
Method Article
単細胞蛍光イメージングによる細菌間競争の定量化
要約
この原稿は、単細胞蛍光顕微鏡を用いて、コカルチャーにおける細菌の競合を可視化し定量する方法を説明する。
要約
細菌間競争はマイクロバイオームの構造と機能に直接影響を与える可能性があります。本研究は、単一細胞レベルで異なる細菌株間の競合的相互作用を可視化し、定量化するために使用できる蛍光顕微鏡アプローチを記述する。ここで説明するプロトコルは、アップライトおよび反転した蛍光顕微鏡、生細胞およびタイムラプスイメージング技術、およびオープンソースソフトウェアFIJIを用いた定量的画像分析の両方で、スライド調製における高度なアプローチの方法を提供する。本稿のアプローチは、安定したプラスミドから異なる蛍光タンパク質を発現している2つのコインキュベート株の時間の経過とともに面積の変化を測定することによって、共生 ビブリオ・フィシェリ 集団間の競争的相互作用の定量化を概説する。別の成長条件を必要とする細菌モデルシステムでこのプロトコルを最適化するための代替方法が記載されています。ここで説明するアッセイは V.フィシェリに最適化された条件を使用していますが、このアプローチは再現性が高く、多様なマイクロバイオームからのカルト性分離株間の競争を研究するために容易に適応することができます。
概要
本稿では、蛍光顕微鏡を用いて単細胞レベルで細菌の競争を定量化する方法を概説する。微生物群集の構造と機能は、微生物間の競合的相互作用によって形成されることが多く、多くの場合、これらの相互作用を特徴付けるには、コインキュベーション1、2、3、4、5、7、8で異なる細菌株を観察する必要があります。 .伝統的に、細菌の競争は、コインキュベーション期間2、9の前後に阻害剤および標的株のコロニー形成単位(CFUs)を数えることによって集団レベルで定量化される。微生物の競争のための機構は細菌間に広く分布しており、標的細胞10、11、12、13、14、15、16、17、18、19を阻害するために拡散または細胞細胞接触のいずれかに依存し得る。
細菌株は集団レベルでコインキュベーションでしばしば観察されるが、この原稿は細菌の競争の単細胞定量化のためのアッセイを概説する。さらに、この研究は、他の細菌種との使用のためにプロトコルを適応するための提案を含む。本稿の特定の技術は、共生菌ビブリオ・フィシェリ20、21、22の株間の接触依存性の非特異的な競争を研究するために使用されるが、それらは多くの生物間の競争のために適応することができる。この記事では、直立顕微鏡と反転型顕微鏡の両方でスライドセットアップの手順を説明し、すべての分析はオープンソースソフトウェアFIJI23を使用して説明され、異なるイメージングセットアップおよび分析プログラムにアクセスできる研究者が使用できるようにします。この方法は、集団レベルと単一細胞レベルの両方で微生物の競争を研究することの重要性を考えると、研究者が競争的相互作用を定量化するための貴重なリソースとなり、特に独自の分析ソフトウェアにアクセスできないものになります。
プロトコル
1. 細菌株の最適化
- 単細胞細菌競争アッセイ用の2つの細菌株を選択してください。ここでは、V.フィシェリの2つの株が使用されている:標的株(ES11424)および染色体II(T6SS2)1の型VI分泌系を用いて標的株を殺すことが知られている阻害剤株(MJ1125)、接触依存性の殺傷機構である。
- 実験に適したコントロールを決定します。この例では、適切な制御は、標的株を用いて野生型およびT6SS変異阻害剤株の両方をインキュベートし、T6SS媒介性殺傷の効果を定量化することである。
注:追加のコントロールは、T6SS依存性の殺傷を防ぐために必要な免疫遺伝子を発現する標的株、またはT6SS活性を回復するためにトランス中の変異遺伝子の野生型コピーを発現するインヒビター変異株を含むことができる1。 - 可能であれば、異なる蛍光タンパク質(GFPまたはRFPなど)の遺伝子をコードする安定したプラスミドを使用して、顕微鏡上の歪みの種類を視覚的に区別するように変換します。ここで、阻害剤株は、GFPコードプラスミド(pVSV102)でタグ付けされ、標的株はdsRedコードプラスミド(pVSV208)26でタグ付けされる。
注:安定したプラスミドを使用することができない場合、蛍光タグは、可視化27、28の細菌染色体に導入することができます。 - 初期最適化期間中、実験中に使用される蛍光フィルターの下でタグ付けされた株の画像クローン培養は、細胞が意図されたチャネルでのみ見えるようにします。たとえば、GFP タグ付きひずみは FITC チャネルでのみ表示されるようにします。
2. アガロースパッドの準備
- 2%低溶融アガロー(w/v)をmPBSに溶解してアガロースパッド溶液を調製します。溶液をマイクロ波と渦中で短時間加熱し、アガロースが完全に溶解するまで加熱します。この溶液は、55°Cの水浴に入れ、使用できる状態になるまで温かんでください。アガロースパッドの準備の詳細については、「 ディスカッション 」セクションを参照してください。
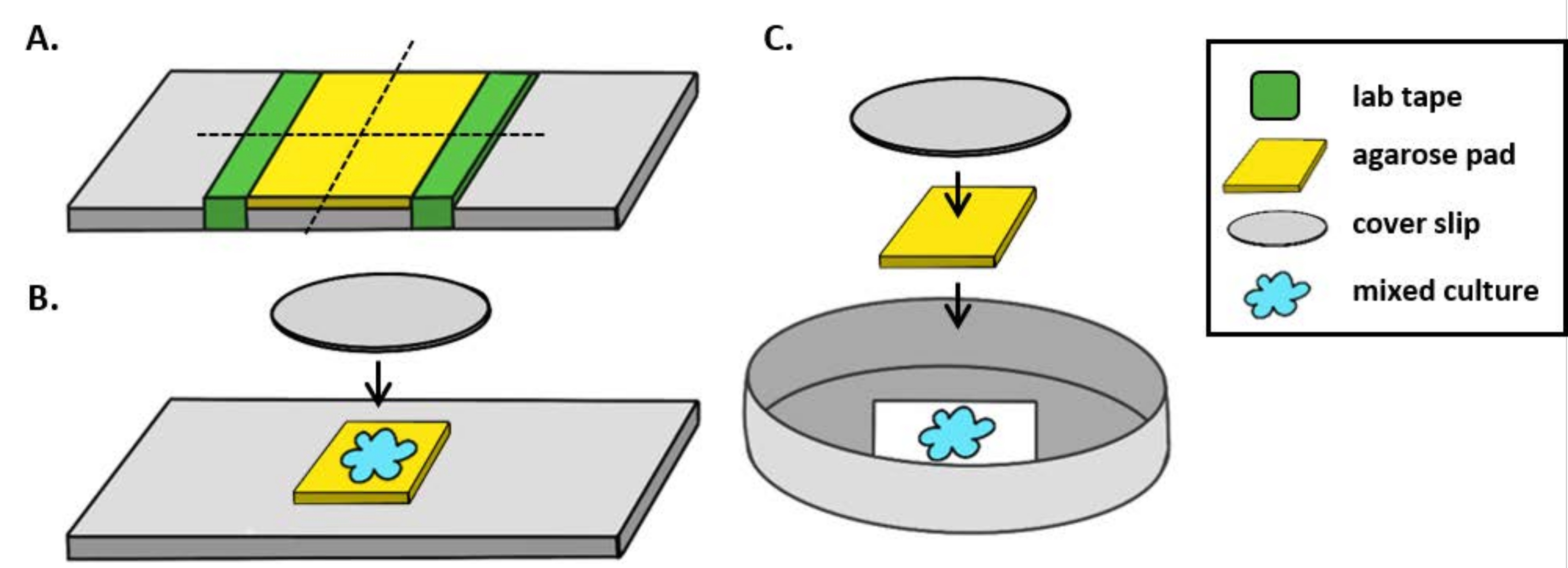
注: ここでは、mPBS は標準の 1x PBS に 20 g/L NaCl を加えて調製しました。 - ラボテープをガラススライドに5回巻き付けます。同じスライドでこのプロセスを 2 回繰り返して、2 つのテープの間隔がカバースリップの幅よりわずかに小さくなるようにします (図 1A)。例えば、25 mm2 カバーリップを使用する場合、テープのピースは約 20 mm 離れた間隔にする必要があります。
メモ:テープがスライドの周りに巻かれる回数は、アガロースパッドの厚さを調整するために変更することができますが、テープの層は、アガロースパッドが平らなままになるように、スライドの両側の同じ高さであることが重要です。 - ピペットは、テープの2つの部分の間にアガロース溶液を温め、すぐにカバースリップで上に置き、テープの破片の上に置きます。これにより、アガロースパッドの表面が平坦なままであることを保証します。このステップでピプレットされたアガロース溶液の体積は、カバースリップが液体と接触し、アガロース溶液内の気泡を押し出すのに十分であるはずです。この特定のセットアップのために、暖かいアガロースの200 μLで十分です。
- コインキュベーションアッセイの前に、アガロースパッドを室温で少なくとも1時間固化させます。ステップ2.2は約20 mm 2のアガロースパッドを作り出す。
- このアガロースパッドをカミソリの刃で4つ、5mm2 パッドに切り、イメージングに使用します。
注:アガロースパッドは、実験の1週間前まで作ることができ、乾燥を防ぐためにパラフィルムで密封された空の無菌ペトリプレートに4°Cで保存することができます。
3. 共孵化のための株を準備する
- 各株を-80°C株からLBS寒天プレートに使用する各株を適切な抗生物質を補い、24°Cで一晩インキュベートする。 この例では、野生型阻害剤株、型VI分泌系変異株、および標的株の3つの株が使用されます。
- 翌日、各株から2つのコロニーを摘み取り、適切な抗生物質を添加したLBS培地で再懸濁し、200rpmで振盪して24°Cで一晩インキュベートすることによって、生物学的複製の一晩培養を開始する。
- 翌朝、サブカルチャーは、各生物学的に抗生物質なしで新鮮なLBS培地に1:1000を複製し、4〜5時間、または細胞が〜1.5のOD600 に達するまで24°Cでインキュベートする。
注: 歩数 3.1、3.2、3.3 のタイミングは、増殖速度が大きく異なる可能性があるため、異なる細菌種に対して最適化する必要があります。このアッセイのために、細胞は、コインキュベーションアッセイの開始時に中ログ段階であることを目的とした。
4. コインキュベート細菌株
- ステップ3.3の中ログ培養から始めて、すべてのサンプルの光密度を600 nm(OD600)で測定および記録します。
- 各サンプルを、LBS培地で培養を希釈して、V.フィシェリの約109 CFU/mLに相当するOD600 = 1.0に正規化します。
- 各正規化された歪みの30 μLをラベル付き1.5 mLチューブに加えることで、体積に基づいて1:1比で2つの競合株を混合します。1〜2sの混合歪培養を渦巻く。
注: 場合によっては、異なる比率での混合が適切な場合があります。例えば、一方の株が他の株よりもはるかに速く成長する場合、競争を観察するために数値的な利点で成長の遅い株を開始する必要があるかもしれません。OD600 が両方の株に対して類似のCFU/mLに対応していない場合、最適化が必要になる場合もあります。 - 生物学的複製および治療ごとにステップ 4.3 を繰り返します。ここに示す例では、これは合計4つの混合株チューブをもたらす:標的株と混合野生型阻害剤株と2つの生物学的複製と、標的株と混合された型VI分泌系変異株との2つの生物学的複製。
- 競合する細胞が寒天パッド上のコインキュベーション中の接触依存性の殺死に十分に密になるように、混合培養物を21,130 x gで1分間標準1.5 mL遠心管に遠心し、上澄みを捨て、各ペレットを20μL LBS培地に再懸濁させて3倍に濃縮します。各サンプルについて繰り返します。
注:一部の細菌細胞は高いrcfで遠心分離によって損傷に敏感です。このような場合、混合培養は4600 xg29で3分間遠心することができる。 さらに、接触依存性の競合を定量化する場合、スライド上で殺しを観察するのに十分な細胞密度を確保することが重要である。今回の記事では、殺死が観察される「混み付き」の治療は、およそ10細胞/20μm2を有していた。詳細については、「ディスカッション」セクションを参照してください。
5. スライドの設定
- 直立した顕微鏡を使用する場合は、標準の1mmガラススライド上に約5mm2 アガロースパッドを置きます。2 μLの混合培養物をアガロースパッドに入れ、#1.5カバースリップ(25mm2)をその場に置きます。例については 、図 1B を参照してください。
- 逆顕微鏡を使用する場合は、混合培養物の2μLを35mmのペトリ皿の#1.5カバースリップ底にスポットし、コインキュベーションスポットの上に約5mm2 アガロースパッドを置きます。アガロースパッドの上に12mmの円形ガラスカバースリップを置きます。例については 、図 1C を参照してください。
- 使用する顕微鏡のセットアップに応じて、ステップ5.1または5.2を繰り返し、残りの3つの混合培養について、4枚のスライドまたは皿を画像化する結果となる。
- ステップ6に進む前に、スライドを約5分間ベンチトップに座らします。これにより、細胞は寒天パッドに落ち着き、イメージングプロセス中の動きを排除します。
6. 蛍光顕微鏡
- まず、白色光(位相コントラストまたはDIC)を使用して細胞に焦点を当て、光の漂白の影響を最小限に抑えます。単一細菌細胞の平均サイズに基づいて、60xまたは100xの油目的を使用する。
- バックグラウンド検出を最小限に抑えて適切なチャネルでセルが表示されるように、各チャネルの露出時間と取得の設定を調整します。
注:異なるチャネルに対して異なる露光時間を使用することは適切ですが、同じ露光時間を、特定のチャネルのすべての生物学的複製および治療で使用する必要があります。 - 各サンプルについて、少なくとも 5 つの視野(FOV)を選択し、ステップ6.2の取得設定を使用して適切なチャネルごとに画像を取得します ( 図 2の例を参照)。最終タイム ポイントで同じ FOV をイメージ化できるように、各 FOV の XY ポイントを保存します。各時点で同じFOVを撮像することは、分析工程中の標的または阻害細胞が占める面積の割合を決定する必要がある。
注: この例では、GFP の蛍光は、励起波長が 467 ~ 498 nm のフィルターと 513 ~ 556 nm のエミッション フィルタを使用して検出され、偽色の緑色です。dsRedの蛍光は、励起波長542-582nmのフィルターと603-678nmの発光フィルタを使用して検出され、偽色のマゼンタである。 - 2時間後、前に保存したXYポイントを使用して、各サンプルについてステップ6.3を繰り返します(図2)。
注:その後の画像のタイミングは、異なる成長率または競合メカニズムを持つ生物に対して最適化する必要があります。
7. フィジーにおける画像解析
- 次の手順に従って、フィジー画像処理ソフトウェアをダウンロードしてインストール https://imagej.net/Fiji/Downloads
- フィジーを開き、分析用の画像ファイルをインポートします。
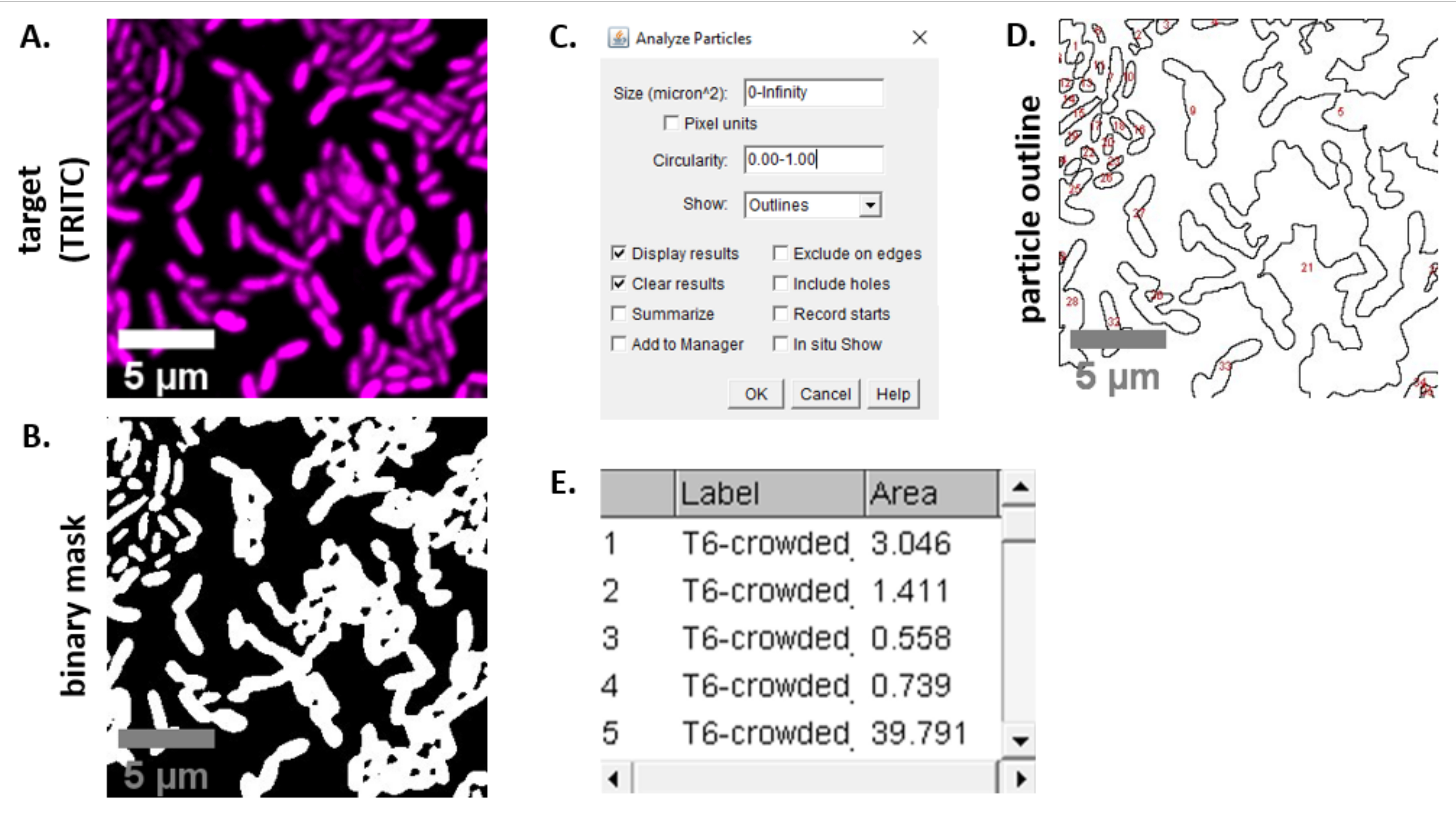
注: ほとんどの場合.TIFF ファイルは画像解析に使用されますが、一部の画像取得ソフトウェアは独自のファイルタイプを使用してエクスポートされます。FIJIは、ほとんどの独自のファイルタイプを認識することができ、画像は次のようにインポートして分析することができます。 - 手順 6.3 および 6.4 で取得した各画像について、画像をグレースケールに変換し、チャンネルを分離し、しきい値 (Ctrl + Shift + T) を作成し、前処理されたイメージのバイナリ マスクを作成します (図3A、B)。
注: ここでは、フィジーのデフォルトのしきい値設定が使用されます。場合によっては、これらの設定を変更する必要がある場合もありますが、その場合は、その実験のすべての画像に同じ設定を使用する必要があります。 - イメージにスケールを設定する (分析|顕微鏡設定23に適切な値を使用して Scale を設定します。
- 測定の設定 (分析|[計測値]を設定し、[ 面積] を選択します。
注: 実験に適している場合は、他の測定値を追加できます。ここで示す解析例では、対象 の面積 測定のみが必要です。 - パーティクルの解析 (|の分析デフォルトの設定(図3C)を使用してパーティクルを解析します。サンプルに破片がある場合は、非細胞粒子を除外するためにサイズまたは円形を調整する必要がある場合があります。 [|を表示] を選択します。 この解析の出力に、分析されたすべてのパーティクルの番号付きアウトラインが含まれるようにアウトラインを作成します (図 3D)。
注: (1) すべてのセルが解析され、(2) すべての破片が解析から除外されるようにするには、最適化手順で 図 3D のアウトラインを初期画像と比較することが特に重要です。 - ステップ 7.4 ( 図3E) の測定値をスプレッドシート ソフトウェアにエクスポートして、さらに分析とグラフ化を行います。
- 実験中に取得したすべてのチャンネルと画像について、手順7.1~7.5を繰り返します。
8. 初期目標領域の時間経過に対する割合の計算
- セクション 7 で分析した各視野について、エクスポートされたファイルに、解析された各パーティクルの個別の面積測定が含まれていることを確認します。ターゲット株の蛍光チャネルから始めて、個々の視野の合計粒子面積を計算します。それぞれ5つのFOVを持つ2つの生物学的複製の場合、これは各時点で治療ごとに10の合計領域をもたらすはずです。
- 各 FOV の初期ターゲット領域の割合を、次の式を使用して計算します。

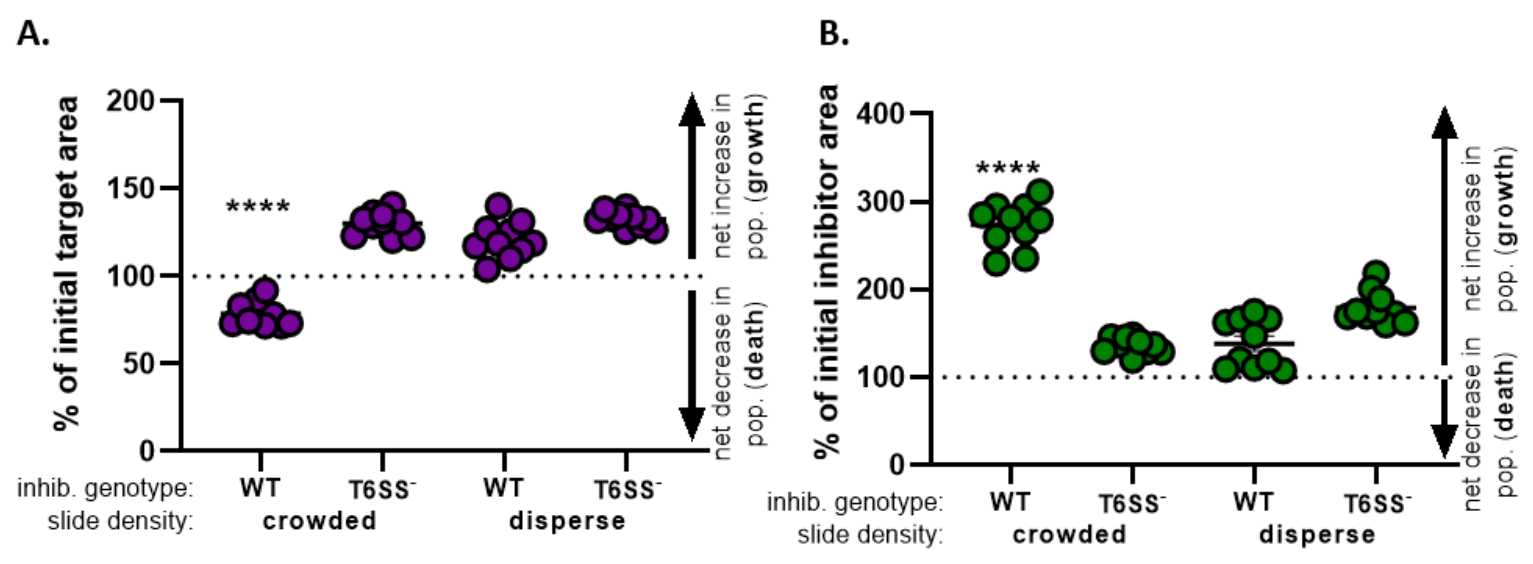
- すべての処理についてこの計算を繰り返し、各処理について初期目標領域のパーセント(ステップ8.2からの式の結果)をグラフ化します(図4A)。
- 対象集団の純増加(成長を示す)、対象集団の純減少(死亡を示す)、または各治療の変化(成長または死を示さない)があるかどうかを判断する。
注: 100 より大きい値を持つ初期ターゲット領域のパーセントは、正味ターゲットの成長を示し、100 より小さい値は正味目標死を示します。100 に残る初期目標値のパーセントは、ターゲットの人口に正味の変化がないことを示します。推奨されるフォローアップ実験については、議論を参照してください。
9. 初期インヒビター領域の時間経過に伴う割合の計算
- ステップ8.1~8.3を繰り返し、今度はセクション7の阻害剤株の蛍光チャネルから収集した測定値を用いて(図4B)。。
- 阻害剤の集団の純増加があったかどうかを判断します (成長);阻害剤集団の純減少(死亡)、または各治療に変化はない。100を超える値は、正味阻害剤の成長を示し、100未満の値は、ネット阻害剤の死亡を示す。
結果
単一細胞レベルでの細菌間の競合相互作用を可視化し定量化するために、確立されたCFUベースのアッセイ1,2を改変することにより、V.fischeri用にプロトコルを開発し最適化しました。この方法は、GFP−およびdsRed符号化安定なプラスミドを利用して、V.フィシェリの異なる株を視覚的に区別する。これらの相互作用の競合的な結果は、オープンソースソフトウェアFIJIを使用して、このアッセイから取得した画像を分析することによって定量化することができます。一例として、以下の実験をV.フィシェリ分離株を用いて行った。阻害剤株は、GFPをコードするプラスミドを収容し、標的株はdsRedをコードするプラスミドを収容した。阻害剤によってコードされるT6SS2が接触依存性の殺し込みメカニズムであることを考えると、細胞が混雑(高細胞接触)または分散(低細胞細胞接触)であった場合に、このアッセイの最終結果に対する実験セットアップの影響を強調する治療が含まれていた。サンプルデータでは、競合株を1:1比で混合し、アガロースパッド上で2時間インキュベートし、初期および最終(2h)画像の両方を撮影した。対照として、T6SS2変異株も、混雑状態と分散条件の両方で標的株を用いてコインキュベートした。各株の培養物を上述のように調製し、コインキュベートし、スライドを図1に示すように調製した。
図2 は、初期および最終時点で同じ視野を持つ各実験治療の代表的な蛍光顕微鏡画像を示す。各処置について、GFPをコードするプラスミドを収容する野生型阻害剤またはT6SS変異株のいずれかを、dsRedコードプラスミドを収容する標的株と1:1比で混合した。この実験セットアップで2hのコインキュベーション期間中に、成長する V.フィシェリ 細胞は1-2の分裂を通過する可能性があります(図2;灰色の矢印)。 図2Aにおいて、細胞細胞接触は、スライド上にスポッティングする前に混合培養物を濃縮することによって標的と阻害剤との間に強制された。複数の標的細胞が2時間の間に丸くなったり消えたりすることが観察され、阻害剤によって排除される標的細胞と一致する(図2;白い矢印)。丸めまたはターゲットセルのリジングの解釈の詳細については、「 議論 」セクションを参照してください。 図2Bでは、同じコインキュベーションがスライド上に発見され、今回は混合培養物を濃縮することなく細胞が分散したままになり、スライド上の株間の接触が最小限であった。ここで、標的細胞が消失または丸くなることは観察されず、この治療において標的株が阻害されないことを示唆している。 図2C および 図2D は、上述した同一の混和処理と分散処理を示し、今回はT6SS変異体を阻害剤株として用いた。標的細胞は、T6SS変異体を混用または分散した状態でコインキュベートした場合に消失または丸くなることは観察されなかったが、いずれの治療でも標的が阻害されないことを再び示唆した。
図 3は、このプロトコルで競合を定量化するために使用される FIJI 分析ワークフローを示しています。ターゲットチャネルの代表的な画像が選択され (図 3A)、フィジーのデフォルトのしきい値設定を使用してバイナリマスクが作成されました (図 3B)。画像スケールは、この顕微鏡の設定に適して設定されました。パーティクルは、サイズパラメータ = 0 - 無限大、円形パラメータ = 0.00 ~ 1.00、および [アウトラインの表示]が選択された状態で分析されました (図 3C)。この粒子分析の結果は、各粒子の番号付きアウトライン(図3D)と、粒子番号、ファイル名(ラベル)、および粒子面積(面積)のカラム(面積)の両方として示される(図3E)。
図4では、図3Eから得られたデータをグラフ化し、分析した。図4Aでは、ステップ8.2に従って、最終時点での初期目標領域の割合が各治療に対して提示される。初期標的領域の割合が100を超える場合、これは目標の正味の増加(すなわち、成長)を表し、標的集団が有意に阻害されない条件で観察される。しかし、初期標的領域の割合が100より低い場合、この結果は標的の正味減少(すなわち、死)を示し、標的集団が有意に阻害される条件で観察される。標的を混入条件下で野生型阻害剤でコインキュベートした場合、データは標的領域の純減少を示す。対照的に、標的を分散条件で野生型阻害剤またはT6SS変異体を混血または分散状態でコインキュベートした場合、データは標的領域の純増加を示す。標的が混雑した状態で野生型阻害剤でコインキュベートされたときの初期標的領域の割合は100未満であり、一方通行のANOVAに従って他のすべての治療法よりも有意に低く、その後、すべての治療にわたるTukeyの複数比較テスト(p < 0.0001)であった。これらのデータは、標的細胞死が阻害剤における機能的T6SSに依存していることを示し、接触依存性殺死機構からの細胞死を検出するために、十分な細胞と細胞接触を可能にする実験的なセットアップの重要性を強調する。
図4B は、各治療の最終時点における初期阻害剤面積の割合を示す。この例では、阻害剤株の純成長は全ての治療にわたって観察された。しかし、野生型阻害剤が一方向のANOVAに従って他のすべての治療法と比較して混雑した状態で標的とコインキュベートされた場合、初期阻害剤領域の割合は有意に高かった(p < 0.0001)。当初、阻害剤領域の正味の増加は、標的細胞が排除されるにつれて成長するために利用可能なスペースの増加によって駆動され得ると考えました。しかし、これと同じ阻害剤増殖の増加は、阻害剤細胞がコインキュベーションの初めから成長する余地があった分散治療では認められなかった。あるいは、この結果は、標的細胞をライシングから放出される栄養素が阻害剤集団のより大きな増加を可能にすることを示唆することができる。これらの結果をまとめて、阻害剤株は、スライド上の細胞を混雑させることによって細胞間の高い接触が強制された場合にのみ、T6SS依存的な方法で標的を排除することを示唆している。

図1:コインキュベーションアッセイ用のアガロースパッド調製とスライドセットアップ(A)2%アガロースパッドを作るためのセットアップ。ラボテープ(緑)の5層は、約20mm離れた2点のカバースリップに巻き付けられます。次に、mPBS(黄色)の2%アガロースをテープの間にピペットで、すぐに25mm2カバースリップで覆い、室温で少なくとも1時間固化させた。カミソリの刃を使用してアガロースパッドを〜5mm2個にカットし、ピンセットを使用してパッドを新しいスライドに移してイメージングします。(B)直立した顕微鏡で撮像する場合は、5mm2アガロースパッドをスライドに直接置き、混合培養(青)と12mm円形#1.5カバースリップに続きます。(C)反転顕微鏡でイメージングする場合は、混合培養物を35mmペトリ皿の#1.5ガラスカバースリップボトムに直接見つけ、培養物の上にアガロースパッドを置き、続いて2番目の12mm円形カバースリップを置いてアガロースパッドを平らにします。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図2:混雑状態または分散状態におけるコインキュベーションスポットのタイムラプス画像(A)は、対象と野生型阻害剤の混合培養がスライド上で発見する前に3倍集中し、株間の細胞細胞接触を強制する初期および最終時点における代表的な画像である。TRITCチャネルの白い矢印は、実験の過程を通して丸めまたはライスする標的細胞の例を示す。(B)標的と野生型阻害剤の混合培養が濃縮されずに発見された代表的な画像で、細胞が分散し、株間の細胞間接触が最小限となる。FITCおよびTRITCチャンネルのグレーの矢印は、実験の過程で細胞分裂の例を示しています。(C)標的とT6SSの混合培養-突然変異体が、株間の細胞細胞接触を強制するためにスライド上でスポッティングする前に3倍濃縮した代表的な画像。(D)標的とT6SSの混合培養が、細胞が分散し、株間の細胞間接触が最小限となるように濃縮せずに変異体を発見した代表的な画像。スケールバー = 5 μm、すべての画像で一貫しています。TRITCチャンネルは偽色のマゼンタであり、FITCチャンネルは偽色の緑色である。デコンボリューションはすべての画像に対して行われました。背景を減算し、すべての画像で明るさ/コントラストが均一に調整されました。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図3:フィジー分析ワークフロー (A)解析の代表的な画像。このワークフローは、ビューとサンプルのすべてのフィールドの両方のチャネルに対して繰り返されます。スケールバー = 5 μm、すべての画像で一貫しています。TRITCチャンネルは偽色のマゼンタであり、FITCチャンネルは偽色の緑色である。(B) フィジーのデフォルト設定を使用して画像をしきい値にすることで作成されたバイナリマスク。(C)この原稿で使用する粒子分析の設定例サイズ範囲 = 0 - 無限大 μm2;循環性 = 0.00 - 1.00;表示 = アウトライン。(D) パーティクル分析の出力として作成された粒子の輪郭 (C).(D)の粒子の輪郭は、元の画像 (A) と比較して、すべての細胞がパーティクル分析で確実にキャプチャされるようにする必要があります。(E) 結果テーブルは、(C)での粒子解析からの出力として作成された。オブジェクト番号(列1)は、パネル(D)で赤で輪郭を描かれ、ラベル付けされた個々の粒子(1つまたは複数のセル)に対応します。ラベル = 分析されたイメージのファイル名。面積 = μm2の総粒子面積。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図4:標的株が阻害されているかどうかを評価するためのサンプルデータ。ターゲット株の最終時点(A)および阻害剤株(B)における初期領域のパーセントを、異なる初期細胞密度で示す。スライド密度は、図2に記載されているように、混雑している開始細胞密度(株間の高い細胞接触)、またはより分散(株間の低い細胞接触)のいずれかを示す。阻害剤遺伝子型は、野生型またはT6SS変異体(T6SS-)株のいずれかを標的株でコインキュベーションしたことを示す。アスタリスクは、すべての治療を比較する変化率の有意差を示しています(一方向の分散分析、それに続くTukeyの多重比較テストがすべての治療を比較します。(p < 0.0001)破線は、初期時間と最終タイムポイントの間のひずみ領域に正味変化がないことを示します。100>%の変化は、純増加(すなわち、成長)を示し、100<の変化率は、純減少(すなわち細胞死)を示す。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
ディスカッション
上述のプロトコルは、単一細胞レベルでの細菌間競争を定量化し、特性を示す強力なツールを提供します。このアッセイは、CFUベースの競争アッセイを寒天プレート1,2に変更することで開発され、V.フィッシャーリ分離物間の単細胞競争の可視化を可能にし、幅広いシステムおよび顕微鏡用セットアップの方法を最適化するための提案が提供されています。ここで説明する方法は、光器官共生V.fischeriのために最適化されたが、それは容易に多くの多様な、化学的な微生物を収容するために変更することができる。競合メカニズムは、温度、多量、および粘度30、31、32、33、34を含む任意の数の環境変数によって制御できることに注意することが重要です。これまでの研究では、V.fischeriが表面30上で活性である接触依存型VI分泌系を使用して競合することを確認しており、このアッセイに記載されている条件は、例株間の競争を研究するのに適している。また、細菌の競合を定量化する際には、スライド上の細胞の初期密度を考慮することも重要です。殺すために標的細胞と阻害剤細胞との接触がしばしば必要であることを考えると、混合培養は細胞細胞の接触が最大化され、細胞がスライド上の単一の平面に残るように濃縮されるべきである。細胞培養は、同様の光学密度(中ログ段階)に成長し、異なる増殖段階における細胞の生理学的変化のために単に高い光学密度に培養を成長させるのではなく、接触を強制するために濃縮されるべきである。他のシステムでは、培養条件および実験セットアップは、競合機構がアクティブであり、コインキュベーション状態で検出できることを確認するために変更する必要があります。
このアッセイで使用されるアガロースパッドは、細胞が自由に動き回らないように安定化を提供し、実験の過程で培養物が乾燥するのを防ぐといういくつかの利点を提供します。さらに、イソプロピルβ-D-チオガラクトシド(IPTG)などの化学誘導剤が実験に必要な場合、アガロース溶液に容易に添加することができます。しかし、アガロース製剤は、異なるシステムのために調整する必要がある可能性が高いことに注意することが重要です。上述した例では、アガロースパッドを2%アガロース(w/v)を20 psu mPBSに溶解して調製した。 さらに、細胞が成長し、より長い実験で競争するために、炭素源をアガロースパッドに加える必要がある場合もあります。このような場合、アガロースパッド中のmPBSは、任意の増殖培地に置き換えることができるが、成長培地中の栄養素は、追加のバックグラウンド蛍光のトレードオフを伴い得る。
独自の画像解析ソフトウェアがなければ、セルとセルの接触が高いときに個々の細胞数を得ることは非常に困難であり、ここで示すように接触依存性の殺害を観察するために必要とされる。このアッセイは、個々の細胞数に依存しない定量化のための代替方法を提供するように設計された。代わりに、各蛍光チャネルの全細胞面積を使用して、コインキュベート株間の殺死の程度を定量化します。この方法は個々のセル数ではなく面積に依存するため、通常、セル全体の面積をアウトライン化するには、既定のしきい値設定で十分です。閾値の精度は、代表的視野の総対象領域をモデル生物の平均セルサイズで割り、この推定セル数を同じ画像の手動セル数と比較することによって検証できます。
1つの阻害剤と1つの標的(非キラー)株との間のコインキュベーションにおいて、阻害剤の純成長が予測される。図4に示すように、標的細胞をライシングすることによって放出される栄養素が阻害剤株をより迅速に成長させるためか、殺しが観察されない治療と比較して、殺しが観察される治療において阻害剤の成長が有意に高い可能性がある。ここに示す例では、T6SS媒介性の競争が標的が物理的に排除される標的細胞のリシスをもたらすため、正味標的死が観察される。しかし、すべての競合メカニズムが標的細胞の物理的な排除をもたらすわけではないことに注意することが重要です。成長阻害を引き起こす毒素によって標的が無力化された場合、ここで概説するプロトコルは、標的細胞が成長しなくなるだけでなく、lyseもなくなるので、目に見える標的集団が時間の経過とともに安定したままになる可能性がある。このような場合、コロニー形成ユニット(CFUs)のめっきなどの標的細胞生存率に関するフォローアップテストと、ヨウ化プロピジウムまたはSYTOXグリーン35,36で染色して生死性アッセイを行うことにより、このアッセイの結果を比較することが適切であろう。
CFUカウントに依存するコインキュベーションアッセイと比較して、このアッセイは、株間の競争の空間構造を観察し、定量化し、時間の経過とともに標的細胞形態の変化を追跡することを可能にする。例えば、T6SSを用いて殺すインヒビター細胞は、標的細胞壁を分解するLysMドメインタンパク質をコードすることが知られており、その結果、初期細胞丸めおよび次に、リシス13が、 図2Aに示す例で観察した。さらに、このプロトコルは、非常に短い時間スケールで高解像度で競合を追跡するために使用することができます。ここに示す例では、細胞が混雑し、細胞と細胞の接触が株間に強制されると、わずか2時間後に標的領域の有意な減少が観察される(図4)。ここで説明する画像解析は共焦点顕微鏡法を用いて行うこともでき、コインキュベート株の空間分布を妨げることなく 、生体内 または複雑なバイオフィルムで細菌の競争を研究することが可能になる。
要約すると、ここで説明するアッセイは、蛍光顕微鏡を用いて単細胞レベルで細菌の競争を可視化し、定量するためのアクセス可能で簡単に変更されたアプローチを提供することを目的としている。この方法は、多様な細菌分離物に適用することができ、宿主またはバイオフィルムマトリックス内のような複雑な環境でも細菌の競争を視覚化するために使用することができる。
開示事項
著者らは開示するものは何もない。
謝辞
A.N.SはNIGMS助成金R35 GM137886によって支援され、S.N.Sは国防科学技術大学院フェローシッププログラムによって支援されました。
資料
| Name | Company | Catalog Number | Comments |
| 1.5 mL Microcentrifuge tube | Fisher | 05-408-129 | |
| 10 uL single channel pipette | |||
| 1000 uL single channel pipette | |||
| 20 uL single channel pipette | |||
| 200 uL single channel pipette | |||
| Agarose | Fisher | BP165-25 | Low melting agarose |
| Calculator | |||
| Cellvis 35 mm Dish | Fisher | NC0409658 | #1.5 cover glass bottom |
| Chloramphenicol | Sigma | C0378 | stock (20 mg/mL in Ethanol); final concentration in media (2 μg /mL LBS) |
| DAPI Nucleic Acid Stain | Fisher | EN62248 | optional (if not using stable plasmids) |
| FIJI image analysis sofware | ImageJ | https://imagej.net/Fiji/Downloads | open-source software |
| Fisherbrand Cover Glasses: Circles | Fisher | 12-545-81P | #1.5 cover glass; 12 mm diameter |
| Kanamycin Sulfate | Fisher | BP906-5 | stock (100 mg/mL in water, filter sterilize); final concentration in media (1 μg/mL LBS) |
| Lens Cleaning Tissue Paper | Fisher | S24530 | |
| Parafilm | Fisher | 13-374-12 | |
| Petri Plates | Fisher | FB0875713 | sterile with lid |
| Razor Blades | Fisher | S65921 | |
| Semi-micro Cuvettes | VWR | 97000-586 | |
| Spectrophotometer | |||
| SYBR Green Nucleic Acid Stain | Fisher | S7563 | optional (if not using stable plasmids) |
| Thermo Scientific Gold Seal Plain Microscope Slides | Fisher | 12-518-100B | |
| Thermo Scientific Richard-Allan Scientific Cover Glass | Fisher | 22-050-235 | #1.5 cover glass, 25 mm2 |
| Type F Immersion Oil | Fisher | NC0297589 | |
| Upright or inverted fluorescence microscope with camera and imaging software | Images in this article were acquired on a Nikon TI-2 inverted fluorescent microscope outfitted with an ORCA-Fusion Digital CMOS camera using NIS-Elements software. | ||
| Vortex | |||
| Water bath | Used to keep agarose warm prior to pipetting | ||
| LBS media | |||
| 1M Tris Buffer (pH ~7.5) | 50 mL 1 M stock buffer (62 mL HCl, 938 mL DI water, 121 g Trizma Base) | ||
| Agar Technical | Fisher | DF0812-17-9 | 15 g (Add only for plates) |
| DI water | 950 mL | ||
| Sodium Chloride | Fisher | S640-3 | 20 g |
| Tryptone | Fisher | BP97265 | 10 g |
| Yeast Extract | Fisher | BP9727-2 | 5 g |
| mPBS (marine PBS) | Phosphate buffered saline with marine salts added; used for making agarose pad | ||
| 10X PBS | Fisher | ICN1960454 | |
| Instant Ocean Sea Salt | Instant Ocean | SS1-160P | Adjust concentration to appropriate salinity; 20 psu used here |
| Sterile Vacuum Filter Units | Fisher | SCGVU01RE | Used to filter-sterilize mPBS |
| Vacuum pump | Used to filter-sterilize mPBS |
参考文献
- Speare, L., et al. Bacterial symbionts use a type VI secretion system to eliminate competitors in their natural host. Proceedings of the National Academy of Sciences. 115 (36), 8528-8537 (2018).
- Speare, L., Septer, A. N. Coincubation assay for quantifying competitive interactions between Vibrio fischeri isolates. Journal of Visualized Experiments. (149), e59759 (2019).
- Frost, I., et al. Cooperation, competition and antibiotic resistance in bacterial colonies. The ISME journal. 12 (6), 1582-1593 (2018).
- Stubbendieck, R. M., Vargas-Bautista, C., Straight, P. D. Bacterial communities: interactions to scale. Frontiers in Microbiology. 7, 1234 (2016).
- Souza, D. P., et al. Bacterial killing via a type IV secretion system. Nature Communications. 6 (1), 1-9 (2015).
- Anderson, M. C., Vonaesch, P., Saffarian, A., Marteyn, B. S., Sansonetti, P. J. Shigella sonnei encodes a functional T6SS used for interbacterial competition and niche occupancy. Cell Host and Microbe. 21 (6), 769-776 (2017).
- Basler, M., Ho, B., Mekalanos, J. Tit-for-tat: Type VI secretion system counterattack during bacterial cell-cell interactions. Cell. 152 (4), 884-894 (2013).
- Guillemette, R., Ushijima, B., Jalan, M., Häse, C. C., Azam, F. Insight into the resilience and susceptibility of marine bacteria to T6SS attack by Vibrio cholerae and Vibrio coralliilyticus. PloS One. 15 (1), 0227864 (2020).
- Hachani, A., Lossi, N. S., Filloux, A. A visual assay to monitor T6SS-mediated bacterial competition. Journal of Visualized Experiments. (73), e50103 (2013).
- Hibbing, M. E., Fuqua, C., Parsek, M. R., Peterson, S. B. Bacterial competition: surviving and thriving in the microbial jungle. Nature Reviews Microbiology. 8 (1), 15-25 (2010).
- Ruhe, Z. C., Low, D. A., Hayes, C. S. Bacterial contact-dependent growth inhibition. Trends in Microbiology. 21 (5), 230-237 (2013).
- Wood, D. W., Pierson, L. S. The phzI gene of Pseudomonas aureofaciens 30-84 is responsible for the production of a diffusible signal required for phenazine antibiotic production. Gene. 168 (1), 49-53 (1996).
- Smith, W. P., et al. The evolution of the type VI secretion system as a disintegration weapon. PLoS Biology. 18 (5), 3000720 (2020).
- Chen, L., Zou, Y., She, P., Wu, Y. Composition, function, and regulation of T6SS in Pseudomonas aeruginosa. Microbiological Research. 172, 19-25 (2015).
- Sana, T. G., Lugo, K. A., Monack, D. M. T6SS: The bacterial "fight club" in the host gut. PLoS Pathogens. 13 (6), 1006325 (2017).
- Basler, M. Type VI secretion system: secretion by a contractile nanomachine. Philosophical Transactions of the Royal Society B: Biological Sciences. 370 (1679), 20150021 (2015).
- Joshi, A., et al. Rules of engagement: the type VI secretion system in Vibrio cholerae. Trends in Microbiology. 25 (4), 267-279 (2017).
- Nadell, C. D., Drescher, K., Foster, K. R. Spatial structure, cooperation and competition in biofilms. Nature Reviews Microbiology. 14 (9), 589-600 (2016).
- Stubbendieck, R. M., Straight, P. D. Multifaceted interfaces of bacterial competition. Journal of Bacteriology. 198 (16), 2145-2155 (2016).
- Septer, A. N. The Vibrio-squid symbiosis as a model for studying interbacterial competition. Msystems. 4 (3), (2019).
- Tischler, A. H., Hodge-Hanson, K. M., Visick, K. L. Vibrio fischeri-squid symbiosis. eLS. , 1-9 (2019).
- Mandel, M. J., Dunn, A. K. Impact and influence of the natural Vibrio-squid symbiosis in understanding bacterial-animal interactions. Frontiers in Microbiology. 7, 1982 (2016).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Boettcher, K., Ruby, E. Depressed light emission by symbiotic Vibrio fischeri of the sepiolid squid Euprymna Scolopes. Journal of Bacteriology. 172 (7), 3701-3706 (1990).
- Doino, J. A., McFall-Ngai, M. J. A transient exposure to symbiosis-competent bacteria induces light organ morphogenesis in the host squid. The Biological Bulletin. 189 (3), 347-355 (1995).
- Dunn, A. K., Millikan, D. S., Adin, D. M., Bose, J. L., Stabb, E. V. New rfp-and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Applied and Environmental Microbiology. 72 (1), 802-810 (2006).
- Lambertsen, L., Sternberg, C., Molin, S. Mini-Tn7 transposons for site-specific tagging of bacteria with fluorescent proteins. Environmental Microbiology. 6 (7), 726-732 (2004).
- Koch, B., Jensen, L. E., Nybroe, O. A panel of Tn7-based vectors for insertion of the gfp marker gene or for delivery of cloned DNA into Gram-negative bacteria at a neutral chromosomal site. Journal of Microbiological Methods. 45 (3), 187-195 (2001).
- Peterson, B. W., Sharma, P. K., Van Der Mei, H. C., Busscher, H. J. Bacterial cell surface damage due to centrifugal compaction. Applied and Environmental Microbiology. 78 (1), 120-125 (2012).
- Speare, L., Smith, S., Salvato, F., Kleiner, M., Septer, A. N. Environmental viscosity modulates interbacterial killing during habitat transition. MBio. 11 (1), (2020).
- Salomon, D., Gonzalez, H., Updegraff, B. L., Orth, K. Vibrio parahaemolyticus type VI secretion system 1 is activated in marine conditions to target bacteria, and is differentially regulated from system 2. PloS One. 8 (4), 61086 (2013).
- Sana, T. G., et al. Salmonella Typhimurium utilizes a T6SS-mediated antibacterial weapon to establish in the host gut. Proceedings of the National Academy of Sciences. 113 (34), 5044-5051 (2016).
- Bachmann, V., et al. Bile salts modulate the mucin-activated type VI secretion system of pandemic Vibrio cholerae. PLoS Neglected Tropical Diseases. 9 (8), 0004031 (2015).
- Ishikawa, T., et al. Pathoadaptive conditional regulation of the type VI secretion system in Vibrio cholerae O1 strains. Infection and Immunity. 80 (2), 575-584 (2012).
- Johnson, M. B., Criss, A. K. Fluorescence microscopy methods for determining the viability of bacteria in association with mammalian cells. Journal of Visualized Experiments. (79), e50729 (2013).
- Stiefel, P., Schmidt-Emrich, S., Maniura-Weber, K., Ren, Q. Critical aspects of using bacterial cell viability assays with the fluorophores SYTO9 and propidium iodide. BMC Microbiology. 15 (1), 36 (2015).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved