
Method Article
ゼブラフィッシュ胚からの単一体節外植片の調製
要約
私たちは、ゼブラフィッシュの胚から単一体節を単離するためのプロトコルを提示し、そのダイナミクスを蛍光タイムラプス顕微鏡法で数時間培養中で追跡できるため、単一細胞の分解能で組織スケールの形状変化を定量化する方法論を提供します。
要約
脊椎動物の胚の体軸は、体節と呼ばれる3次元多細胞単位に周期的に細分されます。遺伝的振動と分子のプレパターンが体節の初期の長さスケールを決定する一方で、その最終的なサイズと形状の設定には機械的プロセスが関与しています。体節の固有の材料特性をよりよく理解するために、ゼブラフィッシュの胚から単一体節外植片を培養する方法が開発されています。単一体節は、最初に胚の皮膚を切除し、次に卵黄を採取し、隣接する組織を順次切除することによって分離されます。トランスジェニック胚を用いて、蛍光タイムラプス顕微鏡で様々な細胞内構造の分布を観察することができます。外植体節のダイナミクスを数時間追跡できるため、単一細胞の解像度で組織スケールの形状変化を研究するための実験フレームワークを提供します。このアプローチにより、体節の直接的な機械的操作が可能になり、組織の材料特性の解剖が可能になります。最後に、ここで概説した技術は、脊索、神経板、側板中胚葉などの他の組織を移植するために容易に拡張できます。
概要
脊椎動物の成体筋骨格系の多くは、胚の体軸に沿って周期的かつリズミカルに形成される胚体節から出現します1,2。体節は、典型的には間葉系細胞の内核とフィブロネクチンに富む細胞外マトリックス3に囲まれた末梢上皮層からなる3次元(3D)多細胞ユニットである。体節の形態、すなわちその大きさと形状は、セグメンテーションクロックと下流の分子プレパターンによって部分的に決定されます。しかし、過去10年間で、機械的な手がかりと力が、体節形成5,6,7を促進し、最初の体節形成8に続く体節の長さの向上を保証するだけでなく、セグメンテーションクロック4の調節にも役割を果たすことが明らかになった。
組織の力学は、新しいツール9の利用可能性により、生体内で直接研究することができるが、物理的プロセスの根底にある全体像を得るためには、組織の固有の材料特性を同時に研究する必要がある。ここで説明するプロトコルは、細胞スケールから組織スケールまでの物理的特性を胚から分離して研究できる単一の体節を調製するための簡単なアプローチを提供します。同様の発達段階10,11,12,13,14で外植片を調製するためのいくつかのプロトコルが存在するが、我々の知る限り、これは単一の体節の単離を説明する最初のプロトコルである。このプロトコルは簡単に実施でき、胚を扱うほとんどのゼブラフィッシュラボで利用可能な基本的な機器のみが必要です。
形態学的体節形成における力学の役割を解明するために、ゼブラフィッシュの胚から単一体節外植片を培養する方法が開発され、体節の固有の材料特性を調べるために使用できるようになった。
プロトコル
このプロトコルには、受精後 1 日未満の生きた脊椎動物の胚の使用が含まれます。すべての実験は、自由に交配する成体に由来する胚を使用して行われたため、スイスのヴォー州のService de la Consommation et des Affaires Vétérinairesによって付与されたEPFLの一般的な動物実験ライセンス(承認番号VD-H23)の対象となります。
1. 解剖前
- 目的のヘテロ接合性トランスジェニック系統の交配から胚を取得します。2〜3個の体節を移植する場合、通常は数個の胚のみが必要です。このプロトコルでは、1つの体節を移植するために1つの胚を使用しました。
- 2つの50倍ストックから1x E3バッファーを調製します:ストック1には0.458 mM Na2HPO4、0.042 mM KH2PO4、4.084 mM NaCl、0.128 mM KClが含まれ、ストック2には0.33 mM CaCl2、0.33 mM MgSO4が含まれます。480 mLの蒸留水と2つの50xストックのそれぞれ10 mLを混合して、500 mLの1x E3バッファーを調製します。
- 同じ日に解剖する場合は、ペトリ皿内の25mLのE3培地で胚を33°Cで3体節になるまで育てます。翌朝に解剖する場合は、E3培地で胚を28°Cのシールドステージまで育ててから、19°Cに移して一晩インキュベートします。
- シールドステージまたは異なる体節ステージを特定するには、約0.67倍から4.5倍までの倍率範囲を調整できる標準的なラボ用実体顕微鏡を使用してください。
- 解剖に必要なツールや試薬を組み立てます。
- ライボビッツのL-15培地を50mLチューブに入れて分注し、冷蔵庫で保存してください。冷蔵庫から出ている同じアリコートを最大2ラウンド使用することをお勧めします。
- マグネチックスターラーを使用し、温度を85°Cに保ちながら、L-15培地に2%アガロース50mLを調製します。 溶解したら、溶液を室温で保存します。
- マグネチックスターラーを使用し、温度を85°Cに保ち、L-15培地に25mLの2%低融点アガロースを調製します。 溶解したら、溶液を1.5 mLチューブに分注し、4°Cで保存します。 これは、イメージングチャンバーの準備に使用されます。
- 溶融した2%アガロースを20mmのシャーレに注ぎ、ディッシュの深さの3分の1が満たされるまで注ぎ、アガロース溶液が固まるまで待ちます。これらのシャーレで解剖が行われます。アガロースコーティングを施したペトリ皿を事前に十分に準備し、4°Cで保存してください。
- 次のツールを組み立てます:胚移植用のパスツールピペット、胚を解剖するための一対の鉗子、胚の皮膚を除去するための一対の細い鉗子、胚を切開するためのマイクロナイフ、外植片をイメージングチャンバーに移すためのファイヤーポリッシュガラスピペット。
- ガラスピペットを100%エタノールで15分間滅菌し、5 mLのL-15培地を上下にピペッティングして2回すすぎます。
- 眉毛のまつげや髪の毛をガラスの毛細管に刺して、まつげツールを準備します。
- 約0.63倍から6.3倍までの倍率範囲を調整できる蛍光ステレオスコープと、標準的なGFPおよびRFPフィルターを使用して、陽性トランスジェニック胚を選別し、E3培地で別のシャーレに移します。
2. シングルソマイト外植片の調製
- 2個または3個の胚を、10mLのL-15培地で満たされたアガロースコーティング皿(ステップ1.5.4で作製)に移します。胚を移植する前に、L-15培地と一緒にディッシュをインキュベーターで28°Cで30分間予温していることを確認してください。
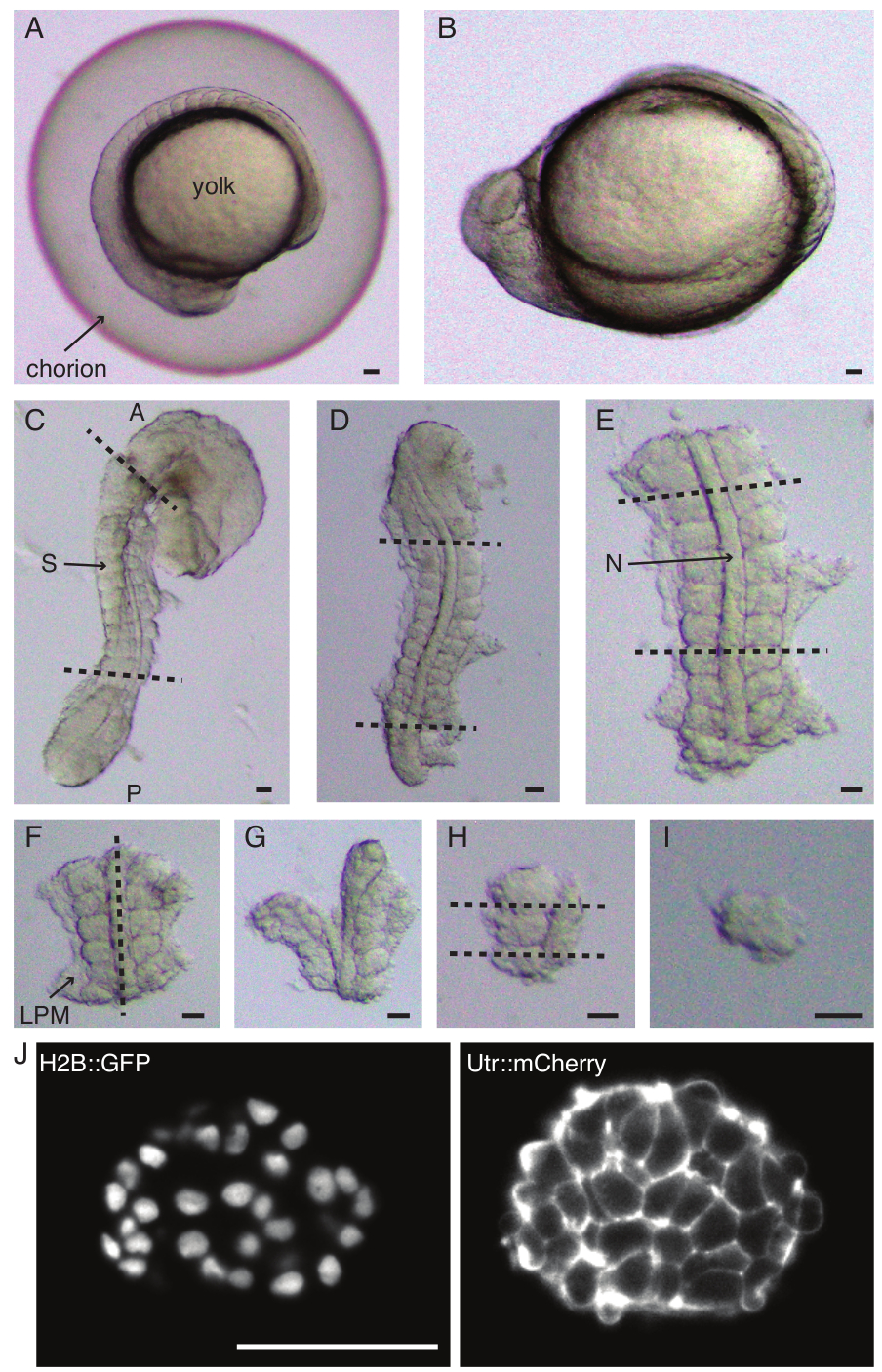
- 一対の鉗子で胚を慎重に脱毛します(図1A、B)。鉗子の1つで絨毛膜を保持し、2番目の鉗子を使用して、最初の鉗子が配置されている場所の近くで絨毛膜をつまんで引っ張ります。
- この手順をもう一度繰り返して絨毛膜を大きく開き、その後、胚を穏やかに押し出すことができます。デコレーション中に胚が損傷したり圧迫されたりしないようにしてください。
- 胚をその側面に向け、細い鉗子の1つで胚の頭と尾の間の皮膚をつかみます。2番目の細い鉗子を使用して、最初の鉗子がまだ位置している場所の近くの皮膚をつまみます。
- 皮膚をつかみながら、2つの鉗子を互いに離して動かすと、大きな開口部ができます。鉗子の1つを皮膚から離し、2番目の鉗子が保持されている場所に近づけ、体節形成領域で皮膚が除去されるまで皮膚除去プロセスを繰り返します。
注:摘出される体節の周囲の皮膚を完全に除去することが重要であり、そうしないと、プロトコルの後のステップでより細かい解剖を行うことが困難になります。 - マイクロナイフを使用して卵黄をこすり落とし(図1C-皮膚を取り除き卵黄をこすり落とした外植片)、次のように一連の切開を行います。
- 胚の最初の形態学的体節の前方に最初のカットがあり、最後に形成された体節の後方に2番目のカットがある状態で、胚をスライスします(図1D)。目的の体節の後方および前方にもう一度スライスします(図1E)。
- マイクロナイフを使用して、胚の外植領域から残っている卵黄をこすり落とします。外植片がマイクロナイフに付着する可能性を減らすために、できるだけ多くの卵黄を取り除くようにしてください。このような場合は、まつげツールを使用して、マイクロナイフを培地に浸したまま、外植片を溶液に戻します。
- 外植片を背側に配置し、脊索を通って前後軸に沿ってスライスし、片側の体節に近づけます(図1F、G)。
- 脊索なしで外植片を取り、体節の近くで側板中胚葉(LPM)をスライスします。
注:このステップは、前のステップと交換することができます、つまり、最初にLPMをスライスし、次にnotochordをスライスするのも同様にうまく機能します。
- 外植する体節を選択し、体節の後部と前方にそれぞれ2つのスライスを作成します(図1H)。
- 最後に、まつげツールを使用して体節を90°回転させ、体節に付着したままの神経板を視覚化します。体節と神経板の間にスライスを作り、1つの体節を放出します(図1I)。
注:ニューラルプレートは、ステップ2.6.4の直後、つまり脊索とLPMを取り外した後、隣接する体節から体節を解剖する前にも取り外すことができます。
3. 単一体節外植片のイメージング
注:ここでは、シングルビューライトシート顕微鏡を使用してシングルソマイト外植片をイメージングする手順について説明します。別の方法として、外植片の単一体節は、より広く利用可能な共焦点顕微鏡で画像化することも、外植片の全体的な形態を追跡することだけに興味がある場合は広視野顕微鏡でイメージングすることもできます。
- 次のようにイメージングチャンバーを準備します。
- 1 mLシリンジにシリコーンゴム製剤を入れます。200μLのピペットチップを口から約1cm離してカットし、シリンジをピペットチップに挿入します。
- メンブレンホルダーを事前にカットされたフッ素化エチレンプロピレン(FEP)メンブレンストリップ(図2A-C)で手動で包み、イメージングチャンバーをメンブレンホルダーに固定します(図2D)。
- チャンバーとメンブレンの交差点に沿ってシリコーンゴム配合物を追加し、メンブレンがチャンバーにくっつくように一晩乾燥させます。外植片実験の1時間前にメンブレンホルダーを取り外してください。
- L-15培地で作製した2%低融点アガロース0.5mLを含むアリコートを75°Cで10分間加熱します。加熱した低融点アガロース160μLをチャンバーの中央に加え、アガロースにイメージングモールドを置き(図2E、F)、アガロースが固まるまで直立させます。
- さらに160μLの加熱した低融点アガロースを金型の両端に加え、ユニット全体を4°Cに30分間移します。
- 600 μLのL-15培地をチャンバーに加え、鉗子でチャンバーから型を取り除きます。L-15培地のチャンバーが室温に戻るまで待ちます。
- 摘出した体節を研磨されたガラスピペットでイメージングチャンバーに移し、まつげツールを使用して外植片をチャンバーの中央に配置します。
- メーカーの指示に従ってイメージングレーザーの位置を合わせ、イメージングチャンバーをインキュベーションチャンバーを備えた顕微鏡に慎重に移し、外植片を28°Cに維持します。
- トランスジェニックラインからの関心領域で2回目のレーザーアライメントを行い、その後、タイムラプスイメージングの目的のzスタック、時間間隔、および期間を設定します。この実験では、2 μm の間隔と 2 分のフレーム間隔で 70 個の z スライスを取得します。
- 561 nm(レーザー出力10%、露光100 ms)および488 nmレーザー(レーザ出力10%、露光100 ms)で外植片の2色ライトシートイメージング(図1J)を実行します。このプロトコルでは、信号を25x/1.1 NA対物レンズで収集し、561/25 nmおよび525/50-25バンドパスフィルターをそれぞれsCMOSカメラに収集しました。
結果
外植片は、大規模な形状変化を3Dで定量化することができます。これを説明するために、Utr::mCherry (Tg(actb2:mCherry-Hsa.UTRN); e119Tg) と H2B::GFP (Tg(h2az2a:h2az2a-GFP); kca6Tg) ヘテロ接合系統の交配から得られた初期のゼブラフィッシュ胚から体節 4 (N = 3) を摘出しました。Utrophinはアクチン結合タンパク質であり、Utr::mCherry系統は糸状アクチン構造の分布を示しています。この導入遺伝子は、ここでは細胞の輪郭のマーカーとして使用されました。H2B::GFPは蛍光タグ付きヒストンであり、その結果、クロマチンの分布をマークし、核の位置と形状の効果的な説明、および細胞分裂を強調する有糸分裂の図を提供します。
イメージング中に外植片プロトコルの成功を評価しました。解剖中に損傷した体節では、体節組織の完全性が損なわれ、多くの細胞が解離して外植体節から押し出されるか、および/または多くの細胞が死滅することが観察されました。成功した外植片は4〜6時間健康を維持し、その後、細胞が外植片から解離して死滅すると、体節の完全性の変化が観察されました。
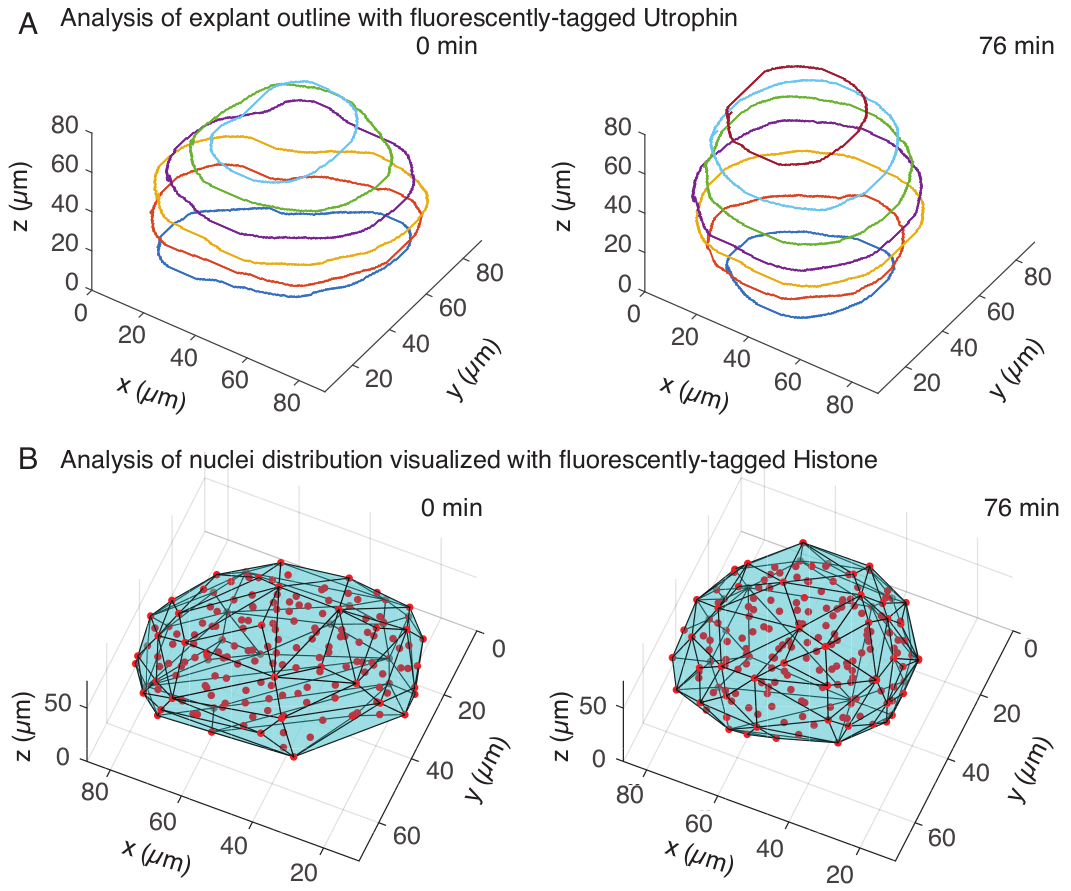
ユートロフィンチャネルでは、10 μm間隔のzスライス上で、MATLAB(R2018b)で外植片の手動セグメンテーションを行いました。セグメンテーション用のカスタムアルゴリズムをMATLABで開発し、無料でダウンロードできます(https://github.com/sundar07/SomSeg)。このアルゴリズムでは、セグメント化するファイル、フレーム、Zスライスを設定し、その後にユーザープロンプトを表示して、関心領域の周囲にアウトラインを手動で描画することができます。これを複数のzスライスに対して繰り返してプロットしたところ、3Dで経時的に外植片が切り上げられることが示されました(図3A)。さらに、核チャネルを用いた組織形状に関する補完的な情報も得られた。このために、FIJI15 プラグインであるMastodon(バージョン1.0.0-beta-19、https://github.com/mastodon-sc/mastodon)を使用して、スポット検出を通じて核の重心位置を取得しました。まず、顕微鏡のtifファイルをFIJIのxml/hdf5形式に変換し、その後、Mastodonプラグインを使用して新しいプロジェクトを開きました。プラグインでは、スポット検出オプションを選択し、外植片をカバーする関心領域を定義し、直径5μm、スポット検出の品質係数25のガウス検出器の差を使用しました。次に、核の重心位置をMATLABに転送し、組み込み関数(convhull)を使用して、外植片の形状を特徴付ける凸包(図3B)を取得しました。これは同様に、経時的な外植片の丸みを示しました(図3B)。さらに、核データにより、3Dで追跡し、細胞数の経時的な変化を追跡することにより、細胞の動きを定量化することができます。一方、ユートロフィンチャネルは、外植片が丸くなるにつれて細胞形状の変化を定量化することができます。これらのパラメータは、体節の固有の材料特性を特徴付けるために価値があり、組織スケールの形状変化の効果的な物理的記述を作成するのに役立ちます。
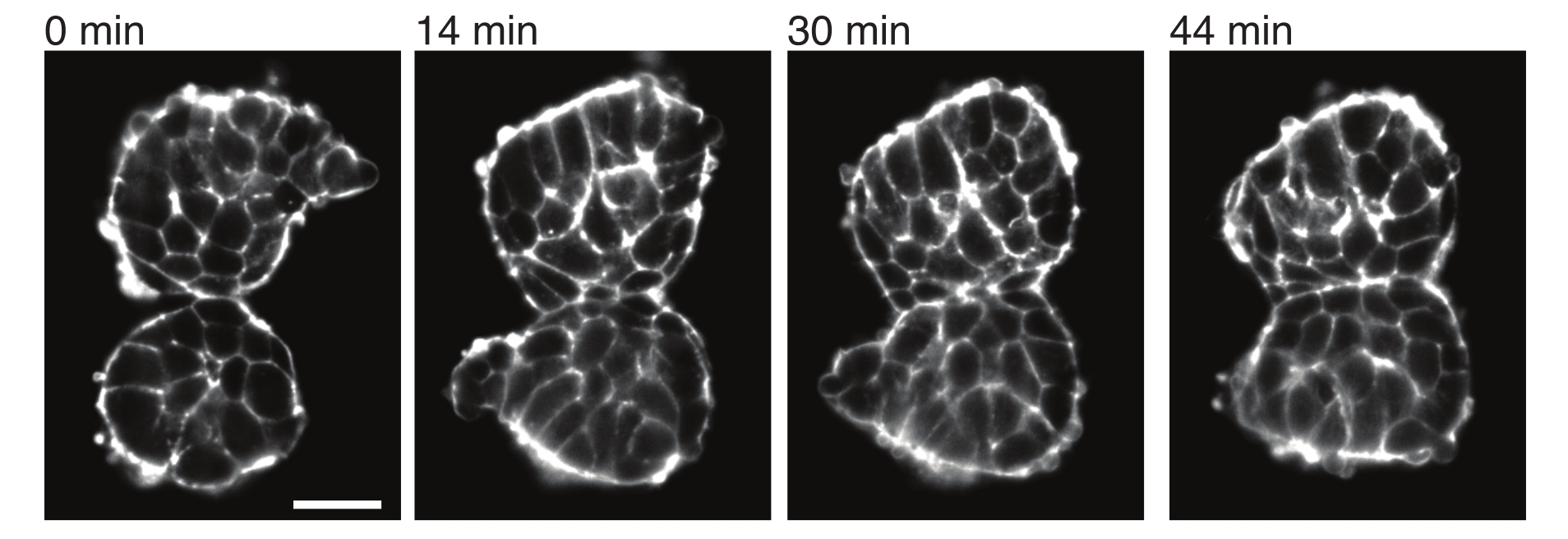
また、外植片は、隣接する組織との接触応力を定量的に特徴付けることも可能にします。これを説明するために、2つの体節を手動で分離し、それらを近接して配置し、それらのダイナミクスを観察しました(N = 2)。この実験では、 in vivo の体軸に対する体節の向きを追跡しませんでした。興味深いことに、時間の経過とともに、外植した体節は1つの表面に沿って互いに付着し、自由表面、つまり接触部位から離れた領域は切り上げられました(図4)。これは、接着力が外植片の接触部位での表面張力によって生成される応力を克服することを示唆しています。これは、上述したように形状変化を追うこと、および2つの組織間の接触角を経時的に3Dで定量化することによって、さらに特徴付けることができる。したがって、外植片は、特定の組織形状につながる作用する競合する力を定量化するための魅力的なシステムを提供し、その意味を in vivoで調査することができます。

図1:単一体節外植片の調製。ゼブラフィッシュの胚(A)は最初にデコレーション化され(B)、続いて皮膚と卵黄が除去されます(C)。次いで、胚の体性体含有領域は、残りの組織(D、E)を除去することによって選択される。次に、目的の体節の周囲の領域を連続的に除去(F-H)して、最終的に単一の体節(I)を分離し、続いてライトシート顕微鏡(ここに示されている体節の中央のzセクション)(J)によるタイムラプスイメージングを行います。略語:S =ソマイト;N =ノトコード;LPM = 側板中胚葉;A =前方;P =後部。破線はカット位置を示します。スケールバー = すべてのパネルで50μm。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:ライトシートイメージングチャンバーの組み立て (A-D) FEPメンブレンストリップ((B)の寸法)をメンブレンホルダーに巻き付けてイメージングチャンバーに取り付け、メンブレンをイメージングチャンバーに接着します。(E-F)翌日、メンブレンホルダーを取り外し、イメージングモールド((E)の寸法)を低融点アガロースのイメージングチャンバーの中央に配置します。ユニット全体を4°Cで30分間保持した後、型を取り外し、トラフ付きのチャンバーを使用して外植片をイメージングします。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:組織形状の3D解析 (A)細胞の輪郭を蛍光タグ付きユートロフィン(Utr::mCherry)で可視化し、核を蛍光タグ付きヒストン(H2B::GFP)で可視化した。外植片のアウトラインは、MATLAB を使用して複数の深さで手動でセグメント化され、ここに示されています。(B)核(赤)は、FIJIのプラグインであるMastodonを使用して同じ外植片で検出され、外植片の形状を通知する凸包(シアン)にかけられました。注:外植片の切り上げは、両方の分析で明らかです。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図4:外植した体節は互いに付着します。 胚から単離され、手動で近接して配置された2つの体節は、時間の経過とともに付着する傾向があります(N = 2)。フレーム間隔が2分の複数のzスライスをライトシート顕微鏡を使用して取得し、選択した時点からの体節の中央切片をここに示しました。細胞の輪郭は、蛍光タグ付きUtrophin(Utr::mCherry)で可視化しました。スケールバー = 25 μm. この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
ディスカッション
体節形成の分野は、胚発生中のセグメントの長さを設定するセグメンテーションクロックの役割に関する研究によって支配されてきました。しかし、最終的な体節の形態を決定する上での組織力学の役割を考慮することも同様に重要です。ここで説明するプロトコルは、単一の体節を移植することを可能にし、その固有の物理的特性を胚から分離して研究することができます。ただし、手動調製では、通常、イメージングセッションごとに準備される体節の数が4〜6に制限されます。このプロトコルでは、組織の除去を容易にするための酵素を使用せずに、胚のいくつかの組織を慎重に細かく解剖するため、解剖プロセスを習得するには数週間の練習が必要になる場合があります。私たちの手では、プロトコルの重要なステップは、外植片から皮膚と卵黄を慎重に除去することを含み、これにより、プロトコルのさまざまな段階で外植片が使用されるツールに付着するのを防ぎ、その結果、体節を取り巻く組織を連続的に除去するための解剖が容易になります。
解剖プロトコルは、周囲の組織の1つだけに付着した体節を得るため、または体節のグループの外植片を得るために、中間ステップで停止することができます。これにより、組織を連続的に剥ぎ取り、体節や隣接組織の形状変化を促進する隣接する組織の影響を研究するための強力な方法が提供されます。一方、このプロトコルを使用すると、個々の外植された組織を接着させ、機械的に自己組織化させることができ、これは、2つの外植体節を近接して配置することによって実証される。
この方法で調製された外植片は、バッファーに成分を追加する必要も、最大数時間の生存を保証するための制約も必要ありません。しかし、ハイドロゲルで培養し、外部制約の存在下でそのダイナミクスを追跡することを想像することはでき、これは体節が 生体内で遭遇する接触ストレスのモデルとして役立つ可能性があります。さらに、外植片は、原子間力顕微鏡、ピペット吸引、またはマイクロロボットツール16を使用して、それらの材料特性を直接調べることを可能にする。最後に、この方法は、脊索、神経板、側板中胚葉など、同様の段階にある他の発生組織の培養と研究に容易に適応できると期待しています。
開示事項
著者は、競合する利益を宣言しません。
謝辞
École polytechnique fédérale de Lausanne (EPFL) のプロトコルと魚の施設についてコメントをいただいた Oates ラボのメンバーに感謝します。特に、解剖プロトコルでの皮膚と卵黄の除去に関する貴重なヒントを提供してくれたLaurel Ann Rohdeに感謝します。Arianne Bercowsky Rama は、Mastodon を通じてライトシートデータセットを処理するための効率的なパイプラインを構築しました。Jean-Yves Tinevez は、オープンソースの Mastodon ソフトウェアを構築しました。Marko Popović データ分析のヒント。クロエ・ジョリヴェ、ギヨーム・ヴァランタン、フロリアン・ラングは、魚の施設で広範なサポートを提供してくれました。Petr Strnad と Andrea Boni は、ライトシート顕微鏡の製作とライトシートイメージングのヒントを提供します。この研究はEPFLの支援を受け、SRNはLong-Term Human Frontier Science Programのポスドクフェローシップ(LT000078/2016)の支援を受けました。
資料
| Name | Company | Catalog Number | Comments |
| Agarose | Sigma | 9012-36-6 | For coating bottom of petri dishes |
| Agarose, low gelling temperature | Sigma | 39346-81-1 | For preparing Viventis imaging chamber |
| Camera | Andor | Andor Zyla 4.2 Plus | For image acquisition in the light-sheet microscope |
| Detection objective | Nikon | Nikon CFI75 Apo LWD 25x/1.1 NA | For imaging explants |
| FEP membrane strip | Lohmann Technologies UK Ltd | Dupont FEP Fluorocarbon film, 200A | For preparing Viventis imaging chamber |
| Fine forceps | Dumont | Dumont 5SF 11252-00 | For removal of skin of embryos |
| Forceps | Dumont | Dumont 55 | For dechorionating embryos |
| Leibovitz's L-15 medium | Gibco | 21083-027 | Explant culture medium |
| Light-sheet microscope | Viventis | LS1 live | For imaging explants |
| Micro knife | Fine Science Tools | 10318-14 | For making incisions in embryos |
| Silicone rubber formulation | Wacker Chemie AG | Silpuran 4200 | For preparing Viventis imaging chamber |
参考文献
- Oates, A. C., Morelli, L. G., Ares, S. Patterning embryos with oscillations: structure, function and dynamics of the vertebrate segmentation clock. Development. 139 (4), Cambridge, England. 625-639 (2012).
- Pourquié, O. Segmentation of the vertebrate spine: From clock to scoliosis. Cell. 145 (5), 650-663 (2011).
- Naganathan, S. R., Oates, A. C. Patterning and mechanics of somite boundaries in zebrafish embryos. Seminars in Cell & Developmental Biology. 107, 170-178 (2020).
- Hubaud, A., Regev, I., Mahadevan, L., Pourquié, O. Excitable Dynamics and Yap-Dependent Mechanical Cues Drive the Segmentation Clock. Cell. 171 (3), 668-682 (2017).
- Dias, A. S., de Almeida, I., Belmonte, J. M., Glazier, J. A., Stern, C. D. Somites without a clock. Science. 343 (6172), New York, N.Y. 791-795 (2014).
- Nelemans, B. K. A., Schmitz, M., Tahir, H., Merks, R. M., Smit, T. H. Somite Division and New Boundary Formation by Mechanical Strain. iScience. 23 (4), 100976(2020).
- Grima, R., Schnell, S. Can tissue surface tension drive somite formation. Developmental Biology. 307 (2), 248-257 (2007).
- Naganathan, S. R., Popovic, M., Oates, A. C. Left–right symmetry of zebrafish embryos requires somite surface tension. Nature. 605, 516-521 (2022).
- Campàs, O. A toolbox to explore the mechanics of living embryonic tissues. Seminars in Cell & Developmental Biology. 55, 119-130 (2016).
- Langenberg, T., Brand, M., Cooper, M. S. Imaging brain development and organogenesis in zebrafish using immobilized embryonic explants. Developmental Dynamics: An Official Publication of The American Association of Anatomists. 228 (3), 464-474 (2003).
- Henry, C. A., Poage, C. T., McCarthy, M. B., Campos-Ortega, J., Cooper, M. S. Regionally autonomous segmentation within zebrafish presomitic mesoderm. Zebrafish. 2 (1), 7-18 (2005).
- Picker, A., Roellig, D., Pourquié, O., Oates, A. C., Brand, M. Tissue micromanipulation in zebrafish embryos. Methods in Molecular Biology. 546, Clifton, N.J. 153-172 (2009).
- Manning, A. J., Kimelman, D. Tbx16 and Msgn1 are required to establish directional cell migration of zebrafish mesodermal progenitors. Developmental Biology. 406 (2), 172-185 (2015).
- Simsek, M. F., Özbudak, E. M. A 3-D Tail Explant Culture to Study Vertebrate Segmentation in Zebrafish. Journal of Visualized Experiments:JoVE. (172), e61981(2021).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Özkale, B., et al. Modular soft robotic microdevices for dexterous biomanipulation. Lab on a Chip. 19 (5), 778-788 (2019).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved