
Method Article
Delivery of the Cas9/sgRNA Ribonucleoprotein Complex in Immortalized and Primary Cells via Virus-like Particles ("Nanoblades")
In This Article
Summary
We have developed a simple and inexpensive protocol to load Cas9/single-guide RNA (sgRNA) ribonucleoprotein complexes within virus-like particles. These particles, called "Nanoblades", allow efficient delivery of the Cas9/sgRNA complex in immortalized and primary cells as well as in vivo.
Abstract
The clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system has democratized genome-editing in eukaryotic cells and led to the development of numerous innovative applications. However, delivery of the Cas9 protein and single-guide RNA (sgRNA) into target cells can be technically challenge. Classical viral vectors, such as those derived from lentiviruses (LVs) or adeno-associated viruses (AAVs), allow for efficient delivery of transgenes coding for the Cas9 protein and its associated sgRNA in many primary cells and in vivo. Nevertheless, these vectors can suffer from drawbacks such as integration of the transgene in the target cell genome, a limited cargo capacity, and long-term expression of the Cas9 protein and guide RNA in target cells.
To overcome some of these problems, a delivery vector based on the murine Leukemia virus (MLV) was developed to package the Cas9 protein and its associated guide RNA in the absence of any coding transgene. By fusing the Cas9 protein to the C-terminus of the structural protein Gag from MLV, virus-like particles (VLPs) loaded with the Cas9 protein and sgRNA (named "Nanoblades") were formed. Nanoblades can be collected from the culture medium of producer cells, purified, quantified, and used to transduce target cells and deliver the active Cas9/sgRNA complex. Nanoblades deliver their ribonucleoprotein (RNP) cargo transiently and rapidly in a wide range of primary and immortalized cells and can be programmed for other applications, such as transient transcriptional activation of targeted genes, using modified Cas9 proteins. Nanoblades are capable of in vivo genome-editing in the liver of injected adult mice and in oocytes to generate transgenic animals. Finally, they can be complexed with donor DNA for "transfection-free" homology-directed repair. Nanoblade preparation is simple, relatively low-cost, and can be easily carried out in any cell biology laboratory.
Introduction
Compared to other programmable nucleases, the CRISPR-Cas system dramatically simplified and democratized the procedure of sequence-specific genome targeting and cleavage in eukaryotic cells. Through the simple expression of a sgRNA, users can program the Cas9 protein (or optimized variants) for almost any cellular locus1. In this scenario, delivery of the Cas9 protein and sgRNA becomes the main limitation when performing site-directed mutagenesis. In immortalized cells, the sgRNA and the Cas protein can be easily expressed from transfected plasmids to achieve efficient genome targeting in most cells. However, constitutive expression of the Cas9/sgRNA complex can increase off-target activity of the Cas9 protein and introduce undesired changes in non-specific loci2. In primary cells, DNA transfection can be technically difficult to achieve and lead to poor expression or a small percentage of transfected cells. Alternatives to classic DNA transfection comprise the use of viral vectors that deliver a transgene coding for the Cas9 and sgRNA or the electroporation of recombinant Cas9 protein coupled to a synthetic sgRNA. However, these approaches can lead to transgene integration within the cell host genome (as is the case for classical retroviral and lentiviral expression vectors), restriction by cellular factors, and lead to constitutive expression of the Cas9 protein and sgRNA.
Electroporation of the Cas9/sgRNA RNP complex can overcome most of these problems and lead to efficient and transient delivery in primary cells and in vivo as well as allow a dose-dependent response. Nevertheless, it usually relies on expensive equipment and reagents and is also difficult to upscale if a large number of cells have to be treated. As an alternative to the above-mentioned techniques, these authors have developed "Nanoblades"-a retroviral delivery vector for the Cas9 protein and sgRNA3 that is conceptually similar to other viral-derived capsid protein delivery systems4,5,6,7,8. Nanoblades exploit the natural capacity of the Gag polyprotein from retroviruses to produce, when expressed alone in cultured cells, VLPs that are released in the extracellular medium9. By fusing the Cas9 protein to the C-terminal end of the murine leukemia virus (MLV) Gag polyprotein and co-expressing the sgRNA and viral envelope glycoproteins, the Cas9 protein can be encapsidated within released VLPs or Nanoblades. Upon purification, the Nanoblades can be incubated with target cells or injected in vivo to mediate rapid, transient, and dose-dependent delivery of the Cas9/sgRNA RNP complex3.
Nanoblades can be programmed with multiple sgRNAs for simultaneous editing at different loci or with Cas9 variants to perform other applications such as target-specific transcriptional activation or repression3. In contrast to protein electroporation, which relies on recombinant expression, newly described Cas variants from the literature can be easily cloned into the Gag fusion expression vector, making it a versatile platform. Nanoblades can be further complexed or loaded with single-stranded and double-stranded oligodeoxynucleotides (ssODNs) to perform homology-directed repair3. Nanoblade production is relatively simple and cheap. Moreover, Nanoblades can be stored at 4 °C for many days or at -80 °C for long-term storage. Typically, Nanoblades mediate efficient, transgene-free genome-editing in most immortalized and primary cultured cells. However, some primary cells might be sensitive to the presence of viral particles, resulting in increased mortality. Cells of the innate immune system can also react to the presence of Nanoblades (because of their viral origin) and become activated. In these cases, the transduction protocol has to be optimized to limit the exposure time to Nanoblades and minimize nonspecific effects. Nanoblades represent a viable and easy-to-implement alternative to other available CRISPR delivery methods.
Protocol
1. sgRNA Design and cloning
NOTE: Guidelines for the design of sgRNAs can be obtained from multiple sources such as https://blog.addgene.org/how-to-design-your-grna-for-crispr-genome-editing or from Hanna and Doench10.
- Once the 20 nucleotide sgRNA sequences have been designed, order the following single-stranded DNA oligonucleotides:
- Forward: 5' caccgNNNNNNNNNNNNNNNNNNNN 3' (N corresponds to the targeted locus without the protospacer-adjacent motif (PAM) sequence)
- Reverse: 5' aaacNNNNNNNNNNNNNNNNNNNNc 3' (N correspond to the reverse-complement of the targeted locus without the PAM sequence)
NOTE: No special modifications are required when ordering the oligonucleotides (no requirement for 5' phosphate).
- Hybridize the two DNA oligonucleotides in a 0.2 mL polymerase chain reaction (PCR) tube by mixing 5 µL of annealing buffer (500 mM NaCl; 100 mM Tris-HCl; 100 mM MgCl2; 10 mM DTT; pH 7.9 at 25 °C), 1 µL of each DNA oligonucleotide (100 µM stock solution in water), and 42 µL of water.
- On a PCR block, incubate samples at 95 °C for 15 s and then decrease the temperature to 20 °C with a ramp of 0.5 °C/s. Keep at room temperature or store at -20 °C.
NOTE: The protocol can be paused here. - Digest 10 µg of the BLADE or SUPERBLADE sgRNA expression plasmids with 10 units of BsmBI-v2 restriction enzyme for 3 h at 55 °C in a total reaction volume of 50 µL.
NOTE: The digested vector should release a DNA insert of ~1.9 kb and a second DNA fragment of ~3.3 kb. - Load the restriction reaction on a 1% agarose gel stained with 5 µg/mL of ethidium bromide (or a safer alternative DNA gel stain).
NOTE: Wear appropriate protection gear when manipulating ethidium bromide, which is suspected of causing genetic defects.- On an ultraviolet (UV) table set at a wavelength of 312 nm (to avoid damaging the DNA), cut the 3.3 kb DNA fragment from the gel, and place it in a 1.5 mL microcentrifuge tube.
NOTE: Wear appropriate protection gear (gloves and UV protection goggles) when manipulating ethidium bromide and working on the UV table. - Extract DNA from the sliced gel containing the 3.3 kb DNA fragment using a dedicated DNA gel extraction kit (see the Table of Materials). Quantify the amount of purified DNA using a spectrophotometer.
NOTE: The protocol can be paused here.
- On an ultraviolet (UV) table set at a wavelength of 312 nm (to avoid damaging the DNA), cut the 3.3 kb DNA fragment from the gel, and place it in a 1.5 mL microcentrifuge tube.
- Ligate the hybridized forward and reverse DNA oligonucleotides from step 1.2 to the BsmB1-digested, gel-purified BLADES or SUPERBLADE vector from step 1.5.2. For this, add 2 µL of T4 DNA ligase buffer, 50 ng of the gel-purified vector (from step 1.5.2), 1 µL of the hybridized DNA oligonucleotides (from step 1.2), water to make up the volume to 19 µL, and 1 µL of T4 DNA ligase. Incubate the reaction at 25 °C for 10 min.
- Transform the ligation product into competent bacteria (see the Table of Materials) as described in11. Plate the transformed bacteria on an ampicillin Luria Bertani agar plate and incubate overnight at 37 °C.
- Select several isolated colonies on the agar plate to perform DNA minipreparation11 (see the Table of Materials), and perform Sanger sequencing using a U6 forward primer (5' GACTATCATATGCTTACCGT 3') to check for correct ligation of the sgRNA variable sequence.
NOTE: Other sgRNA expression plasmids can be used if they do not code for the Cas9 protein, which could interfere with Nanoblade production.
2. Plasmid preparation
- Perform maxipreparation (see the Table of Materials) of all required plasmids, and prepare 10 µg aliquots at 1 µg/mL to store at -20 °C. Avoid repeated freeze/thawing cycles of the plasmids; use aliquots twice before discarding them.
3. Nanoblade preparation
- On Day 1, seed between 3.5 and 4 × 106 HEK293T cells (see the Table of Materials) in 10 mL of Dulbecco's modified Eagle medium (DMEM) containing high glucose, sodium pyruvate, L-glutamine, 10% fetal bovine serum (FBS), and penicillin/streptomycin in a 10 cm cell culture dish. Move the 10 cm plate gently backward and forward, then from right to left (repeat this sequence 5x) to distribute cells homogeneously over the culture dish. Incubate cells at 37 °C in a cell incubator with 5% CO2.
NOTE: All procedures related to the handling of cultured cells and Nanoblades should be performed under a cell culture laminar flow hood to avoid their contamination. - Day 2: Plasmid transfection
- Cells should be 70-80% confluent 24 h after plating (Figure 1A). Replace the medium with 10 mL of fresh DMEM containing high glucose, sodium pyruvate, L-glutamine, 10% FBS (penicillin and streptomycin can be omitted although it is not mandatory) before transfection.
NOTE: At this step, it is important that the cells are not confluent. Otherwise, transfection efficiency as well as particle production could be reduced. - For each 10 cm plate, prepare the following quantities of plasmids in a 1.5 mL tube: 0.3 µg pCMV-VSV-G, 0.7 µg pBaEVRless, 2.7 µg MLV Gag/Pol, 1.7 µg BIC-Gag-Cas9, 4.4 µg of BLADES or SUPERBLADES plasmid encoding the cloned sgRNA (or 2.2 µg each if using two sgRNAs).
- Add 500 µL of transfection buffer (see the Table of Materials), vortex for 10 s, and then centrifuge for 1 s. Add 20 µL of the transfection reagent (see the Table of Materials), vortex the tube for 1 s, and then centrifuge for 1 s.
- Incubate for 10 min at room temperature, and add the entire solution dropwise to the cells in DMEM medium using a P1000 pipettor. Move the 10 cm plate gently backward and forward, then from right to left (repeat this sequence 5x) to uniformly distribute the transfection reagent over the cells. Incubate cells at 37 °C for at least 40 h in a cell incubator with 5% CO2.
NOTE: If desired, medium can be changed 4 h after transfection.
- Cells should be 70-80% confluent 24 h after plating (Figure 1A). Replace the medium with 10 mL of fresh DMEM containing high glucose, sodium pyruvate, L-glutamine, 10% FBS (penicillin and streptomycin can be omitted although it is not mandatory) before transfection.
- On Day 3, check the morphology of the transfected cells under the microscope.
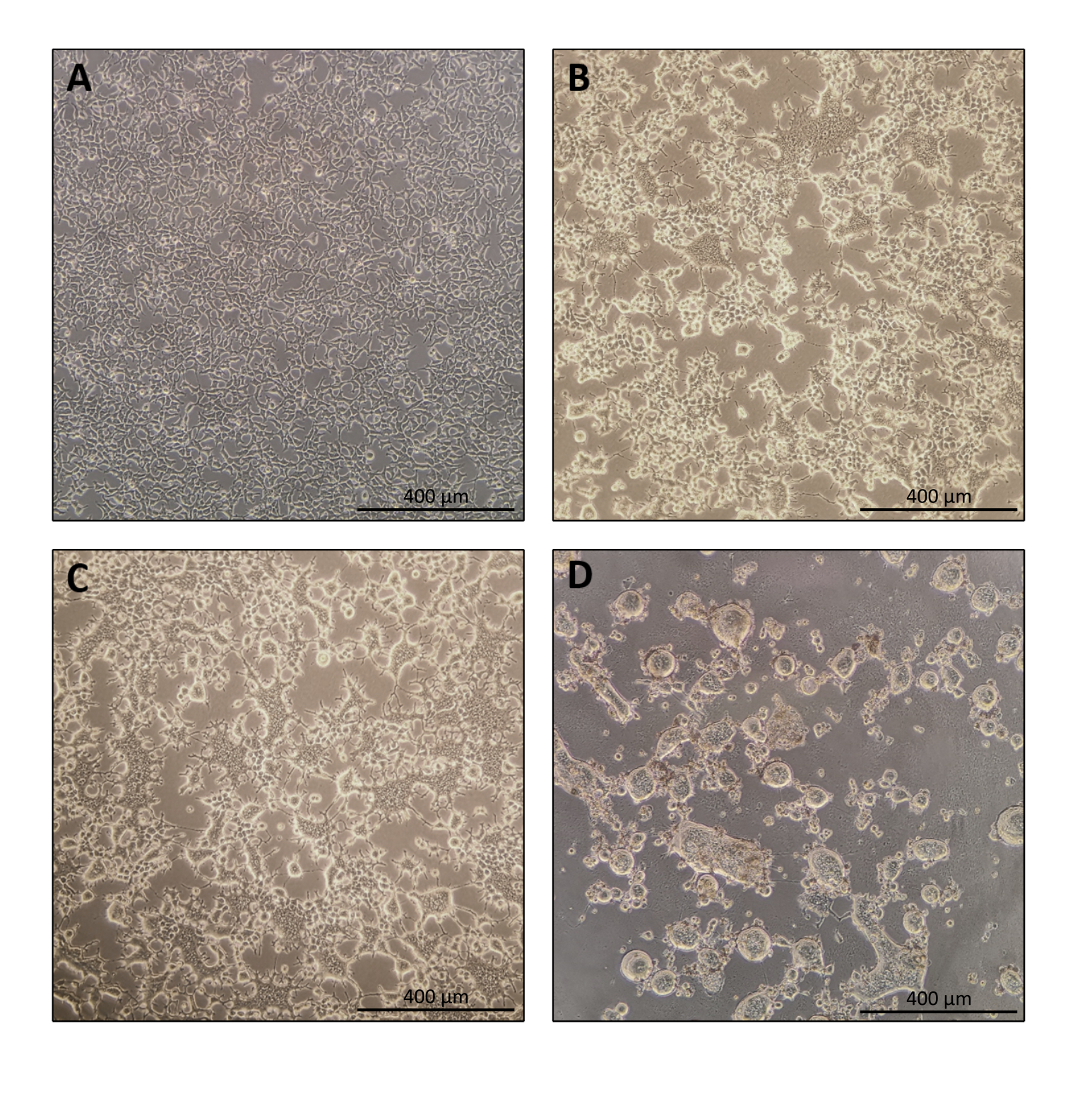
NOTE: Producer cells will begin to fuse. This is a normal occurrence due to the expression of fusogenic viral envelopes (Figure 1B,C). - Day 4: Harvesting Nanoblades
NOTE: At least 40 h after transfection, the cells would have fused together because of expression of the fusogenic viral envelopes, and sometimes, the cells are completely detached from the plate support (Figure 1D).- Collect 9 mL of the culture medium supernatant using a 10 mL pipette.
NOTE: Nanoblades are VLPs capable of delivering the Cas9 protein and its associated sgRNA into primary cells and in vivo. Although they are not considered genetically modified organisms as they are devoid of genetic material, they can induce genetic changes. Therefore, they must be manipulated with caution to avoid any contact with users (especially if they are programmed to target tumor suppressor genes). Users are advised to follow their local safety guidelines for the manipulation of retroviral vectors and work in a BSL-2 level laboratory when preparing VLPs and performing transduction experiments. Nanoblades can be inactivated with 70% ethanol or 0.5% of sodium hypochlorite. It is also advisable to treat all plastic waste (pipette tips, tissue culture plates, centrifugation tubes) with 0.5% sodium hypochlorite for at least 10 min to inactivate the Nanoblades. - Centrifuge the collected supernatant at 500 × g for 5 min to remove cellular debris and recover the supernatant without disturbing the cell pellet.
NOTE: If Nanoblades are meant to be used on primary cells, filter the supernatant using a 0.45 µm or 0.8 µm filter. Be aware that this step drastically reduces the Nanoblade titer as a significant fraction will be blocked in the filter membrane. - Pellet the Nanoblades overnight (12-16 h) in a swinging bucket rotor at 4,300 × g or at 209,490 × g in an ultracentrifuge for 75 min at 4 °C (see the Table of Materials).
NOTE: If target cells can grow in DMEM, it is possible to incubate them directly with the supernatant obtained after step 3.4.2 without concentrating the Nanoblades.
- Collect 9 mL of the culture medium supernatant using a 10 mL pipette.
- Day 5: Resuspension and storage of Nanoblades
- After centrifugation, slowly aspirate the medium and resuspend the white pellet with 100 µL of cold 1x phosphate-buffered saline (PBS). Cover the tube with parafilm, and incubate for 1 h at 4 °C with gentle agitation before resuspending the pellet by pipetting up and down.
NOTE: A white viscous material may appear upon resuspension; this is normal and does not significantly affect the efficiency of transduction. - Store the Nanoblades at 4 °C if planning on using them within four weeks. Otherwise, snap-freeze the Nanoblades in liquid nitrogen and store them at -80 °C.
NOTE: Wear protection goggles and cryogenic gloves when manipulating liquid nitrogen. Snap-freezing and storage at -80 °C leads to a significant decrease in Nanoblade efficiency. Moreover, thawed Nanoblades should not be frozen again. The protocol can be paused here.
- After centrifugation, slowly aspirate the medium and resuspend the white pellet with 100 µL of cold 1x phosphate-buffered saline (PBS). Cover the tube with parafilm, and incubate for 1 h at 4 °C with gentle agitation before resuspending the pellet by pipetting up and down.
4. Concentration of Nanoblades on a sucrose-cushion
NOTE: As an alternative to overnight centrifugation or ultracentrifugation (step 3.4.3), the Nanoblades can be concentrated on a sucrose cushion. This yields a purer fraction of Nanoblades, although the total amount recovered will be lower.
- Prepare a 10% sucrose solution (weight to volume) in 1x PBS, and filter it through a 0.2 µm syringe filter (see the Table of Materials).
- Begin the process of concentrating the Nanoblades on the sucrose cushion.
- Place 9 mL of VLP-containing sample (from step 3.4.3) into an ultracentrifuge tube (see the Table of Materials). Using a 3 mL syringe and cannula, slowly layer 2.5 mL of the 10% sucrose under the sample, trying not to mix the VLP-containing sample and the sucrose solution.
- Alternatively, place 2.5 mL of 10% sucrose into an ultracentrifuge tube (see the Table of Materials). Tilt the tube and slowly add the 9 mL of VLP-containing sample (from step 3.4.3) with a low-speed pipettor. During this operation, progressively raise the tube to a vertical position.
- Centrifuge the samples at 209,490 × g in an ultracentrifuge for 90 min at 4 °C.
NOTE: This technique can be adapted for low-speed centrifugation (4,300 × g) overnight as described in 12. - After centrifugation, remove the supernatant carefully and place the tube upside down on tissue paper to remove any remaining liquid. After 1 min, add 100 µL of 1x PBS and place the tube at 4 °C with a parafilm cover in a tube holder on an agitation table for 1 h (see the Table of Materials) before resuspending the pellet by pipetting up and down.
NOTE: The protocol can be paused here.
5. Monitoring Cas9 loading within Nanoblades by dot-blot
- Prepare the dilution buffer by adding 1 volume of lysis buffer containing a non-ionic surfactant (see the Table of Materials) in 4 volumes of 1x PBS. Dilute 2 µL of concentrated Nanoblades in 50 µL of dilution buffer, vortex briefly, and transfer 25 µL of this mixture into a new tube containing 25 µL of dilution buffer. Repeat this operation to have 4 tubes of Nanoblade dilutions (2-fold dilution steps).
- For the standard controls, dilute 2 µL of recombinant Cas9 nuclease (see the Table of Materials) into 50 µL of dilution buffer, vortex briefly, and proceed to make eight serial dilutions (2-fold dilution for each step).
- Carefully spot 2.5 µL of each VLP dilution and 2.5 µL of each standard onto a nitrocellulose membrane with a multichannel pipet (a larger volume may result in overlapping spots).
NOTE: A methanol-treated polyvinyldifluoride membrane may also be used. - Once the particles are absorbed onto the membrane, block the membrane with 1x Tris-buffered saline containing a non-ionic surfactant (TBS-T) supplemented with non-fat dry-milk (5% w/v) for 45 min at room temperature.
NOTE: The protocol can be paused here, and the membrane stored at 4 °C in 1x TBS-T. - Discard the 1x TBST supplemented with non-fat dry-milk, and incubate the membrane overnight at 4 °C with the Cas9-horseradish peroxidase antibody (1/1000 dilution in 1x TBST, 5% milk). Wash the membrane 3x with TBS-T, and visualize the signal using an enhanced chemiluminescent substrate kit.
- Quantify the dot intensity for the Nanoblades and recombinant Cas9 standard dilutions using the proprietary software provided with the gel imaging station or imageJ13. Define a linear curve linking dot intensity to the Cas9 concentration. Using the function of the obtained curve, extrapolate the Cas9 content in each preparation.
NOTE: The amount of recombinant Cas9 protein control can saturate the reading for the most concentrated samples of the standard dilution set (Figure 2). It is therefore advised, when defining the linear curve, to remove the reading from the undiluted samples (and sometimes that of the first dilution steps) if they are not in the linear range with respect to the known concentration of Cas9 that was spotted. Similarly, when extrapolating the amount of Cas9 within the Nanoblade samples, only use the readings that are within the linear range of the standard curve.
6. Transduction of target cells with Nanoblades (procedure for transduction in a 12-well plate)
- In a 12-well plate, seed 100,000-200,000 cells (either primary or immortalized adherent cells) per well in 1 mL of the appropriate cell culture medium. Allow the cells to adhere to the plate surface before transduction.
- In a 1.5 mL microcentrifuge tube, add 5-20 µL of concentrated Nanoblades (from step 3.5.1 or 4.4) to 500 µL of cell culture medium, and mix by pipetting up and down with a P1000 pipettor. Remove the medium from cells, and replace it with the 500 µL of this Nanoblade mixture.
NOTE: Transduction must be optimized for each cell type. It is important to use the smallest possible volume of medium (while avoiding drying of the target cells) so that the Nanoblades remain highly concentrated. Adherent cells must be transduced directly while attached to the plate (do not transduce in suspension as this will significantly decrease transduction efficiency). Some cells tolerate prolonged exposure to Nanoblades (24-48 h) while others are very sensitive and may form small syncytia. In this case, Nanoblades must be incubated with cells only for 4-6 h before replacing the medium. Spinoculation14 can also improve transduction for cells grown in suspension. Adjuvants such as cationic polymers (see the Table of Materials) can also improve transduction efficiency in some cell types. - After 4-6 h of cell incubation in a low volume of medium containing Nanoblades, increase the volume of medium to the normal amount (1 mL if working with a 12-well plate), or replace it with fresh medium if the cells are sensitive to VLPs.
NOTE: Cell medium containing Nanoblades must be inactivated with 0.5% sodium hypochlorite for 10 min before discarding it. Use gloves and protective goggles when manipulating sodium hypochlorite. If Nanoblades induce cell death, adapt the amount and total time of exposure to reduce cell mortality.
7. Measuring CRISPR efficiency at the targeted locus by T7 endonuclease assay
- Design PCR primers to amplify a 400-700 base-pair (bp) region encompassing the CRISPR-cleavage site.
NOTE: The cleavage site should be distant from the amplicon edge by at least 200 bp and should be slightly shifted from the center of the amplicon so that upon T7 endonuclease cleavage, 2 fragments of different sizes will be released. - Extract genomic DNA from cells treated with Nanoblades targeting the gene of interest and from control cells treated with Nanoblades programmed with a control sgRNA (see the Table of Materials).
NOTE: The protocol can be paused here. - Using 150 ng of genomic DNA as a template, program a PCR reaction of 30 µL volume (final volume) by following the manufacturer's protocol. Check that the PCR amplification yields a single amplicon of the expected size by running a 2% agarose gel stained with 5 µg/mL of ethidium bromide (or a safer alternative DNA gel stain).
NOTE: Wear appropriate protection gear when manipulating ethidium bromide, which is suspected of causing genetic defects. The protocol can be paused here. - Heteroduplex generation and digestion
- In a 0.2 mL PCR tube, add 5 µL of the enzyme buffer (provided with the T7 endonuclease I), 20 µL of water, and 24 µL of the PCR product from step 7.3. Allow heteroduplex formation by heating the samples to 94 °C over 3 min and then by decreasing the temperature (2 °C per min) to reach 40 °C.
- Add 0.5 µL of T7-endonuclease I at room temperature to each heteroduplex tube, including the control. Incubate at 37 °C for 15 min. Load the resulting reaction in a 2.5% (weight/volume) agarose gel stained by ethidium bromide. After migration, image the gel on a UV transilluminator.
NOTE: Wear appropriate protection gear when manipulating ethidium bromide, which is suspected of causing genetic defects. Use UV-protection goggles when using the UV transilluminator. - Measure cleavage efficiencies by analyzing the image resulting from the digestion reaction to quantify the intensity of each band with appropriate software (see the Table of Materials).
8. Measuring CRISPR efficiency at the targeted locus by Sanger sequencing and TIDE analysis
NOTE: As an alternative to the T7 endonuclease assay, CRISPR efficiency can be monitored by analysis and deconvolution of Sanger sequencing traces based on the TIDE protocol15.
- Perform Sanger sequencing of PCR amplicons from step 7.3 (include a control condition corresponding to untreated cells) using either the forward or reverse PCR primer.
- Analyze the Sanger sequencing traces of the control condition (untreated cells) and Nanoblade-treated samples using the TIDE server (https://tide.nki.nl) and following their analysis guidelines.
9. Nanoblade complex-formation with ssODN donors for homology-directed repair (procedure for transduction in a 12-well plate)
NOTE: Guidelines for the design of ssODN for efficient homology-directed repair mediated editing have been described previously16.
- In a 12-well plate, seed 100,000-200,000 cells per well in 1 mL of the appropriate cell culture medium. Allow the cells to adhere to the plate surface before transduction.
- Prepare 100 µL of a solution of the cationic polymer (see the Table of Materials) at 8 µg/mL in 1x PBS.
- Mix 19 µL of the cationic polymer solution with 100 pmol of the ssODN template. Add 20 µL of concentrated Nanoblades (from step 3.5.1 or 4.4), and incubate for 15 min on ice.
- Remove the complexed Nanoblades/ssODN from ice, and add 500 µL of cell culture medium (at 37 °C). Remove the medium from target cells (from step 9.1), and add the 500 µL of medium containing the complexed Nanoblades/ssODN. Allow the cells to proliferate for 48 h before genotyping.
- Extract genomic DNA from a fraction of the cell population using a dedicated extraction kit (see the Table of Materials).
- Design PCR primers to amplify a 400-700 bp region encompassing the knock-in site.
NOTE: PCR primers should not overlap with the homology arms of the ssODN to avoid false-positive results resulting from the PCR amplification of any residual ssODN still present within target cells. - Using 150 ng of genomic DNAs from control cells (untreated) or Nanoblade-treated-cells as a template, program a 30 µL PCR reaction following the manufacturer's protocol.
NOTE: ssODN traces may be present in the cell medium several days after transduction with the complex. This ssODN may serve as a partial template for PCR assays attempting to screen for the correct integration. Hence, it is advisable to passage the cells at least twice after transduction, to avoid eventual false-positive assays. - Load 5 µL of the control and Nanoblade-treated PCR reactions in a 1% (weight/volume) agarose gel stained by ethidium bromide. After migration, image the gel on a UV-transilluminator.
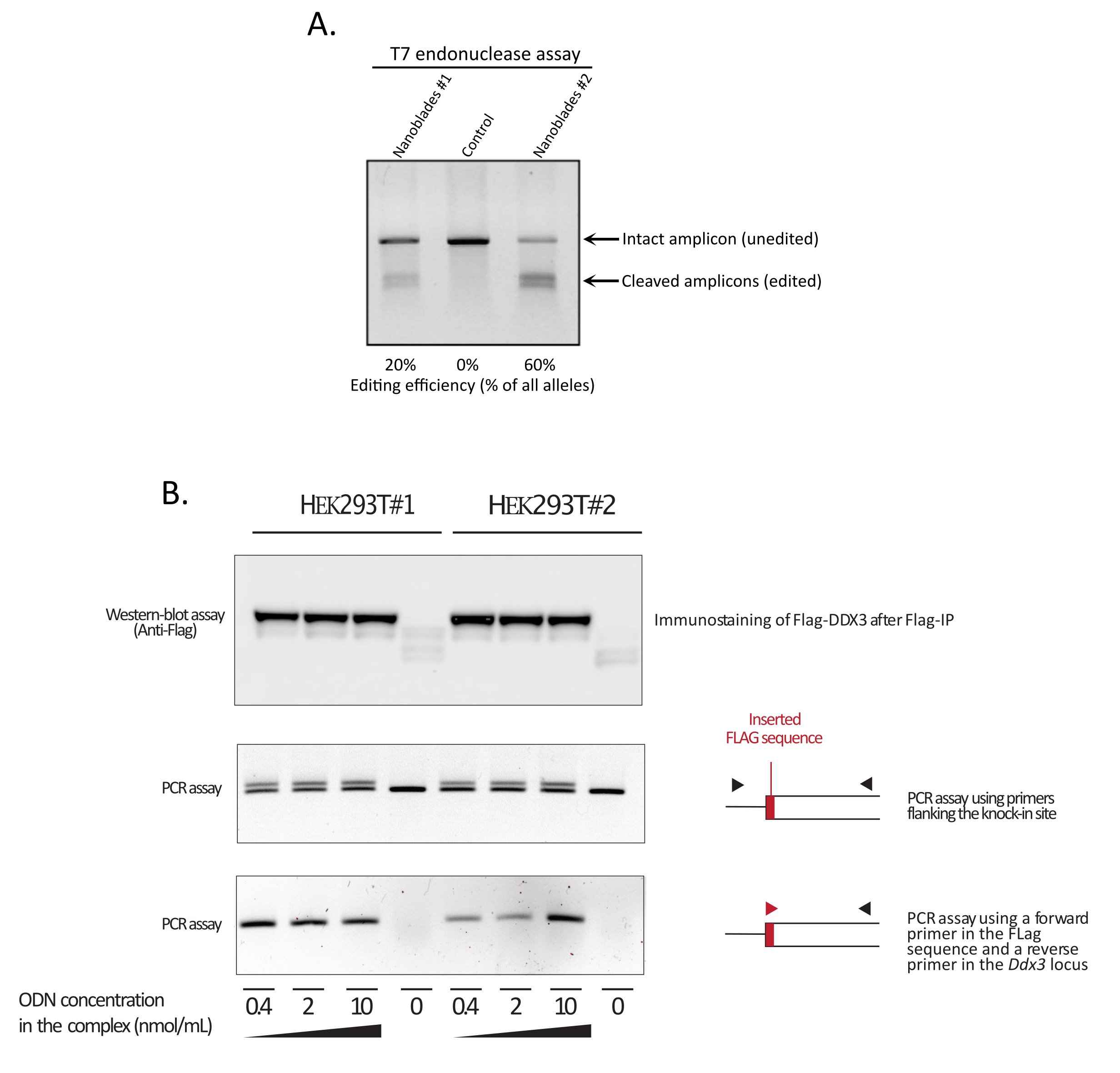
NOTE: If homology recombination is successful and corresponds to the insertion of more than 1 bp of genetic material, there should be a difference in the molecular weight of the PCR amplicons between the control and the Nanoblade-treated sample. As the efficiency of HDR does not reach 100%, two bands should be visible in the Nanoblade-treated sample (one of similar size to the control PCR amplicon corresponding to the unedited allele and one of higher molecular weight corresponding to the knock-in allele, see Figure 3B middle panel).
- Design PCR primers to amplify a 400-700 bp region encompassing the knock-in site.
- Perform Sanger sequencing of the control and Nanoblade-treated PCR amplicons.
- Quantify knock-in efficiency using the TIDER protocol17.
10. Nanoblade delivery in vivo
- Deliver up to 25 µL of concentrated Nanoblades from step 3.5.1 through retro-orbital injection or up to 100 µL through tail vein injection, as described in18, if working with mice.
NOTE: All procedures involving animal experimentation (including Nanoblade injections for genome editing purposes) require an approved protocol from a local ethics committee. - For the generation of transgenic mice, use a micro-injector to deliver from 1 pL to 10 pL of concentrated Nanoblades from step 4.4 into the perivitelline space of mouse oocytes as described previously18.
NOTE: For perivitelline injection, it is essential to purify and concentrate Nanoblades on a sucrose cushion to avoid clogging of the micro-injector.
Results
The protocol for Nanoblade preparation is fairly straightforward and requires simple laboratory equipment besides access to a tissue culture hood, a CO2 incubator, and a swinging bucket centrifuge or an ultracentrifuge. However, some steps require particular attention such as the source and handling of producer cells, as well as transduction conditions. As shown in Figure 1A, it is important to seed cells so that they are homogeneously distributed in the plate and reach ~70-80% confluence on the day of transfection (avoid having clumps of cells). Twenty-four hours after transfection (Figure 1B,C), producer cells will form syncytia leading to cells of larger size with multiple nuclei. Forty hours after transfection (Figure 1D), most cells in the plate will have formed syncytia and start detaching from the plate.
This is perfectly normal and is caused by the expression of the envelope glycoprotein, which induces fusion between neighboring cells. Upon concentration by centrifugation (or even straight from the supernatant of producer cells), the amount of Cas9 loaded within Nanoblades can be quantified in an absolute manner by dot-blot on a nitrocellulose membrane using recombinant Cas9 as reference (Figure 2). This step is important to determine the correct amount of Nanoblades to use for transduction of target cells. When performing the dot-blot assay, it is important to consider only the readings that fall within the linear range of the standard curve. However, independently of the amount of Cas9 present within Nanoblades, it is essential to test the efficiency of genome editing directly on target cells using the T7 endonuclease assay (Figure 3) or Sanger sequencing.
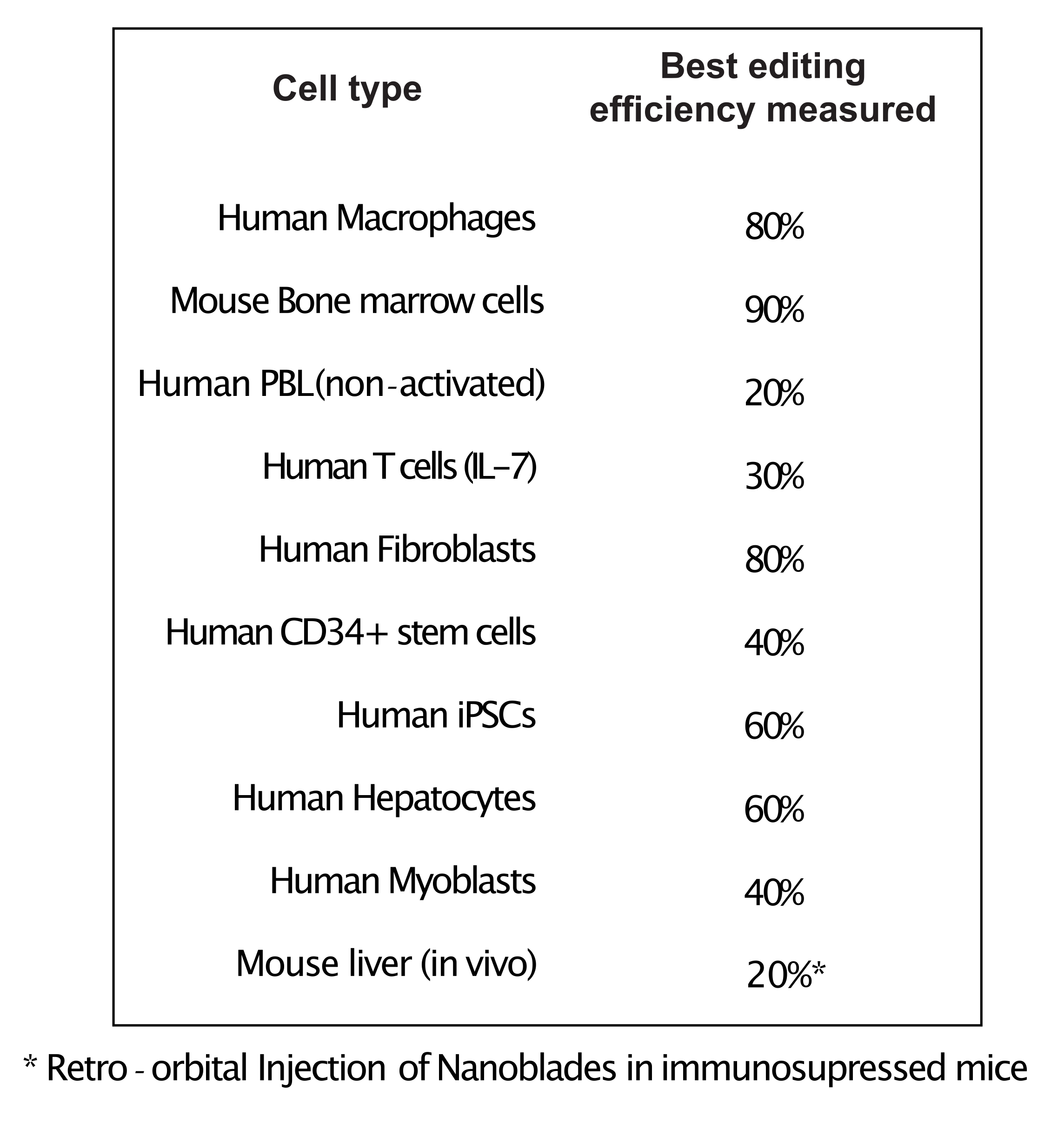
As shown in Figure 3, the efficiency of Nanoblades can differ from batch to batch although it is usually correlated to the amount of Cas9. In the example shown in Figure 3, the batch from lane 1 leads to 20% overall editing efficiency while the batch from lane 3 leads to 60% efficiency. In this case, it is possible to increase the volume of Nanoblades used from batch 1 to achieve an editing efficiency similar to that from batch 3. Figure 4 shows the maximum editing efficiency obtained using Nanoblades in different types of primary cells. It is important to note that the efficiency may vary depending on the sequence of the sgRNA used and the target accessibility.

Figure 1: Morphology of producer cells during Nanoblade production. (A) HEK293T cells at 70-80% confluence 24 h after plating. (B and C) HEK293T cell morphology 24 h after transfection. (D) HEK293T cell morphology 40 h after transfection. Scale bars = 400 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Quantification of Cas9 loading within Nanoblades by dot-blot. (A) Recombinant Cas9 or 100x concentrated (by ultracentrifugation) Nanoblade samples (#1, #2, and #3) are diluted 2-fold sequentially and spotted on a nitrocellulose membrane before incubating with anti-Cas9 HRP-coupled antibodies. Signal is revealed by enhanced chemiluminescence. (B) Chemiluminescence signal is acquired and quantified for the recombinant Cas9 dilutions and signal intensity plotted against the known amount of Cas9 spotted on the nitrocellulose membrane. A regression curve is calculated for the dilutions that are within the linear range (see blue crosses), excluding all concentrations that are outside of the linear range (see red crosses). (C) Cas9 concentration (nM) in each Nanoblade preparation was extrapolated using the equation from the linear regression obtained in (B). For this, it is important to only use the quantified signal from the Nanoblade dilutions that fall within the linear range of the regression curve. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Monitoring editing efficiency upon transduction. (A) T7 endonuclease assay measuring cleavage efficiency in Nanoblade-treated cells. Cells transduced with Nanoblades targeting the EMX1 gene were analyzed by T7 endonuclease assay. Lane 1: Nanoblade preparation batch #1 (20% cleavage efficiency); Lane 2: Control cells; Lane 3: Nanoblade preparation batch #2 (60% cleavage efficiency). (B) Knock-in of the Flag-tag sequence within the DDX3 open reading frame. Concentrated Nanoblades programmed with an sgRNA targeting the DDX3 locus were produced from different HEK293T clones (#1, #2) and complexed with increasing doses of a Flag-DDX3 ssODN template and the obtained complexes used for the transduction of HEK293T target cells. Upon transduction, cells were grown for three days before collecting them to extract genomic DNA and total proteins. Flag-DDX3 proteins were immunoprecipitated using anti-Flag agarose beads followed by western-blot analysis of the recovered proteins using an anti-Flag antibody (top panel). Site-directed insertion of the Flag-tag in the Ddx3 locus was also assayed by PCR using either primers flanking the insertion site (middle panel), or using a forward primer that recognizes the Flag-tag sequence and a reverse primer specific to the Ddx3 locus downstream of the Flag insertion site (bottom panel). Abbreviations: EMX1 = Empty Spiracles Homeobox 1; DDX3 = DEAD-box RNA helicase 3; PCR = polymerase chain reaction; ODN = oligodeoxynucleotide; ssODN = sing-stranded ODN; sgRNA = single-guide RNA; IP = immunoprecipitation. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Editing efficiency achieved in different primary cell types using Nanoblades. Abbreviations: PBL = peripheral blood lymphocyte; IL = interleukin; CD = cluster of differentiation; iPSC = induced pluripotent stem cell. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Nanoblades allow for rapid and dose-dependent delivery of the Cas9/sgRNA RNP complex in cell lines and primary cells. In contrast to classical transfection and other viral delivery vectors, but like protein electroporation, Nanoblades have the advantage of transient delivery of the Cas9/sgRNA RNP in a transgene-free manner. Nanoblades offer a highly versatile, simple, and inexpensive platform for protein delivery that can be easily and rapidly adapted to the ever-expanding family of CRISPR variants. Nanoblades can be produced in the HEK293T cell line or its derivatives. HEK293T cell lines used here have been developed to maximize retroviral and lentiviral particle production (see the Table of Materials). However, although other sources of HEK293T cells may be suitable, users must test and compare HEK293T cells from different sources as major differences in particle production have been observed depending on the HEK293T cell-source. Cells have also to be checked for Mycoplasma contamination frequently and passaged every three days (classically ⅛ dilution) to avoid overconfluence, which has a negative impact on particle production.
Cells should not be maintained for over 20 passages. DMEM supplemented with glucose, penicillin/streptomycin, glutamine, and 10% decomplemented fetal bovine serum was used for cell culture. As serum origin may affect the quality of the Nanoblade preparation, different batches of serum should be tested before large-scale production. Nanoblades can be efficiently produced in other media such as RPMI or serum-free modifications of minimum essential medium that can replace DMEM on the day after transfection. As indicated below, although medium replacement after transfection with some DNA-transfection reagents is optional, it may be beneficial to modify the medium into which the VLPs are released, especially to limit serum traces in the particle preparation. However, cultivation of cells in reduced-serum minimum essential medium on the day before transfection has not yet been attempted.
As mentioned, Nanoblades are produced upon overexpression of a mixture of plasmids in producer cells. The overexpression appears to be required for optimal production. Indeed, this laboratory developed a producer cell line where the Gag-Pol-expressing construct was stabilized by antibiotic selection; however, this system failed to produce significant amounts of Nanoblades. A similar observation was made when the sgRNA-coding construct was stably integrated into the genome of producer cells. As described for other particle production systems, a stable cell line expressing at least some constructs involved in Nanoblades production may be useful; however, this would certainly require the processing of large volumes of supernatant and an appropriate technique to purify particles. The above protocol outlines the preferred procedure to produce Nanoblades that exploits specific transfection reagents (see the Table of Materials).
Although transfection reagents from other manufacturers have also been tested with success, the vast majority of this group's results with Nanoblades follow the procedure described herein. Low-cost transfection can be achieved using calcium phosphate reagents and yield good production efficiency; however, this method absolutely requires the replacement of transfection medium on the day after transfection and may leave calcium phosphate residues in the sedimented particle preparation. Consistent with the necessity of high expression levels for Nanoblade components within producer cells is the observation that the amount of sgRNAs associated with the Cas9 protein can be a limiting factor for efficient genome-editing. To improve sgRNA loading, two technical approaches have been recently developed by independent groups using protein delivery vectors similar to Nanoblades. These rely on the use of T7 polymerase-dependent cytoplasmic expression of sgRNA6 or through the addition of a retroviral encapsidation signal to the sgRNA sequence to mediate binding to the Gag polyprotein6. These approaches could indeed improve sgRNA loading within Nanoblades although they have not been tested yet.
Transduction of target cells is a critical step in the procedure. In most immortalized cell lines, transduction with Nanoblades has little or no cytopathic effect. However, in primary cells, toxicity can be an issue. Transduction has therefore to be optimized for each cell type. Specifically, the exposure time to Nanoblades is an important factor to modify when optimizing the transduction protocol. For sensitive cells such as primary neurons or bone marrow cells, 4-6 h of incubation with Nanoblades before replacing the medium allows for efficient delivery of the Cas9 protein while minimizing cell toxicity. Furthermore, adjuvants such as cationic polymers, among others, can significantly improve the efficiency of transduction in some cells (see the Table of Materials). It is important to note that Nanoblades are VLPs and can induce an immunogenic response. This can be a limitation if working with certain types of primary cells, such as macrophages or dendritic cells, in which incubation with Nanoblades could induce important changes in gene expression and the phenotype of the cells. If macrophages and dendritic cells are derived from hematopoietic stem cell precursors (such as mouse bone marrow cells), it is preferable to transduce cells with the Nanoblades before they are fully differentiated to avoid inducing a cellular response against the Nanoblades. Otherwise, Cas9 protein electroporation could represent a viable alternative when working with differentiated immune cells.
Nanoblades can be used in vivo to transduce mouse zygotes or embryos to generate transgenic animals. Similar to classical retroviral or lentiviral vectors, they can also be injected directly into tissues from adult animals. However, Nanoblades (similar to retroviral and lentiviral vectors) can be inactivated by the immune response of the host animal; hence, the dose to be injected has to be optimized for each application. This immune response can also limit the distribution of functional VLPs to tissues close to the injection site. Finally, unlike lentiviral vectors, Nanoblades are transgene-free and deliver the Cas9 in a restricted timeframe. Therefore, they cannot be used to perform genome-wide functional screenings that require high-throughput sequencing of sgRNAs upon selection of cells. Nanoblades are useful when rapid, dose-dependent, and transgene-free genome-editing is required20. Furthermore, similar to protein electroporation, Nanoblades lead to fewer off-target effects than prolonged expression of Cas9/sgRNA through DNA transfection or classical viral vectors3. Future development of Nanoblades is focused on incorporating Cas9 variants for different technological applications such as base-editing and RNA targeting.
Disclosures
Philippe E. Mangeot and Emiliano P. Ricci are named as inventors on a patent relating to the Nanoblades technology (patent applicants: Institut National de la Santé et de la Recherche Médicale (INSERM), Centre National de la Recherche Scientifique (CNRS), Ecole Normale Superieure de Lyon, Université Claude Bernard Lyon 1, Villeurbanne Cedex; application number: WO 2017/068077 Al; patent status: published, 27th April 2017; all aspects of the manuscript are covered by the patent application. The remaining authors declare no competing interests.
Acknowledgements
This work was funded by Labex Ecofect (ANR-11-LABX-0048) of the Université de Lyon, within the program Investissements d'Avenir (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR), Fondation FINOVI, Agence Nationale des Recherches sur le SIDA et les Hépatites Virales (ANRS-ECTZ3306) and by the European Research Council (ERC-StG-LS6-805500 to E.P.R.) under the European Union's Horizon 2020 research and innovation programs.
Materials
| Name | Company | Catalog Number | Comments |
| 13.2 mL, Thinwall Polypropylene Tubes, 14 x 89 mm - 50Pk | Beckman Coulter Life Sciences | 331372 | Ultracentrifugation tubes for Nanoblades purification |
| Amersham Protran Premium Western blotting membranes, nitrocellulose | Merck | GE10600004 | Nitrocellulose membrane for quantifying Cas9 levels within purified Nanoblades |
| BIC-Gag-CAS9 | Addgene | 119942 | Encodes a GAG (F-MLV)-CAS9(sp) fusion. Allows the production of GAG-CAS9 Virus like particles from producer cells in association with over expressed gRNA(s) and appropriate envelopes |
| BICstim-Gag-dCAS9-VPR | Addgene | 120922 | Encodes a GAG-dCAS9-VPR fusion for targeted transcriptional activation |
| BLADE | Addgene | 134912 | Empty backbone for cloning sgRNA sequence to be used in Nanoblades system |
| BsmBI-v2 | New England Biolabs | R0739S | Restriction enzyme to digest the BLADE and SUPERBLADES vectors for sgRNA cloning |
| Cas9 (7A9-3A3) Mouse mAb (HRP Conjugate) #97982 | Cell Signaling Technology | 97982S | Anti-Cas9 antibody for Cas9 quantification by dot-blot |
| Cas9 Nuclease, S. pyogenes | New England Biolabs | M0386T | Recombinant Cas9 protein to be used as a reference for absolute quantification of the amount of Cas9 loaded within Nanoblades |
| Ethidium bromide solution (10 mg/mL in H2O) | Sigma-Aldrich | E1510-10ML | For staining agarose gels and visualize DNA |
| Fisherbrand Wave Motion Shakers | Fisher Scientific | 88-861-028 | Agitation table to resuspend Nanoblades upon centrifugation |
| gelAnalyzer | http://www.gelanalyzer.com; quantifying band intensity after digestion | ||
| Gesicle Producer 293T | Takara | 632617 | Nanoblades producer cell line |
| Gibco DMEM, high glucose, pyruvate | ThermoFisher Scientific | 41966052 | Cell culture medium for Gesicle Producer 293T cells |
| GoTaq G2 DNA Polymerase | Promega | M7848 | Taq polymerase for amplification of genomic DNA before T7 endonuclease assays |
| jetPRIME Transfection Reagent kit for DNA and DNA/siRNA | Polyplus | POL114-15 | Transfection reagent for Nanoblade production in Gesicle Producer 293T cells |
| Millex-AA, 0.80 µm, syringe filter | Millipore | SLAA033SS | Syringe filter to remove cellular debris before concentration of Nanoblades |
| Millex-GS, 0.22 µm, syringe filter | Millipore | SLGS033SS | Syringe filter to sterilise the sucrose cushion solution |
| Millex-HP, 0.45 µm, polyethersulfone, syringe filter | Millipore | SLHP033RS | Syringe filter to remove cellular debris before Nanoblades concentration |
| Monarch DNA Gel Extraction Kit | New England Biolabs | T1020L | DNA gel extraction kit for purification of the pBLADES or pSUPERBLADES plasmid fragment upon digestion with BsmBI |
| NEB Stable Competent E. coli (High Efficiency) | New England Biolabs | C3040I | Competent bacteria for plasmid transformation and amplification |
| NucleoBond Xtra Midi kit for transfection-grade plasmid DNA | Macherey-Nagel | 740410.50 | Maxipreparation kit for purification of plasmid DNA from cultured bacteria |
| Nucleospin gDNA extraction kit | Macherey-Nagel | 740952.50 | Extraction of genomic DNA from transduced cells |
| NucleoSpin Plasmid, Mini kit for plasmid DNA | Macherey-Nagel | 740588.50 | Minipreparation kit for purification of plasmid DNA from cultured bacteria |
| NucleoSpin Tissue, Mini kit for DNA from cells and tissue | Macherey-Nagel | 740952.5 | Genomic DNA extraction kit |
| Optima XE-90 | Beckman Coulter Life Sciences | A94471 | Ultracentrifuge |
| pBaEVRless | Els Verhoeyen (Inserm U1111) | Personnal requests have to be sent to: els.verhoyen@ens-lyon.fr | Baboon Endogenous retrovirus Rless glycoprotein described in Girard-Gagnepain, A. et al. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood 124, 1221–1231 (2014) |
| pBS-CMV-gagpol | Addgene | 35614 | Enocdes the Murine Leukemia Virus gag and pol genes |
| pCMV-VSV-G | Addgene | 8454 | Envelope protein for producing lentiviral and MuLV retroviral particles |
| Phosphate-Buffered Saline (PBS) | ThermoFisher Scientific | 14200091 | 10X PBS to dilute in millipore water |
| Polybrene Transfection Reagent | Millipore Sigma | TR-1003-G | Cationic polymer that enhances the efficiency of retroviral transduction in specific mammalian cells. It can also allow viral-dependent entry of an Oligodeoxynucleotide (ODN) for homology-directed repair |
| Sucrose,for molecular biology, ≥99.5% (GC) | Sigma-Aldrich | S0389-5KG | Sucrose to prepare a cushion for Nanoblade purification through ultracentrifugation |
| SUPERBLADE5 | Addgene | 134913 | Empty backbone for cloning sgRNA sequence to be used in nanoblades system (Optimized for increased genome editing efficiency via Chen B et al., 2013) |
| SuperSignal West Dura Extended Duration Substrate | ThermoFisher Scientific | 34076 | Enhanced chemiluminescence (ECL) HRP substrate for Cas9 dot blots |
| SW 41 Ti Swinging-Bucket Rotor | Beckman Coulter Life Sciences | 331362 | Rotor for ultracentrifugation |
| SYBR Safe DNA Gel Stain | ThermoFisher Scientific | S33102 | Alternative to ethidium bromide for staining agarose gels and visualize DNA |
| T4 DNA Ligase | New England Biolabs | M0202S | DNA ligase to ligate the BLADE or SUPERBLADES vectors with the duplexed DNA oligos corresponding to the variable region of the sgRNA |
| T7 Endonuclease I | New England Biolabs | M0302S | T7 Endonuclease I recognizes and cleaves non-perfectly matched DNA. Allows to monitor the extent of genome editing at a specific locus |
| Triton-containing lysis buffer | Promega | E291A | Lysis buffer to disrupt Nanoblades and allow Cas9 quantification |
| TWEEN 20 | Sigma-Aldrich | P9416 | For the preparation of TBST |
References
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Zuris, J. A., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome-editing in vitro and in vivo. Nature Biotechnology. 33, 73-80 (2015).
- Mangeot, P. E., et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nature Communications. 10 (1), 45 (2019).
- Cai, Y., Bak, R. O., Mikkelsen, J. G. Targeted genome editing by lentiviral protein transduction of zinc-finger and TAL-effector nucleases. eLife. 3, 01911 (2014).
- Choi, J. G., et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Therapy. 23 (7), 627-633 (2016).
- Gee, P., et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nature Communications. 11, 1334 (2020).
- Indikova, I., Indik, S. Highly efficient 'hit-and-run' genome editing with unconcentrated lentivectors carrying Vpr.Prot.Cas9 protein produced from RRE-containing transcripts. Nucleic Acids Research. 48 (14), 8178-8187 (2020).
- Lyu, P., Javidi-Parsijani, P., Atala, A., Lu, B. Delivering Cas9/sgRNA ribonucleoprotein (RNP) by lentiviral capsid-based bionanoparticles for efficient 'hit-and-run' genome editing. Nucleic Acids Research. 47 (17), 99 (2019).
- Gheysen, D., Jacobs, E., de Foresta, F., Thiriart, C. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell. 59 (1), 103-112 (1989).
- Hanna, R. E., Doench, J. G. Design and analysis of CRISPR-Cas experiments. Nature Biotechnology. 38 (7), 813-823 (2020).
- Sambrook, J. . Molecular cloning a laboratory manual. Third edition. , (2001).
- Jiang, W., et al. An optimized method for high-titer lentivirus preparations without ultracentrifugation. Scientific Reports. 5, 13875 (2015).
- Rueden, C. T., et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics. 18 (1), 529 (2017).
- O'Doherty, U., Swiggard, W. J., Malim, M. H. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. Journal of Virology. 74 (21), 10074-10080 (2000).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Bollen, Y., Post, J., Koo, B. -. K., Snippert, H. J. G. How to create state-of-the-art genetic model systems: strategies for optimal CRISPR-mediated genome editing. Nucleic Acids Research. 46 (13), 6435-6454 (2018).
- Brinkman, E. K., et al. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Research. 46 (10), 58 (2018).
- Dussaud, S., Pardanaud-Glavieux, C., Sauty-Colace, C., Ravassard, P. Lentiviral mediated production of transgenic mice: a simple and highly efficient method for direct study of founders. Journal of Visualized Experiments. (140), e57609 (2018).
- Montagna, C., et al. VSV-G-enveloped vesicles for traceless delivery of CRISPR-Cas9. Molecular Therapy. Nucleic Acids. 12, 453-462 (2018).
- Marnef, A., et al. A cohesin/HUSH- and LINC-dependent pathway controls ribosomal DNA double-strand break repair. Genes & Development. 33 (17-18), 1175-1190 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved