
Method Article
Präparation von Einzelsomiten-Explantaten aus Zebrafischembryonen
In diesem Artikel
Zusammenfassung
Wir stellen ein Protokoll zur Isolierung einzelner Somiten aus Zebrafischembryonen vor, dessen Dynamik in Kultur über mehrere Stunden durch Fluoreszenz-Zeitraffermikroskopie verfolgt werden kann, und liefern damit eine Methodik zur Quantifizierung von Formveränderungen auf Gewebeebene mit Einzelzellauflösung.
Zusammenfassung
Die Körperachse von Wirbeltierembryonen wird periodisch in mehrzellige 3D-Einheiten unterteilt, die Somiten genannt werden. Während genetische Oszillationen und molekulare Vormuster die anfängliche Längenskala von Somiten bestimmen, sind mechanische Prozesse an der Festlegung ihrer endgültigen Größe und Form beteiligt. Um die intrinsischen Materialeigenschaften von Somiten besser zu verstehen, wird eine Methode entwickelt, um einzelnes Somiten-Explantat aus Zebrafischembryonen zu kultivieren. Einzelne Somiten werden isoliert, indem zuerst die Haut der Embryonen entfernt wird, gefolgt von der Entfernung des Eigelbs und der anschließenden Exzision benachbarter Gewebe. Anhand transgener Embryonen kann die Verteilung verschiedener subzellulärer Strukturen mittels fluoreszierender Zeitraffermikroskopie beobachtet werden. Die Dynamik explantierter Somiten kann über mehrere Stunden verfolgt werden, was einen experimentellen Rahmen für die Untersuchung von Formänderungen im Gewebemaßstab mit Einzelzellauflösung bietet. Dieser Ansatz ermöglicht die direkte mechanische Manipulation von Somiten, wodurch die Materialeigenschaften des Gewebes zerlegt werden können. Schließlich kann die hier skizzierte Technik leicht für die Explantation anderer Gewebe wie der Chorda, der Neuralplatte und des Mesoderms der lateralen Platte erweitert werden.
Einleitung
Ein Großteil des Bewegungsapparates der adulten Wirbeltiere geht aus embryonalen Somiten hervor, die sich periodisch und rhythmisch entlang der Körperachse der Embryonen bilden 1,2. Somiten sind dreidimensionale (3D) mehrzellige Einheiten, die typischerweise aus einem inneren Kern mesenchymaler Zellen und einer peripheren Epithelschicht bestehen, die von einer fibronektinreichen extrazellulären Matrixumgeben ist 3. Die Morphologie der Somiten, d.h. ihre Größe und Form, wird zum Teil durch die Segmentierungsuhr und nachgeschaltete molekulare Vormuster bestimmt. In den letzten zehn Jahren hat sich jedoch herausgestellt, dass mechanische Signale und Kräfte auch eine Rolle bei der Regulierung der Segmentierungsuhrspielen 4, zusätzlich zur Erleichterung der Somitenbildung 5,6,7 und zur Gewährleistung einer erhöhten Präzision der Somitenlängen nach der anfänglichen Somitenbildung8.
Die Gewebemechanik kann mit der Verfügbarkeit neuer Werkzeuge direkt in vivo untersucht werden9, aber um ein vollständiges Bild der physikalischen Prozesse zu erhalten, müssen gleichzeitig die intrinsischen Materialeigenschaften von Geweben untersucht werden. Das hier beschriebene Protokoll bietet einen einfachen Ansatz zur Herstellung einzelner Somiten, deren physikalische Eigenschaften von zellulären bis hin zu Gewebemaßstäben isoliert vom Embryo aus untersucht werden können. Während es mehrere Protokolle für die Herstellung von Explantaten in ähnlichen Entwicklungsstadiengibt 10,11,12,13,14, ist dies unseres Wissens nach das erste Protokoll, das die Isolierung einzelner Somiten beschreibt. Das Protokoll ist einfach zu implementieren und erfordert nur eine Grundausstattung, die in den meisten Zebrafischlaboren, die mit Embryonen arbeiten, verfügbar ist.
Um die Rolle der Mechanik bei der morphologischen Somitenbildung zu entschlüsseln, wird eine Methode entwickelt, um einzelnes Somiten-Explantat aus Zebrafischembryonen zu kultivieren, mit dem die intrinsischen Materialeigenschaften von Somiten untersucht werden können.
Protokoll
Dieses Protokoll beinhaltet die Verwendung von lebenden Wirbeltierembryonen, die jünger als 1 Tag nach der Befruchtung sind. Alle Versuche wurden mit Embryonen durchgeführt, die von frei paarenden adulten Tieren stammen, und fallen somit unter die allgemeine Tierversuchslizenz der EPFL, die vom Service de la Consommation et des Affaires Vétérinaires des Kantons Waadt - Schweiz erteilt wurde (Zulassungsnummer VD-H23).
1. Vor der Sektion
- Gewinnung von Embryonen aus einer Kreuzung heterozygoter transgener Linien von Interesse. Für die Explantation von zwei bis drei Somiten werden in der Regel nur wenige Embryonen benötigt. In diesem Protokoll wurde ein Embryo für die Explantation eines Somiten verwendet.
- 1x E3-Puffer aus zwei 50er-Stammstoffen herstellen: Stamm 1 enthält 0,458 mM Na2HPO4, 0,042 mMKH2PO4, 4,084 mM NaCl, 0,128 mM KCl und Stamm 2 enthält 0,33 mM CaCl2, 0,33 mM MgSO4. Bereiten Sie 500 mL 1x E3-Puffer vor, indem Sie 480 mL destilliertes Wasser mit je 10 mL der beiden 50x Brühen mischen.
- Wenn die Embryonen am selben Tag präpariert werden, werden die Embryonen in 25 ml E3-Medium in einer Petrischale bei 33 °C bis zum Drei-Somiten-Stadium aufgezogen. Wenn Sie am nächsten Morgen präparieren, ziehen Sie die Embryonen in E3-Medium bei 28 °C bis zum Schildstadium auf, bevor Sie für die Inkubation über Nacht auf 19 °C übertragen werden.
- Zur Identifizierung des Abschirmtisches oder der verschiedenen Somitenstufen verwenden Sie ein Standard-Laborstereomikroskop mit einem einstellbaren Vergrößerungsbereich von ca. 0,67x bis 4,5x.
- Stellen Sie Werkzeuge und Reagenzien zusammen, die für das Sezieren benötigt werden.
- Aliquotieren Sie das L-15-Medium von Leibovitz in 50-ml-Röhrchen und lagern Sie es im Kühlschrank. Wir empfehlen, das gleiche Aliquot aus dem Kühlschrank für maximal zwei Runden zu verwenden.
- Bereiten Sie 50 mL 2%ige Agarose in L-15-Medium vor, indem Sie einen Magnetrührer verwenden und die Temperatur auf 85 °C halten. Sobald sich die Lösung aufgelöst hat, lagern Sie sie bei Raumtemperatur.
- Bereiten Sie 25 ml 2%ige niedrigschmelzende Agarose in L-15-Medium zu, indem Sie einen Magnetrührer verwenden und die Temperatur auf 85 °C halten. Nach dem Auflösen aliquotieren Sie die Lösung in 1,5-ml-Röhrchen und lagern sie bei 4 °C. Diese wird für die Vorbereitung der Bildgebungskammer verwendet.
- Gießen Sie geschmolzene 2%ige Agarose in eine 20 mm Petrischale, bis ein Drittel der Tiefe der Schale gefüllt ist, und lassen Sie die Agaroselösung fest werden. In diesen Petrischalen werden Sezierungen durchgeführt. Die Petrischalen mit Agarosebeschichtung rechtzeitig vorbereiten und bei 4 °C lagern.
- Stellen Sie folgende Werkzeuge zusammen: Pasteurpipette für den Transfer von Embryonen, eine Pinzette für die Dechorionierung von Embryonen, eine feine Pinzette für die Entnahme der Haut von Embryonen, ein Mikromesser für die Schnitte in Embryonen, eine Pipette aus feuerpoliertem Glas für den Transfer von Explantaten in die Bildgebungskammer.
- Sterilisieren Sie die Glaspipette 15 Minuten lang in 100 % Ethanol und spülen Sie sie zweimal aus, indem Sie 5 ml L-15-Medium auf und ab pipettieren.
- Bereiten Sie ein Wimpernwerkzeug vor, indem Sie eine Wimper oder ein Haar von der Augenbraue auf eine Glaskapillare kleben.
- Verwenden Sie ein fluoreszierendes Stereoskop mit einem einstellbaren Vergrößerungsbereich von ca. 0,63x bis 6,3x sowie Standard-GFP- und RFP-Filter, um positive transgene Embryonen zu sortieren und in eine separate Petrischale mit E3-Medium zu übertragen.
2. Vorbereitung des Single-Somit-Explantats
- Übertragen Sie 2 oder 3 Embryonen in eine mit Agarose überzogene Schale (hergestellt in Schritt 1.5.4), die mit 10 ml L-15-Medium gefüllt ist. Stellen Sie sicher, dass die Schale zusammen mit L-15-Medium 30 Minuten lang in einem Inkubator auf 28 °C vorgewärmt wird, bevor Sie die Embryonen übertragen.
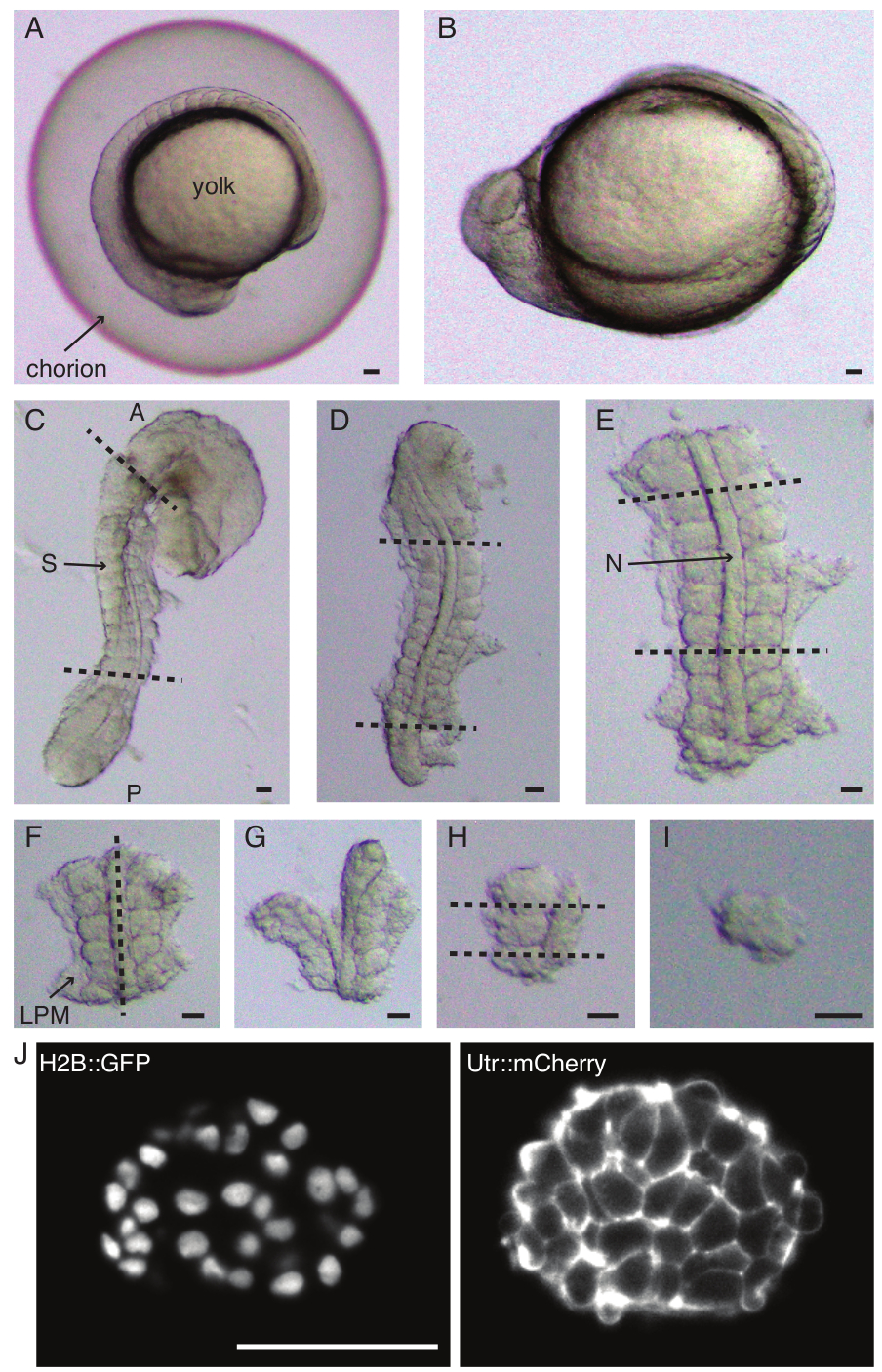
- Dechorionieren Sie einen Embryo vorsichtig mit einer Pinzette (Abbildung 1 A,B). Halten Sie das Chorion mit einer der Pinzetten fest und verwenden Sie die zweite Pinzette, um das Chorion in der Nähe der Position der ersten Pinzette zu kneifen und daran zu ziehen.
- Wiederholen Sie diesen Schritt noch einmal, um das Chorion weit zu öffnen, wonach der Embryo vorsichtig herausgedrückt werden kann. Stellen Sie sicher, dass der Embryo während der Dechorionation nicht beschädigt oder gequetscht wird.
- Richten Sie den Embryo auf die seitliche Seite und halten Sie ihn mit einer der feinen Pinzetten zwischen Kopf und Schwanz des Embryos an der Haut fest. Kneifen Sie mit der zweiten feinen Pinzette auf die Haut in der Nähe der Stelle, an der sich die erste Pinzette noch befindet.
- Bewegen Sie die beiden Pinzetten voneinander weg, während Sie sich an der Haut festhalten, wodurch eine große Öffnung entsteht. Lösen Sie eine der Pinzetten von der Haut und bewegen Sie sie in die Nähe der Stelle, an der die zweite Pinzette gehalten wird, und wiederholen Sie den Hautentfernungsvorgang, bis die Haut in den somitbildenden Bereichen entfernt wird.
HINWEIS: Es ist wichtig, die Haut, die die zu explantierenden Somiten umgibt, vollständig zu entfernen, da es sonst schwierig wird, in den späteren Schritten des Protokolls feinere Dissektionen durchzuführen. - Verwenden Sie ein Mikromesser, um das Eigelb abzukratzen (Abbildung 1C - Explantat mit entfernter Haut und abgekratztem Eigelb) und machen Sie eine Reihe von Schnitten wie folgt:
- Schneiden Sie den Embryo mit dem ersten Schnitt vor dem ersten morphologischen Somiten im Embryo und dem zweiten Schnitt hinter dem zuletzt gebildeten Somiten durch den Embryo (Abbildung 1D). Schneiden Sie noch einmal posterior und anterior zu den interessierenden Somiten (Abbildung 1E).
- Kratzen Sie mit dem Mikromesser das restliche Eigelb von den explantierten Regionen des Embryos ab. Versuchen Sie, so viel Eigelb wie möglich zu entfernen, um die Möglichkeit zu verringern, dass Explantate am Mikromesser haften bleiben. Verwenden Sie in diesem Fall ein Wimpernwerkzeug, um das Explantat wieder in die Lösung zu bringen, während Sie das Mikromesser in das Medium eintauchen lassen.
- Positionieren Sie das Explantat dorsal und machen Sie einen Schnitt entlang der anteroposterioren Achse durch die Chorda, auf einer Seite in der Nähe der Somiten (Abbildung 1F, G).
- Nehmen Sie das Explantat ohne Notochord und schneiden Sie das laterale Plattenmesoderm (LPM) in der Nähe der Somiten durch.
HINWEIS: Dieser Schritt kann mit dem vorherigen Schritt ausgetauscht werden, d.h. es funktioniert genauso gut, zuerst durch das LPM zu schneiden und dann durch die Chorda zu schneiden.
- Wählen Sie den Somiten, der explantiert werden soll, und machen Sie jeweils zwei Scheiben hinter und vor dem Somiten (Abbildung 1H).
- Drehen Sie schließlich den Somiten mit dem Wimpernwerkzeug um 90°, um die Neuralplatte zu visualisieren, die noch mit dem Somiten verbunden ist. Machen Sie einen Schnitt zwischen dem Somiten und der Neuralplatte, wodurch ein einzelner Somite freigesetzt wird (Abbildung 1I).
HINWEIS: Die Neuralplatte kann auch unmittelbar nach Schritt 2.6.4 entfernt werden, d.h. nach der Entfernung des Notochords und des LPM und vor dem Präparieren eines Somiten von benachbarten Somiten.
3. Bildgebung von Einzel-Somiten-Explantaten
HINWEIS: Hier wird das Verfahren zur Abbildung von Einzel-Somiten-Explantaten mit einem Einsicht-Lichtblattmikroskop beschrieben. Alternativ können die explantierten Einzelsomiten auch in einem Konfokalmikroskop abgebildet werden, das weiter verbreitet ist, oder sogar in einem Weitfeldmikroskop, wenn es ausschließlich darum geht, die Gesamtmorphologie der Explantate zu verfolgen.
- Bereiten Sie eine Bildgebungskammer wie folgt vor:
- Füllen Sie eine 1-ml-Spritze mit Silikonkautschuk-Formulierung. Schneiden Sie eine 200 μL Pipettenspitze ca. 1 cm vom Mund entfernt ab und führen Sie die Spritze in die Pipettenspitze ein.
- Umwickeln Sie den Membranhalter manuell mit einem vorgeschnittenen Membranstreifen aus fluoriertem Ethylenpropylen (FEP) (Abbildung 2A-C) und befestigen Sie die Bildgebungskammer an den Membranhaltern, die eng ineinander passen (Abbildung 2D).
- Fügen Sie die Silikonkautschukformulierung entlang der Schnittstelle zwischen der Kammer und der Membran hinzu und trocknen Sie sie über Nacht, damit die Membran an der Kammer haften bleibt. Entfernen Sie den Membranhalter 1 h vor dem Explantatexperiment.
- Ein Aliquot mit 0,5 ml 2 % niedrigschmelzender Agarose, hergestellt mit L-15-Medium, wird 10 min lang bei 75 °C erhitzt. Geben Sie 160 μl der erhitzten, niedrigschmelzenden Agarose in die Mitte der Kammer und platzieren Sie eine bildgebende Form in der Agarose (Abbildung 2E, F) und halten Sie sie aufrecht, bis die Agarose fest wird.
- Geben Sie weitere 160 μl erhitzte niedrigschmelzende Agarose auf die beiden Enden der Form und stellen Sie die gesamte Einheit für 30 min auf 4 °C um.
- Geben Sie 600 μl L-15-Medium in die Kammer und entfernen Sie die Form mit einer Pinzette aus der Kammer. Warten Sie, bis die Kammer mit L-15-Medium wieder Raumtemperatur erreicht hat.
- Übertragen Sie den explantierten Somiten mit einer Pipette aus poliertem Glas, indem Sie ihn in die Bildgebungskammer pipettieren und verwenden Sie ein Wimpernwerkzeug, um das Explantat in der Mitte der Kammer zu positionieren.
- Richten Sie die bildgebenden Laser gemäß den Anweisungen des Herstellers aus und übertragen Sie die Bildgebungskammer vorsichtig in das Mikroskop, das mit einer Inkubationskammer ausgestattet ist, um die Explantate auf 28 °C zu halten.
- Führen Sie eine zweite Runde der Laserausrichtung mit den interessierenden Regionen der transgenen Linie durch, gefolgt von der Einrichtung des gewünschten Z-Stapels, des Zeitintervalls und der Dauer für die Zeitrafferbildgebung. Für dieses Experiment werden 70 Z-Schichten mit einem Abstand von 2 μm und einem Bildintervall von 2 min aufgenommen.
- Führen Sie eine zweifarbige Lichtblatt-Bildgebung (Abbildung 1J) der Explantate mit 561 nm (10 % Laserleistung, 100 ms Belichtung) und 488 nm (10 % Laserleistung, 100 ms Belichtung) durch. In diesem Protokoll wurde das Signal mit einem 25x/1.1 NA-Objektiv und durch 561/25 nm bzw. 525/50-25 Bandpassfilter auf eine sCMOS-Kamera gesammelt.
Ergebnisse
Explantate ermöglichen die Quantifizierung großräumiger Formänderungen in 3D. Um dies zu veranschaulichen, explantierten wir Somitte vier (N = 3) aus frühen Zebrafischembryonen, die aus einer Kreuzung zwischen Utr::mCherry (Tg(actb2:mCherry-Hsa.UTRN); e119Tg) und H2B::GFP (Tg(h2az2a:h2az2a-GFP); kca6Tg) heterozygoten Linien gewonnen wurden. Utrophin ist ein Aktin-bindendes Protein, und die Utr::mCherry-Linie zeigt die Verteilung der filamentösen Aktinstrukturen. Dieses Transgen wurde hier als Marker für Zellumrisse verwendet. H2B::GFP ist ein fluoreszenzmarkiertes Histon und markiert daher die Verteilung des Chromatins, was eine effektive Beschreibung der Position und Form des Kerns sowie mitotische Figuren liefert, die die Zellteilung hervorheben.
Wir untersuchten den Erfolg des Explantatprotokolls während der Bildgebung. Wir beobachteten, dass bei einem Somiten, der während der Dissektion geschädigt wurde, entweder die Integrität des Somitengewebes beeinträchtigt ist, wobei viele Zellen sich vom explantierten Somiten ablösen und extrudieren und/oder viele Zellen absterben, was durch das Vorhandensein von fragmentierten Kernen im Kernkanal festgestellt werden kann. Erfolgreiche Explantate blieben 4-6 Stunden lang gesund, danach wurden Veränderungen der Somitenintegrität beobachtet, wobei sich die Zellen vom Explantat dissoziierten und abstarben.
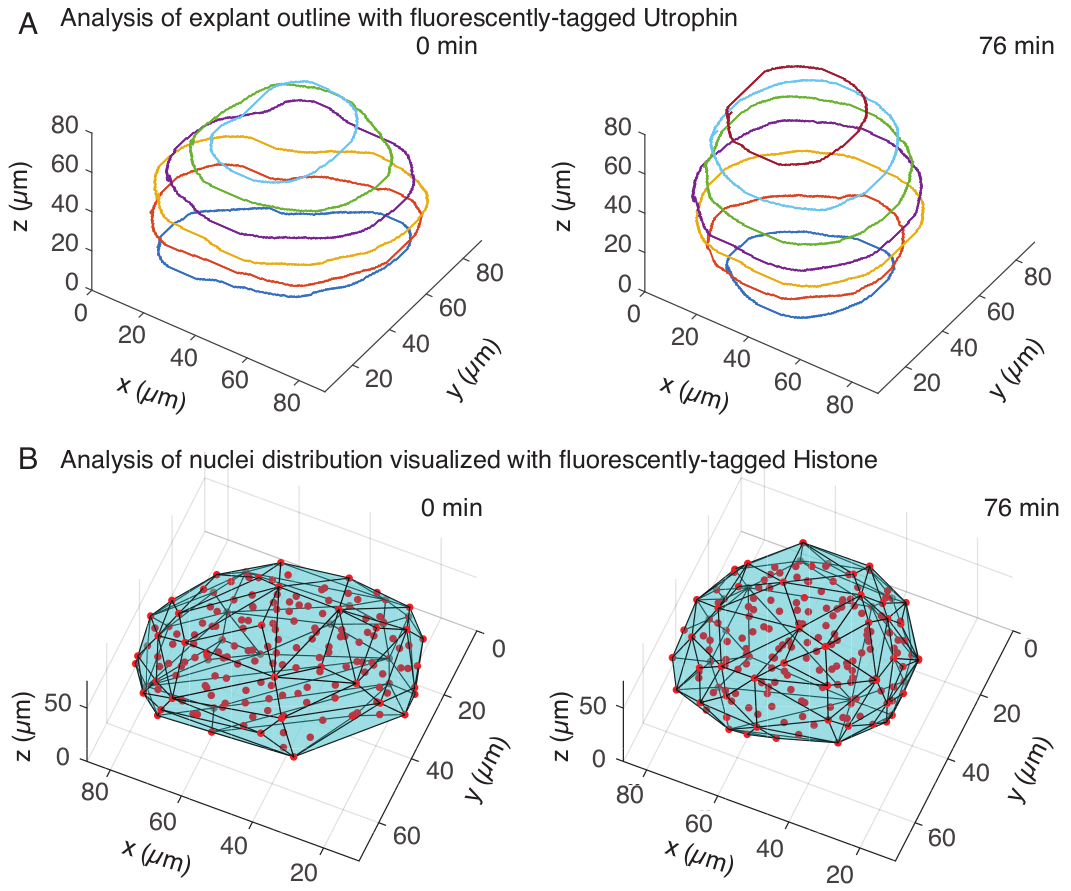
Im Utrophin-Kanal führten wir eine manuelle Segmentierung von Explantaten in MATLAB (R2018b) auf z-Schichten im Abstand von 10 μm durch. Wir haben einen benutzerdefinierten Algorithmus in MATLAB für die Segmentierung entwickelt, der kostenlos zum Download zur Verfügung steht (https://github.com/sundar07/SomSeg). Im Algorithmus können die Datei, der Rahmen und die Z-Scheibe eingestellt werden, die segmentiert werden sollen, gefolgt von einer Benutzeraufforderung, die das manuelle Zeichnen eines Umrisses um den interessierenden Bereich ermöglicht. Dies wurde für mehrere Z-Schichten wiederholt und geplottet, was eine Aufrundung der Explantate im Laufe der Zeit in 3D zeigte (Abbildung 3A). Darüber hinaus wurden ergänzende Informationen über die Gewebeform unter Verwendung des Kernkanals gewonnen. Zu diesem Zweck haben wir Mastodon (Version 1.0.0-beta-19, https://github.com/mastodon-sc/mastodon), ein FIJI15-Plugin , verwendet, um die Positionen der Kernschwerpunkte durch Spot-Detektion zu erhalten. Zuerst haben wir die tif-Dateien auf den Fidschi-Inseln vom Mikroskop in das xml/hdf5-Format konvertiert, woraufhin ein neues Projekt mit dem Mastodon-Plugin eröffnet wurde. Im Plugin wählten wir die Option Spot-Detektion, bei der wir einen Bereich of Interest definierten, der das Explantat abdeckte, und die Differenz des Gauß-Detektors mit einem Durchmesser von 5 μm und einem Qualitätsfaktor von 25 für die Erkennung von Spots verwendeten. Anschließend übertrugen wir die Positionen der Kernschwerpunkte auf MATLAB und verwendeten eine eingebaute Funktion (convhull), um eine konvexe Hülle zu erhalten (Abbildung 3B), die die Geometrie des Explantats charakterisierte. Dies zeigte in ähnlicher Weise eine Rundung der Explantate im Laufe der Zeit (Abbildung 3B). Die Zellkerndaten ermöglichen zusätzlich die Quantifizierung von Zellbewegungen durch Verfolgung in 3D und eine Veränderung der Zellzahl im Laufe der Zeit. Auf der anderen Seite ermöglicht der Utrophinkanal die Quantifizierung von Veränderungen in der Zellform, wenn die Explantate runder werden. Zusammen sind diese Parameter wertvoll für die Charakterisierung der intrinsischen Materialeigenschaften von Somiten, was bei der Entwicklung effektiver physikalischer Beschreibungen von Formänderungen im Gewebemaßstab hilft.
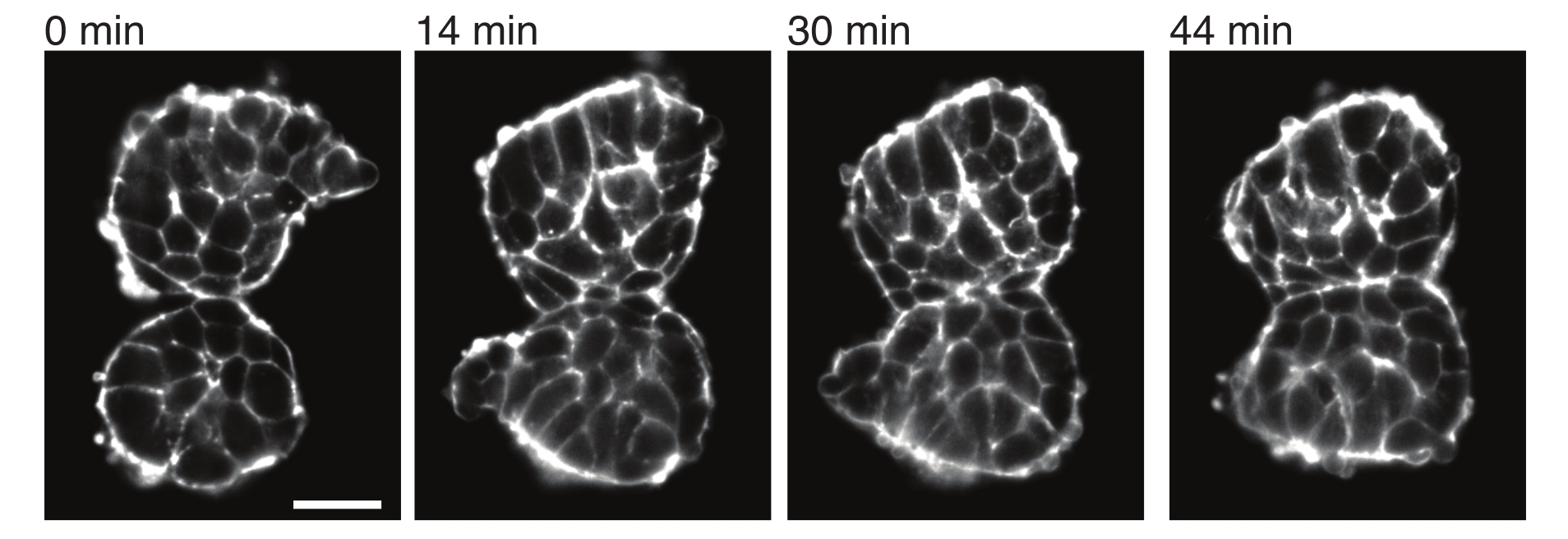
Explantate ermöglichen es auch, Kontaktspannungen mit benachbarten Geweben quantitativ zu charakterisieren. Um dies zu veranschaulichen, haben wir zwei Somiten manuell isoliert und in unmittelbarer Nähe platziert und ihre Dynamik beobachtet (N = 2). Für dieses Experiment wurde die Orientierung der Somiten in Bezug auf die in vivo Körperachsen nicht verfolgt. Interessanterweise hafteten die explantierten Somiten im Laufe der Zeit entlang einer Oberfläche aneinander, während die freien Oberflächen, d.h. die von der Kontaktstelle entfernten Regionen, aufgerundet wurden (Abbildung 4). Dies deutet darauf hin, dass Adhäsionskräfte Spannungen überwinden, die durch Oberflächenspannung an Kontaktstellen in Explantaten erzeugt werden. Dies kann weiter charakterisiert werden, indem Formänderungen wie oben beschrieben verfolgt und die Kontaktwinkel zwischen den beiden Geweben im Laufe der Zeit in 3D quantifiziert werden. Somit bieten die Explantate ein attraktives System zur Quantifizierung konkurrierender Kräfte, die zu spezifischen Gewebeformen führen, deren Implikationen dann in vivo erforscht werden können.

Abbildung 1: Präparation von Einzel-Somiten-Explantaten. Der Zebrafischembryo (A) wird zuerst dechorioniert (B), gefolgt von der Entfernung von Haut und Eigelb (C). Die somithaltige Region des Embryos wird dann ausgewählt, indem der Rest des Gewebes entfernt wird (D, E). Die Regionen um den interessierenden Somiten werden dann seriell entfernt (F-H), um schließlich einen einzelnen Somiten (I) zu isolieren, gefolgt von einer Zeitrafferbildgebung mit einem Lichtblattmikroskop (mittlerer Z-Schnitt des Somiten hier gezeigt) (J). Abkürzungen: S = Somit; N = Chord; LPM = laterale Platte Mesoderm; A = anterior; P = posterior. Gestrichelte Linien zeigen Schnittpositionen an. Maßstabsleiste = 50 μm in allen Panels. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Aufbau der Lichtblatt-Bildgebungskammer. (A-D) Ein FEP-Membranstreifen (Abmessungen in (B)) wird um den Membranhalter gewickelt, auf die Bildgebungskammer aufgesetzt und die Membran mit der Bildgebungskammer verklebt. (E-F) Am nächsten Tag wird der Membranhalter entfernt und eine bildgebende Form (Abmessungen in (E)) in die Mitte der Bildkammer in niedrigschmelzende Agarose gesetzt. Die gesamte Einheit wird 30 Minuten lang bei 4 °C gehalten, danach wird der Schimmel entfernt und die Kammer mit dem Trog für die Bildgebung von Explantaten verwendet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: 3D-Analyse von Gewebeformen. (A) Die Zellumrisse wurden mit fluoreszenzmarkiertem Utrophin (Utr::mCherry) visualisiert und die Zellkerne wurden mit fluoreszenzmarkiertem Histon (H2B::GFP) visualisiert. Die Umrisse des Explantats wurden mit MATLAB manuell in mehreren Tiefen segmentiert und hier gezeigt. (B) Kerne (rot) wurden im selben Explantat mit Mastodon, einem FIJI-Plugin, nachgewiesen und einer konvexen Hülle (cyan) ausgesetzt, die über die Geometrie des Explantats informiert. Hinweis: Die Aufrundung der Explantate ist in beiden Analysen offensichtlich. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Explantierte Somiten haften aneinander. Zwei Somiten, die aus Embryonen isoliert und manuell in unmittelbarer Nähe platziert wurden, neigen dazu, im Laufe der Zeit zu haften (N = 2). Mehrere Z-Schichten mit einem Bildintervall von 2 min wurden mit einem Lichtblattmikroskop aufgenommen und Mittelschnitte von Somiten aus ausgewählten Zeitpunkten sind hier zu sehen. Die Zellumrisse wurden mit fluoreszenzmarkiertem Utrophin (Utr::mCherry) sichtbar gemacht. Maßstabsleiste = 25 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Das Gebiet der Somitogenese wurde von Studien über die Rolle der Segmentierungsuhr bei der Festlegung der Segmentlängen während der Embryonalentwicklung dominiert. Es ist jedoch ebenso wichtig, die Rolle der Gewebemechanik bei der Bestimmung der endgültigen Somitenmorphologien zu berücksichtigen. Das hier beschriebene Protokoll ermöglicht die Explantation einzelner Somiten, deren intrinsische physikalische Eigenschaften isoliert vom Embryo untersucht werden können. Die manuelle Präparation begrenzt jedoch die Anzahl der Somiten, die typischerweise präpariert werden, auf vier bis sechs pro Bildgebungssitzung. Da das Protokoll eine sorgfältige Feindissektion mehrerer Gewebe in Embryonen ohne den Einsatz von Enzymen zur Erleichterung der Gewebeentnahme beinhaltet, kann es einige Wochen Übung erfordern, den Präparierprozess zu beherrschen. In unseren Händen beinhalten die kritischen Schritte im Protokoll die sorgfältige Entfernung von Haut und Eigelb von den Explantaten, wodurch verhindert wird, dass Explantate in den verschiedenen Stadien des Protokolls an den verwendeten Werkzeugen haften bleiben, was wiederum einfachere Dissektionen ermöglicht, um das Gewebe, das ein Somiten umgibt, seriell zu entfernen.
Das Dissektionsprotokoll kann in Zwischenschritten gestoppt werden, um Somiten zu erhalten, die nur an eines der umgebenden Gewebe gebunden sind, oder um Explantate von Gruppen von Somiten zu erhalten. Dies bietet eine leistungsfähige Methode, um Gewebe seriell zu zerlegen und den Einfluss benachbarter Gewebe auf die Erleichterung von Formveränderungen in Somiten oder in den benachbarten Geweben zu untersuchen. Auf der anderen Seite kann mit diesem Protokoll einzelnen explantierten Geweben erlaubt werden, zu haften und sich mechanisch selbst zu organisieren, wie durch die Platzierung von zwei explantierten Somiten in unmittelbarer Nähe gezeigt wird.
Explantate, die nach dieser Methode hergestellt werden, erfordern keine zusätzlichen Bestandteile im Puffer oder Einschränkungen, um das Überleben über einen Zeitraum von bis zu mehreren Stunden zu gewährleisten. Man kann sich jedoch vorstellen, sie in Hydrogelen zu kultivieren und ihre Dynamik in Gegenwart äußerer Einschränkungen zu verfolgen, was als Modell für die Kontaktspannungen dienen könnte, denen Somiten in vivo ausgesetzt sind. Darüber hinaus ermöglichen Explantate die direkte Sondierung ihrer Materialeigenschaften durch Rasterkraftmikroskopie, Pipettenaspiration oder durch den Einsatz von mikrorobotischen Werkzeugen16. Schließlich gehen wir davon aus, dass diese Methode leicht für die Kultivierung und Untersuchung anderer Entwicklungsgewebe in ähnlichen Stadien wie der Notochord, der Neuralplatte und des Mesoderms der lateralen Platte angepasst werden kann.
Offenlegungen
Die Autoren erklären, dass keine konkurrierenden Interessen bestehen.
Danksagungen
Wir danken den Mitgliedern des Labors Oates für ihre Kommentare zum Protokoll und zur Fischanlage der École polytechnique fédérale de Lausanne (EPFL). Insbesondere danken wir Laurel Ann Rohde für wertvolle Tipps zur Haut- und Dotterentfernung im Sezierprotokoll; Arianne Bercowsky Rama für den Aufbau einer effizienten Pipeline für die Verarbeitung von Light-Sheet-Datensätzen durch Mastodon; Jean-Yves Tinevez für die Entwicklung der Open-Source-Software Mastodon; Marko Popović für Tipps zur Datenanalyse; Chloé Jollivet, Guillaume Valentin und Florian Lang für die umfangreiche Unterstützung in der Fischanlage; Petr Strnad und Andrea Boni für den Bau des Lichtblattmikroskops und für Tipps zur Lichtblattbildgebung. Diese Arbeit wurde von der EPFL unterstützt und S.R.N. wurde durch ein Postdoktorandenstipendium des Long-Term Human Frontier Science Program (LT000078/2016) unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Agarose | Sigma | 9012-36-6 | For coating bottom of petri dishes |
| Agarose, low gelling temperature | Sigma | 39346-81-1 | For preparing Viventis imaging chamber |
| Camera | Andor | Andor Zyla 4.2 Plus | For image acquisition in the light-sheet microscope |
| Detection objective | Nikon | Nikon CFI75 Apo LWD 25x/1.1 NA | For imaging explants |
| FEP membrane strip | Lohmann Technologies UK Ltd | Dupont FEP Fluorocarbon film, 200A | For preparing Viventis imaging chamber |
| Fine forceps | Dumont | Dumont 5SF 11252-00 | For removal of skin of embryos |
| Forceps | Dumont | Dumont 55 | For dechorionating embryos |
| Leibovitz's L-15 medium | Gibco | 21083-027 | Explant culture medium |
| Light-sheet microscope | Viventis | LS1 live | For imaging explants |
| Micro knife | Fine Science Tools | 10318-14 | For making incisions in embryos |
| Silicone rubber formulation | Wacker Chemie AG | Silpuran 4200 | For preparing Viventis imaging chamber |
Referenzen
- Oates, A. C., Morelli, L. G., Ares, S. Patterning embryos with oscillations: structure, function and dynamics of the vertebrate segmentation clock. Development. 139 (4), Cambridge, England. 625-639 (2012).
- Pourquié, O. Segmentation of the vertebrate spine: From clock to scoliosis. Cell. 145 (5), 650-663 (2011).
- Naganathan, S. R., Oates, A. C. Patterning and mechanics of somite boundaries in zebrafish embryos. Seminars in Cell & Developmental Biology. 107, 170-178 (2020).
- Hubaud, A., Regev, I., Mahadevan, L., Pourquié, O. Excitable Dynamics and Yap-Dependent Mechanical Cues Drive the Segmentation Clock. Cell. 171 (3), 668-682 (2017).
- Dias, A. S., de Almeida, I., Belmonte, J. M., Glazier, J. A., Stern, C. D. Somites without a clock. Science. 343 (6172), New York, N.Y. 791-795 (2014).
- Nelemans, B. K. A., Schmitz, M., Tahir, H., Merks, R. M., Smit, T. H. Somite Division and New Boundary Formation by Mechanical Strain. iScience. 23 (4), 100976(2020).
- Grima, R., Schnell, S. Can tissue surface tension drive somite formation. Developmental Biology. 307 (2), 248-257 (2007).
- Naganathan, S. R., Popovic, M., Oates, A. C. Left–right symmetry of zebrafish embryos requires somite surface tension. Nature. 605, 516-521 (2022).
- Campàs, O. A toolbox to explore the mechanics of living embryonic tissues. Seminars in Cell & Developmental Biology. 55, 119-130 (2016).
- Langenberg, T., Brand, M., Cooper, M. S. Imaging brain development and organogenesis in zebrafish using immobilized embryonic explants. Developmental Dynamics: An Official Publication of The American Association of Anatomists. 228 (3), 464-474 (2003).
- Henry, C. A., Poage, C. T., McCarthy, M. B., Campos-Ortega, J., Cooper, M. S. Regionally autonomous segmentation within zebrafish presomitic mesoderm. Zebrafish. 2 (1), 7-18 (2005).
- Picker, A., Roellig, D., Pourquié, O., Oates, A. C., Brand, M. Tissue micromanipulation in zebrafish embryos. Methods in Molecular Biology. 546, Clifton, N.J. 153-172 (2009).
- Manning, A. J., Kimelman, D. Tbx16 and Msgn1 are required to establish directional cell migration of zebrafish mesodermal progenitors. Developmental Biology. 406 (2), 172-185 (2015).
- Simsek, M. F., Özbudak, E. M. A 3-D Tail Explant Culture to Study Vertebrate Segmentation in Zebrafish. Journal of Visualized Experiments:JoVE. (172), e61981(2021).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Özkale, B., et al. Modular soft robotic microdevices for dexterous biomanipulation. Lab on a Chip. 19 (5), 778-788 (2019).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten