
Method Article
Quantifizierung der interbakteriellen Konkurrenz mittels Einzelzell-Fluoreszenzbildgebung
In diesem Artikel
Zusammenfassung
Dieses Manuskript beschreibt eine Methode zur Verwendung der Einzelzellfluoreszenzmikroskopie zur Visualisierung und Quantifizierung der bakteriellen Konkurrenz in der Kokultur.
Zusammenfassung
Interbakterielle Konkurrenz kann sich direkt auf die Struktur und Funktion von Mikrobiomen auswirken. Diese Arbeit beschreibt einen Fluoreszenzmikroskopie-Ansatz, mit dem kompetitive Wechselwirkungen zwischen verschiedenen Bakterienstämmen auf Einzelzellebene visualisiert und quantifiziert werden können. Das hier beschriebene Protokoll bietet Methoden für fortgeschrittene Ansätze in der Objektträgervorbereitung auf aufrechten und invertierten Epifluoreszenzmikroskopen, Lebendzell- und Zeitrafferbildgebungsverfahren sowie quantitative Bildanalyse mit der Open-Source-Software FIJI. Der Ansatz in diesem Manuskript skizziert die Quantifizierung kompetitiver Interaktionen zwischen symbiotischen Vibrio fischeri-Populationen, indem die Flächenänderung im Laufe der Zeit für zwei koincubatierte Stämme gemessen wird, die verschiedene fluoreszierende Proteine aus stabilen Plasmiden exprimieren. Es werden alternative Methoden zur Optimierung dieses Protokolls in bakteriellen Modellsystemen beschrieben, die unterschiedliche Wachstumsbedingungen erfordern. Obwohl der hier beschriebene Assay für V. fischerioptimierte Bedingungen verwendet, ist dieser Ansatz hochgradig reproduzierbar und kann leicht angepasst werden, um den Wettbewerb zwischen kultivierbaren Isolaten aus verschiedenen Mikrobiomen zu untersuchen.
Einleitung
Dieser Artikel beschreibt eine Methode zur Quantifizierung der bakteriellen Konkurrenz auf Einzelzellebene mittels Fluoreszenzmikroskopie. Die Struktur und Funktion mikrobieller Gemeinschaften wird oft durch kompetitive Interaktionen zwischen Mikroben geprägt, und in vielen Fällen erfordert die Charakterisierung dieser Interaktionen die Beobachtung verschiedener Bakterienstämme in Kokubation1,2,3,4,5,6,7,8 . Traditionell wird die bakterielle Konkurrenz auf Populationsebene quantifiziert, indem koloniebildende Einheiten (CFUs) von Inhibitoren und Zielstämmen vor und nach einer Kokubationszeit2,9gezählt werden. Mechanismen für den mikrobiellen Wettbewerb sind breit zwischen Bakterien verteilt und können entweder auf Diffusion oder Zell-Zell-Kontakt beruhen, um Zielzellen zu hemmen10,11,12,13,14,15,16,17,18,19.
Obwohl Bakterienstämme häufig bei der Kokubation auf Populationsebene beobachtet werden, skizziert dieses Manuskript einen Assay zur Einzelzellquantifizierung der bakteriellen Konkurrenz. Darüber hinaus enthält diese Arbeit Vorschläge zur Anpassung des Protokolls für die Verwendung mit anderen Bakterienarten. Während die spezifischen Techniken in diesem Artikel verwendet werden, um die kontaktabhängige intraspezifische Konkurrenz zwischen Stämmen des symbiotischen Bakteriums Vibrio fischeri20,21,22zu untersuchen, können sie für die Konkurrenz zwischen vielen Organismen angepasst werden. Dieser Artikel enthält Anweisungen für die Objektträgereinrichtung sowohl auf aufrechten als auch auf invertierten Mikroskopen, und alle Analysen werden mit der Open-Source-Software FIJI23 beschrieben, so dass die Methode von Forschern mit Zugriff auf verschiedene Bildgebungs-Setups und Analyseprogramme verwendet werden kann. Angesichts der Bedeutung der Untersuchung des mikrobiellen Wettbewerbs sowohl auf Bevölkerungs- als auch auf Einzelzellebene wird diese Methode eine wertvolle Ressource für Forscher sein, um kompetitive Interaktionen zu quantifizieren, insbesondere solche, die keinen Zugang zu proprietärer Analysesoftware haben.
Protokoll
1. Optimierung von Bakterienstämmen
- Wählen Sie zwei Bakterienstämme für einzellige Bakterienkonkurrenztests. Hier werden zwei Stämme von V. fischeri verwendet: ein Zielstamm (ES11424) und ein Inhibitorstamm (MJ1125), von dem bekannt ist, dass er den Zielstamm mit dem Typ-VI-Sekretionssystem auf Chromosom II (T6SS2)1abtöten kann, was ein kontaktabhängiger Tötungsmechanismus ist.
- Bestimmen Sie die geeigneten Steuerelemente für das Experiment. In diesem Beispiel besteht die geeignete Kontrolle darin, sowohl den Wildtyp- als auch den T6SS-Mutanteninhibitorstamm mit dem Zielstamm zu inkubieren, um die Wirkung der T6SS-vermittelten Tötung zu quantifizieren.
HINWEIS: Zusätzliche Kontrollen können einen Zielstamm umfassen, der die notwendigen Immunitätsgene exprimiert, um eine T6SS-abhängige Tötung zu verhindern, oder einen Inhibitor-Mutantenstamm, der Wildtyp-Kopien der mutierten Gene in trans exprimiert, um die T6SS-Aktivitätwiederherzustellen 1. - Wenn möglich, transformieren Sie Stämme mit stabilen Plasmiden, die Gene für verschiedene fluoreszierende Proteine (z. B. GFP oder RFP) kodieren, um Stammtypen auf dem Mikroskop visuell zu unterscheiden. Hier wird der Inhibitorstamm mit einem GFP-kodierenden Plasmid (pVSV102) und der Zielstamm mit einem dsRed-kodierenden Plasmid (pVSV208)26markiert.
HINWEIS: Wenn es nicht möglich ist, stabile Plasmide zu verwenden, können fluoreszierende Tags zur Visualisierung auf das bakterielle Chromosom27,28eingeführt werden. - Bilden Sie während der anfänglichen Optimierungsphase klonale Kulturen der markierten Stämme unter jedem der fluoreszierenden Filter ab, die während des Experiments verwendet werden, um sicherzustellen, dass Zellen nur im beabsichtigten Kanal sichtbar sind. Stellen Sie beispielsweise sicher, dass eine GFP-markierte Dehnung nur im FITC-Kanal sichtbar ist.
2. Agarose Pad Vorbereitung
- Bereiten Sie die Agarose-Pad-Lösung vor, indem Sie 2% schmelzarme Agarose (w / v) in mPBS auflösen. Erhitzen Sie die Lösung kurz in der Mikrowelle und im Wirbel, bis die Agarose vollständig aufgelöst ist. Halten Sie diese Lösung warm, indem Sie sie in ein 55 ° C warmes Wasserbad geben, bis sie einsatzbereit ist. Weitere Informationen zur Vorbereitung von Agarose-Pads finden Sie im Abschnitt Diskussion.
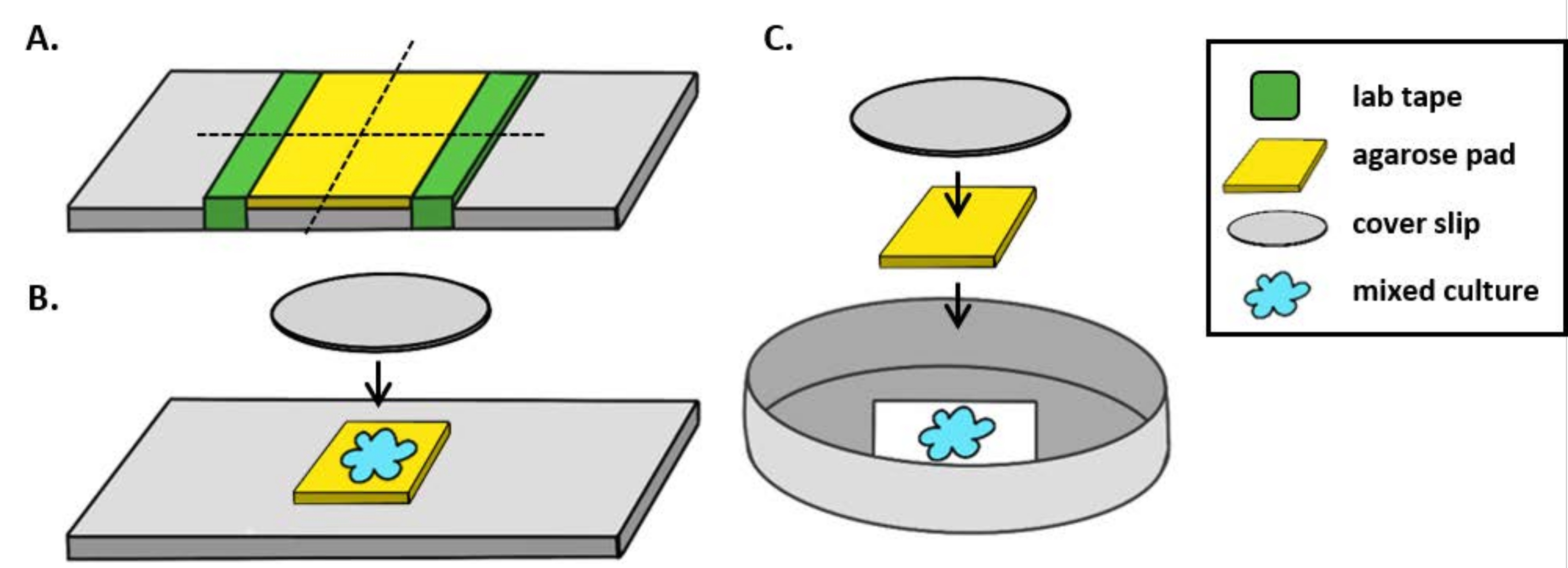
HINWEIS: Hier wurde mPBS durch Zugabe von 20 g / L NaCl zu Standard 1x PBS hergestellt. - Wickeln Sie fünfmal ein Stück Laborband um einen Glasträger. Wiederholen Sie diesen Vorgang ein zweites Mal auf demselben Dia, sodass der Abstand zwischen den beiden Bandstücken etwas kleiner ist als die Breite eines Abdeckzettels (Abbildung 1A). Wenn Sie beispielsweise 25 mm2 Abdeckstücke verwenden, sollten die Bandstücke etwa 20 mm voneinander entfernt sein.
HINWEIS: Während die Häufigkeit, mit der das Band um den Dia gewickelt wird, geändert werden kann, um die Dicke des Agarose-Pads einzustellen, ist es wichtig, dass die Bandschichten auf beiden Seiten des Dias gleich hoch sind, damit das Agarose-Pad flach bleibt. - Warme Agaroselösung zwischen den beiden Bandstücken pipetten und sofort mit einem Abdeckeln aufdecken, so dass sie auf den Bandstücken aufliegt. Dadurch wird sichergestellt, dass die Oberfläche des Agarose-Pads flach bleibt. Das Volumen der in diesem Schritt pipetierten Agaroselösung sollte ausreichen, damit der Abdeckklip mit der Flüssigkeit in Kontakt kommt und Blasen in der Agaroselösung herausdrückt. Für diesen speziellen Aufbau sind 200 μL warme Agarose ausreichend.
- Lassen Sie das Agarose-Pad vor dem Kozifizierungstest mindestens 1 h lang bei Raumtemperatur erstarren. Schritt 2.2 wird ein Agarose-Pad von ca. 20 mm2 erzeugen.
- Schneiden Sie dieses Agarose-Pad mit einer Rasierklinge in vier, 5 mm2 Pads, die für die Bildgebung verwendet werden.
HINWEIS: Agarose-Pads können bis zu einer Woche vor dem Experiment hergestellt und bei 4 ° C in einer leeren, sterilen Petriplatte gelagert werden, die mit Parafilm versiegelt ist, um das Austrocknen zu verhindern.
3. Stämme für die Co-Inkubation vorbereiten
- Streichen Sie jeden Stamm, der im Kozubationstest verwendet werden soll, aus -80 °C-Beständen auf LBS-Agarplatten, die mit den entsprechenden Antibiotika ergänzt sind, und inkubieren Sie sie über Nacht bei 24 °C. Für dieses Beispiel werden drei Stämme verwendet: der Wildtyp-Inhibitor-Stamm, die Typ-VI-Sekretsystem-Mutante und der Zielstamm.
- Beginnen Sie am nächsten Tag Kulturen über Nacht in biologischen Duplikaten, indem Sie zwei Kolonien aus jedem Stamm auswählen und sie in LBS-Medium, ergänzt mit den entsprechenden Antibiotika, erneut inspazieren und über Nacht bei 24 ° C mit Schütteln bei 200 U / min inkubieren.
- Am nächsten Morgen repliziert jede biologische Subkultur 1:1000 in frisches LBS-Medium ohne Antibiotika und inkubiert bei 24°C mit Schütteln für 4-5 h oder bis die Zellen einen OD600 von ~1,5 erreichen.
HINWEIS: Der Zeitpunkt der Schritte 3.1, 3.2 und 3.3 muss möglicherweise für verschiedene Bakterienarten optimiert werden, da ihre Wachstumsrate erheblich variieren kann. Für diesen Assay sollten sich die Zellen zu Beginn des Coincubation-Assays in der Mid-Log-Phase befinden.
4. Bakterienstämme koinkubieren
- Beginnend mit Mid-Log-Kulturen ab Schritt 3.3 messen und zeichnen Sie die optische Dichte bei 600 nm (OD600)für alle Proben auf.
- Normalisieren Sie jede Probe auf einen OD600 = 1,0, was ungefähr 109 KBE/ml für V. fischerientspricht, indem Sie die Kultur mit LBS-Medium verdünnen.
- Mischen Sie die beiden konkurrierenden Stämme in einem Volumenverhältnis von 1:1 zusammen, indem Sie 30 μL jeder normalisierten Dehnung in ein markiertes 1,5-ml-Röhrchen geben. Wirbeln Sie die Mischsortekultur für 1-2 s durch.
HINWEIS: In einigen Fällen kann es angebracht sein, Kokulturen in verschiedenen Verhältnissen zu mischen. Wenn zum Beispiel eine Sorte viel schneller wächst als die andere, kann es notwendig sein, die langsamer wachsende Sorte mit einem numerischen Vorteil zu beginnen, um die Konkurrenz zu beobachten. Eine Optimierung kann auch erforderlich sein, wenn OD600 nicht für beide Stämme einer ähnlichen CFU/ml entspricht. - Wiederholen Sie Schritt 4.3 für jede biologische Replikation und Behandlung. Im hier gezeigten Beispiel ergeben sich daraus insgesamt vier Mischstammröhrchen: zwei biologische Replikate mit dem Wildtyp-Inhibitorstamm gemischt mit dem Zielstamm und zwei biologische Replikate mit dem Typ-VI-Sekretionssystem-Mutantenstamm gemischt mit dem Zielstamm.
- Um sicherzustellen, dass die konkurrierenden Zellen ausreichend dicht für die kontaktabhängige Tötung in der Kokubation auf dem Agarpad sind, konzentrieren Sie jede Mischkultur 3-fach, indem Sie die Mischkultur in einem Standard-1,5-ml-Zentrifugenröhrchen für 1 minute bei 21.130 x gzentrifugieren, den Überstand entsorgen und jedes Pellet in 20 μL LBS-Medium wieder aufwenden. Wiederholen Sie diesen Vorgang für jedes Beispiel.
HINWEIS: Einige Bakterienzellen reagieren empfindlich auf Schäden durch Zentrifugation bei hohem rcf; in solchen Fällen kann die Mischkultur für 3 min bei 4600 x g29zentrifugiert werden. Darüber hinaus ist es bei der Quantifizierung der kontaktabhängigen Konkurrenz wichtig, eine ausreichende Zelldichte auf dem Objektträger sicherzustellen, um die Tötung zu beobachten. In diesem Artikel hatten "überfüllte" Behandlungen, bei denen eine Tötung beobachtet wird, etwa 10 Zellen / 20 μm2; Weitere Informationen finden Sie im Abschnitt Diskussion.
5. Folieneinrichtung
- Wenn Sie ein aufrechtes Mikroskop verwenden, legen Sie ein ~ 5 mm2 Agarose-Pad auf einen Standard-1-mm-Glasträger. Erkennen Sie 2 μL einer Mischkultur auf das Agarose-Pad und legen Sie einen #1,5-Coverlip (25 mm2)über den Spot. Ein Beispiel finden Sie in Abbildung 1B.
- Wenn Sie ein invertiertes Mikroskop verwenden, entdecken Sie 2 μL einer Mischkultur auf den # 1,5 Coverlip-Boden einer 35-mm-Petrischale und legen Sie ein ~ 5 mm2 Agarose-Pad über den Kozubationsfleck. Legen Sie einen 12 mm runden Glasdeckel über das Agarose-Pad. Ein Beispiel finden Sie in Abbildung 1C.
- Wiederholen Sie Schritt 5.1 oder 5.2, je nach verwendeter Mikroskopaufbau, für die verbleibenden drei Mischkulturen, was zu vier Dias oder Schalen führt, die abgebildet werden sollen.
- Lassen Sie die Rutschen ca. 5 Minuten auf der Tischplatte sitzen, bevor Sie mit Schritt 6 fortfahren. Dadurch können sich die Zellen auf dem Agarpad ansiedeln und bewegungen während des Bildgebungsprozesses eliminiert werden.
6. Fluoreszenzmikroskopie
- Beginnen Sie mit der Fokussierung auf Zellen mit weißem Licht (Phasenkontrast oder DIC), um die Auswirkungen des Photobleichens zu minimieren. Basierend auf der durchschnittlichen Größe einer einzelnen Bakterienzelle verwenden Sie ein 60x oder 100x Ölobjektiv.
- Passen Sie die Belichtungszeit und die Aufnahmeeinstellungen für jeden Kanal so an, dass Zellen im entsprechenden Kanal mit minimaler Hintergrunderkennung sichtbar sind.
HINWEIS: Es ist angemessen, unterschiedliche Expositionszeiten für verschiedene Kanäle zu verwenden, aber die gleiche Expositionszeit sollte für alle biologischen Replikate und Behandlungen für einen bestimmten Kanal verwendet werden. - Wählen Sie für jedes Beispiel mindestens fünf Sichtfelder (FOV) aus und erfassen Sie Bilder in jedem geeigneten Kanal mithilfe der Erfassungseinstellungen aus Schritt 6.2 (siehe Beispiele in Abbildung 2). Speichern Sie die XY-Punkte aus jedem Sichtfeld, damit dasselbe Sichtfeld während des letzten Zeitpunkts abgebildet werden kann. Die Abbildung desselben Sichtfelds zu jedem Zeitpunkt ist notwendig, um den Anteil der Fläche zu bestimmen, die von Ziel- oder Inhibitorzellen während der Analyseschritte belegt wird.
HINWEIS: In diesem Beispiel wird die Fluoreszenz von GFP mit einem Filter mit einer Anregungswellenlänge von 467 - 498 nm und einem Emissionsfilter von 513 - 556 nm detektiert und ist falsch grün. Die Fluoreszenz von dsRed wird mit einem Filter mit einer Anregungswellenlänge von 542 - 582 nm und einem Emissionsfilter von 603 - 678 nm detektiert und ist falsch gefärbtes Magenta. - Wiederholen Sie nach 2 h Schritt 6.3 für jede Probe mit den zuvor gespeicherten XY-Punkten (Abb. 2).
HINWEIS: Das Timing der nachfolgenden Bilder muss möglicherweise für Organismen mit unterschiedlichen Wachstumsraten oder Wettbewerbsmechanismen optimiert werden.
7. Bildanalyse in FIJI
- Laden Sie die FIJI-Bildverarbeitungssoftware herunter und installieren Sie sie, indem Sie die hier aufgeführten Anweisungen befolgen: https://imagej.net/Fiji/Downloads
- Öffnen Sie FIJI und importieren Sie Bilddateien zur Analyse.
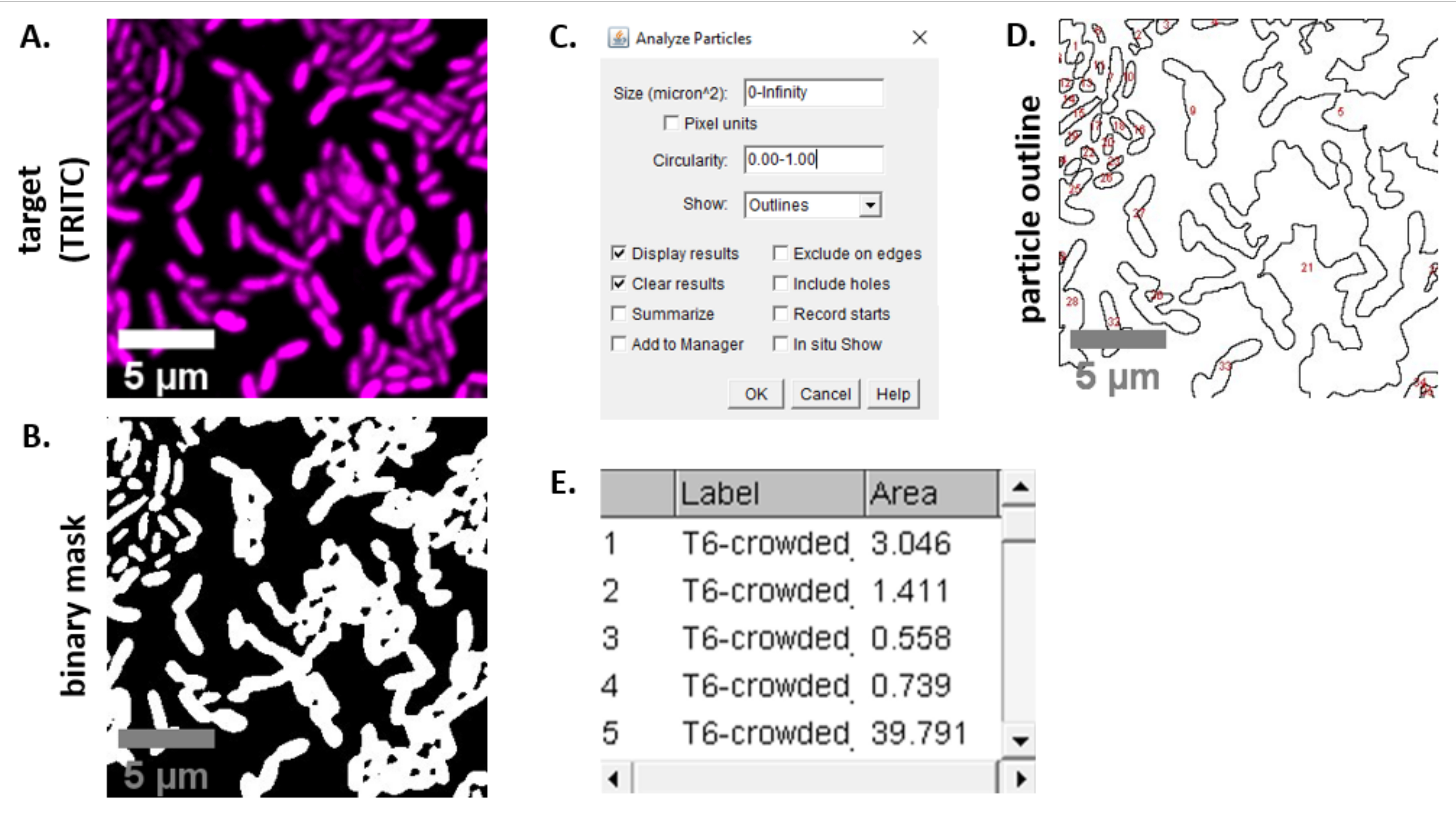
HINWEIS: In den meisten Fällen . TIFF-Dateien werden für die Bildanalyse verwendet, obwohl einige Bilderfassungssoftware mit proprietären Dateitypen exportiert. FIJI kann die meisten proprietären Dateitypen erkennen und Bilder können wie folgt importiert und analysiert werden. - Konvertieren Sie für jedes in den Schritten 6.3 und 6.4 erfasste Bild das Bild in Graustufen, trennen Sie die Kanäle und beginnen Sie mit dem Schwellenwert (Strg + Umschalt + T) und erstellen Sie eine binäre Maske des vorverarbeiteten Bildes (Abbildung 3A, B).
HINWEIS: Hier werden die Standardschwellenwerteinstellungen in FIJI verwendet. In einigen Fällen kann es notwendig sein, diese Einstellungen zu ändern, in diesem Fall sollten die gleichen Einstellungen für alle Bilder in diesem Experiment verwendet werden. - Festlegen der Skalierung des Bildes (Analysieren | Setzen Sie Scale) unter Verwendung der entsprechenden Werte für den Mikroskopieaufbau23.
- Festlegen von Messungen (Analysieren | Maße einstellen) und Fläche auswählen.
HINWEIS: Andere Messungen können hinzugefügt werden, wenn sie für das Experiment geeignet sind. Für die hier gezeigte Beispielanalyse wird nur die Objektflächenmessung benötigt. - Partikel analysieren (| analysieren Partikel analysieren) mit den Standardeinstellungen (Abbildung 3C). Wenn sich Ablagerungen in der Probe befinden, kann es erforderlich sein, die Größe oder Zirkularität anzupassen, um Nicht-Zellpartikel herauszufiltern. Wählen Sie | anzeigen aus. Umrisse, so dass das Ergebnis dieser Analyse einen nummerierten Umriss aller analysierten Partikel enthält (Abbildung 3D).
HINWEIS: Der Vergleich der Kontur in Abbildung 3D mit dem Ausgangsbild ist im Optimierungsschritt besonders wichtig, um sicherzustellen, dass (1) alle Zellen analysiert werden und (2) alle Trümmer von der Analyse ausgeschlossen werden. - Exportieren Sie die Messungen aus Schritt 7.4 (Abbildung 3E) in eine Tabellenkalkulationssoftware zur weiteren Analyse und Grafischen Darstellung.
- Wiederholen Sie die Schritte 7.1 - 7.5 für alle Kanäle und Bilder, die während des Experiments aufgenommen wurden.
8. Berechnung des Prozentsatzes der ursprünglichen Zielfläche im Zeitverlauf
- Stellen Sie für jedes in Abschnitt 7 analysierte Sichtfeld sicher, dass die exportierte Datei eine individuelle Flächenmessung für jedes analysierte Partikel enthält. Beginnend mit dem Fluoreszenzkanal des Zielstamms berechnen Sie die Summe der Partikelfläche für jedes einzelne Sichtfeld. Bei zwei biologischen Replikaten mit jeweils fünf FOV sollte dies zu zehn Summenflächen pro Behandlung zu jedem Zeitpunkt führen.
- Berechnen Sie den Prozentsatz der anfänglichen Zielfläche im Zeitverlauf für jedes Sichtfeld mit der folgenden Gleichung: (
 )
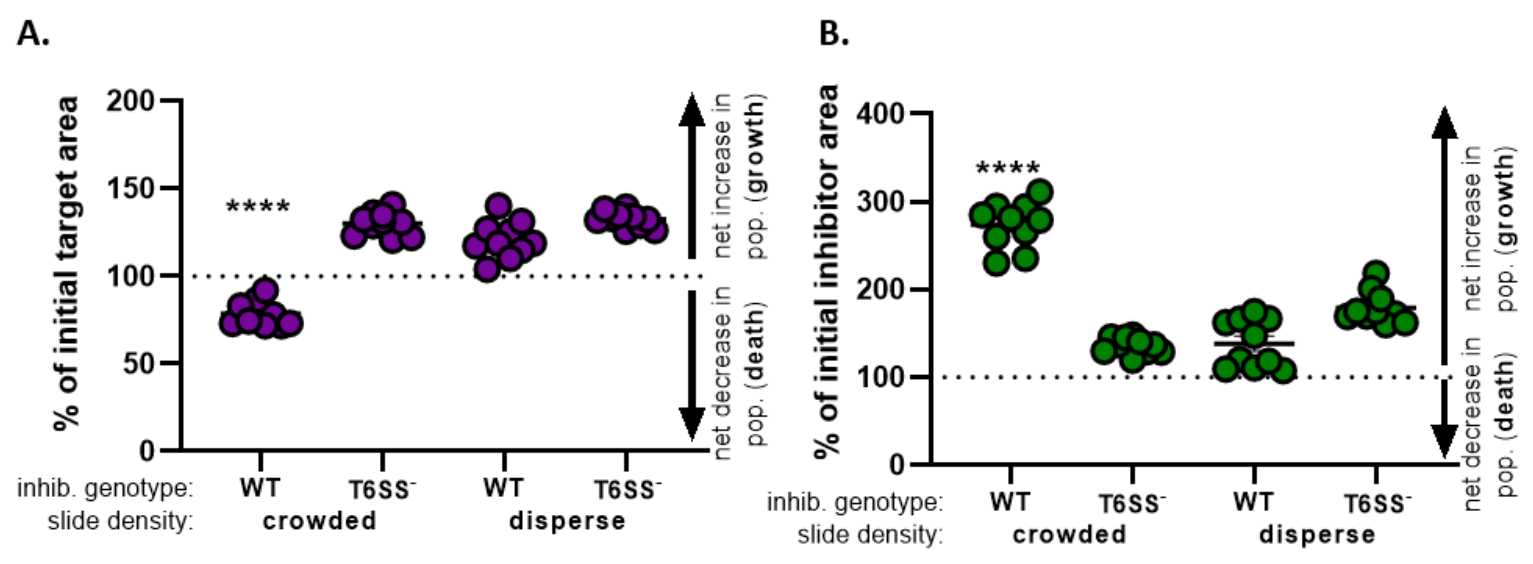
) - Wiederholen Sie diese Berechnung für alle Behandlungen und stellen Sie den Prozentsatz der anfänglichen Zielfläche (Ergebnis der Gleichung aus Schritt 8.2) für jede Behandlung grafisch her (Abbildung 4A).
- Bestimmen Sie, ob es für jede Behandlung einen Nettoanstieg der Zielpopulation (was auf Wachstum hinweist), einen Nettorückgang der Zielpopulation (der auf den Tod hinweist) oder keine Veränderung (ohne Wachstum oder Tod) vorliegt.
HINWEIS: Prozent der anfänglichen Zielfläche mit Werten größer als 100 zeigen ein Nettozielwachstum an, und Werte unter 100 zeigen den Nettozieltod an. Prozentuale Werte der ursprünglichen Zielwerte, die bei 100 bleiben, zeigen keine Nettoveränderung der Zielpopulation an. Siehe Diskussion für vorgeschlagene Folgeexperimente.
9. Berechnung des Prozentsatzes der anfänglichen Inhibitorfläche im Laufe der Zeit
- Wiederholen Sie die Schritte 8.1 bis 8.3, diesmal unter Verwendung der Messungen, die vom Fluoreszenzkanal des Inhibitorstamms in Abschnitt 7(Abbildung 4B) gesammelt wurden.
- Bestimmen Sie, ob es einen Nettoanstieg der Inhibitorpopulation gab (Wachstum); eine Nettoabnahme der Inhibitorpopulation (Tod) oder keine Veränderung für jede Behandlung. Werte größer als 100 zeigen das Nettowachstum des Inhibitors an, und Werte unter 100 zeigen den Nettotod des Inhibitors an.
Ergebnisse
Um kompetitive Interaktionen zwischen Bakterien auf Einzelzellebene zu visualisieren und zu quantifizieren, wurde ein Protokoll für V. fischeri entwickelt und optimiert, indem unser etablierter CFU-basierter Assay1,2geändert wurde. Diese Methode verwendet GFP- und dsRed-kodierende stabile Plasmide, um verschiedene Stämme von V. fischerivisuell zu unterscheiden. Das Wettbewerbsergebnis dieser Interaktionen kann quantifiziert werden, indem die aus diesem Assay gewonnenen Bilder mit der Open-Source-Software FIJI analysiert werden. Als Beispiel wurde das folgende Experiment mit V. fischeri-Isolaten durchgeführt. Ein Inhibitorstamm beherbergte ein Plasmid, das GFP kodiert, und ein Zielstamm beherbergte ein Plasmid, das dsRed kodiert. Da das vom Inhibitor kodierte T6SS2 ein kontaktabhängiger Tötungsmechanismus ist, wurden Behandlungen eingeschlossen, bei denen die Zellen entweder überfüllt waren (hoher Zell-Zell-Kontakt) oder sich auf einem Objektträger dispergierten (niedriger Zell-Zell-Kontakt), um den Einfluss des Experimentellenaufbaus auf die Endergebnisse dieses Assays hervorzuheben. In den Probendaten wurden konkurrierende Stämme im Verhältnis 1:1 gemischt und auf einem Agarose-Pad für 2 h inkubiert, und sowohl anfängliche als auch endgültige (2 h) Bilder wurden aufgenommen. Zur Kontrolle wurde auch ein T6SS2-Mutantenstamm mit dem Zielstamm sowohl unter überfüllten als auch unter dispersen Bedingungen koziert. Kulturen jeder Sorte wurden wie oben beschrieben vorbereitet und kokubatiert, und Dias wurden wie in Abbildung 1gezeigt vorbereitet.
Abbildung 2 zeigt repräsentative Fluoreszenzmikroskopiebilder jeder experimentellen Behandlung mit dem gleichen Sichtfeld, das zu einem anfangsen und letzten Zeitpunkt abgebildet wurde. Für jede Behandlung wurde entweder ein Wildtyp-Inhibitor oder ein T6SS-Mutantenstamm, der ein GFP-kodierendes Plasmid beherbergte, im Verhältnis 1:1 gemischt, wobei der Zielstamm ein dsRed-kodierendes Plasmid beherbergte. Während einer Kokubationszeit von 2 h mit diesem Versuchsaufbau können wachsende V. fischeri-Zellen 1-2 Divisionen durchlaufen (Abbildung 2; graue Pfeile). In Abbildung 2Awurde der Zell-Zell-Kontakt zwischen dem Target und dem Inhibitor erzwungen, indem die Mischkultur konzentriert wurde, bevor sie auf den Objektträger entdeckt wurde. Es wird beobachtet, dass mehrere Zielzellen im Laufe von 2 Stunden abgerundet werden und/oder verschwinden, was mit den Zielzellen übereinstimmt, die vom Inhibitor eliminiert werden (Abbildung 2; weiße Pfeile). Weitere Informationen zum Interpretieren von Rundungen oder Lysingzielzellen finden Sie im Abschnitt Diskussion. In Abbildung 2Bwurde die gleiche Kokubation auf einem Objektträger gesichtet, diesmal ohne die Mischkultur zu konzentrieren, so dass die Zellen dispergiert blieben und es nur einen minimalen Kontakt zwischen den Stämmen auf dem Objektträger gab. Hier werden keine Zielzellen beobachtet, die verschwinden oder sich runden, was darauf hindeutet, dass der Zielstamm bei dieser Behandlung nicht gehemmt wurde. Abbildung 2C und Abbildung 2D zeigen die gleichen überfüllten und dispergierten Behandlungen, die oben beschrieben wurden, diesmal mit einer T6SS-Mutante als Inhibitorstamm. Es wurde nicht beobachtet, dass Zielzellen verschwinden oder sich runden, wenn sie mit einer T6SS-Mutante unter überfüllten oder dispersen Bedingungen zusammenfallen, was wiederum darauf hindeutet, dass das Ziel bei keiner der Behandlungen gehemmt wurde.
Abbildung 3 zeigt den FIJI-Analyseworkflow, der zur Quantifizierung des Wettbewerbs in diesem Protokoll verwendet wird. Ein repräsentatives Bild aus dem Zielkanal wurde ausgewählt (Abbildung 3A) und eine binäre Maske wurde mit den Standardschwellenwerteinstellungen in FIJI erstellt (Abbildung 3B). Der Bildmaßstab wurde passend für diesen Mikroskopieaufbau eingestellt. Partikel wurden mit dem Größenparameter = 0 - unendlich, Zirkularitätsparameter = 0,00 - 1,00 analysiert und Umrisse anzeigen ausgewählt (Abbildung 3C). Die Ergebnisse dieser Partikelanalyse werden sowohl als nummerierte Umrisse jedes Partikels (Abbildung 3D) als auch als Tabelle mit Spalten für die Partikelnummer, den Dateinamen (Etikett) und die Partikelfläche in μm2 (Fläche) angezeigt (Abbildung 3E).
In Abbildung 4werden die aus Abbildung 3E erhaltenen Daten grafisch dargestellt und analysiert. In Abbildung 4Aist der Prozentsatz der anfänglichen Zielfläche zum endgültigen Zeitpunkt für jede Behandlung gemäß Schritt 8.2 dargestellt. Wenn der Prozentsatz der ursprünglichen Zielfläche größer als 100 ist, stellt dies eine Nettoerhöhung des Ziels (d. h. wachstum) dar und wird unter Bedingungen beobachtet, unter denen die Zielpopulation nicht signifikant gehemmt wird. Wenn der Prozentsatz der ursprünglichen Zielfläche jedoch unter 100 liegt, deutet dieses Ergebnis auf eine Nettoabnahme des Ziels (d. H. Tod) hin und wird unter Bedingungen beobachtet, unter denen die Zielpopulation signifikant gehemmt ist. Wenn das Ziel unter überfüllten Bedingungen mit einem Wildtyp-Inhibitor koziert wurde, zeigen die Daten eine Nettoabnahme im Zielgebiet. Im Gegensatz dazu, wenn das Ziel entweder mit einem Wildtyp-Inhibitor unter dispersen Bedingungen oder einer T6SS-Mutante unter überfüllten oder dispersen Bedingungen kokubatiert wurde, zeigen die Daten einen Nettoanstieg im Zielgebiet. Der Prozentsatz des ursprünglichen Zielgebiets, in dem das Ziel unter überfüllten Bedingungen mit einem Wildtyp-Inhibitor kokubatiert wurde, lag unter 100 und signifikant niedriger als bei allen anderen Behandlungen nach einer Einweg-ANOVA, gefolgt von einem Tukey-Mehrfachvergleichstest über alle Behandlungen hinweg (p < 0,0001). Diese Daten deuten darauf hin, dass der Zielzelltod von einem funktionellen T6SS im Inhibitor abhängt und unterstreichen die Bedeutung eines experimentellen Aufbaus, der einen ausreichenden Zell-Zell-Kontakt ermöglicht, um den Zelltod durch einen kontaktabhängigen Tötungsmechanismus zu erkennen.
Abbildung 4B zeigt den Prozentsatz der anfänglichen Inhibitorfläche zum letzten Zeitpunkt für jede Behandlung. In diesem Beispiel wurde ein Nettowachstum des Inhibitorstamms über alle Behandlungen hinweg beobachtet. Der Prozentsatz der anfänglichen Inhibitorfläche war jedoch signifikant höher, wenn ein Wildtyp-Inhibitor unter überfüllten Bedingungen mit dem Ziel verglichen wurde, verglichen mit allen anderen Behandlungen nach einer Einweg-ANOVA, gefolgt von einem Tukey-Mehrfachvergleichstest über alle Behandlungen hinweg (S. < 0,0001). Anfangs dachten wir, dass die Nettozunahme der Inhibitorfläche durch die Zunahme des verfügbaren Raums zum Wachsen getrieben werden könnte, wenn Zielzellen eliminiert werden. Die gleiche Zunahme des Inhibitorwachstums wurde jedoch nicht bei dispersen Behandlungen beobachtet, bei denen Inhibitorzellen von Beginn der Kozubation an Raum zum Wachsen hatten. Alternativ könnte dieses Ergebnis darauf hindeuten, dass Nährstoffe, die aus lysierenden Zielzellen freigesetzt werden, eine größere Zunahme der Inhibitorpopulation ermöglichen. Zusammengenommen deuten diese Ergebnisse darauf hin, dass der Inhibitorstamm das Ziel nur dann T6SS-abhängig eliminiert, wenn ein hoher Zell-Zell-Kontakt durch Dasein von Zellen auf dem Objektträger erzwungen wird.

Abbildung 1: Vorbereitung von Agarose-Pads und Slide-Setup für Coincubation-Assays. (A) Setup für die Herstellung von 2% Agarose-Pads. Fünf Schichten Laborband (grün) werden an zwei Stellen im Abstand von etwa 20 mm um einen Deckzettel gewickelt. Als nächstes wird warme 2% Agarose in mPBS (gelb) zwischen den Bandstücken pipetiert und sofort mit einem 25 mm2 Abdeckungsschlupf bedeckt und für mindestens 1 h bei Raumtemperatur erstarren gelassen. Verwenden Sie eine Rasierklinge, um das Agarose-Pad in ~ 5 mm2 Stücke zu schneiden, und verwenden Sie eine Pinzette, um das Pad auf einen neuen Dia für die Bildgebung zu übertragen. (B) Bei der Bildgebung auf einem aufrechten Mikroskop das 5 mm2 Agarose-Pad direkt auf den Objektträger legen, gefolgt von der Mischkultur (blau) und einem 12 mm kreisförmigen 1,5-Abdeckungsschlupf. (C) Bei der Bildgebung an einem invertierten Mikroskop erkennen Sie die Mischkultur direkt auf dem Glasdeckel -Boden einer 35-mm-Petrischale mit der Nr. 1,5 und legen Sie ein Agarose-Pad auf die Kultur, gefolgt von einem zweiten 12-mm-runden Deckrutsch, um das Agarose-Pad abzuflachen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Zeitrafferbilder von Kokubationsflecken unter überfüllten oder dispersen Bedingungen. (A) Repräsentative Bilder zu Anfangs- und Endzeitpunkten, bei denen eine Mischkultur aus Ziel- und Wildtyp-Inhibitor 3x konzentriert wurde, bevor sie auf dem Objektträger entdeckt wurden, um den Zellzellkontakt zwischen stämmen zu erzwingen. Weiße Pfeile im TRITC-Kanal zeigen Beispiele von Zielzellen an, die sich im Laufe des Experiments runden oder lysieren. (B) Repräsentative Bilder, auf denen eine Mischkultur aus Ziel- und Wildtypinhibitor ohne Konzentration entdeckt wurde, so dass die Zellen verteilt sind und es nur einen minimalen Zell-Zell-Kontakt zwischen den Stämmen gibt. Graue Pfeile in FITC- und TRITC-Kanälen zeigen Beispiele für die Zellteilung im Laufe des Experiments an. (C) Repräsentative Bilder, bei denen die Mischkultur von Target- und T6SS-Mutante 3x konzentriert wurde, bevor sie auf dem Objektträger entdeckt wurde, um den Zell-Zell-Kontakt zwischen den Stämmen zu erzwingen. (D) Repräsentative Bilder, auf denen eine Mischkultur aus Target- und T6SS-Mutante ohne Konzentration entdeckt wurde, so dass die Zellen dispergiert sind und es nur einen minimalen Zell-Zell-Kontakt zwischen den Stämmen gibt. Maßstabsbalken = 5 μm und sind über alle Bilder hinweg konsistent; TRITC-Kanal ist falsch farbig magenta, FITC-Kanal ist falsch gefärbt grün. Die Dekonvolution wurde auf allen Bildern durchgeführt; Hintergrund wurde subtrahiert und Helligkeit/Kontrast gleichmäßig über alle Bilder hinweg angepasst. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3:FIJI-Analyse-Workflow. (A) Repräsentatives Bild für die Analyse. Dieser Workflow wird für beide Kanäle über alle Sichtfelder und Beispiele hinweg wiederholt. Maßstabsbalken = 5 μm und sind über alle Bilder hinweg konsistent; TRITC-Kanal ist falsch farbig magenta, FITC-Kanal ist falsch gefärbt grün. (B) Binäre Maske, die durch Festlegen der Schwellenwerte für das Bild mit den Standardeinstellungen in FIJI erstellt wird. (C) Beispiel für Einstellungen für die Partikelanalyse, die in diesem Manuskript verwendet werden. Größenbereich = 0 - unendlich μm2; Zirkularität = 0,00 - 1,00; show = Umrisse. (D) Partikelumriss, der als Ausgabe der Partikelanalyse in (C) erstelltwurde. Der Partikelumriss in (D) sollte mit dem Originalbild (A) verglichen werden, um sicherzustellen, dass alle Zellen in der Partikelanalyse erfasst wurden. (E) Ergebnistabelle erstellt als Ausgabe der Partikelanalyse in (C). Die Objektnummer (Spalte 1) entspricht einzelnen Partikeln (eine oder mehrere Zellen), die in Feld (D) rot umrandet und markiert sind. Label = Dateiname des analysierten Bildes; Fläche = Gesamtpartikelfläche in μm2. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Beispieldaten zur Beurteilung, ob der Zielstamm gehemmt ist. Der Prozentsatz der Anfangsfläche zum endzeitpunkt für den Zielstamm (A) und den Inhibitorstamm (B) bei unterschiedlichen Anfangszelldichten. Die Objektträgerdichte gibt entweder eine Anfangszelldichte an, die überfüllt ist (hoher Zellkontakt zwischen Stämmen) oder mehr dispers (geringer Zellkontakt zwischen Stämmen), wie in Abbildung 2beschrieben. Der Inhibitor-Genotyp weist darauf hin, dass entweder ein Wildtyp- oder der T6SS-Mutantenstamm (T6SS-)mit dem Zielstamm koincubatiert wurde. Sternchen zeigen einen signifikanten Unterschied in der prozentualen Veränderung im Vergleich aller Behandlungen an (Einweg-ANOVA gefolgt von einem Tukey-Mehrfachvergleichstest, der alle Behandlungen vergleicht; (S. < 0,0001). Die gestrichelte Linie zeigt keine Nettoänderung der Dehnungsfläche zwischen dem anfangsen und dem letzten Zeitpunkt an; eine % Veränderung > 100 bedeutet Nettoanstieg (d. h. Wachstum) und prozentuale Veränderung < 100 bedeutet Nettoabnahme (d. h. Zelltod). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Das oben beschriebene Protokoll bietet ein leistungsfähiges Werkzeug zur Quantifizierung und Charakterisierung der interbakteriellen Konkurrenz auf Einzelzellebene. Dieser Assay, der durch Modifikation unseres CFU-basierten Wettbewerbstests auf den Agarplatten1,2entwickelt wurde, ermöglichte die Visualisierung der Einzelzellkonkurrenz zwischen V. fischeri-Isolaten und es werden Vorschläge zur Optimierung der Methode für eine Vielzahl von Systemen und Mikroskopie-Setups gegeben. Obwohl die hier beschriebene Methode für den Lichtorgan-Symbionten V. fischerioptimiert wurde, kann sie leicht modifiziert werden, um viele verschiedene, kultivierbare Mikroben aufzunehmen. Es ist wichtig zu beachten, dass Wettbewerbsmechanismen durch eine beliebige Anzahl von Umweltvariablen reguliert werden können, einschließlich Temperatur, Salzgehalt und Viskosität30,31,32,33,34. Frühere Arbeiten haben bestätigt, dass V. fischeri mit einem kontaktabhängigen Typ-VI-Sekretionssystem konkurriert, das auf Oberflächen30aktiv ist, wodurch die in diesem Assay beschriebenen Bedingungen für die Untersuchung des Wettbewerbs zwischen den Beispielstämmen geeignet sind. Es ist auch wichtig, die anfängliche Dichte der Zellen auf dem Objektträger bei der Quantifizierung der bakteriellen Konkurrenz zu berücksichtigen. Da der Kontakt zwischen Ziel- und Inhibitorzellen häufig für die Tötung erforderlich ist, sollte die Mischkultur so konzentriert werden, dass der Zell-Zell-Kontakt maximiert wird und die Zellen in einer einzigen Ebene auf dem Objektträger verbleiben. Zellkulturen sollten auf eine ähnliche optische Dichte (Mid-Log-Phase) gezüchtet und dann konzentriert werden, um den Kontakt zu erzwingen, anstatt einfach Kulturen aufgrund der physiologischen Veränderungen von Zellen in verschiedenen Wachstumsphasen auf eine höhere optische Dichte zu züchten. In anderen Systemen müssen die Kulturbedingungen und der Versuchsaufbau möglicherweise modifiziert werden, um sicherzustellen, dass der Wettbewerbsmechanismus aktiv ist und im Kozubationszustand nachgewiesen werden kann.
Die in diesem Assay verwendeten Agarose-Pads bieten mehrere Vorteile: Sie bieten eine Stabilisierung, so dass sich die Zellen nicht frei bewegen, und sie verhindern, dass die Kultur im Laufe des Experiments austrocknet. Wenn chemische Induktoren wie Isopropyl-β-D-Thiogalactosid (IPTG) für das Experiment benötigt werden, können sie der Agaroselösung leicht zugesetzt werden. Es ist jedoch wichtig zu beachten, dass das Agarosepräparat wahrscheinlich für verschiedene Systeme angepasst werden muss. Im oben beschriebenen Beispiel wurde das Agarose-Pad hergestellt, indem 2% Agarose (w/v) in 20 psu mPBS aufgelöst wurden, was der Standardsalzgehalt ist, der in V. fischeri Wachstumsmedium verwendet wird. Darüber hinaus muss in einigen Fällen dem Agarose-Pad möglicherweise eine Kohlenstoffquelle hinzugefügt werden, damit Zellen wachsen und über längere Experimente konkurrieren können. In einem solchen Fall kann das mPBS in Agarose-Pads durch jedes Wachstumsmedium ersetzt werden, obwohl die Nährstoffe im Wachstumsmedium mit dem Kompromiss einer zusätzlichen Hintergrundfluoreszenz einhergehen können.
Ohne proprietäre Bildanalysesoftware kann es sehr schwierig sein, einzelne Zellzahlen zu erhalten, wenn der Zell-Zell-Kontakt hoch ist, was, wie wir hier zeigen, erforderlich ist, um kontaktabhängige Tötungen zu beobachten. Dieser Assay wurde entwickelt, um eine alternative Methode zur Quantifizierung bereitzustellen, die nicht auf einzelnen Zellzahlen angewiesen ist. Stattdessen wird die gesamtzellfläche für jeden Fluoreszenzkanal verwendet, um das Ausmaß der Tötung zwischen coincubated Stämmen zu quantifizieren. Da diese Methode auf der Fläche und nicht auf der Anzahl einzelner Zellen basiert, reichen die Standardschwellenwerteinstellungen in der Regel aus, um die gesamte Zellfläche zu skizzieren. Die Genauigkeit der Schwellenwerte kann überprüft werden, indem die gesamte Objektfläche für ein repräsentatives Sichtfeld durch die durchschnittliche Zellgröße für den Modellorganismus dividiert und diese geschätzte Zellzahl mit einer manuellen Zellzahl für dasselbe Bild verglichen wird.
In Kokubationen zwischen einem Inhibitor und einem Zielstamm (Nicht-Killer) wird das Nettowachstum des Inhibitors vorhergesagt. Wie in Abbildung 4zu sehen ist, kann das Inhibitorwachstum bei Behandlungen, bei denen eine Tötung beobachtet wird, signifikant höher sein als bei Behandlungen, bei denen keine Tötung beobachtet wird, vielleicht weil Nährstoffe, die durch Lysing-Zielzellen freigesetzt werden, es dem Inhibitorstamm ermöglichen, schneller zu wachsen. Im hier gezeigten Beispiel wird der Nettozieltod beobachtet, da T6SS-vermittelter Wettbewerb zu einer Zielzelllyse führt, bei der das Ziel physisch eliminiert wird. Es ist jedoch wichtig zu beachten, dass nicht alle Konkurrenzmechanismen zur physischen Eliminierung von Zielzellen führen. Wenn ein Ziel durch ein Toxin, das eine Wachstumshemmung verursacht, außer Gefecht gesetzt wird, kann das hier beschriebene Protokoll dazu führen, dass die sichtbare Zielpopulation im Laufe der Zeit stabil bleibt, da die Zielzellen nicht mehr wachsen, aber auch nicht lysieren. In einem solchen Fall wäre es angebracht, die Ergebnisse dieses Assays mit Follow-up-Tests auf Lebensfähigkeit der Zielzelle zu vergleichen, wie z. B. die Beschichtung für koloniebildende Einheiten (CFUs) oder durch Durchführung von Live-Dead-Assays durch Färben mit Propidiiumiodid oder SYTOX-Grün35,36.
Im Vergleich zu Coincubation-Assays, die auf CFU-Zählungen basieren, ermöglicht dieser Assay die Beobachtung und Quantifizierung der räumlichen Struktur des Wettbewerbs zwischen Stämmen und die Verfolgung von Veränderungen in der Zielzellmorphologie im Laufe der Zeit. Zum Beispiel ist bekannt, dass Inhibitorzellen, die mit einem T6SS töten, LysM-Domänenproteine kodieren, die die Zielzellwand abbauen, was zu einer anfänglichen Zellrundung und dann zu einer Lyse13führt, die wir im Beispiel in Abbildung 2Abeobachtet haben. Darüber hinaus kann dieses Protokoll verwendet werden, um den Wettbewerb mit hoher Auflösung über sehr kurze Zeitskalen zu verfolgen. Im hier gezeigten Beispiel wird eine signifikante Abnahme des Zielbereichs bereits nach zwei Stunden beobachtet, wenn die Zellen überfüllt sind und der Zell-Zell-Kontakt zwischen den Stämmen erzwungen wird (Abbildung 4). Die hier beschriebene Bildanalyse könnte auch mittels konfokaler Mikroskopie durchgeführt werden, die es ermöglichen würde, bakterielle Konkurrenz in vivo oder in komplexen Biofilmen zu untersuchen, ohne die räumliche Verteilung der kokubatierten Stämme zu stören.
Zusammenfassend zielt der hier beschriebene Assay darauf ab, einen zugänglichen und leicht zu modifizierenden Ansatz zur Visualisierung und Quantifizierung der bakteriellen Konkurrenz auf Einzelzellebene mittels Fluoreszenzmikroskopie zu bieten. Diese Methode kann auf verschiedene Bakterienisolate angewendet werden und kann verwendet werden, um die bakterielle Konkurrenz auch in komplexen Umgebungen wie innerhalb einer Wirts- oder Biofilmmatrix zu visualisieren.
Offenlegungen
Die Autoren haben nichts preiszugeben.
Danksagungen
A.N.S wurde durch den NIGMS-Zuschuss R35 GM137886 und S.N.S durch das National Defense Science and Engineering Graduate Fellowship Program unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| 1.5 mL Microcentrifuge tube | Fisher | 05-408-129 | |
| 10 uL single channel pipette | |||
| 1000 uL single channel pipette | |||
| 20 uL single channel pipette | |||
| 200 uL single channel pipette | |||
| Agarose | Fisher | BP165-25 | Low melting agarose |
| Calculator | |||
| Cellvis 35 mm Dish | Fisher | NC0409658 | #1.5 cover glass bottom |
| Chloramphenicol | Sigma | C0378 | stock (20 mg/mL in Ethanol); final concentration in media (2 μg /mL LBS) |
| DAPI Nucleic Acid Stain | Fisher | EN62248 | optional (if not using stable plasmids) |
| FIJI image analysis sofware | ImageJ | https://imagej.net/Fiji/Downloads | open-source software |
| Fisherbrand Cover Glasses: Circles | Fisher | 12-545-81P | #1.5 cover glass; 12 mm diameter |
| Kanamycin Sulfate | Fisher | BP906-5 | stock (100 mg/mL in water, filter sterilize); final concentration in media (1 μg/mL LBS) |
| Lens Cleaning Tissue Paper | Fisher | S24530 | |
| Parafilm | Fisher | 13-374-12 | |
| Petri Plates | Fisher | FB0875713 | sterile with lid |
| Razor Blades | Fisher | S65921 | |
| Semi-micro Cuvettes | VWR | 97000-586 | |
| Spectrophotometer | |||
| SYBR Green Nucleic Acid Stain | Fisher | S7563 | optional (if not using stable plasmids) |
| Thermo Scientific Gold Seal Plain Microscope Slides | Fisher | 12-518-100B | |
| Thermo Scientific Richard-Allan Scientific Cover Glass | Fisher | 22-050-235 | #1.5 cover glass, 25 mm2 |
| Type F Immersion Oil | Fisher | NC0297589 | |
| Upright or inverted fluorescence microscope with camera and imaging software | Images in this article were acquired on a Nikon TI-2 inverted fluorescent microscope outfitted with an ORCA-Fusion Digital CMOS camera using NIS-Elements software. | ||
| Vortex | |||
| Water bath | Used to keep agarose warm prior to pipetting | ||
| LBS media | |||
| 1M Tris Buffer (pH ~7.5) | 50 mL 1 M stock buffer (62 mL HCl, 938 mL DI water, 121 g Trizma Base) | ||
| Agar Technical | Fisher | DF0812-17-9 | 15 g (Add only for plates) |
| DI water | 950 mL | ||
| Sodium Chloride | Fisher | S640-3 | 20 g |
| Tryptone | Fisher | BP97265 | 10 g |
| Yeast Extract | Fisher | BP9727-2 | 5 g |
| mPBS (marine PBS) | Phosphate buffered saline with marine salts added; used for making agarose pad | ||
| 10X PBS | Fisher | ICN1960454 | |
| Instant Ocean Sea Salt | Instant Ocean | SS1-160P | Adjust concentration to appropriate salinity; 20 psu used here |
| Sterile Vacuum Filter Units | Fisher | SCGVU01RE | Used to filter-sterilize mPBS |
| Vacuum pump | Used to filter-sterilize mPBS |
Referenzen
- Speare, L., et al. Bacterial symbionts use a type VI secretion system to eliminate competitors in their natural host. Proceedings of the National Academy of Sciences. 115 (36), 8528-8537 (2018).
- Speare, L., Septer, A. N. Coincubation assay for quantifying competitive interactions between Vibrio fischeri isolates. Journal of Visualized Experiments. (149), e59759(2019).
- Frost, I., et al. Cooperation, competition and antibiotic resistance in bacterial colonies. The ISME journal. 12 (6), 1582-1593 (2018).
- Stubbendieck, R. M., Vargas-Bautista, C., Straight, P. D. Bacterial communities: interactions to scale. Frontiers in Microbiology. 7, 1234(2016).
- Souza, D. P., et al. Bacterial killing via a type IV secretion system. Nature Communications. 6 (1), 1-9 (2015).
- Anderson, M. C., Vonaesch, P., Saffarian, A., Marteyn, B. S., Sansonetti, P. J. Shigella sonnei encodes a functional T6SS used for interbacterial competition and niche occupancy. Cell Host and Microbe. 21 (6), 769-776 (2017).
- Basler, M., Ho, B., Mekalanos, J. Tit-for-tat: Type VI secretion system counterattack during bacterial cell-cell interactions. Cell. 152 (4), 884-894 (2013).
- Guillemette, R., Ushijima, B., Jalan, M., Häse, C. C., Azam, F. Insight into the resilience and susceptibility of marine bacteria to T6SS attack by Vibrio cholerae and Vibrio coralliilyticus. PloS One. 15 (1), 0227864(2020).
- Hachani, A., Lossi, N. S., Filloux, A. A visual assay to monitor T6SS-mediated bacterial competition. Journal of Visualized Experiments. (73), e50103(2013).
- Hibbing, M. E., Fuqua, C., Parsek, M. R., Peterson, S. B. Bacterial competition: surviving and thriving in the microbial jungle. Nature Reviews Microbiology. 8 (1), 15-25 (2010).
- Ruhe, Z. C., Low, D. A., Hayes, C. S. Bacterial contact-dependent growth inhibition. Trends in Microbiology. 21 (5), 230-237 (2013).
- Wood, D. W., Pierson, L. S. The phzI gene of Pseudomonas aureofaciens 30-84 is responsible for the production of a diffusible signal required for phenazine antibiotic production. Gene. 168 (1), 49-53 (1996).
- Smith, W. P., et al. The evolution of the type VI secretion system as a disintegration weapon. PLoS Biology. 18 (5), 3000720(2020).
- Chen, L., Zou, Y., She, P., Wu, Y. Composition, function, and regulation of T6SS in Pseudomonas aeruginosa. Microbiological Research. 172, 19-25 (2015).
- Sana, T. G., Lugo, K. A., Monack, D. M. T6SS: The bacterial "fight club" in the host gut. PLoS Pathogens. 13 (6), 1006325(2017).
- Basler, M. Type VI secretion system: secretion by a contractile nanomachine. Philosophical Transactions of the Royal Society B: Biological Sciences. 370 (1679), 20150021(2015).
- Joshi, A., et al. Rules of engagement: the type VI secretion system in Vibrio cholerae. Trends in Microbiology. 25 (4), 267-279 (2017).
- Nadell, C. D., Drescher, K., Foster, K. R. Spatial structure, cooperation and competition in biofilms. Nature Reviews Microbiology. 14 (9), 589-600 (2016).
- Stubbendieck, R. M., Straight, P. D. Multifaceted interfaces of bacterial competition. Journal of Bacteriology. 198 (16), 2145-2155 (2016).
- Septer, A. N. The Vibrio-squid symbiosis as a model for studying interbacterial competition. Msystems. 4 (3), (2019).
- Tischler, A. H., Hodge-Hanson, K. M., Visick, K. L. Vibrio fischeri-squid symbiosis. eLS. , 1-9 (2019).
- Mandel, M. J., Dunn, A. K. Impact and influence of the natural Vibrio-squid symbiosis in understanding bacterial-animal interactions. Frontiers in Microbiology. 7, 1982(2016).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Boettcher, K., Ruby, E. Depressed light emission by symbiotic Vibrio fischeri of the sepiolid squid Euprymna Scolopes. Journal of Bacteriology. 172 (7), 3701-3706 (1990).
- Doino, J. A., McFall-Ngai, M. J. A transient exposure to symbiosis-competent bacteria induces light organ morphogenesis in the host squid. The Biological Bulletin. 189 (3), 347-355 (1995).
- Dunn, A. K., Millikan, D. S., Adin, D. M., Bose, J. L., Stabb, E. V. New rfp-and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Applied and Environmental Microbiology. 72 (1), 802-810 (2006).
- Lambertsen, L., Sternberg, C., Molin, S. Mini-Tn7 transposons for site-specific tagging of bacteria with fluorescent proteins. Environmental Microbiology. 6 (7), 726-732 (2004).
- Koch, B., Jensen, L. E., Nybroe, O. A panel of Tn7-based vectors for insertion of the gfp marker gene or for delivery of cloned DNA into Gram-negative bacteria at a neutral chromosomal site. Journal of Microbiological Methods. 45 (3), 187-195 (2001).
- Peterson, B. W., Sharma, P. K., Van Der Mei, H. C., Busscher, H. J. Bacterial cell surface damage due to centrifugal compaction. Applied and Environmental Microbiology. 78 (1), 120-125 (2012).
- Speare, L., Smith, S., Salvato, F., Kleiner, M., Septer, A. N. Environmental viscosity modulates interbacterial killing during habitat transition. MBio. 11 (1), (2020).
- Salomon, D., Gonzalez, H., Updegraff, B. L., Orth, K. Vibrio parahaemolyticus type VI secretion system 1 is activated in marine conditions to target bacteria, and is differentially regulated from system 2. PloS One. 8 (4), 61086(2013).
- Sana, T. G., et al. Salmonella Typhimurium utilizes a T6SS-mediated antibacterial weapon to establish in the host gut. Proceedings of the National Academy of Sciences. 113 (34), 5044-5051 (2016).
- Bachmann, V., et al. Bile salts modulate the mucin-activated type VI secretion system of pandemic Vibrio cholerae. PLoS Neglected Tropical Diseases. 9 (8), 0004031(2015).
- Ishikawa, T., et al. Pathoadaptive conditional regulation of the type VI secretion system in Vibrio cholerae O1 strains. Infection and Immunity. 80 (2), 575-584 (2012).
- Johnson, M. B., Criss, A. K. Fluorescence microscopy methods for determining the viability of bacteria in association with mammalian cells. Journal of Visualized Experiments. (79), e50729(2013).
- Stiefel, P., Schmidt-Emrich, S., Maniura-Weber, K., Ren, Q. Critical aspects of using bacterial cell viability assays with the fluorophores SYTO9 and propidium iodide. BMC Microbiology. 15 (1), 36(2015).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten