
Method Article
ウイルス様粒子(「ナノブレード」)を介した不死化細胞および初代細胞におけるCas9/sgRNAリボヌクレオタンパク質複合体の送達
要約
我々は、Cas9/シングルガイドRNA(sgRNA)リボヌクレオタンパク質複合体をウイルス様粒子内にロードするためのシンプルで安価なプロトコルを開発しました。「ナノブレード」と呼ばれるこれらの粒子は、不死化および初代細胞ならびにインビボでCas9/sgRNA複合体の効率的な送達を可能にする。
要約
クラスター化された規則的に間隔をあけた短い回文反復(CRISPR)-Casシステムは、真核細胞におけるゲノム編集を民主化し、数多くの革新的なアプリケーションの開発につながった。しかしながら、Cas9タンパク質およびシングルガイドRNA(sgRNA)の標的細胞への送達は、技術的に困難な場合がある。レンチウイルス(LV)またはアデノ随伴ウイルス(AAV)に由来するものなどの古典的なウイルスベクターは、Cas9タンパク質およびその関連sgRNAをコードする導入遺伝子を多くの初代細胞およびインビボで効率的に送達することを可能にする。それにもかかわらず、これらのベクターは、標的細胞ゲノムにおける導入遺伝子の組み込み、限られた貨物容量、および標的細胞におけるCas9タンパク質およびガイドRNAの長期発現などの欠点に苦しむ可能性がある。
これらの問題のいくつかを克服するために、マウス白血病ウイルス(MLV)に基づく送達ベクターが開発され、コード導入遺伝子の非存在下でCas9タンパク質およびそれに関連するガイドRNAをパッケージ化した。MLV由来の構造タンパク質GagのC末端にCas9タンパク質を融合させることにより、Cas9タンパク質とsgRNA(ナノブレードと命名)をロードしたウイルス様粒子(VLP)が形成された。ナノブレードは、産生細胞の培養培地から収集し、精製、定量し、標的細胞を形質導入し、活性Cas9/sgRNA複合体を送達するために使用することができる。ナノブレードは、リボヌクレオタンパク質(RNP)の貨物を広範囲の初代および不死化細胞に一過性かつ迅速に送達し、修飾Cas9タンパク質を使用して、標的遺伝子の一過性転写活性化などの他の用途向けにプログラムすることができます。ナノブレードは、注射された成体マウスの肝臓および卵母細胞においてインビボゲノム編集が可能であり、トランスジェニック動物を生成することができる。最後に、ドナーDNAと複合化して、「トランスフェクションフリー」相同性指向修復を行うことができます。ナノブレード調製は、簡単で、比較的低コストであり、そして任意の細胞生物学実験室で容易に実施することができる。
概要
他のプログラム可能なヌクレアーゼと比較して、CRISPR-Casシステムは、真核細胞における配列特異的ゲノムターゲティングおよび切断の手順を劇的に簡素化し、民主化しました。sgRNAの単純な発現により、ユーザーはほぼすべての細胞遺伝子座に対してCas9タンパク質(または最適化されたバリアント)をプログラムできます1。このシナリオでは、Cas9タンパク質およびsgRNAの送達が、部位特異的変異誘発を行う際の主な制限となる。不死化細胞では、sgRNAおよびCasタンパク質をトランスフェクトプラスミドから容易に発現させ、ほとんどの細胞で効率的なゲノム標的化を達成することができる。しかし、Cas9/sgRNA複合体の構成的発現は、Cas9タンパク質のオフターゲット活性を増加させ、非特異的遺伝子座2に望ましくない変化をもたらす可能性がある。初代細胞では、DNAトランスフェクションを達成することは技術的に困難であり、発現不良またはトランスフェクトされた細胞のわずかな割合につながる可能性がある。古典的なDNAトランスフェクションに代わるものは、Cas9およびsgRNAをコードする導入遺伝子を送達するウイルスベクターの使用、または合成sgRNAに結合された組換えCas9タンパク質のエレクトロポレーションを含む。しかしながら、これらのアプローチは、細胞宿主ゲノム内での導入遺伝子組込み(古典的なレトロウイルスおよびレンチウイルス発現ベクターの場合と同様)、細胞因子による制限、およびCas9タンパク質およびsgRNAの構成的発現をもたらし得る。
Cas9/sgRNP 複合体のエレクトロポレーションは、これらの問題のほとんどを克服し、初代細胞およびインビボでの効率的かつ一過性の送達をもたらし、用量依存性応答を可能にすることができる。それにもかかわらず、それは通常高価な装置と試薬に依存しており、多数の細胞を処理する必要がある場合、高級化することも困難です。上記の技術の代替として、これらの著者らは、他のウイルス由来のカプシドタンパク質送達系と概念的に類似したCas9タンパク質およびsgRNA3用のレトロウイルス送達ベクターである「ナノブレード」を開発しました4,5,6,7,8。ナノブレードは、レトロウイルス由来のGagポリタンパク質の自然な能力を利用して、培養細胞で単独で発現すると、細胞外培地中に放出されるVLPを産生します9。Cas9タンパク質をマウス白血病ウイルス(MLV)Gagポリタンパク質のC末端末端に融合させ、sgRNAおよびウイルスエンベロープ糖タンパク質を共発現させることにより、Cas9タンパク質を放出されたVLPまたはナノブレード内に包埋することができる。精製時に、ナノブレードを標的細胞と共にインキュベートするか、インビボで注射して、Cas9/sgRNA RNP複合体の迅速、一過性、および用量依存的な送達を媒介することができます3。
ナノブレードは、複数のsgRNAでプログラムして異なる遺伝子座で同時に編集したり、Cas9バリアントを使用して標的特異的転写活性化や抑制などの他のアプリケーションを実行することができます3。組換え発現に依存するタンパク質エレクトロポレーションとは対照的に、文献から新たに記載されたCas変異体は、Gag融合発現ベクターに容易にクローニングすることができ、汎用性の高いプラットフォームとなる。ナノブレードをさらに複合化または一本鎖および二本鎖オリゴデオキシヌクレオチド(ssODN)と装填して、相同性指向性修復を行うことができます3。ナノブレードの生産は比較的簡単で安価です。さらに、ナノブレードは、4°Cで何日間も保管することも、-80°Cで長期保管することもできます。通常、ナノブレードは、ほとんどの不死化および初代培養細胞において、効率的で導入遺伝子を含まないゲノム編集を媒介します。しかし、一部の初代細胞はウイルス粒子の存在に敏感である可能性があり、その結果、死亡率が増加する。自然免疫系の細胞はまた、ナノブレードの存在に反応し(ウイルス起源のため)、活性化され得る。このような場合、形質導入プロトコルを最適化して、ナノブレードへの曝露時間を制限し、非特異的な影響を最小限に抑える必要があります。ナノブレードは、他の利用可能なCRISPR送達方法に代わる実行可能で実装が容易な代替手段です。
プロトコル
1. sgRNAの設計とクローニング
注:sgRNAの設計に関するガイドラインは、https://blog.addgene.org/how-to-design-your-grna-for-crispr-genome-editing などの複数の情報源、またはHannaとDoench10から入手できます。
- 20ヌクレオチドのsgRNA配列を設計したら、以下の一本鎖DNAオリゴヌクレオチドを注文する:
- 前方:5' caccgNNNNNNNNNNNNNNNNNNNNNNNNNN 3' (Nはプロトスペーサー隣接モチーフ(PAM)配列を持たない標的遺伝子座に対応する)
- 逆: 5' aaacNNNNNNNNNNNNNNNNNNNNNNNN 3' (N は PAM 配列を持たない標的遺伝子座の逆補体に対応する)
注:オリゴヌクレオチドを注文する際に特別な変更は必要ありません(5'リン酸は必要ありません)。
- 5 μL のアニーリングバッファー (500 mM NaCl; 100 mM Tris-HCl; 100 mM MgCl2; 10 mM DTT; pH 7.9 at 25°C)、各 DNA オリゴヌクレオチド 1 μL (水中の 100 μM ストック溶液)、および 42 μL の水を混合して、2 つの DNA オリゴヌクレオチドを 0.2 mL ポリメラーゼ連鎖反応 (PCR) チューブでハイブリダイズさせます。
- PCR ブロック上で、サンプルを 95 °C で 15 秒間インキュベートした後、0.5 °C/s のランプで温度を 20 °C に下げます。室温で保存するか、-20°Cで保存してください。
メモ: プロトコルはここで一時停止できます。 - 10 μg の BLADE または SUPERBLADE sgRNA 発現プラスミドを 10 ユニットの BsmBI-v2 制限酵素と共に、全反応量 50 μL で 55 °C で 3 時間消化します。
注:消化されたベクターは、〜1.9 kbのDNAインサートと〜3.3 kbの第2のDNA断片を放出する必要があります。 - 5 μg/mL の臭化エチジウムで染色した 1% アガロースゲル (またはより安全な代替 DNA ゲル染色) に制限反応をロードします。
注:遺伝的欠陥を引き起こす疑いのある臭化エチジウムを操作するときは、適切な保護具を着用してください。- 波長 312 nm に設定した紫外線 (UV) テーブル上で (DNA の損傷を避けるため)、ゲルから 3.3 kb DNA 断片を切り取り、1.5 mL マイクロ遠心分離管に入れます。
メモ:エチジウムブロマイドを操作し、UVテーブルで作業するときは、適切な保護具(手袋とUV保護ゴーグル)を着用してください。 - 専用のDNAゲル抽出キットを使用して、3.3 kb DNA断片を含むスライスゲルからDNAを抽出 します(材料表を参照)。分光光度計を用いて精製DNAの量を定量する。
メモ: プロトコルはここで一時停止できます。
- 波長 312 nm に設定した紫外線 (UV) テーブル上で (DNA の損傷を避けるため)、ゲルから 3.3 kb DNA 断片を切り取り、1.5 mL マイクロ遠心分離管に入れます。
- ステップ1.2からハイブリダイズした順方向および逆方向DNAオリゴヌクレオチドを、ステップ1.5.2からBsmB1消化、ゲル精製BLADESまたはSUPERBLADEベクターにライゲーションする。このために、2 μLのT4 DNAリガーゼバッファー、50 ngのゲル精製ベクター(ステップ1.5.2から)、1 μLのハイブリダイズDNAオリゴヌクレオチド(ステップ1.2から)、19 μLまでの容量を構成する水、および1 μLのT4 DNAリガーゼを加える。反応物を25°Cで10分間インキュベートする。
- ライゲーション生成物を、in11に記載されているようにコンピテントバクテリア(材料表を参照)に変換します。形質転換細菌をアンピシリン・ルリア・ベルタニ寒天プレート上に平板し、37°Cで一晩インキュベートした。
- 寒天プレート上で単離されたコロニーをいくつか選択してDNAミニ調製11 を行い( 材料表を参照)、U6フォワードプライマー(5' GACTATCATATGCTTACCGT 3')を用いてサンガーシーケンシングを行い、sgRNA可変配列の正しいライゲーションを確認します。
注:他のsgRNA発現プラスミドは、ナノブレード産生を妨げる可能性のあるCas9タンパク質をコードしていない場合は、使用することができます。
2. プラスミド調製
- 必要なすべてのプラスミドのマキシプロプレージング( 材料表を参照)を行い、10 μgアリコートを1 μg/mLで調製して-20 °Cで保存します。 プラスミドの繰り返し凍結/融解サイクルを避けます;アリコートを破棄する前に 2 回使用します。
3. ナノブレードの調製
- 1日目に、高グルコース、ピルビン酸ナトリウム×、L-グルタミン、10%ウシ胎児血清(FBS)、およびペニシリン/ストレプトマイシンを含むダルベッコの改変イーグル培地(DMEM)の10mLに106 個のHEK293T細胞(材料表を参照)を10cmの細胞培養皿に3.5〜4個( 材料表を参照)を播種した。10cmプレートを前後に静かに動かし、次いで右から左に(このシーケンスを5倍繰り返す)培養皿上に細胞を均質に分配する。5%CO2の細胞インキュベーター内で37°Cで細胞をインキュベートする。
注:培養細胞およびナノブレードの取り扱いに関連するすべての手順は、それらの汚染を避けるために、細胞培養層流フードの下で実行する必要があります。 - 2日目: プラスミドトランスフェクション
- 細胞は、プレーティング後24時間で70~80%コンフルエントでなければなりません(図1A)。トランスフェクション前に、高グルコース、ピルビン酸ナトリウム、L-グルタミン、10%FBS(ペニシリンおよびストレプトマイシンは必須ではありませんが省略できます)を含む10 mLの新鮮なDMEMで培地を交換してください。
注: このステップでは、セルがコンフルエントでないことが重要です。そうしないと、トランスフェクション効率と粒子生産が低下する可能性があります。 - 10 cm プレートごとに、1.5 mL チューブに次の量のプラスミドを調製します: 0.3 μg pCMV-VSV-G, 0.7 μg pBaEVRless, 2.7 μg MLV Gag/Pol, 1.7 μg BIC-Gag-Cas9, 4.4 μg の BLADES または SUPERBLADES プラスミド クローニングされた sgRNA をコードするプラスミド (または 2 つの sgRNA を使用する場合は、それぞれ 2.2 μg)。
- 500 μL のトランスフェクションバッファー ( 材料表を参照) を加え、ボルテックスを 10 秒間、遠心分離機で 1 秒間行います。20 μL のトランスフェクション試薬 ( 材料表を参照) を加え、チューブを 1 秒間ボルテックスし、次に 1 秒間遠心分離します。
- 室温で10分間インキュベートし、P1000ピペッターを用いて溶液全体をDMEM培地中の細胞に滴下して加えた。10 cm プレートを前後に静かに動かし、次に右から左に (このシーケンスを 5 回繰り返す)、トランスフェクション試薬を細胞上に均一に分配します。細胞を5%CO2の細胞インキュベーター内で37°Cで少なくとも40時間インキュベートする。
注:必要に応じて、培地はトランスフェクションの4時間後に交換することができます。
- 細胞は、プレーティング後24時間で70~80%コンフルエントでなければなりません(図1A)。トランスフェクション前に、高グルコース、ピルビン酸ナトリウム、L-グルタミン、10%FBS(ペニシリンおよびストレプトマイシンは必須ではありませんが省略できます)を含む10 mLの新鮮なDMEMで培地を交換してください。
- 3日目に、顕微鏡下でトランスフェクトされた細胞の形態を確認する。
注:生産者細胞は融合し始めます。これは、フソゲンウイルスエンベロープの発現に起因する正常な発生である(図1B、C)。 - 4日目:ナノブレードの収穫
注:トランスフェクションの少なくとも40時間後には、融合原性ウイルスエンベロープの発現のために細胞が融合し、時には細胞がプレート支持体から完全に剥離することもあります(図1D)。- 10 mLピペットを用いて9 mLの培養液上清を集める。
注:ナノブレードは、Cas9タンパク質およびそれに関連するsgRNAを初代細胞およびインビボに送達することができるVLPである。遺伝物質が存在しないため、遺伝子組み換え生物とは見なされませんが、遺伝的変化を誘発する可能性があります。したがって、ユーザーとの接触を避けるために(特に腫瘍抑制遺伝子を標的にするようにプログラムされている場合)、慎重に操作する必要があります。ユーザーは、レトロウイルスベクターの操作に関する現地の安全ガイドラインに従い、VLPを準備して形質導入実験を行う際にBSL-2レベルの実験室で働くことをお勧めします。ナノブレードは、70%エタノールまたは0.5%の次亜塩素酸ナトリウムで不活性化することができる。また、すべてのプラスチック廃棄物(ピペットチップ、組織培養プレート、遠心チューブ)を0.5%次亜塩素酸ナトリウムで少なくとも10分間処理して、ナノブレードを不活性化することもお勧めします。 - 回収した上清を500× g で5分間遠心分離し、細胞破片を除去し、細胞ペレットを乱すことなく上清を回収した。
注: ナノブレードを初代細胞で使用する場合は、0.45 μm または 0.8 μm のフィルターを使用して上清をろ過します。このステップは、かなりの画分がフィルター膜でブロックされるため、ナノブレードの力価を大幅に低下させることに注意してください。 - ナノブレードを4,300 × g または209,490 × gの超遠心分離 機でスイング バケットローターで一晩(12〜16時間)4°Cで75分間ペレット化します( 材料表を参照)。
注:標的細胞がDMEM中で増殖できる場合、ナノブレードを濃縮することなく、ステップ3.4.2の後に得られた上清と直接インキュベートすることが可能です。
- 10 mLピペットを用いて9 mLの培養液上清を集める。
- 5日目:ナノブレードの再懸濁と保管
- 遠心分離後、培地をゆっくりと吸引し、白色ペレットを100μLの冷たい1xリン酸緩衝生理食塩水(PBS)で再懸濁する。チューブをパラフィルムで覆い、穏やかな攪拌で4°Cで1時間インキュベートしてから、ペレットを上下にピペッティングして再懸濁させる。
注:再懸濁時に白色の粘性材料が現れることがあります。これは正常であり、形質導入の効率に大きく影響しません。 - ナノブレードを4週間以内に使用する予定がある場合は、4°Cで保管してください。それ以外の場合は、ナノブレードを液体窒素でスナップフリーズし、-80°Cで保存します。
注:液体窒素を操作するときは、保護ゴーグルと極低温手袋を着用してください。スナップ凍結と-80°Cでの保存は、ナノブレード効率の大幅な低下につながります。さらに、解凍したナノブレードは再び凍結しないでください。プロトコルはここで一時停止できます。
- 遠心分離後、培地をゆっくりと吸引し、白色ペレットを100μLの冷たい1xリン酸緩衝生理食塩水(PBS)で再懸濁する。チューブをパラフィルムで覆い、穏やかな攪拌で4°Cで1時間インキュベートしてから、ペレットを上下にピペッティングして再懸濁させる。
4. スクロースクッションへのナノブレードの濃縮
注:一晩遠心分離または超遠心分離(ステップ3.4.3)の代わりに、ナノブレードをスクロースクッションに濃縮することができます。これにより、ナノブレードのより純粋な画分が得られますが、回収される総量は少なくなります。
- 1x PBS中の10%スクロース溶液(重量対体積)を調製し、0.2μmシリンジフィルターでろ過する( 材料表を参照)。
- ナノブレードをスクロースクッションに集中させるプロセスを開始します。
- 9 mL の VLP 含有サンプル (ステップ 3.4.3 から) を超遠沈管に入れます ( 材料表を参照)。3 mLシリンジおよびカニューレを用いて、試料の下に10%スクロースの2.5 mLをゆっくりと層状にし、VLP含有試料とスクロース溶液とを混合しないようにする。
- あるいは、2.5 mLの10%スクロースを超遠心管に入れます( 材料表を参照)。チューブを傾け、9 mLのVLP含有サンプル(ステップ3.4.3から)を低速ピペッターでゆっくりと加えます。この操作の間、チューブを垂直位置まで徐々に持ち上げる。
- サンプルを超遠心機で209,490 × g で4°Cで90分間遠心分離する。
注:この技術は、12に記載されているように、一晩の低速遠心分離(4,300× g)に適合させることができます。 - 遠心分離後、上清を慎重に取り除き、チューブをティッシュペーパーの上に逆さまに置き、残りの液体を除去します。1分後、1x PBSを100 μL加え、パラフィルムカバーを撹拌テーブル上のチューブホルダーに1時間入れてチューブを4°Cに置き( 材料表を参照)、ペレットを上下にピペッティングして再懸濁します。
メモ: プロトコルはここで一時停止できます。
5. ドットブロットによるナノブレード内のCas9負荷の監視
- 非イオン性界面活性剤を含む溶解緩衝液( 材料表を参照)を4倍量の1x PBSに1倍量加えて希釈緩衝液を調製する。濃縮ナノブレード2 μLを50 μLの希釈バッファーで希釈し、ボルテックスで短時間行い、この混合物25 μLを25 μLの希釈バッファーを含む新しいチューブに移します。この操作を繰り返して、4本のチューブのナノブレード希釈液(2倍希釈ステップ)を有する。
- 標準対照の場合、2 μLの組換えCas9ヌクレアーゼ(材料表を参照)を50 μLの希釈バッファーに希釈し、ボルテックスを短時間行い、8つの段階希釈(各ステップで2倍希釈)を行います。
- 各VLP希釈液2.5 μLおよび各標準物質2.5 μLをマルチチャンネルピペットでニトロセルロース膜上に注意深くスポットします(容量が大きいほど、スポットが重なる可能性があります)。
注:メタノール処理されたポリビニルジフルオライド膜も使用することができる。 - 粒子が膜に吸収されたら、非イオン性界面活性剤(TBS-T)を含む1xトリス緩衝生理食塩水で膜をブロックし、無脂ドライミルク(5% w / v)を室温で45分間補充します。
注:プロトコルはここで一時停止することができ、メンブレンは1x TBS-Tで4°Cで保存されます。 - 無脂ドライミルクを添加した1x TBSTを捨て、Cas9-西洋ワサビペルオキシダーゼ抗体(1x TBSTで1/1000希釈、5%ミルク)と共に膜を4°Cで一晩インキュベートした。メンブレン3xをTBS-Tで洗浄し、増強化学発光基質キットを用いてシグナルを可視化した。
- ナノブレードおよび組換えCas9標準希釈液のドット強度は、ゲルイメージングステーションまたはimageJ13に付属の独自のソフトウェアを使用して定量化します。ドット強度をCas9濃度にリンクする直線曲線を定義します。得られた曲線の関数を用いて、各調製物中のCas9含有量を外挿する。
注:組換えCas9タンパク質コントロールの量は、標準希釈セットの最も濃縮されたサンプルの測定値を飽和させる可能性があります(図2)。したがって、線形曲線を定義する際には、スポットされたCas9の既知の濃度に対して線形範囲にない場合、希釈されていないサンプル(および時には最初の希釈ステップの測定値)から読み取り値を削除することをお勧めします。同様に、ナノブレードサンプル内のCas9の量を外挿する場合は、標準曲線の直線範囲内にある測定値のみを使用します。
6. ナノブレードによる標的細胞の形質導入(12穴プレートでの形質導入手順)
- 12ウェルプレートにおいて、1ウェル当たり100,000〜200,000個の細胞(初代または不死化接着細胞のいずれか)を1mLの適切な細胞培養培地中に種子化する。形質導入前に細胞がプレート表面に接着するのを許す。
- 1.5 mL の微量遠心チューブで、5 ~ 20 μL の濃縮ナノブレード (ステップ 3.5.1 または 4.4 から) を 500 μL の細胞培養培地に追加し、P1000 ピペッターで上下にピペッティングして混合します。細胞から培地を取り出し、このナノブレード混合物の500μLと交換する。
注: 形質導入は、細胞タイプごとに最適化する必要があります。ナノブレードが高度に濃縮されたままであるように、(標的細胞の乾燥を避けながら)可能な限り最小の量の培地を使用することが重要です。付着細胞は、プレートに付着した状態で直接形質導入する必要があります(形質導入効率が著しく低下するため、懸濁状態で形質導入しないでください)。いくつかの細胞はナノブレードへの長期曝露(24〜48時間)に耐えるが、他の細胞は非常に敏感であり、小さな合胞体を形成する可能性がある。この場合、ナノブレードは、培地を交換する前に4〜6時間だけ細胞とインキュベートしなければならない。Spinoculation14 はまた、懸濁液中で増殖した細胞に対する形質導入を改善することができる。カチオン性ポリマー( 材料表を参照)などのアジュバントもまた、いくつかの細胞型において形質導入効率を改善することができる。 - ナノブレードを含む少量の培地で4~6時間の細胞インキュベーションを行った後、培地の体積を通常の量(12ウェルプレートで作業する場合は1mL)まで増やすか、細胞がVLPに敏感な場合は新鮮な培地と交換してください。
注:ナノブレードを含む細胞培地は、廃棄する前に0.5%次亜塩素酸ナトリウムで10分間不活性化する必要があります。次亜塩素酸ナトリウムを操作するときは、手袋と保護ゴーグルを使用してください。ナノブレードが細胞死を誘導する場合は、曝露の量と合計時間を適応させて細胞死亡率を低下させる。
7. T7エンドヌクレアーゼアッセイによる標的遺伝子座におけるCRISPR効率の測定
- CRISPR切断部位を包含する400~700塩基対(bp)領域を増幅するようにPCRプライマーを設計します。
注:切断部位は、アンプリコン縁から少なくとも200bp離れるべきであり、T7エンドヌクレアーゼ切断時に異なるサイズの2つの断片が放出されるように、アンプリコンの中心からわずかにずらすべきである。 - 目的の遺伝子を標的とするナノブレードで処理した細胞、およびコントロールsgRNAでプログラムしたナノブレードで処理したコントロール細胞からゲノムDNAを抽出します( 材料表を参照)。
メモ: プロトコルはここで一時停止できます。 - 150 ng のゲノム DNA を鋳型として使用し、メーカーのプロトコールに従って 30 μL 容量 (最終容量) の PCR 反応をプログラムします。5 μg/mL のエチジウムブロマイドで染色した 2% アガロースゲル (またはより安全な代替 DNA ゲル染色) を実行して、PCR 増幅で期待されるサイズの単一アンプリコンが得られることを確認します。
注:遺伝的欠陥を引き起こす疑いのある臭化エチジウムを操作するときは、適切な保護具を着用してください。プロトコルはここで一時停止できます。 - ヘテロデュプレックスの生成と消化
- 0.2 mL PCR チューブに、5 μL の酵素バッファー (T7 エンドヌクレアーゼ I を付属)、20 μL の水、およびステップ 7.3 の PCR 産物 24 μL を加えます。サンプルを3分間かけて94°Cに加熱し、次に温度を下げて40°Cに達するまでヘテロ二重鎖の形成を可能にします。
- 対照を含む各ヘテロ二本鎖チューブに室温で0.5 μLのT7-エンドヌクレアーゼIを加える。37°Cで15分間インキュベートする。得られた反応物を、臭化エチジウムで染色した2.5%(重量/体積)のアガロースゲルにロードする。移行後、UVトランスイルミネーターでゲルを画像化します。
注:遺伝的欠陥を引き起こす疑いのある臭化エチジウムを操作するときは、適切な保護具を着用してください。UVトランスイルミネーターを使用する場合は、UV保護ゴーグルを使用してください。 - 消化反応から生じる画像を解析し、適切なソフトウェアで各バンドの強度を定量化することにより、切断効率を測定します( 材料表を参照)。
8. サンガーシーケンシングとTIDE解析による標的遺伝子座でのCRISPR効率の測定
注: T7 エンドヌクレアーゼアッセイの代替として、CRISPR 効率は、TIDE プロトコルに基づくサンガーシーケンシングトレースの分析およびデコンボリューションによって監視できます15。
- ステップ7.3からのPCRアンプリコンのサンガーシークエンシング(未処理細胞に対応する対照条件を含む)をフォワードまたはリバースPCRプライマーのいずれかを用いて行う。
- 対照条件(未処理細胞)およびナノブレード処理サンプルのサンガーシーケンシングトレースを、TIDEサーバー(https://tide.nki.nl)を使用して分析し、それらの分析ガイドラインに従います。
9. 相同性指向修復のためのssODNドナーとのナノブレード複合体形成(12ウェルプレートにおける形質導入手順)
注: 効率的な相同性指向修復媒介編集のための ssODN の設計に関するガイドラインは、以前に説明されています16。
- 12ウェルプレートに、1ウェル当たり100,000〜200,000個の細胞を1mLの適切な細胞培養培地でシードする。形質導入前に細胞がプレート表面に接着するのを許す。
- カチオン性ポリマーの溶液100 μL( 材料表を参照)を1x PBS中に8 μg/mLで調製する。
- 19 μL のカチオン性ポリマー溶液を 100 pmol の ssODN テンプレートと混合します。濃縮ナノブレード20μL(ステップ3.5.1または4.4から)を加え、氷上で15分間インキュベートする。
- 複合化したナノブレード/ssODNを氷から取り出し、500 μLの細胞培養培地(37°C)を加えます。標的細胞から培地を取り出し(ステップ9.1から)、複合化されたナノブレード/ssODNを含む500μLの培地を添加する。ジェノタイピングの前に細胞が48時間増殖するのを許してください。
- 専用の抽出キットを使用して細胞集団の画分からゲノムDNAを抽出する( 材料表を参照)。
- PCRプライマーを設計して、ノックイン部位を包含する400〜700 bp領域を増幅する。
注: PCR プライマーは、標的細胞内にまだ存在する残留 ssODN の PCR 増幅に起因する偽陽性の結果を避けるために、ssODN の相同性アームと重複してはなりません。 - コントロール細胞(未処理)またはナノブレード処理細胞からの150 ngのゲノムDNAを鋳型として使用し、メーカーのプロトコールに従って30 μL PCR反応をプログラムします。
注:ssODNトレースは、複合体との形質導入の数日後に細胞培地中に存在し得る。このssODNは、正しい統合をスクリーニングしようとするPCRアッセイのための部分的なテンプレートとして役立つ可能性がある。したがって、最終的な偽陽性アッセイを避けるために、形質導入後に少なくとも2回細胞を継代することをお勧めします。 - コントロールおよびナノブレード処理したPCR反応5 μLを、臭化エチジウムで染色した1%(重量/容量)のアガロースゲルにロードします。移行後、UVトランスイルミネーターでゲルを画像化します。
注:相同性組換えが成功し、1 bpを超える遺伝物質の挿入に対応する場合、コントロールとナノブレード処理サンプルの間でPCRアンプリコンの分子量に差があるはずです。HDRの効率は100%に達しないため、ナノブレード処理サンプル中に2つのバンドが見えるはずです(1つは未編集対立遺伝子に対応する対照PCRアンプリコンと同様のサイズで、もう1つはノックイン対立遺伝子に対応する高分子量の1つは、 図3B中央パネルを参照)。
- PCRプライマーを設計して、ノックイン部位を包含する400〜700 bp領域を増幅する。
- コントロールおよびナノブレード処理PCRアンプリコンのサンガーシーケンシングを行う。
- TIDERプロトコル17を使用してノックイン効率を定量化する。
10. ナノブレードの生体内送達
- ステップ3.5.1から眼窩後注射で最大25 μL、またはin18で説明したように尾静脈注射で最大100 μLの濃縮ナノブレードをマウスで作業する場合は、送達します。
注:動物実験を含むすべての手順(ゲノム編集目的のナノブレード注射を含む)には、地元の倫理委員会から承認されたプロトコルが必要です。 - トランスジェニックマウスの作製には、マイクロインジェクターを使用して、ステップ4.4からマウス卵母細胞のペリビテリン空間に1pLから10pLの濃縮ナノブレードを送達する18。
注:ペリビテリン注入の場合、マイクロインジェクターの目詰まりを避けるために、ナノブレードを精製してスクロースクッションに集中させることが不可欠です。
結果
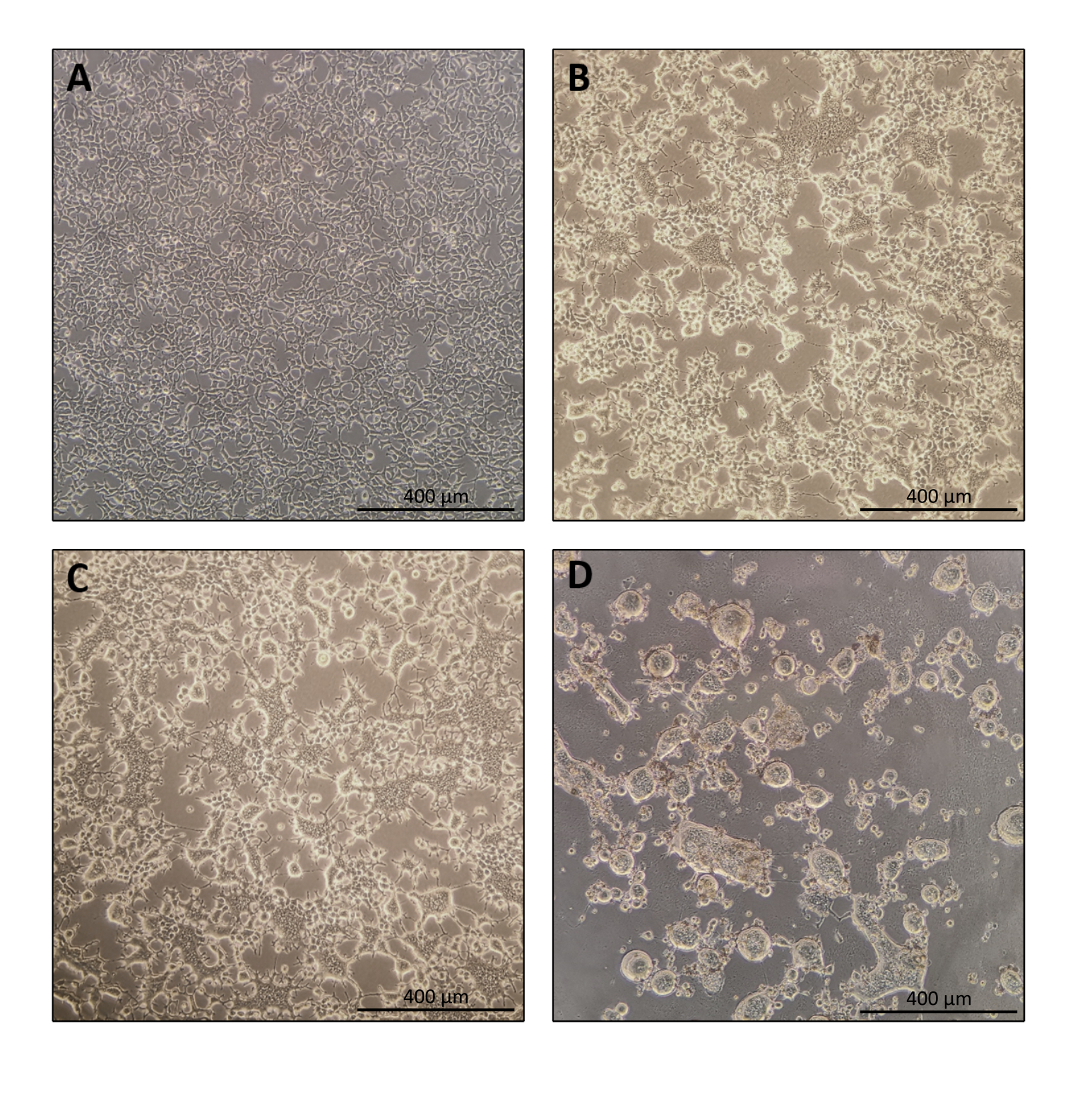
ナノブレード調製のプロトコルは非常に簡単で、組織培養フード、CO2インキュベーター、スイングバケット遠心分離機または超遠心分離機へのアクセスに加えて、簡単な実験装置が必要です。しかしながら、いくつかのステップは、産生細胞の供給源および取り扱い、ならびに形質導入条件など、特に注意を必要とする。図 1A に示すように、細胞をプレート内に均質に分布させ、トランスフェクション当日に約 70 ~ 80% の合流点に達するように細胞をシードすることが重要です (細胞の凝集塊は避けてください)。トランスフェクションの24時間後(図1B、C)、産生細胞は合胞体を形成し、複数の核を持つより大きなサイズの細胞につながります。トランスフェクションの40時間後(図1D)、プレート内のほとんどの細胞はシンシチアを形成し、プレートから剥離し始めます。
これは完全に正常であり、隣接する細胞間の融合を誘導するエンベロープ糖タンパク質の発現によって引き起こされる。遠心分離による濃縮(または生産者細胞の上清からの直接)により、ナノブレード内に装填されたCas9の量は、組換えCas9を基準として使用してニトロセルロース膜上のドットブロットによって絶対的に定量することができる(図2)。このステップは、標的細胞の形質導入に使用するナノブレードの正しい量を決定するために重要です。ドットブロットアッセイを行う場合、標準曲線の直線範囲内にある読み取り値のみを考慮することが重要です。しかし、ナノブレード内に存在するCas9の量とは無関係に、T7エンドヌクレアーゼアッセイ(図3)またはサンガーシーケンシングを使用して、標的細胞上で直接ゲノム編集の効率をテストすることが不可欠です。
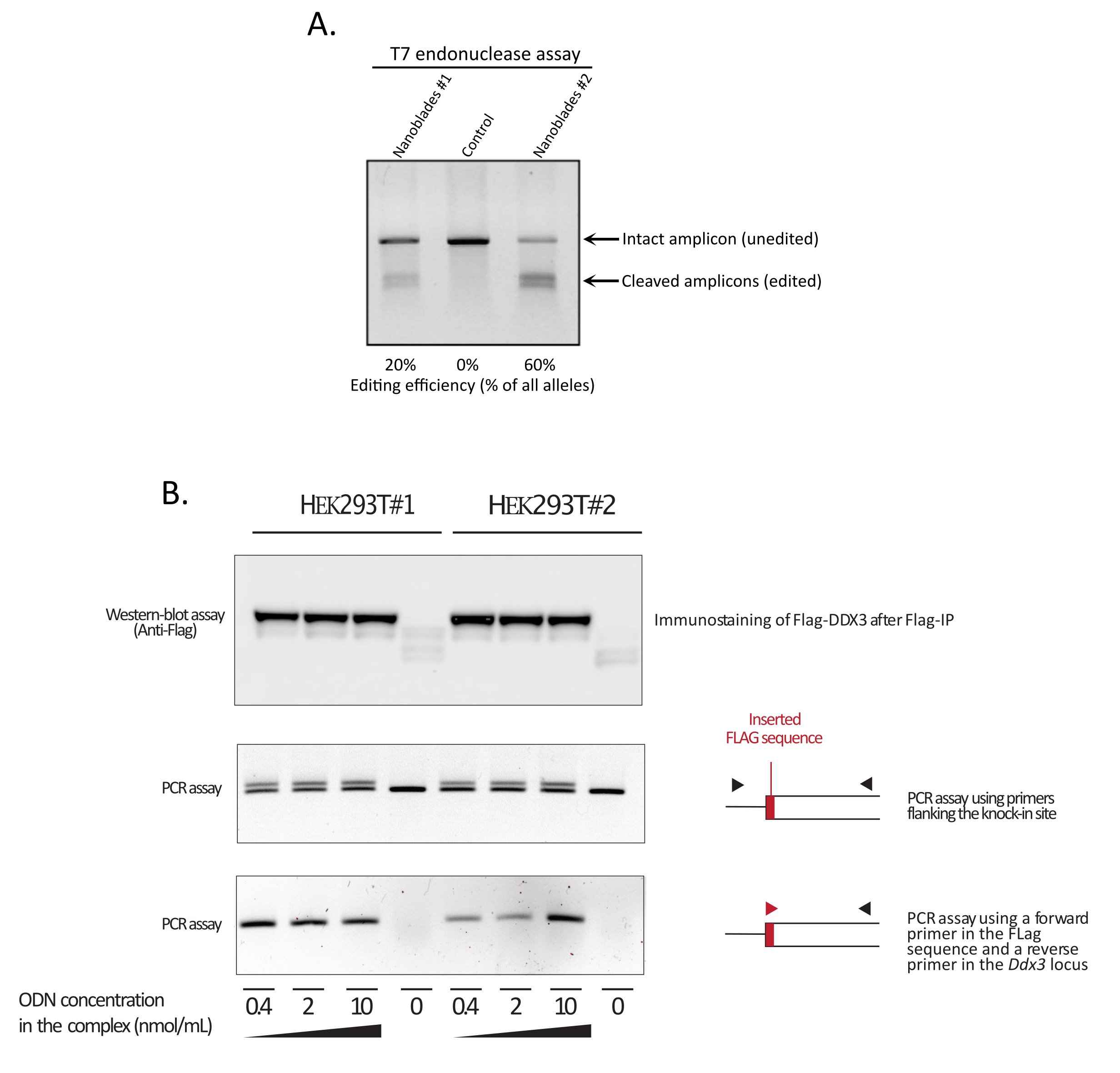
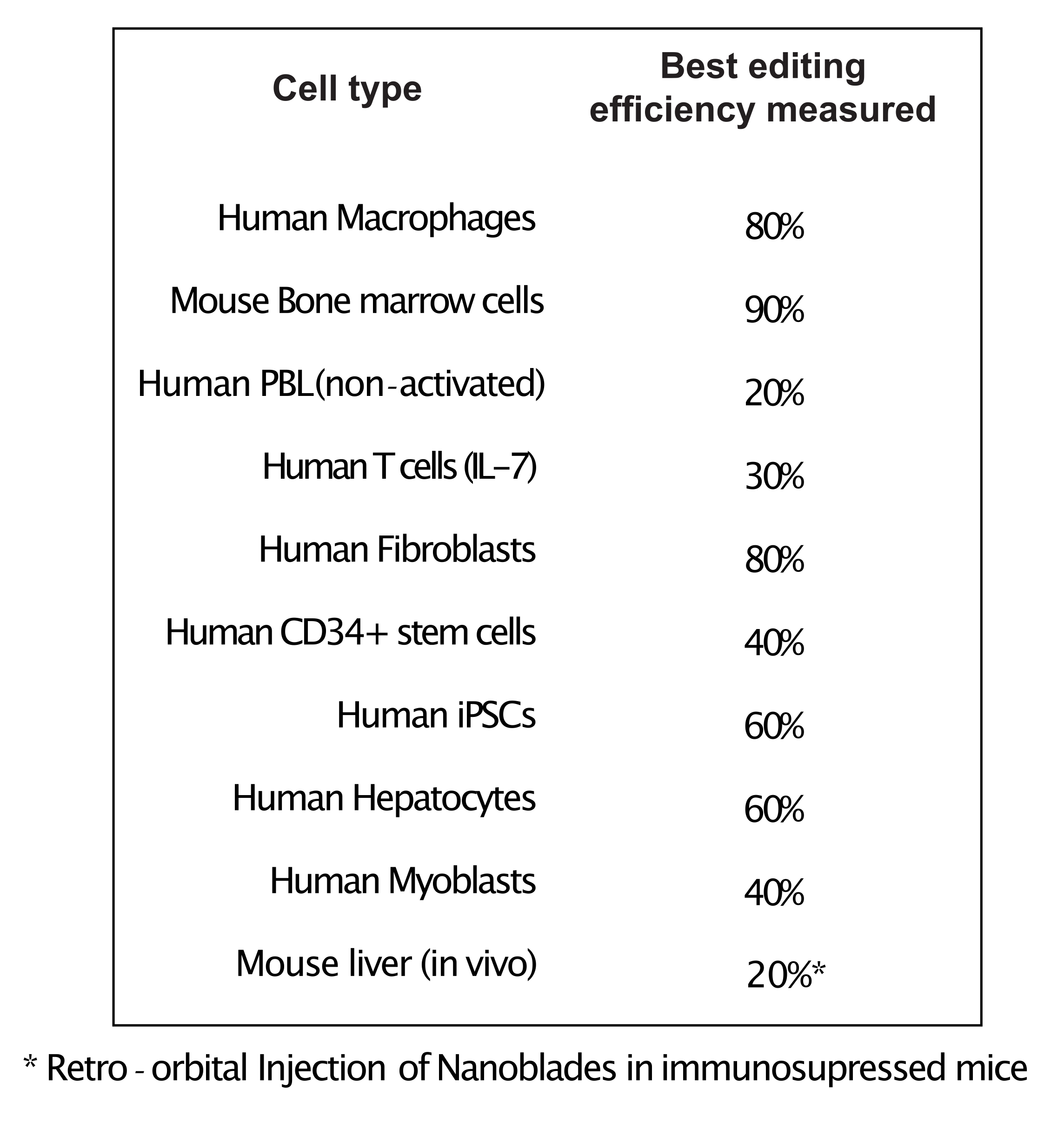
図3に示すように、ナノブレードの効率はバッチごとに異なる可能性がありますが、通常はCas9の量と相関しています。図 3 に示す例では、レーン 1 からのバッチは全体の編集効率が 20% になり、レーン 3 からのバッチは 60% の効率になります。この場合、バッチ1から使用されるナノブレードの体積を増やして、バッチ3からの編集と同様の編集効率を達成することができる。図4は、異なるタイプの初代細胞においてナノブレードを用いて得られた最大編集効率を示す。効率は、使用されるsgRNAの配列および標的のアクセシビリティに応じて変化し得ることに注意することが重要です。

図1:ナノブレード生産中の産生細胞の形態 。 (A)めっき後24時間で70〜80%コンフルエントでのHEK293T細胞。(B および C)トランスフェクション後24時間後のHEK293T細胞形態。(d)トランスフェクション後40時間後のHEK293T細胞形態。スケール バー = 400 μm。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

(A)組換えCas9または100倍濃縮(超遠心分離による)ナノブレードサンプル(#1、#2、および#3)は、2倍順次希釈され、抗Cas9 HRP結合抗体とインキュベートされる前にニトロセルロース膜上にスポットされる。シグナルは、増強された化学発光によって明らかにされる。(b)化学発光シグナルを取得し、ニトロセルロース膜上にスポットした既知量のCas9に対してプロットした組換えCas9希釈物およびシグナル強度について定量化する。回帰曲線は、線形範囲内にある希釈(青い十字を参照)について、線形範囲外にあるすべての濃度(赤い十字を参照)を除いて計算されます。(c)各ナノブレード調製物におけるCas9濃度(nM)は、(B)で得られた線形回帰から式を用いて外挿した。このためには、回帰曲線の線形範囲内にあるナノブレード希釈からの定量化された信号のみを使用することが重要です。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:形質導入時の編集効率のモニタリング。 (A)ナノブレード処理細胞における切断効率を測定するT7エンドヌクレアーゼアッセイ。 EMX1 遺伝子を標的とするナノブレードで形質導入された細胞を、T7エンドヌクレアーゼアッセイによって分析した。レーン1:ナノブレード調製バッチ#1(20%の切断効率);レーン2:コントロールセル;レーン3:ナノブレード調製バッチ#2(60%の切断効率)。(B) DDX3 オープンリーディングフレーム内のフラグタグシーケンスのノックイン。 DDX3 遺伝子座を標的とするsgRNAでプログラムされた濃縮ナノブレードを、異なるHEK293Tクローン(#1、#2)から作製し、Flag-DDX3 ssODNテンプレートの用量の増加と、HEK293T標的細胞の形質導入に使用される得られた複合体とを複合化した。形質導入時に、細胞を3日間増殖させてから、それらを収集してゲノムDNAおよび総タンパク質を抽出した。Flag-DDX3タンパク質を抗Flagアガロースビーズを用いて免疫沈降させ、続いて抗Flag抗体を用いて回収したタンパク質のウェスタンブロット解析を行った(トップパネル)。 Ddx3 遺伝子座におけるFlagタグの部位特異的挿入も、挿入部位に隣接するプライマー(中央パネル)、またはFlagタグ配列を認識するフォワードプライマーおよびFlag挿入部位の下流の Ddx3 遺伝子座に特異的なリバースプライマー(下部パネル)を用いたPCRによってアッセイされた。略語: EMX1 = 空のスパイラクルホメオボックス 1;DDX3 = デッドボックスRNAヘリカーゼ3;PCR = ポリメラーゼ連鎖反応;ODN = オリゴデオキシヌクレオチド;ssODN = 独り立ち ODN;sgRNA = シングルガイドRNA;IP = 免疫沈降。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図4:ナノブレードを使用して、異なる一次細胞タイプで達成された編集効率。 略語:PBL =末梢血リンパ球;IL = インターロイキン;CD = 分化のクラスター;iPSC = 人工多能性幹細胞。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
ディスカッション
ナノブレードは、細胞株および初代細胞におけるCas9/sgRNA RNP複合体の迅速かつ用量依存的な送達を可能にする。従来のトランスフェクションおよび他のウイルス送達ベクターとは対照的に、タンパク質エレクトロポレーションと同様に、ナノブレードは、導入遺伝子を含まない方法でCas9/sgRNA RNAを一過性送達するという利点を有する。ナノブレードは、拡大し続けるCRISPRバリアントファミリーに簡単かつ迅速に適合させることができる、タンパク質送達のための非常に汎用性が高く、シンプルで安価なプラットフォームを提供します。ナノブレードは、HEK293T細胞株またはその誘導体において製造することができる。ここで使用されるHEK293T細胞株は、レトロウイルスおよびレンチウイルス粒子産生を最大化するために開発されている( 材料表を参照のこと)。しかしながら、HEK293T細胞の他の供給源が適しているかもしれないが、HEK293T細胞源によって粒子産生における大きな違いが観察されているため、ユーザーは異なる供給源からのHEK293T細胞を試験および比較しなければならない。また、細胞はマイコプラズマ汚染を頻繁にチェックし、粒子産生に悪影響を及ぼす過剰合流を避けるために3日ごとに継代(古典的には1/8希釈)する必要があります。
細胞は20回以上の継代にわたって維持されるべきではない。グルコース、ペニシリン/ストレプトマイシン、グルタミン、および10%脱相補ウシ胎児血清を添加したDMEMを細胞培養に使用した。血清起源はナノブレード製剤の品質に影響を与える可能性があるため、大規模生産の前に異なるバッチの血清を試験する必要があります。ナノブレードは、RPMIなどの他の培地、またはトランスフェクションの翌日にDMEMを置き換えることができる最小限の必須培地の無血清修飾で効率的に製造することができます。以下に示すように、トランスフェクション後の培地置換はいくつかのDNAトランスフェクション試薬による任意であるが、VLPが放出される培地を改変することは、特に粒子調製物中の血清微量を制限するために有益であり得る。しかしながら、トランスフェクションの前日に低血清最小必須培地で細胞を培養することは未だ試みられていない。
前述のように、ナノブレードは、産生細胞におけるプラスミドの混合物の過剰発現時に産生される。過剰発現は、最適な生産のために必要であるように思われる。実際、この研究室では、Gag-Pol発現コンストラクトが抗生物質選択によって安定化された産生細胞株を開発しました。しかし、このシステムは大量のナノブレードを生産できませんでした。sgRNAコード構築物が産生細胞のゲノムに安定に組み込まれた場合にも同様の観察が行われた。他の粒子産生系について記載されるように、ナノブレード産生に関与する少なくともいくつかの構築物を発現する安定な細胞株が有用であり得る;しかし、これには確かに大量の上清の処理と粒子を精製するための適切な技術が必要です。上記のプロトコルは、特定のトランスフェクション試薬を利用するナノブレードを製造するための好ましい手順を概説しています( 材料表を参照)。
他のメーカーのトランスフェクション試薬も成功裏にテストされていますが、ナノブレードを使用したこのグループの結果の大部分は、ここに記載されている手順に従います。リン酸カルシウム試薬を使用して低コストのトランスフェクションを達成し、良好な生産効率を得ることができます。しかしながら、この方法は、トランスフェクションの翌日にトランスフェクション培地の交換を絶対に必要とし、沈降粒子調製物中にリン酸カルシウム残基を残す可能性がある。産生細胞内でのナノブレード成分の高発現レベルの必要性と一致するのは、Cas9タンパク質に関連するsgRNAの量が効率的なゲノム編集のための制限因子となり得るという観察である。sgRNAローディングを改善するために、ナノブレードに類似したタンパク質送達ベクターを使用する独立したグループによって、2つの技術的アプローチが最近開発されている。これらは、sgRNA6のT7ポリメラーゼ依存性細胞質発現の使用、またはGagポリタンパク質6への結合を媒介するsgRNA配列へのレトロウイルス捕捉シグナルの付加に依存している。これらのアプローチは、ナノブレード内のsgRNAローディングを実際に改善する可能性がありますが、まだテストされていません。
標的細胞の形質導入は、この手順における重要なステップである。ほとんどの不死化細胞株において、ナノブレードによる形質導入は、細胞変性効果をほとんどまたは全く有さない。しかし、初代細胞では、毒性が問題になる可能性があります。したがって、形質導入は細胞タイプごとに最適化されなければならない。具体的には、ナノブレードへの曝露時間は、形質導入プロトコルを最適化する際に変更する重要な要素である。初代ニューロンや骨髄細胞などの敏感な細胞の場合、培地を交換する前にNanobladesで4〜6時間インキュベーションすることで、細胞毒性を最小限に抑えながらCas9タンパク質を効率的に送達できます。さらに、カチオン性ポリマーなどのアジュバントは、とりわけ、いくつかの細胞における形質導入の効率を有意に改善することができる( 材料表を参照のこと)。ナノブレードはVLPであり、免疫原性応答を誘導することができることに注意することが重要です。これは、マクロファージや樹状細胞などの特定のタイプの初代細胞で作業する場合、ナノブレードとのインキュベーションが遺伝子発現および細胞の表現型に重要な変化を誘導する場合、制限となり得る。マクロファージおよび樹状細胞が造血幹細胞前駆体(マウス骨髄細胞など)に由来する場合、ナノブレードに対する細胞応答を誘導することを避けるために、細胞が完全に分化する前にナノブレードで形質導入することが好ましい。さもなければ、Cas9タンパク質エレクトロポレーションは、分化した免疫細胞を扱う際に実行可能な代替手段を表すことができる。
ナノブレードは、インビボでマウス接合体または胚を形質導入してトランスジェニック動物を生成するために使用することができる。古典的なレトロウイルスまたはレンチウイルスベクターと同様に、成体動物由来の組織に直接注射することもできる。しかしながら、ナノブレード(レトロウイルスおよびレンチウイルスベクターに類似)は、宿主動物の免疫応答によって不活性化され得る。したがって、注射する用量は、アプリケーションごとに最適化する必要があります。この免疫応答はまた、注射部位に近い組織への機能的VLPの分布を制限することができる。最後に、レンチウイルスベクターとは異なり、ナノブレードは導入遺伝子フリーであり、限られた時間枠でCas9を送達する。したがって、細胞の選択時にsgRNAのハイスループットシーケンシングを必要とするゲノムワイドな機能スクリーニングの実行には使用できません。ナノブレードは、迅速で用量依存的で、導入遺伝子を含まないゲノム編集が必要な場合に有用です20。さらに、タンパク質エレクトロポレーションと同様に、ナノブレードは、DNAトランスフェクションまたは古典的なウイルスベクターを介したCas9/sgRNAの長期発現よりもオフターゲット効果が少なくなります3。ナノブレードの将来の開発は、塩基編集やRNAターゲティングなどのさまざまな技術アプリケーション向けにCas9バリアントを組み込むことに焦点を当てています。
開示事項
Philippe E. MangeotとEmiliano P. Ricciは、ナノブレード技術に関する特許の発明者として指名されています(特許出願人:国立サンテ研究所(INSERM)、国立科学研究センター(CNRS)、リヨンノルマルスーパーリュール、クロードベルナールリヨン1大学、ヴィルルバンヌセデックス、出願番号:WO 2017/068077 Al、特許ステータス: 2017年4月27日発行。原稿のすべての側面は特許出願によってカバーされています。残りの著者は、競合する利益を宣言していません。
謝辞
この研究は、フランス国立研究庁(ANR)、フィノヴィ財団、Agence Nationale des Recherches sur SIDA et les Hépatites Viloles(ANRS-ECTZ3306)、および欧州連合のHorizon 2020研究およびイノベーションプログラムの下での欧州研究評議会(ERC-StG-LS6-805500からE.P.R.)が運営するプログラムInvestissements d'Avenir(ANR-11-IDEX-0007)内のLabex Ecofect(ANR-11-LABX-0048)によって資金提供されました。
資料
| Name | Company | Catalog Number | Comments |
| 13.2 mL, Thinwall Polypropylene Tubes, 14 x 89 mm - 50Pk | Beckman Coulter Life Sciences | 331372 | Ultracentrifugation tubes for Nanoblades purification |
| Amersham Protran Premium Western blotting membranes, nitrocellulose | Merck | GE10600004 | Nitrocellulose membrane for quantifying Cas9 levels within purified Nanoblades |
| BIC-Gag-CAS9 | Addgene | 119942 | Encodes a GAG (F-MLV)-CAS9(sp) fusion. Allows the production of GAG-CAS9 Virus like particles from producer cells in association with over expressed gRNA(s) and appropriate envelopes |
| BICstim-Gag-dCAS9-VPR | Addgene | 120922 | Encodes a GAG-dCAS9-VPR fusion for targeted transcriptional activation |
| BLADE | Addgene | 134912 | Empty backbone for cloning sgRNA sequence to be used in Nanoblades system |
| BsmBI-v2 | New England Biolabs | R0739S | Restriction enzyme to digest the BLADE and SUPERBLADES vectors for sgRNA cloning |
| Cas9 (7A9-3A3) Mouse mAb (HRP Conjugate) #97982 | Cell Signaling Technology | 97982S | Anti-Cas9 antibody for Cas9 quantification by dot-blot |
| Cas9 Nuclease, S. pyogenes | New England Biolabs | M0386T | Recombinant Cas9 protein to be used as a reference for absolute quantification of the amount of Cas9 loaded within Nanoblades |
| Ethidium bromide solution (10 mg/mL in H2O) | Sigma-Aldrich | E1510-10ML | For staining agarose gels and visualize DNA |
| Fisherbrand Wave Motion Shakers | Fisher Scientific | 88-861-028 | Agitation table to resuspend Nanoblades upon centrifugation |
| gelAnalyzer | http://www.gelanalyzer.com; quantifying band intensity after digestion | ||
| Gesicle Producer 293T | Takara | 632617 | Nanoblades producer cell line |
| Gibco DMEM, high glucose, pyruvate | ThermoFisher Scientific | 41966052 | Cell culture medium for Gesicle Producer 293T cells |
| GoTaq G2 DNA Polymerase | Promega | M7848 | Taq polymerase for amplification of genomic DNA before T7 endonuclease assays |
| jetPRIME Transfection Reagent kit for DNA and DNA/siRNA | Polyplus | POL114-15 | Transfection reagent for Nanoblade production in Gesicle Producer 293T cells |
| Millex-AA, 0.80 µm, syringe filter | Millipore | SLAA033SS | Syringe filter to remove cellular debris before concentration of Nanoblades |
| Millex-GS, 0.22 µm, syringe filter | Millipore | SLGS033SS | Syringe filter to sterilise the sucrose cushion solution |
| Millex-HP, 0.45 µm, polyethersulfone, syringe filter | Millipore | SLHP033RS | Syringe filter to remove cellular debris before Nanoblades concentration |
| Monarch DNA Gel Extraction Kit | New England Biolabs | T1020L | DNA gel extraction kit for purification of the pBLADES or pSUPERBLADES plasmid fragment upon digestion with BsmBI |
| NEB Stable Competent E. coli (High Efficiency) | New England Biolabs | C3040I | Competent bacteria for plasmid transformation and amplification |
| NucleoBond Xtra Midi kit for transfection-grade plasmid DNA | Macherey-Nagel | 740410.50 | Maxipreparation kit for purification of plasmid DNA from cultured bacteria |
| Nucleospin gDNA extraction kit | Macherey-Nagel | 740952.50 | Extraction of genomic DNA from transduced cells |
| NucleoSpin Plasmid, Mini kit for plasmid DNA | Macherey-Nagel | 740588.50 | Minipreparation kit for purification of plasmid DNA from cultured bacteria |
| NucleoSpin Tissue, Mini kit for DNA from cells and tissue | Macherey-Nagel | 740952.5 | Genomic DNA extraction kit |
| Optima XE-90 | Beckman Coulter Life Sciences | A94471 | Ultracentrifuge |
| pBaEVRless | Els Verhoeyen (Inserm U1111) | Personnal requests have to be sent to: els.verhoyen@ens-lyon.fr | Baboon Endogenous retrovirus Rless glycoprotein described in Girard-Gagnepain, A. et al. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood 124, 1221–1231 (2014) |
| pBS-CMV-gagpol | Addgene | 35614 | Enocdes the Murine Leukemia Virus gag and pol genes |
| pCMV-VSV-G | Addgene | 8454 | Envelope protein for producing lentiviral and MuLV retroviral particles |
| Phosphate-Buffered Saline (PBS) | ThermoFisher Scientific | 14200091 | 10X PBS to dilute in millipore water |
| Polybrene Transfection Reagent | Millipore Sigma | TR-1003-G | Cationic polymer that enhances the efficiency of retroviral transduction in specific mammalian cells. It can also allow viral-dependent entry of an Oligodeoxynucleotide (ODN) for homology-directed repair |
| Sucrose,for molecular biology, ≥99.5% (GC) | Sigma-Aldrich | S0389-5KG | Sucrose to prepare a cushion for Nanoblade purification through ultracentrifugation |
| SUPERBLADE5 | Addgene | 134913 | Empty backbone for cloning sgRNA sequence to be used in nanoblades system (Optimized for increased genome editing efficiency via Chen B et al., 2013) |
| SuperSignal West Dura Extended Duration Substrate | ThermoFisher Scientific | 34076 | Enhanced chemiluminescence (ECL) HRP substrate for Cas9 dot blots |
| SW 41 Ti Swinging-Bucket Rotor | Beckman Coulter Life Sciences | 331362 | Rotor for ultracentrifugation |
| SYBR Safe DNA Gel Stain | ThermoFisher Scientific | S33102 | Alternative to ethidium bromide for staining agarose gels and visualize DNA |
| T4 DNA Ligase | New England Biolabs | M0202S | DNA ligase to ligate the BLADE or SUPERBLADES vectors with the duplexed DNA oligos corresponding to the variable region of the sgRNA |
| T7 Endonuclease I | New England Biolabs | M0302S | T7 Endonuclease I recognizes and cleaves non-perfectly matched DNA. Allows to monitor the extent of genome editing at a specific locus |
| Triton-containing lysis buffer | Promega | E291A | Lysis buffer to disrupt Nanoblades and allow Cas9 quantification |
| TWEEN 20 | Sigma-Aldrich | P9416 | For the preparation of TBST |
参考文献
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Zuris, J. A., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome-editing in vitro and in vivo. Nature Biotechnology. 33, 73-80 (2015).
- Mangeot, P. E., et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nature Communications. 10 (1), 45 (2019).
- Cai, Y., Bak, R. O., Mikkelsen, J. G. Targeted genome editing by lentiviral protein transduction of zinc-finger and TAL-effector nucleases. eLife. 3, 01911 (2014).
- Choi, J. G., et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Therapy. 23 (7), 627-633 (2016).
- Gee, P., et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nature Communications. 11, 1334 (2020).
- Indikova, I., Indik, S. Highly efficient 'hit-and-run' genome editing with unconcentrated lentivectors carrying Vpr.Prot.Cas9 protein produced from RRE-containing transcripts. Nucleic Acids Research. 48 (14), 8178-8187 (2020).
- Lyu, P., Javidi-Parsijani, P., Atala, A., Lu, B. Delivering Cas9/sgRNA ribonucleoprotein (RNP) by lentiviral capsid-based bionanoparticles for efficient 'hit-and-run' genome editing. Nucleic Acids Research. 47 (17), 99 (2019).
- Gheysen, D., Jacobs, E., de Foresta, F., Thiriart, C. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell. 59 (1), 103-112 (1989).
- Hanna, R. E., Doench, J. G. Design and analysis of CRISPR-Cas experiments. Nature Biotechnology. 38 (7), 813-823 (2020).
- Sambrook, J. . Molecular cloning a laboratory manual. Third edition. , (2001).
- Jiang, W., et al. An optimized method for high-titer lentivirus preparations without ultracentrifugation. Scientific Reports. 5, 13875 (2015).
- Rueden, C. T., et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics. 18 (1), 529 (2017).
- O'Doherty, U., Swiggard, W. J., Malim, M. H. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. Journal of Virology. 74 (21), 10074-10080 (2000).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Bollen, Y., Post, J., Koo, B. -. K., Snippert, H. J. G. How to create state-of-the-art genetic model systems: strategies for optimal CRISPR-mediated genome editing. Nucleic Acids Research. 46 (13), 6435-6454 (2018).
- Brinkman, E. K., et al. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Research. 46 (10), 58 (2018).
- Dussaud, S., Pardanaud-Glavieux, C., Sauty-Colace, C., Ravassard, P. Lentiviral mediated production of transgenic mice: a simple and highly efficient method for direct study of founders. Journal of Visualized Experiments. (140), e57609 (2018).
- Montagna, C., et al. VSV-G-enveloped vesicles for traceless delivery of CRISPR-Cas9. Molecular Therapy. Nucleic Acids. 12, 453-462 (2018).
- Marnef, A., et al. A cohesin/HUSH- and LINC-dependent pathway controls ribosomal DNA double-strand break repair. Genes & Development. 33 (17-18), 1175-1190 (2019).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved