
Method Article
Valutazione dell'impatto della fratturazione idraulica sui flussi utilizzando le firme molecolari microbiche
In questo articolo
Riepilogo
Qui, presentiamo un protocollo per studiare gli impatti della fratturazione idraulica sui corsi d'acqua vicini analizzando le loro comunità microbiche di acqua e sedimenti.
Abstract
La fratturazione idraulica (HF), comunemente chiamata "fracking", utilizza una miscela di acqua ad alta pressione, sabbia e sostanze chimiche per fratturare le rocce, rilasciando petrolio e gas. Questo processo ha rivoluzionato l'industria energetica statunitense, in quanto dà accesso a risorse che prima erano introvabili e ora produce due terzi del gas naturale totale negli Stati Uniti. Sebbene il fracking abbia avuto un impatto positivo sull'economia degli Stati Uniti, diversi studi hanno evidenziato i suoi effetti ambientali dannosi. Di particolare preoccupazione è l'effetto del fracking sui corsi d'acqua, che sono particolarmente importanti a causa del loro impatto sproporzionatamente grande sulla salute dell'intero spartiacque. I batteri all'interno di questi flussi possono essere utilizzati come indicatori della salute del flusso, poiché ci si aspetterebbe che i batteri presenti e la loro abbondanza in un flusso disturbato differiscano da quelli in un flusso altrimenti comparabile ma indisturbato. Pertanto, questo protocollo mira a utilizzare la comunità batterica per determinare se i flussi sono stati influenzati dal fracking. A tal fine, devono essere raccolti sedimenti e campioni d'acqua provenienti da corsi d'acqua vicino al fracking (potenzialmente colpiti) e a monte o in un diverso spartiacque dell'attività di fracking (non impressionato). Questi campioni vengono quindi sottoposti all'estrazione di acido nucleico, alla preparazione della libreria e al sequenziamento per studiare la composizione della comunità microbica. L'analisi correlazionale e i modelli di apprendimento automatico possono successivamente essere impiegati per identificare quali caratteristiche sono esplicative della variazione nella comunità, nonché l'identificazione di biomarcatori predittivi per l'impatto del fracking. Questi metodi possono rivelare una varietà di differenze nelle comunità microbiche tra i flussi di sorgenti, in base alla vicinanza al fracking, e servire come base per future indagini sull'impatto ambientale delle attività di fracking.
Introduzione
La fratturazione idraulica (HF), o "fracking", è un metodo di estrazione del gas naturale, che è diventato sempre più diffuso man mano che la domanda di combustibili fossili continua ad aumentare. Questa tecnica consiste nell'utilizzare attrezzature di perforazione ad alta potenza per iniettare una miscela di acqua, sabbia e sostanze chimiche in depositi di scisto ricchi di metano, di solito per rilasciare gas intrappolati1.
Poiché queste tecniche di raccolta non convenzionali sono relativamente nuove, è importante studiare gli effetti di tali pratiche sui corsi d'acqua vicini. Le attività di fracking impongono la bonifica di vaste aree di terra per il trasporto di attrezzature e la costruzione di pozzi. Circa 1,2-1,7 ettari di terreno devono essere disboscati per ogni pozzopad 2, potenzialmente influenzando il deflusso e la qualità dell'acqua del sistema3. C'è una mancanza di trasparenza che circonda l'esatta composizione chimica del fluido di fracking, compresi i biocidi utilizzati. Inoltre, le acque reflue del fracking tendono ad essere altamente saline2. Inoltre, le acque reflue possono contenere metalli e sostanze radioattive presenti in natura2. Pertanto, la possibilità di perdite e fuoriuscite di fluido di fracking a causa di errori umani o malfunzionamenti delle apparecchiature è preoccupante.
Gli ecosistemi dei torrenti sono noti per essere molto sensibili ai cambiamenti nei paesaggi circostanti4 e sono importanti per mantenere la biodiversità5 e il corretto ciclo dei nutrienti6 all'interno dell'intero spartiacque. I microbi sono gli organismi più abbondanti nei flussi d'acqua dolce e, quindi, sono essenziali per il ciclo dei nutrienti, la biodegradazione e la produzione primaria. La composizione e la funzione della comunità microbica servono come ottimi strumenti per ottenere informazioni sull'ecosistema a causa della loro sensibilità alle perturbazioni, e recenti ricerche hanno mostrato cambiamenti distinti negli assemblaggi batterici osservati in base alla vicinanza all'attività di fracking7,8. Ad esempio, Beijerinckia, Burkholderiae Methanobacterium sono stati identificati come arricchiti in flussi vicino al fracking mentre Pseudonocardia, Nitrospirae Rhodobacter sono stati arricchiti nei flussi non vicino al fracking7.
Il sequenziamento di nuova generazione del gene dell'RNA ribosomiale 16S (rRNA) è un metodo economico per determinare la composizione della comunità batterica che è più veloce ed economico rispetto agli approcci di sequenziamento dell'intero genoma9. Una pratica comune nel campo dell'ecologia molecolare è quella di utilizzare la regione V4 altamente variabile del gene rRNA 16S per la risoluzione del sequenziamento, spesso fino al livello del genere con un ampio ambito di identificazione9, in quanto è ideale per campioni ambientali imprevedibili. Questa tecnica è stata ampiamente implementata in studi pubblicati ed è stata utilizzata con successo per identificare l'impatto delle operazioni di fracking sugli ambientiacquatici 7,8. Tuttavia, vale la pena notare che i batteri hanno numeri di copie variabili del gene rRNA 16S, che influisce sulle loro abbondanze rilevate10. Ci sono alcuni strumenti per spiegare questo, ma la loro efficacia è discutibile10. Un'altra pratica che sta rapidamente crescendo in prevalenza e manca di questa debolezza è il sequenziamento metatrasccrittomico, in cui tutto l'RNA viene sequenziato, consentendo ai ricercatori di identificare sia i batteri attivi che la loro espressione genica.
Pertanto, a differenza dei metodi negli studi precedentemente pubblicati7,8,11,12,questo protocollo copre anche la raccolta, la conservazione, l'elaborazione e l'analisi dei campioni per indagare la funzione della comunità microbica (metatranscriptomics). I passaggi qui descritti consentono ai ricercatori di vedere quale impatto, se del caso, il fracking ha avuto sui geni e sui percorsi espressi dai microbi nei loro flussi, compresi i geni di resistenza antimicrobica. Inoltre, il livello di dettaglio presentato per la raccolta dei campioni è migliorato. Sebbene molti dei passaggi e delle note possano sembrare ovvi per i ricercatori esperti, potrebbero essere inestimabili per coloro che hanno appena iniziato la ricerca.
Qui descriviamo i metodi per la raccolta e l'elaborazione dei campioni per generare dati genetici batterici come mezzo per studiare l'impatto del fracking sui flussi vicini sulla base dei diversi anni di esperienza dei nostri laboratori. Questi dati possono essere utilizzati nelle applicazioni a valle per identificare le differenze corrispondenti allo stato di fracking.
Protocollo
1. Raccolta di campioni di sedimenti per l'estrazione di acidi nucleici
- Immergere un tubo conico sterile da 50 ml nell'acqua del torrente. Indossare guanti durante la raccolta dei campioni per evitare di introdurre contaminazioni umane indesiderate. Eseguire questo passaggio dalla riva o rivolto a monte se in acqua.
- Mentre il tubo conico è sommerso, rimuovere il cappuccio e utilizzarlo per raccogliere circa 3 ml di sedimento da una profondità di 1 a 3 cm nel tubo conico.
- Rimuovere il tubo conico dall'acqua e scaricare tutta l'acqua, ad eccezione di uno strato sottile che copre il campione di sedimento (circa 1 ml).
- Utilizzando una pipetta da 1000 μL e le punte appropriate per pipette, aggiungere 3 mL di conservante DNA/RNA (vedere Tabella dei materiali per le specifiche dei conservanti) al campione raccolto. Conservare le punte delle pipette in una scatola sterile per la punta della pipetta e fissarle solo immediatamente prima dell'uso e scartate dopo l'uso. Invertire il tubo conico tappato 10 volte per garantire che il conservante e il campione siano accuratamente miscelati.
NOTA Il passo 1.4 non è necessario, ma è fortemente raccomandato se l'RNA deve essere estratto dai sedimenti in un secondo momento. - Posizionare i campioni sul ghiaccio per il resto della raccolta dei campioni. Al ritorno dalla raccolta, conservare in un congelatore a -20 °C se i campioni devono essere utilizzati per l'analisi 16S (DNA), o -70 °C, se devono essere utilizzati per l'analisi metatrascrittomica (RNA).
2. Raccolta del filtro per l'estrazione degli acidi nucleici
- Rimuovere il tappo di un flacone sterile da 1 L. Mentre sei rivolto a monte o dalla riva, riempi la bottiglia con acqua di ruscello verso l'alto e poi scaricala fuori. Ripeti questo processo altre due volte per condizionare la bottiglia. Riempire l'intera bottiglia una quarta volta e tapparla.
NOTA: Se si riutilizza un flacone da 1 L, può essere sterilizzato risciacquando con candeggina al 10% per 2 min, seguito da risciacquo tre volte con acqua deionizzata e poi una volta con etanolo al 70%, e infine autoclave con impostazioni: tempo di esposizione di 30 min a 121,1 °C e tempo di asciugatura di 15 min. Durante l'autoclave, il tappo sulla bottiglia deve essere molto allentato per evitare che la bottiglia venga compressa nel processo. - Una volta su una superficie stabile, utilizzare una siringa sterile Luer lock e tirare su un volume pieno. Quindi collegare la siringa a un filtro in polietersolfone sterile e privo di DNA/RNA di 1,7 cm di diametro con una dimensione dei pori di 0,22 μm e spingere l'intero volume attraverso il filtro premendo lo stantuffo fino in fondo. Ripetere questo processo fino a quando il volume totale raccolto nella bottiglia (1 L) viene spinto attraverso il filtro.
NOTA: il volume della siringa può essere variabile, se viene tracciata la quantità totale di acqua spinta attraverso il filtro. Tuttavia, generalmente, è preferibile 60 ml. Mentre 1 L è l'ideale, aneddoticamente, un volume di almeno 200 ml probabilmente raccoglierebbe ancora abbastanza biomassa (assumendo ~ 20.000 cellule per mL) per l'estrazione di DNA e RNA. - Rimuovere l'acqua in eccesso dal filtro prelevando circa 20 ml di aria nella siringa e spingendola attraverso il filtro.
NOTA: Questo aiuterà a prevenire la perdita del conservante se viene eseguito il passaggio 2.4. - Utilizzando una micropipetta P1000, aggiungere 2 mL di un conservante DNA/RNA scaricandolo attraverso l'apertura più grande del filtro (dove è stato attaccato alla siringa) tenendo il filtro orizzontalmente. La punta della pipetta deve essere all'interno della canna del filtro quando la pipetta viene premuta per garantire che il conservante entri nel filtro. Cambiare la punta dopo ogni utilizzo.
NOTA: Come per la raccolta dei sedimenti, questo passaggio non è necessario, ma è fortemente raccomandato per aumentare la resa di acido nucleico in seguito, specialmente per l'RNA. - Staccare un quadrato di pellicola di paraffina e avvolgerlo strettamente attorno a ciascuna apertura / estremità del filtro per sigillare. Posizionare il filtro avvolto con pellicola di paraffina in un sacchetto di campioni sterile e quindi posizionare l'intero sacchetto sul ghiaccio durante la raccolta.
NOTA: Assicurarsi che il lato utilizzato per avvolgere il filtro sia sterile, cioè non precedentemente esposto all'ambiente. - Al ritorno dal campionamento, conservare i filtri a -20 °C per 16S o -70 °C per la meta-trascrittomica.
3. Estrazione e quantificazione degli acidi nucleici
- Pulire l'area di lavoro con il 10% di candeggina e il 70% di etanolo prima di iniziare il trasferimento del campione.
- Per i sedimenti (dal passaggio 1.5), generalmente, utilizzare ~ 0,25 g di campione. La fiamma sterilizza un utensile metallico immergendolo in un becher di etanolo al 70% e bruciando l'etanolo tra i campioni.
- Per i filtri (dal passaggio 2.6), spostare la carta da filtro in un tubo sterile per l'estrazione. Per fare ciò, segui i passaggi seguenti.
- Creare una superficie sterile, priva di DNA e RNA piegando un foglio di alluminio in modo che la parte interna della piega non sia esposta all'ambiente esterno e autoclavando il pezzo piegato con le impostazioni: 121,1 °C e 5 minuti di asciugatura.
- Sterilizzare una morsa con etanolo al 70% e fiamma libera. Quindi utilizzare l'impugnatura della morsa per aprire l'involucro del filtro sulla superficie sterile e rimuovere il nucleo dall'involucro.
- Utilizzare un bisturi sterile per tagliare la carta da filtro lontano dal nucleo affettando in alto e in basso e poi lungo la cucitura. Piegare la carta da filtro con una pinzetta sterile e poi tagliare il filtro a pezzetti usando il bisturi.
- Posizionare i pezzi filtranti in un tubo microcentrifuga per l'estrazione. Assicurarsi che la carta da filtro non entri in contatto con superfici non sterilizzate o che potrebbero avere acido nucleico presente, in quanto ciò porterebbe a una contaminazione indesiderata del campione.
- Eseguire l'isolamento del DNA come descritto in precedenza13 o utilizzando un kit basato su colonne disponibile in commercio (vedere Tabella dei materiali). I passaggi per il kit commerciale elencati sono brevemente descritti di seguito.
- Lisire le cellule all'interno del campione trasferendolo in un tubo di perle e sottoponendolo a un disgregatore cellulare ad alta velocità per almeno 5 minuti. Centrifugare e trasferire il surnatante in un tubo microcentrifuga sterile.
- Aggiungere il buffer di lisi al surnatante (volume 1:1) e trasferire al filtro fornito (giallo). Centrifugare il filtro.
- Trasferire il filtro in un nuovo tubo microcentrifuga sterile. Aggiungere il tampone di preparazione (400 μL), centrifugare ed eliminare il flusso.
- Aggiungere il tampone di lavaggio (700 μL), centrifugare ed eliminare il flusso. Quindi aggiungere il tampone di lavaggio (400 μL), centrifugare e scartare nuovamente il flusso.
- Trasferire il filtro in un nuovo tubo microcentrifuga sterile. Eluire con 50 μL di acqua priva di DNasi/RNasi e lasciare riposare per 5 minuti a temperatura ambiente prima della centrifugazione.
- Durante il periodo di cubazione, preparare il filtro III-HRC posizionandolo in un tubo di raccolta e aggiungendovi la soluzione di preparazione HRC (600 μL), seguita da una fase di centrifugazione di 3 minuti a 8.000 x g.
- Spostare il filtro preparato su un tubo microcentrifuga sterile. Trasferire il DNA eluito dal passo 3.4.5 a questo filtro e centrifugare a 16.000 x g per 3 min. Il flusso attraverso contiene il DNA estratto.
- Conservare gli estratti di DNA sia per i sedimenti che per i filtri a -20 °C.
NOTA: gli estratti di DNA possono essere conservati per circa 8 anni a -20 °C assumendo una temperatura stabile, un'esposizione limitata alla luce e nessun contaminante dannoso14. - Eseguire l'isolamento dell'RNA secondo il protocollo del produttore. Conservare gli estratti di RNA a -80 °C.
- Lisire le cellule all'interno del campione trasferendolo in un tubo di perle e sottoponendolo a un distruttore cellulare ad alta velocità per almeno cinque minuti. Centrifugare e trasferire il surnatante in un tubo microcentrifuga sterile.
- Aggiungere il buffer di lisi al surnatante (volume 1:1) e trasferirli nella colonna fornita (giallo). Centrifugare la colonna.
- Aggiungere un volume uguale di etanolo al 95-100% al flusso e mescolare convogliando su e giù cinque volte.
- Posizionare la colonna IICG (verde) su un tubo microcentrifuga sterile. Trasferire la soluzione mista alla colonna e alla centrifuga.
- Aggiungere il tampone di lavaggio (400 μL), centrifugare ed eliminare il flusso.
- Aggiungere 5 μL di DNasi I e 75 μL di tampone per la digestione del DNA alla colonna e incubare a temperatura ambiente per 15 minuti.
- Aggiungere il buffer di preparazione (400 μL), centrifugare ed eliminare il flusso.
- Aggiungere il tampone di lavaggio (700 μL), centrifugare ed eliminare il flusso. Quindi aggiungere il tampone di lavaggio (400 μL), centrifugare e scartare nuovamente il flusso.
- Trasferire la colonna in un nuovo tubo microcentrifuga sterile. Eluire con 50 μL di acqua priva di DNasi/RNasi e lasciare riposare per 5 minuti prima della centrifugazione.
- Durante il periodo di incubazione, preparare il filtro III-HRC posizionandolo in un tubo di raccolta e aggiungendovi la soluzione di preparazione HRC (600 μL), seguita da una fase di centrifugazione di 3 minuti a 8.000 x g.
- Spostare il filtro preparato su un tubo microcentrifuga sterile. Trasferire l'RNA eluito dallo step 3.6.9 a questo filtro e centrifugare a 16.000 x g per 3 min. Il flusso attraverso contiene l'RNA estratto.
NOTA: gli estratti di RNA possono essere conservati solo per un anno prima che inizino a degradarsi15. Sia gli estratti di DNA che di RNA sono degradati da ripetuti congelamenti-scongelamento. Alcuni protocolli consentono l'estrazione sia del DNA che dell'RNA dallo stesso campione16,17.
- Quantificare i campioni di DNA e RNA estratti utilizzando un fluorometro o uno spettrofotometro. Vedere tabella 1 per esempio i valori di concentrazione del DNA del fluorometro. Per un esempio di protocollo di quantificazione dello spettrofotometro, vedere il riferimento18. I valori di concentrazione del DNA dei sedimenti con il kit elencato nella Tabella dei materiali variano generalmente da 1 a 40 ng/ μL, mentre i valori di concentrazione del DNA del filtro tendono a variare da 0,5 a 10 ng / μL. I valori di concentrazione dell'RNA del sedimento con il kit elencato nella Tabella dei materiali variano generalmente da circa 1 a 20 ng / μL, mentre i valori di concentrazione dell'RNA del filtro tendono ad essere inferiori, in genere vanno da 0,5 a 5 ng/μL.
4. Creazione della libreria rRNA 16S
- Pulire l'area di lavoro con il 10% di candeggina e il 70% di etanolo. L'area di lavoro deve essere uno spazio chiuso in grado di produrre condizioni di flusso laminare (cappa a flusso laminare).
- Utilizzare gli estratti di DNA (dal passaggio 3.5) e preparare campioni per il sequenziamento dell'amplicon rRNA 16S con un protocollo PCR standard, come quello descritto sul sito Web di Earth Microbiome che amplifica la regione ipervariabile V4 di 16S rRNA19 in condizioni di flusso laminare.
- Preparare un gel di acarosio al 2% come descritto in precedenza e lasciarlo solidificare17. Miscelare 7 μL di prodotto PCR e 13 μL di acqua priva di DNasi. Aggiungere un colorante di carico gel ad una concentrazione finale di 1x. Una volta che l'agarose è solidificato, caricare questo mix di prodotti PCR su un gel di agarose al 2%.
NOTA: In alternativa, è possibile utilizzare un gel prefabbricato, poiché questi gel funzionano più velocemente e vengono prefabbricati. - Eseguire il gel a 90 V per 60-90 minuti per verificare la dimensione della banda di 386 come amplificazione di successo per gli ampliconi 16S rRNA V4, utilizzando il protocollo Earth Microbiome.
5. Purificazione della libreria di rRNA DNA 16S
- Pool 10 μL di prodotti PCR per i campioni che hanno prodotto bande luminose e 13 μL per i campioni che hanno prodotto bande deboli in un tubo microcentrifuga sterile di dimensioni appropriate.
- Controllare la concentrazione del pool risultante utilizzando un fluorometro o uno spettrofotometro e preparare un gel di agarose al 2% come prima. Idealmente, il pool dovrebbe avere una concentrazione di almeno 10 ng / μL e la maggior parte dei campioni dovrebbe avere una concentrazione di circa 25 ng / μL.
- Concentrazione e volume permettendo, carico di circa 150-200 ng in un pozzo di gel di agarose al 2%.
- Eseguire il gel per 60-90 minuti a 90 volt.
- Purificare la libreria in pool eseguendo un gel di acarosio al 2%.
- Asportare la banda di DNA da 386 bp dal gel e purificare la libreria in pool utilizzando un kit disponibile in commercio come descritto in precedenza20. Eluire il DNA purificato con 30 μL di 10 mM Tris-Cl (pH 8,5). Esegui questo passaggio in un'area diversa dall'estrazione di DNA o RNA per prevenire future contaminazioni, poiché il taglio del gel diffonderà ampliconi PCR sia sullo sperimentatore che sull'area circostante.
- Controllare la concentrazione della piscina purificata utilizzando un fluorometro o uno spettrofotometro. Se la purificazione è andata bene, la sua concentrazione dovrebbe essere almeno la metà di quella della piscina non purificata. Generalmente, la concentrazione finale dovrebbe variare da 5 a 20 ng/μL.
- Invia le librerie purificate per il sequenziamento di nuova generazione. Assicurarsi che siano mantenuti freddi durante il trasporto includendo ghiaccio secco nel contenitore di spedizione.
6. Creazione e purificazione della libreria di RNA
- Diversi kit commerciali possono essere utilizzati per creare librerie di RNA. Per qualsiasi cosa venga utilizzata, seguire il protocollo del produttore come scritto mentre si lavora in un ambiente sterile a flusso laminare. Una versione molto riassuntiva del protocollo per il kit nella Tabella dei materiali è presentata diseguito 21.
- Fare il primo filamento di sintesi del cDNA master mix (8 μL di acqua priva di nucleasi e 2 μL di First Strand Synthesis Enyzme Mix) e aggiungerlo al campione. Posizionare il campione nel termociclatore con le condizioni specificate nel protocollo.
- Fare il secondo filamento di sintesi del cDNA master mix (8 μL di Second Strand Synthesis Reaction Buffer, 4 μL Second Strand Synthesis Enzyme Mix e 48 μL di acqua priva di nucleasi) su ghiaccio e aggiungerlo al campione. Mettere in un termociclatore impostato a 16 °C per un'ora.
- Purificare la reazione aggiungendo le perle fornite (144 μL) ed eseguendo due lavaggi di etanolo all'80% (200 μL).
- Eluire con il tampone TE fornito (53 μL) e trasferire 50 μL del surnatante in un tubo PCR pulito. Posizionare il tubo PCR sul ghiaccio.
- Preparare la miscela master di preparazione finale (7 μL di End Prep Reaction Buffer e 3 μL di End Prep Enzyme Mix) su ghiaccio e aggiungerla al tubo PCR. Posizionare il tubo PCR in un termociclatore con le condizioni specificate nel protocollo.
- Mescolare le soluzioni di Adattatore Diluito (2,5 μL), Ligation Master Mix (30 μL) e Ligation Enhancer (1 μL) su ghiaccio. Aggiungere le soluzioni miscelate al campione e metterle in un termociclatore per 15 minuti a 20 °C.
- Purificare la reazione aggiungendo le perle fornite (87 μL) ed eseguendo lavaggi di etanolo (200 μL) ed eluizione come prima, tranne aggiungere solo 17 μL di TE.
- Aggiungere gli indici (10 μL) e la soluzione Q5 Master Mix (25 μL) e posizionarli in un termociclatore con le condizioni descritte nel protocollo.
- Purificare la reazione aggiungendo le perle fornite (45 μL) ed eseguendo un'aggiunta di due lavaggi di etanolo (200 μL) ed eluire con 23 μL di TE. Trasferire 20 μL in un tubo PCR pulito.
- Controllare le librerie per le concentrazioni rilevabili di RNA utilizzando un bioanalizzatore, fluorometro o spettrofotometro.
- Raggruppate le librerie metatrascrittomiche in un rapporto approssimativamente equimolare.
- Purificare la biblioteca seguendo lo stesso protocollo per la depurazione della libreria 16S, ad eccezione dei frammenti di accisa tra 250 e 400 bp. Mentre la libreria 16S aveva una banda distinta che rappresentava la regione amplificata, il risultato qui è una macchia.
- Controllare la concentrazione della libreria purificata come prima.
- Spedire la libreria purificata con ghiaccio secco a una struttura di sequenziamento.
NOTA: In alternativa, gli estratti di RNA possono essere inviati a un'università o a un'azienda privata per la preparazione e il sequenziamento della biblioteca.
7. Analisi della comunità microbica
- Una volta completata la sequenziazione, accedere ai dati di esempio. Scaricalo su un computer utilizzabile.
NOTA: idealmente, il dispositivo dovrebbe avere almeno 16 gigabyte di RAM. Per una discussione sui requisiti di elaborazione (per Qiime2), vedere https://forum.qiime2.org/t/recommended-specifications-to-run-qiime2/9808. - Utilizzare software, come mothur, QIIME2 e R, per analizzare i dati rRNA 16S. Vedi qui https://docs.qiime2.org/2020.11/tutorials/moving-pictures/ per un esempio di tutorial di analisi QIIME2 16S.
- Per i dati di metatrascrittomica (RNA), utilizzare HUMAnN2 e ATLAS per determinare quali geni e percorsi sono presenti nei campioni.
NOTA: nel file di informazioni supplementari viene presentato un esempio di pipeline di metatrascrittomica che culmina nell'analisi della diversità e della foresta casuale. Tutti i comandi vengono eseguiti tramite la riga di comando, ad esempio Terminale per utenti Mac.
Risultati
Il successo delle estrazioni di DNA e RNA può essere valutato utilizzando una varietà di apparecchiature e protocolli. Generalmente, qualsiasi concentrazione rilevabile di uno dei due è considerata sufficiente per concludere che l'estrazione ha avuto successo. Esaminando la Tabella 1 quindi, tutte le estrazioni, tranne una, sarebbero soprannominate riuscite. Il fallimento in questa fase è spesso dovuto alla bassa biomassa iniziale, alla scarsa conservazione del campione o all'errore umano durante l'estrazione. Nel caso dei filtri, l'estrazione può aver avuto successo anche se la concentrazione è inferiore al rilevamento. Se questi estratti non producono bande per la PCR (se si fa 16S) o una concentrazione rilevabile dopo la preparazione della libreria (metatrascrittomica), allora probabilmente hanno davvero fallito.
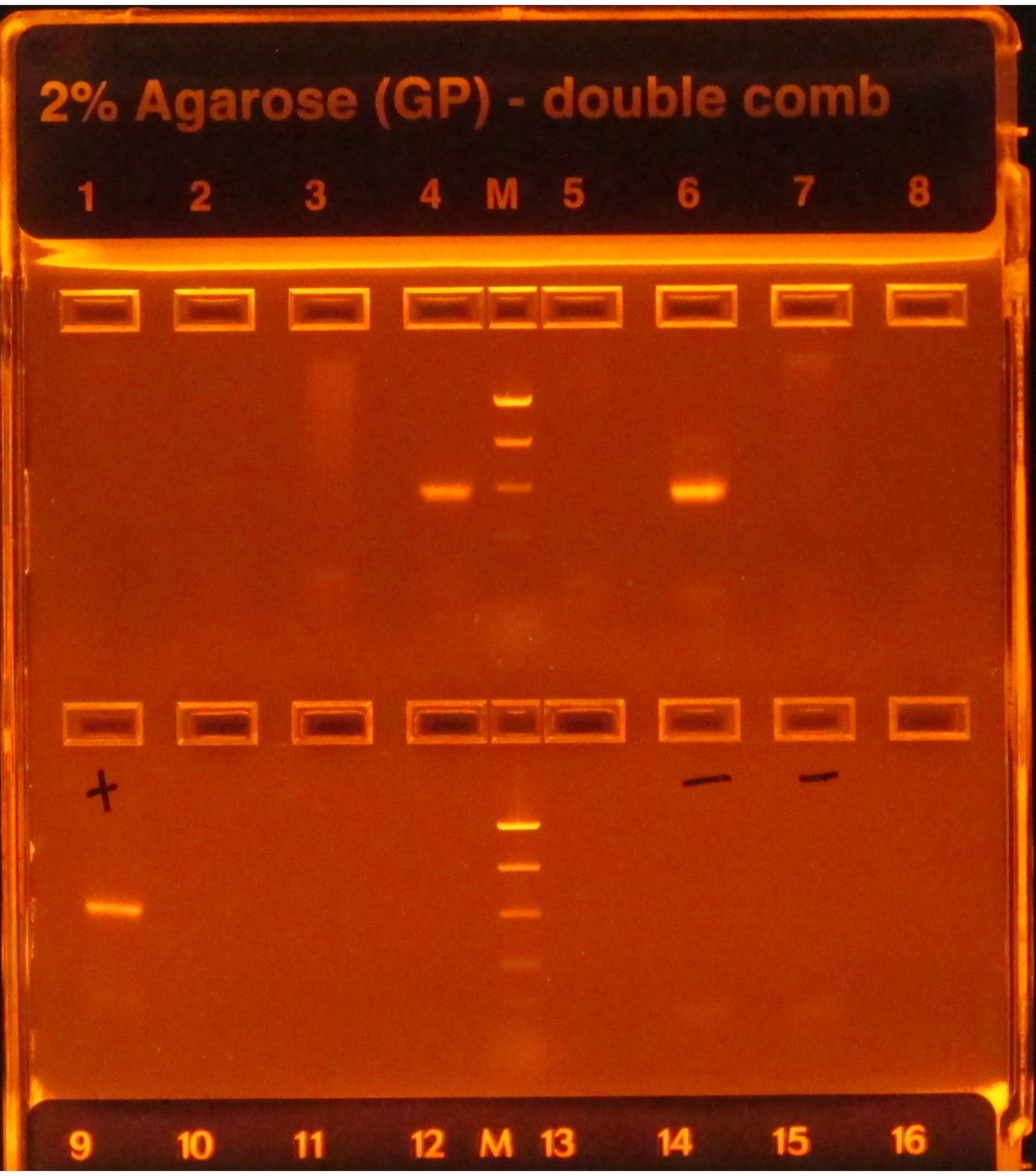
Se si segue il protocollo 16S, le bande luminose successive all'amplificazione PCR, come si vede nei pozzette 4 e 6 in Figura 1,indicano il successo, mentre la mancanza di bande, come si vede negli altri pozzelli nella riga superiore, indica il fallimento. Inoltre, una banda luminosa nella corsia del gel che contiene un controllo PCR negativo indicherebbe anche un guasto poiché sarebbe rischioso presumere che la contaminazione che influisce sui controlli negativi non abbia influenzato i campioni.
Sia per la 16S che per la metatranscriptomics, il successo del sequenziamento può essere valutato osservando il numero di sequenze ottenute (Figura 2). I campioni 16S dovrebbero avere un minimo di 1.000 sequenze, con almeno 5.000 ideali (Figura 2A). Allo stesso modo, i campioni di metatrascrittomica dovrebbero avere un minimo di 500.000 sequenze, con almeno 2.000.000 di essere ideali (Figura 2B). I campioni con meno sequenze di quei minimi non dovrebbero essere utilizzati per le analisi, in quanto potrebbero non rappresentare accuratamente la loro comunità batterica. Tuttavia, i campioni che rientrano tra il minimo e l'ideale possono ancora essere utilizzati, anche se i risultati dovrebbero essere interpretati con maggiore cautela se molti campioni rientrano in tale intervallo.
Il successo della successiva analisi a valle può essere determinato semplicemente sulla base del fatto che i file di output attesi siano stati ottenuti o meno. In ogni caso, programmi, come QIIME2 e R (Figura 3), dovrebbero consentire la valutazione di potenziali differenze significative tra le comunità batteriche basate sul fracking. I dati per la Figura 3 sono stati ottenuti raccogliendo campioni di sedimenti da ventuno siti diversi in tredici diversi flussi per l'analisi 16S e metatrascrittomica. Di questi ventuno siti, dodici di essi erano a valle dell'attività di fracking e classificati come HF+, e nove di essi erano a monte dell'attività di fracking o in uno spartiacque in cui il fracking non si stava verificando; questi flussi sono stati classificati come HF-. Oltre alla presenza di attività di fracking, i flussi erano altrimenti comparabili.
Tali differenze potrebbero assumere la forma di cambiamenti compositici coerenti basati sullo stato di fracking. Se così fosse, ci si aspetterebbe che i campioni HF+ e HF- si raggruppassero l'uno dall'altro in un grafico PCoA, come nel caso della Figura 3A e della Figura 3B. Per confermare che questi apparenti cambiamenti non sono solo un artefatto del metodo di ordinazione, sono necessarie ulteriori analisi statistiche. Ad esempio, un test PERMANOVA22 sulla matrice di distanza su cui si basano la Figura 3A e la Figura 3B ha rivelato un clustering significativo basato sullo stato del fracking, il che significa che la separazione osservata nella trama è coerente con le differenze tra le comunità batteriche dei campioni, invece di un artefatto di ordinazione. Un risultato significativo di PERMANOVA o ANOSIM è una forte indicazione di differenze coerenti tra i campioni HF + e HF-, che indicherebbe che i campioni HF + sono stati influenzati dal fracking, mentre un alto valore p indicherebbe che i campioni non sono stati influenzati. Anche i dati metatrascrittomici possono essere visualizzati e valutati utilizzando gli stessi metodi.
L'esame delle caratteristiche differenziali (microbi o funzioni) può rivelare prove che anche i campioni sono stati influenzati. Un metodo per determinare le feature differenziali consiste nel creare un modello di foresta casuale. Il modello di foresta casuale può essere utilizzato per vedere quanto bene lo stato di fracking dei campioni può essere classificato correttamente. Se il modello funziona meglio del previsto per caso, questa sarebbe un'ulteriore prova delle differenze dipendenti dallo stato di fracking. Inoltre, i predittori più importanti rivelerebbero quali caratteristiche sono più importanti per differenziare correttamente i campioni (Figura 3C). Anche queste caratteristiche avrebbero avuto valori costantemente diversi in base allo stato di fracking. Una volta determinate queste caratteristiche differenziali, la letteratura può essere rivista per vedere se sono state precedentemente associate al fracking. Tuttavia, può essere difficile trovare studi che determinino le funzioni differenziali, poiché la maggior parte ha utilizzato solo dati compositici sull'rRNA 16S. Pertanto, per valutare le implicazioni delle funzioni differenziali, un possibile metodo sarebbe quello di vedere se sono state precedentemente associate a una potenziale resistenza ai biocidi comunemente usati nel fluido di fracking o se potrebbero aiutare a tollerare condizioni altamente saline. Inoltre, l'esame del profilo funzionale di un taxon di interesse potrebbe rivelare prove dell'impatto del fracking (Figura 3D). Ad esempio, se un taxon è identificato come differenziale dal modello di foresta casuale, il suo profilo di resistenza antimicrobica nei campioni HF + potrebbe essere confrontato con il suo profilo nei campioni HF- e se differiscono notevolmente, ciò potrebbe suggerire che il fluido di fracking contenente biocidi è entrato nel flusso.
| SampleID | Concentrazione (ng/μL) |
| 1 | 1.5 |
| 2 | 1.55 |
| 3 | 0.745 |
| 4 | 0.805 |
| 5 | 7.82 |
| 6 | 0.053 |
| 7 | 0.248 |
| 8 | 0.945 |
| 9 | 1.82 |
| 10 | 0.804 |
| 11 | 0.551 |
| 12 | 1.69 |
| 13 | 4.08 |
| 14 | Below_Detection |
| 15 | 7.87 |
| 16 | 0.346 |
| 17 | 2.64 |
| 18 | 1.15 |
| 19 | 0.951 |
Tabella 1: Esempio di concentrazioni di DNA basate sul fluorometro 1x DS DNA ad alta sensibilità. Le estrazioni per tutti questi campioni, ad eccezione di 14, sarebbero considerate di successo a causa della quantità rilevabile di DNA.

Figura 1: Esempio di e-gel con prodotti PCR. Il gel è stato pre-macchiato e visualizzato sotto una luce UV, causando il bagliore di qualsiasi DNA presente su di esso. La PCR ha funzionato per i campioni nei pozzi 4 e 6 della prima fila, poiché entrambi avevano una singola banda luminosa della dimensione prevista (in base alla scala). La PCR per i campioni negli altri sei pozzi fallì, in quanto non produssero alcuna bande. Il controllo positivo (primo pozzo, seconda fila) aveva una banda luminosa, che indicava che la PCR era stata eseguita correttamente, e i controlli negativi (pozzi 6 e 7, seconda fila) non avevano bande, indicando che i campioni non erano contaminati. Se un negativo avesse una banda luminosa come i campioni, la PCR sarebbe stata considerata un fallimento poiché sarebbe stato rischioso presumere che i campioni avessero ampliconi che non erano solo il risultato della contaminazione. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Conteggi di sequenze di esempio. (A) Conteggi di sequenze di esempio 16S. Quasi tutti questi campioni 16S avevano oltre 1.000 sequenze. I pochissimi che avevano meno di 1.000 sequenze dovrebbero essere esclusi dalle analisi a valle, in quanto avevano sequenze insufficienti per rappresentare accuratamente le loro comunità batteriche. Diverse sequenze avevano tra le 1.000 e le 5.000 sequenze; sebbene non ideali, sarebbero comunque utilizzabili poiché superano il minimo indispensabile e la maggior parte dei campioni supera anche il minimo ideale di 5.000. (B) Conteggi di esempio di metatrascrizione. Tutti i campioni hanno superato sia il numero minimo (500.000) che il numero minimo ideale (2.000.000) di sequenze. Pertanto, il sequenziamento ha avuto successo per tutti loro e potrebbero essere tutti utilizzati nell'analisi a valle. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Analisi di esempio. (A) Grafico PCoA basato su coordinate calcolate con una matrice di distanza unifrac ponderata creata e visualizzata tramite QIIME2. (B) Grafico PCoA basato su coordinate calcolate con la matrice di distanza unifrac ponderata esportata da QIIME2. Le coordinate sono state visualizzate utilizzando i pacchetti Phyloseq e ggplot2 in R. I vettori di metadati sono stati montati sulla trama utilizzando il pacchetto Vegan. Ogni punto rappresenta la comunità batterica di un campione, con punti più vicini che indicano composizioni di comunità più simili. È stato osservato un clustering basato sullo stato di fracking per questi campioni di sedimenti 16S (PERMANOVA, p = 0,001). Inoltre, i vettori rivelano che i campioni HF + tendevano ad avere livelli più elevati di bario, bromuro, nichel e zinco, che corrispondevano a una diversa composizione della comunità batterica rispetto ai campioni HF- . (C)Grafico dei migliori predittori per un modello di foresta casuale che ha testato dove l'abbondanza batterica potrebbe essere utilizzata per prevedere lo stato del fracking tra i campioni. Il modello di foresta casuale è stato creato tramite R utilizzando il pacchetto randomForest. Vengono mostrati i primi 20 predittori e le conseguenti diminuzioni di impurità (misura del numero di campioni HF + e HF- raggruppati insieme) sotto forma di diminuzione media dell'indice di Gini quando vengono utilizzati per separare i campioni. (D) Grafico a torta che mostra il profilo di resistenza antimicrobica del profilo burkholderiales basato su dati metatrascrittomici. Le sequenze sono state prima annotate con Kraken2 per determinare a quali taxa appartenevano. BLAST è stato quindi utilizzato con quelle sequenze annotate e il database MEGARes 2.0 per determinare quali geni di resistenza antimicrobica (sotto forma di "MEG_ #") venivano espressi attivamente. I geni di resistenza antimicrobica espressi dai membri di Burkholderiales sono stati quindi estratti per vedere quali erano più diffusi tra quei taxa. Sebbene più costosa e dispendiosa in termini di tempo, la metatranscriptomics consente analisi funzionali, come questa che non può essere eseguita con i dati 16S. In particolare, Kraken2 è stato utilizzato per questa analisi di esempio, invece di HUMAnN2. Kraken2 è più veloce di HUMAnN2; tuttavia, produce solo informazioni compositive, invece di composizione, contributo e funzioni (geni) e percorsi come fa HUMAnN2. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
File supplementare: un esempio di pipeline metatranscriptomics. Fare clic qui per scaricare questo file.
Discussione
I metodi descritti in questo articolo sono stati sviluppati e perfezionati nel corso di diversi studi pubblicati dal nostro gruppo tra il 2014 e il 20187,8,10 e sono stati impiegati con successo in un progetto collaborativo per indagare gli impatti del fracking sulle comunità acquatiche in un progetto triennale che presto presenterà un documento per la pubblicazione. Questi metodi continueranno ad essere utilizzati nel corso del resto del progetto. Inoltre, altra letteratura attuale che studia l'impatto del fracking su flussi ed ecosistemi descrive metodi simili per la raccolta, l'elaborazione e l'analisi dei campioni7,8,10,11. Tuttavia, nessuno di questi articoli ha utilizzato l'analisi metatrascrittomica, rendendo questo documento il primo a descrivere come tali analisi possono essere utilizzate per chiarire l'impatto del fracking sui corsi d'acqua vicini. Inoltre, i metodi qui presentati per la raccolta dei campioni sono più dettagliati, così come le misure adottate per evitare la contaminazione.
Uno dei passaggi più importanti del nostro protocollo è la raccolta e la conservazione iniziale dei campioni. Il campionamento e la raccolta sul campo comportano alcune sfide, poiché il mantenimento di un ambiente asettico o sterile durante la raccolta può essere difficile. Durante questa fase, è fondamentale evitare di contaminare i campioni. Per fare questo, i guanti devono essere indossati e solo i contenitori e gli strumenti sterili devono essere autorizzati a entrare in contatto con i campioni. I campioni devono anche essere immediatamente posti sul ghiaccio dopo la raccolta per mitigare la degradazione dell'acido nucleico. L'aggiunta di un conservante commerciale dell'acido nucleico al momento della raccolta può anche aumentare la resa dell'acido nucleico e consentire la conservazione dei campioni per periodi di tempo più lunghi dopo la raccolta. Ogni volta che viene eseguita l'estrazione dell'acido nucleico, è importante utilizzare la quantità appropriata di campione, troppo può intasare i filtri di spin utilizzati per l'estrazione (per quei protocolli che ne fanno uso) ma troppo poco può portare a basse rese. Assicurati di seguire le istruzioni per qualsiasi kit venga utilizzato.
Analogamente alla raccolta sul campo, evitare o ridurre al minimo la contaminazione è importante anche durante l'estrazione dell'acido nucleico e la preparazione del campione, specialmente quando si lavora con campioni a bassa resa di acido nucleico, come campioni di sedimenti subottimali (campioni contenenti una grande quantità di ghiaia o rocce) o campioni di acqua. Pertanto, come per la raccolta dei campioni, i guanti devono essere indossati durante tutti questi passaggi per ridurre la contaminazione. Inoltre, tutte le superfici di lavoro utilizzate durante le procedure di laboratorio devono essere preventivamente sterilizzate pulendo con una soluzione di candeggina al 10%, seguita da una soluzione di etanolo al 70%. Per le fasi di pipettaggio (3-6), le punte del filtro devono essere utilizzate per evitare la contaminazione dovuta alla pipetta stessa, con le punte che vengono cambiate ogni volta che toccano una superficie non sterile. Tutti gli strumenti utilizzati per il lavoro di laboratorio, comprese le pipette, devono essere puliti prima e dopo con le soluzioni di candeggina ed etanolo. Per valutare la contaminazione, gli spazi vuoti e negativi di estrazione (liquido sterile) devono essere inclusi durante ogni serie di estrazioni di acidi nucleici e reazioni PCR. Se la quantificazione dopo le estrazioni rivela una quantità rilevabile di DNA / RNA nei negativi, le estrazioni possono essere ripetute se rimane un campione sufficiente. Se i campioni negativi per la PCR mostrano amplificazione, è necessario eseguire la risoluzione dei problemi per determinare la sorgente e quindi eseguire nuovamente i campioni. Per tenere conto dei bassi livelli di contaminazione, si raccomanda di sequenziare gli spazi vuoti di estrazione e i negativi PCR in modo che i contaminanti possano essere identificati e rimossi, se necessario, durante l'analisi computazionale. Al contrario, l'amplificazione PCR potrebbe anche fallire a causa di una varietà di cause. Per i campioni ambientali, l'inibizione della reazione PCR è spesso il colpevole, che può essere dovuto a una varietà di sostanze che interferiscono con la Taq polimerasi23. Se si sospetta l'inibizione, l'acqua di grado PCR (vedi Tabella dei materiali)può essere utilizzata per diluire gli estratti di DNA.
Questo protocollo ha alcune notevoli limitazioni e potenziali difficoltà. La raccolta dei campioni può essere difficile sia per i campioni di acqua che di sedimenti. Per ottenere abbastanza biomassa, idealmente 1 L di acqua di flusso deve essere spinto attraverso un filtro. I pori del filtro devono essere piccoli per catturare i microbi, ma possono anche intrappolare i sedimenti. Se molti sedimenti sono nell'acqua a causa delle recenti precipitazioni, il filtro può ostruirsi rendendo difficile spingere l'intero volume attraverso il filtro. Per la raccolta dei sedimenti, può essere difficile stimare la profondità dei sedimenti durante la raccolta. Inoltre, è importante assicurarsi che il sedimento raccolto sia prevalentemente suolo, poiché ciottoli e rocce porteranno a una minore resa di acido nucleico e potrebbero non essere una rappresentazione accurata della comunità microbica. Infine, è anche fondamentale che i campioni siano conservati sul ghiaccio dopo la raccolta, soprattutto se non viene utilizzato un conservante.
Sebbene questo protocollo copra sia la metatrascrittomica che i protocolli di laboratorio 16S, va sottolineato che questi due metodi sono molto diversi sia nel processo che nel tipo di dati che forniscono. Il gene rRNA 16S è una regione comunemente mirata, altamente conservata in batteri e archei e utile per caratterizzare la comunità batterica in un campione. Sebbene si tratti di un approccio mirato e specifico, la risoluzione a livello di specie è spesso irraggiungibile e caratterizzare specie o ceppi di nuova divergenza è difficile. Al contrario, la metatrascrittomica è un approccio più ampio che cattura tutti i geni attivi e i microbi presenti all'interno di un campione. Mentre 16S fornisce solo dati per l'identificazione, la metatrascrittomica può fornire dati funzionali come geni espressi e vie metaboliche. Entrambi sono preziosi e, se combinati, possono rivelare quali batteri sono presenti e quali geni stanno esprimendo.
Questo documento descrive i metodi per la raccolta sul campo e l'elaborazione dei campioni sia per l'rRNA 16S che per le analisi metatrascrittomiche nel contesto dello studio del fracking. Inoltre, descrive in dettaglio i metodi di raccolta per DNA / RNA di alta qualità da campioni di biomassa bassa e per la conservazione a lungo termine. I metodi qui descritti sono il culmine delle nostre esperienze con la raccolta e l'elaborazione dei campioni nei nostri sforzi per imparare come il fracking influisce sui flussi vicini attraverso l'esame della struttura e della funzione delle loro comunità microbiche. I microbi rispondono rapidamente ai disturbi e, di conseguenza, quali microbi sono presenti e i geni che esprimono possono fornire informazioni sugli effetti del fracking sugli ecosistemi. Nel complesso, questi metodi potrebbero essere preziosi per la nostra comprensione di come il fracking influisce su questi importanti ecosistemi.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Gli autori vorrebbero riconoscere le fonti di finanziamento per i progetti che hanno portato allo sviluppo di questi metodi, con quelle fonti che sono: l'Howard Hughes Medical Institute (http://www.hhmi.org) attraverso il Precollege and Undergraduate Science Education Program, nonché dalla National Science Foundation (http://www.nsf.gov) attraverso i premi NSF DBI-1248096 e CBET-1805549.
Materiali
| Name | Company | Catalog Number | Comments |
| 200 Proof Ethanol | Thermo Fisher Scientific | A4094 | 400 mL need to be added to Buffer PE (see Qiagen QIAQuck Gel Extraction kit protocol) and 96 mL needs to be added to the DNA/RNA Wash Buffer (see ZymoBIOMICS DNA/RNA Miniprep kit protocol). Additional ethanol is needed for the ZymoBIOMICS DNA/RNA Miniprep and NEBNext® Ultra™ II RNA Library Prep with Sample Purification Beads kits. |
| Agarose | Thermo Fisher Scientific | BP1356-100 | 100 g per bottle. 0.6 g of agarose would be needed to make one 2% 30 mL gel. |
| Disinfecting Bleach | Walmart (Clorox) | No catalog number | Use a 10% bleach solution for cleaning the work area before and after lab procedures |
| DNA gel loading dye | Thermo Fisher Scientific | R0611 | Each user-made (i.e. non-e-gel) should include loading dye with all of the samples in the ratio of 1 µL dye to 5 µL sample |
| DNA ladder | MilliporeSigma | D3937-1VL | A ladder should be run on every gel/e-gel |
| DNA/RNA Shield (2x) | Zymo Research | R1200-125 | 3 mL per sediment sample (50 mL conical) and 2 mL per water sample (filter) |
| Ethidium bromide | Thermo Fisher Scientific | BP1302-10 | Used for staining user-made e-gels |
| Forward Primer | Integrated DNA Technologies (IDT) | 51-01-19-06 | 0.5 µL per PCR reaction |
| Isopropanol | MilliporeSigma | 563935-1L | Generally less than 2 mL per library. Volume needed varies by mass of excised gel fragment (see Qiagen QIAQuick Gel Extraction kit protocol). |
| PCR-grade water | MilliporeSigma | 3315932001 | 13 µL per PCR reaction (assuming 1 µL of sample DNA template is used) |
| Platinum Hot Start PCR Master Mix (2x) | Thermo Fisher Scientific | 13000012 | 10 µL per PCR reaction |
| Reverse Primer | Integrated DNA Technologies (IDT) | 51-01-19-07 | 0.5 µL per PCR reaction |
| TBE Buffer (Tris-borate-EDTA) | Thermo Fisher Scientific | B52 | 1 L of 10x TBE buffer (30 mL of 1x TBE buffer would be needed to make one 30 mL gel) |
| 1 L bottle | Thermo Fisher Scientific | 02-893-4E | One needed per stream (the same bottle can be used for multiple streams if it is sterilized between uses) |
| 1.5 mL Microcentrifuge tubes | MilliporeSigma | BR780400-450EA | 5 microcentrifuge tubes are needed per DNA extraction and an additional 3 are needed to purify RNA (see ZymoBIOMICS DNA/RNA Miniprep kit protocol) |
| 2% Agarose e-gel | Thermo Fisher Scientific | G401002 | Each gel can run 10 samples (so 9 with a PCR negative and 8 if the extraction negative is run on the same gel) |
| 50 mL Conicals | CellTreat | 229421 | 1 50 mL conical needed per sediment samples |
| 500 mL Beaker | MilliporeSigma | Z740580 | Only 1 needed (for flame sterilization) |
| Aluminum foil | Walmart (Reynolds KITCHEN) | No number | Aluminum foil can be folded and autoclaved. The part not exposed to the environment can then be used as a sterile, DNA and RNA free surface for processing filters (one folded piece per filter to avoid cross-contamination) |
| Autoclave | Gettinge | LSS 130 | Only one needed |
| Centrifuge | MilliporeSigma | EP5404000138-1EA | Only 1 needed |
| Cooler | ULINE | S-22567 | Just about any cooler can be used. This one is listed due to being made of foam, making it lighter and thus easier to take along for field sampling. |
| Disruptor Genie | Bio-Rad | 3591456 | Only one needed |
| Electrophoresis chamber | Bio-Rad | 1664000EDU | Only 1 needed |
| Electrophoresis power supply | Bio-Rad | 1645050 | Only 1 needed |
| Freezer (-20 C) | K2 SCIENTIFIC | K204SDF | One needed to store DNA extracts |
| Freezer (-80 C) | K2 SCIENTIFIC | K205ULT | One needed to store RNA extracts |
| Gloves | Thermo Fisher Scientific | 19-020-352 | The catalog number is for Medium gloves. |
| Heat block | MilliporeSigma | Z741333-1EA | Only one needed |
| Lab burner | Sterlitech | 177200-00 | Only one needed |
| Laminar Flow Hood | AirClean Systems | AC624LFUV | Only 1 needed |
| Library purification kit | Qiagen | 28704 | One kit has enough for 50 reactions |
| Magnet Plate | Alpaqua | A001219 | Only one needed |
| Microcentrifuge | Thermo Fisher Scientific | 75004061 | Only one needed |
| Micropipette (1000 µL volume) | Pipette.com | L-1000 | Only 1 needed |
| Micropipette (2 µL volume) | Pipette.com | L-2 | Only 1 needed |
| Micropipette (20 µL volume) | Pipette.com | L-20 | Only 1 needed |
| Micropipette (200 µL volume) | Pipette.com | L-200R | Only 1 needed |
| NEBNext Ultra II RNA Library Prep with Sample Purification Beads | New England BioLabs Inc. | E7775S | One kit has enough reagents for 24 samples. |
| Parafilm | MilliporeSigma | P7793-1EA | 2 1" x 1" squares are needed per filter |
| PCR Tubes | Thermo Fisher Scientific | AM12230 | One tube needed per reaction |
| Pipette tips (for 1000 µL volume) | Pipette.com | LF-1000 | Pack of 576 tips |
| Pipette tips (for 20 µL volume) | Pipette.com | LF-20 | Pack of 960 tips |
| Pipette tips (for 200 µL volume) | Pipette.com | LF-250 | Pack of 960 tips |
| PowerWulf ZXR1+ computer cluster | PSSC Labs | No number | This is just an example of a supercomputer powerful enough to perform metatranscriptomics analysis in a timely manner. Only one needed. |
| Qubit fluorometer starter kit | Thermo Fisher Scientific | Q33239 | Comes with a Qubit 4 fluorometer, enough reagent for 100 DNA assays, and 500 Qubit tubes |
| Scoopula | Thermo Fisher Scientific | 14-357Q | Only one needed |
| Sterile blades | AD Surgical | A600-P10-0 | One needed per filter |
| Sterivex-GP Pressure Filter Unit | MilliporeSigma | SVGP01050 | 1 filter needed per water sample |
| Thermocycler | Bio-Rad | 1861096 | Only one needed |
| Vise-grip | Irwin | 2078500 | Only one needed (for cracking open the filters) |
| Vortex-Genie 2 | MilliporeSigma | Z258415-1EA | Only 1 needed |
| WHIRL-PAK bags | ULINE | S-22729 | 1 needed per filter |
| ZymoBIOMICS DNA/RNA Miniprep kit | Zymo Research | R2002 | One kit has enough reagents for 50 samples. |
Riferimenti
- The process of unconventional natural gas production. US EPA Available from: https://www.epa.gov/uog/process-unconventional-natural-gas-production (2013)
- Brittingham, M. C., Maloney, K. O., Farag, A. M., Harper, D. D., Bowen, Z. H. Ecological risks of shale oil and gas development to wildlife, aquatic resources, and their habitats. Environmental Science & Technology. 48 (19), 11034-11047 (2014).
- McBroom, M., Thomas, T., Zhang, Y. Soil erosion and surface water quality impacts of natural gas development in East Texas, USA. Water. 4 (4), 944-958 (2012).
- Maloney, K. O., Weller, D. E. Anthropogenic disturbance, and streams: land use and land-use change affect stream ecosystems via multiple pathways. Freshwater Biology. 56 (3), 611-626 (2011).
- Meyer, J. L., et al. The contribution of headwater streams to biodiversity in river networks1. JAWRA Journal of the American Water Resources Association. 43 (1), 86-103 (2007).
- Alexander, R. B., Boyer, E. W., Smith, R. A., Schwarz, G. E., Moore, R. B. The role of headwater streams in downstream water quality. Journal of the American Water Resources Association. 43 (1), 41-59 (2007).
- Ulrich, N., et al. Response of aquatic bacterial communities to hydraulic fracturing in Northwestern Pennsylvania: A five-year study. Scientific Reports. 8 (1), 5683 (2018).
- Chen See, J. R., et al. Bacterial biomarkers of Marcellus shale activity in Pennsylvania. Frontiers in Microbiology. 9, 1697 (2018).
- Rausch, P., et al. Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome. 7 (1), 133 (2019).
- Louca, S., Doebeli, M., Parfrey, L. W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome. 6 (1), 41 (2018).
- Trexler, R., et al. Assessing impacts of unconventional natural gas extraction on microbial communities in headwater stream ecosystems in Northwestern Pennsylvania. Frontiers in Microbiology. 5, 522 (2014).
- Mumford, A. C., et al. Shale gas development has limited effects on stream biology and geochemistry in a gradient-based, multiparameter study in Pennsylvania. Proceedings of the National Academy of Sciences. 117 (7), 3670-3677 (2020).
- JoVE Core Biology DNA Isolation. Journal of Visualized Experiments Available from: https://www.jove.com/cn/science-education/10814/dna-isolation (2020)
- Oxford Gene Technology DNA Storage and Quality. OGT Available from: https://www.ogt.com/resources/literature/403_dna_storage_and_quality (2011)
- ThermoFisher SCIENTIFIC Technical Bulletin #159: Working with RNA. Thermoscientific Available from: https://www.thermofisher.com/us/en/home/references/ambion-tech-support/nuclease-enzymes/general-articles/working-with-rna.html (2020)
- QIAGEN AllPrep DNA/RNA Mini Kit. Qiagen Available from: https://www.qiagen.com/us/products/discovery-and-translational-research/dna-rna-purification/multianalyte-and-virus/allprep-dnarna-mini-kit/#orderinginformation (2020)
- ZymoBIOMICS DNA/RNA Miniprep Kit. Zymo Research Available from: https://www.zymoresearch.com/products/zymobiomics-dna-rna-miniprep-kit (2020)
- Desjardins, P., Conklin, D. NanoDrop microvolume quantitation of nucleic acids. Journal of Visualized Experiments. (45), e2565 (2010).
- 16S Illumina amplicon protocol: Earth microbiome project. Earth microbiome project Available from: https://earthmicrobiome.org/protocols-and-standards/16s/ (2018)
- Gel Purification: Binding, washing and eluting a sample | Protocol. Journal of Visualized Experiments Available from: https://www.jove.com/v/5063/gel-purification (2020)
- New England Biolabs protocol for the use with NEBNext Poly(A) mRNA magnetic isolation module (E7490) and NEBNext Ultra II RNA library prep kit for Illumina (E7770, E7775). New England Biolabs Available from: https://www.neb.com/protocols/2017/03/04/protocol-for-use-with-purified-mrna-or-rrna-depleted-rna-and-nebnext-ultra-ii-rna-library-prep-ki (2020)
- Anderson, M. J. Permutational multivariate analysis of variance (PERMANOVA). Wiley StatsRef: Statistics Reference Online. , 1-15 (2017).
- Schrader, C., Schielke, A., Ellerbroek, L., Johne, R. PCR inhibitors - occurrence, properties and removal. Journal of Applied Microbiology. 113 (5), 1014-1026 (2012).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati