrated DNA Technologies (IDT) | 51-01-19-07 | 0.5 µL per PCR reaction |
| TBE Buffer (Tris-borate-EDTA) | Thermo Fisher Scientific | B52 | 1 L of 10x TBE buffer (30 mL of 1x TBE buffer would be needed to make one 30 mL gel) |
| 1 L bottle | Thermo Fisher Scientific | 02-893-4E | One needed per stream (the same bottle can be used for multiple streams if it is sterilized between uses) |
| 1.5 mL Microcentrifuge tubes | MilliporeSigma | BR780400-450EA | 5 microcentrifuge tubes are needed per DNA extraction and an additional 3 are needed to purify RNA (see ZymoBIOMICS DNA/RNA Miniprep kit protocol) |
| 2% Agarose e-gel | Thermo Fisher Scientific | G401002 | Each gel can run 10 samples (so 9 with a PCR negative and 8 if the extraction negative is run on the same gel) |
| 50 mL Conicals | CellTreat | 229421 | 1 50 mL conical needed per sediment samples |
| 500 mL Beaker | MilliporeSigma | Z740580 | Only 1 needed (for flame sterilization) |
| Aluminum foil | Walmart (Reynolds KITCHEN) | No number | Aluminum foil can be folded and autoclaved. The part not exposed to the environment can then be used as a sterile, DNA and RNA free surface for processing filters (one folded piece per filter to avoid cross-contamination) |
| Autoclave | Gettinge | LSS 130 | Only one needed |
| Centrifuge | MilliporeSigma | EP5404000138-1EA | Only 1 needed |
| Cooler | ULINE | S-22567 | Just about any cooler can be used. This one is listed due to being made of foam, making it lighter and thus easier to take along for field sampling. |
| Disruptor Genie | Bio-Rad | 3591456 | Only one needed |
| Electrophoresis chamber | Bio-Rad | 1664000EDU | Only 1 needed |
| Electrophoresis power supply | Bio-Rad | 1645050 | Only 1 needed |
| Freezer (-20 C) | K2 SCIENTIFIC | K204SDF | One needed to store DNA extracts |
| Freezer (-80 C) | K2 SCIENTIFIC | K205ULT | One needed to store RNA extracts |
| Gloves | Thermo Fisher Scientific | 19-020-352 | The catalog number is for Medium gloves. |
| Heat block | MilliporeSigma | Z741333-1EA | Only one needed |
| Lab burner | Sterlitech | 177200-00 | Only one needed |
| Laminar Flow Hood | AirClean Systems | AC624LFUV | Only 1 needed |
| Library purification kit | Qiagen | 28704 | One kit has enough for 50 reactions |
| Magnet Plate | Alpaqua | A001219 | Only one needed |
| Microcentrifuge | Thermo Fisher Scientific | 75004061 | Only one needed |
| Micropipette (1000 µL volume) | Pipette.com | L-1000 | Only 1 needed |
| Micropipette (2 µL volume) | Pipette.com | L-2 | Only 1 needed |
| Micropipette (20 µL volume) | Pipette.com | L-20 | Only 1 needed |
| Micropipette (200 µL volume) | Pipette.com | L-200R | Only 1 needed |
| NEBNext Ultra II RNA Library Prep with Sample Purification Beads | New England BioLabs Inc. | E7775S | One kit has enough reagents for 24 samples. |
| Parafilm | MilliporeSigma | P7793-1EA | 2 1" x 1" squares are needed per filter |
| PCR Tubes | Thermo Fisher Scientific | AM12230 | One tube needed per reaction |
| Pipette tips (for 1000 µL volume) | Pipette.com | LF-1000 | Pack of 576 tips |
| Pipette tips (for 20 µL volume) | Pipette.com | LF-20 | Pack of 960 tips |
| Pipette tips (for 200 µL volume) | Pipette.com | LF-250 | Pack of 960 tips |
| PowerWulf ZXR1+ computer cluster | PSSC Labs | No number | This is just an example of a supercomputer powerful enough to perform metatranscriptomics analysis in a timely manner. Only one needed. |
| Qubit fluorometer starter kit | Thermo Fisher Scientific | Q33239 | Comes with a Qubit 4 fluorometer, enough reagent for 100 DNA assays, and 500 Qubit tubes |
| Scoopula | Thermo Fisher Scientific | 14-357Q | Only one needed |
| Sterile blades | AD Surgical | A600-P10-0 | One needed per filter |
| Sterivex-GP Pressure Filter Unit | MilliporeSigma | SVGP01050 | 1 filter needed per water sample |
| Thermocycler | Bio-Rad | 1861096 | Only one needed |
| Vise-grip | Irwin | 2078500 | Only one needed (for cracking open the filters) |
| Vortex-Genie 2 | MilliporeSigma | Z258415-1EA | Only 1 needed |
| WHIRL-PAK bags | ULINE | S-22729 | 1 needed per filter |
| ZymoBIOMICS DNA/RNA Miniprep kit | Zymo Research | R2002 | One kit has enough reagents for 50 samples. |
References
- The process of unconventional natural gas production. US EPA Available from: https://www.epa.gov/uog/process-unconventional-natural-gas-production (2013)
- Brittingham, M. C., Maloney, K. O., Farag, A. M., Harper, D. D., Bowen, Z. H. Ecological risks of shale oil and gas development to wildlife, aquatic resources, and their habitats. Environmental Science & Technology. 48 (19), 11034-11047 (2014).
- McBroom, M., Thomas, T., Zhang, Y. Soil erosion and surface water quality impacts of natural gas development in East Texas, USA. Water. 4 (4), 944-958 (2012).
- Maloney, K. O., Weller, D. E. Anthropogenic disturbance, and streams: land use and land-use change affect stream ecosystems via multiple pathways. Freshwater Biology. 56 (3), 611-626 (2011).
- Meyer, J. L., et al. The contribution of headwater streams to biodiversity in river networks1. JAWRA Journal of the American Water Resources Association. 43 (1), 86-103 (2007).
- Alexander, R. B., Boyer, E. W., Smith, R. A., Schwarz, G. E., Moore, R. B. The role of headwater streams in downstream water quality. Journal of the American Water Resources Association. 43 (1), 41-59 (2007).
- Ulrich, N., et al. Response of aquatic bacterial communities to hydraulic fracturing in Northwestern Pennsylvania: A five-year study. Scientific Reports. 8 (1), 5683 (2018).
- Chen See, J. R., et al. Bacterial biomarkers of Marcellus shale activity in Pennsylvania. Frontiers in Microbiology. 9, 1697 (2018).
- Rausch, P., et al. Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome. 7 (1), 133 (2019).
- Louca, S., Doebeli, M., Parfrey, L. W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome. 6 (1), 41 (2018).
- Trexler, R., et al. Assessing impacts of unconventional natural gas extraction on microbial communities in headwater stream ecosystems in Northwestern Pennsylvania. Frontiers in Microbiology. 5, 522 (2014).
- Mumford, A. C., et al. Shale gas development has limited effects on stream biology and geochemistry in a gradient-based, multiparameter study in Pennsylvania. Proceedings of the National Academy of Sciences. 117 (7), 3670-3677 (2020).
- JoVE Core Biology DNA Isolation. Journal of Visualized Experiments Available from: https://www.jove.com/cn/science-education/10814/dna-isolation (2020)
- Oxford Gene Technology DNA Storage and Quality. OGT Available from: https://www.ogt.com/resources/literature/403_dna_storage_and_quality (2011)
- ThermoFisher SCIENTIFIC Technical Bulletin #159: Working with RNA. Thermoscientific Available from: https://www.thermofisher.com/us/en/home/references/ambion-tech-support/nuclease-enzymes/general-articles/working-with-rna.html (2020)
- QIAGEN AllPrep DNA/RNA Mini Kit. Qiagen Available from: https://www.qiagen.com/us/products/discovery-and-translational-research/dna-rna-purification/multianalyte-and-virus/allprep-dnarna-mini-kit/#orderinginformation (2020)
- ZymoBIOMICS DNA/RNA Miniprep Kit. Zymo Research Available from: https://www.zymoresearch.com/products/zymobiomics-dna-rna-miniprep-kit (2020)
- Desjardins, P., Conklin, D. NanoDrop microvolume quantitation of nucleic acids. Journal of Visualized Experiments. (45), e2565 (2010).
- 16S Illumina amplicon protocol: Earth microbiome project. Earth microbiome project Available from: https://earthmicrobiome.org/protocols-and-standards/16s/ (2018)
- Gel Purification: Binding, washing and eluting a sample | Protocol. Journal of Visualized Experiments Available from: https://www.jove.com/v/5063/gel-purification (2020)
- New England Biolabs protocol for the use with NEBNext Poly(A) mRNA magnetic isolation module (E7490) and NEBNext Ultra II RNA library prep kit for Illumina (E7770, E7775). New England Biolabs Available from: https://www.neb.com/protocols/2017/03/04/protocol-for-use-with-purified-mrna-or-rrna-depleted-rna-and-nebnext-ultra-ii-rna-library-prep-ki (2020)
- Anderson, M. J. Permutational multivariate analysis of variance (PERMANOVA). Wiley StatsRef: Statistics Reference Online. , 1-15 (2017).
- Schrader, C., Schielke, A., Ellerbroek, L., Johne, R. PCR inhibitors - occurrence, properties and removal. Journal of Applied Microbiology. 113 (5), 1014-1026 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved