
Method Article
Préparation d’explants de poisson zèbre à partir d’embryons de poisson-zèbre
Dans cet article
Résumé
Nous présentons un protocole pour isoler des somites uniques d’embryons de poisson-zèbre, dont la dynamique peut être suivie en culture pendant plusieurs heures par microscopie à fluorescence, fournissant ainsi une méthodologie pour quantifier les changements de forme à l’échelle tissulaire à une résolution unicellulaire.
Résumé
L’axe corporel des embryons de vertébrés est périodiquement subdivisé en unités multicellulaires 3D appelées somites. Alors que les oscillations génétiques et les prémotifs moléculaires déterminent l’échelle de longueur initiale des somites, des processus mécaniques ont été impliqués dans la définition de leur taille et de leur forme finales. Pour mieux comprendre les propriétés matérielles intrinsèques des somites, une méthode est développée pour cultiver des explants uniques à partir d’embryons de poisson-zèbre. Les somites uniques sont isolés en enlevant d’abord la peau des embryons, puis en enlevant le vitellus et en excisant séquentiellement les tissus voisins. À l’aide d’embryons transgéniques, la distribution de diverses structures subcellulaires peut être observée par microscopie à fluorescence. La dynamique des somites explantés peut être suivie pendant plusieurs heures, fournissant ainsi un cadre expérimental pour étudier les changements de forme à l’échelle tissulaire à la résolution d’une seule cellule. Cette approche permet une manipulation mécanique directe des somites, ce qui permet de disséquer les propriétés matérielles du tissu. Enfin, la technique décrite ici peut être facilement étendue pour l’explantation d’autres tissus tels que la notochorde, la plaque neurale et le mésoderme de la plaque latérale.
Introduction
Une grande partie du système musculo-squelettique adulte des vertébrés émerge des somites embryonnaires, qui se forment de manière périodique et rythmique le long de l’axe corporel des embryons 1,2. Les somites sont des unités multicellulaires tridimensionnelles (3D) généralement constituées d’un noyau interne de cellules mésenchymateuses et d’une couche épithéliale périphérique entourée d’une matrice extracellulaire riche en fibronectine3. La morphologie des somites, c’est-à-dire leur taille et leur forme, est en partie déterminée par l’horloge de segmentation et les prémodèles moléculaires en aval. Cependant, au cours de la dernière décennie, il est apparu que les indices et les forces mécaniques jouent également un rôle dans la régulation de l’horloge de segmentation4, en plus de faciliter la formation de somites 5,6,7 et d’assurer une précision accrue des longueurs de somite après la formation initiale de somite8.
La mécanique tissulaire peut être étudiée directement in vivo avec la disponibilité de nouveaux outils9, cependant, pour obtenir une image complète sous-jacente aux processus physiques, les propriétés intrinsèques des matériaux des tissus doivent être étudiées simultanément. Le protocole décrit ici fournit une approche simple pour préparer des somites uniques, dont les propriétés physiques, de l’échelle cellulaire à l’échelle tissulaire, peuvent être étudiées isolément de l’embryon. Bien qu’il existe plusieurs protocoles pour la préparation d’explants à des stades de développement similaires 10,11,12,13,14, à notre connaissance, il s’agit du premier protocole qui décrit l’isolement de somites uniques. Le protocole est simple à mettre en œuvre et ne nécessite que l’équipement de base disponible dans la plupart des laboratoires de poissons-zèbres travaillant avec des embryons.
Pour aider à démêler le rôle de la mécanique dans la formation morphologique des somites, une méthode est développée pour cultiver des explants de somemites uniques à partir d’embryons de poissons-zèbres, qui peuvent être utilisés pour sonder les propriétés intrinsèques des matériaux des somites.
Protocole
Ce protocole implique l’utilisation d’embryons de vertébrés vivants moins d’un jour après la fécondation. Toutes les expériences ont été réalisées à l’aide d’embryons issus d’adultes en libre accouplement, et sont donc couvertes par la licence générale d’expérimentation animale de l’EPFL délivrée par le Service de la consommation et des affaires vétérinaires du canton de Vaud - Suisse (numéro d’autorisation VD-H23).
1. Avant la dissection
- Obtenir des embryons à partir d’un croisement de lignées transgéniques hétérozygotes d’intérêt. Pour explanter deux à trois somites, seuls quelques embryons sont généralement nécessaires. Dans ce protocole, un embryon a été utilisé pour l’explantation d’un somite.
- Préparez 1 tampon E3 à partir de deux stocks 50x : la matière 1 contient 0,458 mM Na2HPO4, 0,042 mM KH2PO4, 4,084 mM NaCl, 0,128 mM KCl et la matière 2 contient 0,33 mM de CaCl2, 0,33 mM de MgSO4. Préparez 500 ml de 1 tampon E3 en mélangeant 480 ml d’eau distillée avec 10 ml chacun des deux 50 tampons.
- Si vous disséquez le même jour, faites cuire les embryons dans 25 ml de milieu E3 dans une boîte de Pétri à 33 °C jusqu’au stade de trois somemites. Si vous dissectionnez le lendemain matin, élever les embryons dans un milieu E3 à 28 °C jusqu’au stade de bouclier avant de les transférer à 19 °C pour une incubation d’une nuit.
- Pour identifier l’étage du bouclier ou les différents étages du somite, utilisez un stéréomicroscope de laboratoire standard avec une plage de grossissement réglable d’environ 0,67x à 4,5x.
- Assembler les outils et les réactifs nécessaires à la dissection.
- Aliquote Leibovitz’s L-15 medium en tubes de 50 mL et conserver au réfrigérateur. Nous vous recommandons d’utiliser la même aliquote du réfrigérateur pendant un maximum de deux tours.
- Préparez 50 ml d’agarose à 2 % dans un milieu L-15 à l’aide d’un agitateur magnétique et en maintenant la température à 85 °C. Une fois dissoute, conservez la solution à température ambiante.
- Préparez 25 mL d’agarose à bas point de fusion à 2 % dans un milieu L-15 à l’aide d’un agitateur magnétique et en maintenant la température à 85 °C. Une fois dissoute, aliquote la solution dans des tubes de 1,5 mL et conserver à 4 °C. Celui-ci sera utilisé pour préparer la chambre d’imagerie.
- Versez l’agarose fondue à 2 % dans une boîte de Pétri de 20 mm jusqu’à ce qu’un tiers de la profondeur de la boîte soit rempli et laissez la solution d’agarose se solidifier. Des dissections seront effectuées dans ces boîtes de Pétri. Préparez les boîtes de Pétri avec enrobage d’agarose bien à l’avance et conservez-les à 4 °C.
- Assemblez les outils suivants : une pipette Pasteur pour le transfert des embryons, une paire de pinces pour la déchorionation des embryons, une paire de pinces fines pour l’ablation de la peau des embryons, un micro-couteau pour faire des incisions dans les embryons, une pipette en verre poli au feu pour le transfert des explants dans la chambre d’imagerie.
- Stérilisez la pipette en verre avec de l’éthanol à 100 % pendant 15 minutes et rincez deux fois en pipetant 5 ml de milieu L-15 de haut en bas.
- Préparez un outil pour cils en collant un cil ou un cheveu du sourcil à un capillaire en verre.
- Utilisez un stéréoscope fluorescent avec une plage de grossissement réglable d’environ 0,63x à 6,3x et des filtres GFP et RFP standard pour trier les embryons transgéniques positifs et les transférer dans une boîte de Pétri séparée avec un milieu E3.
2. Préparation d’explants d’un seul somite
- Transférez 2 ou 3 embryons dans une boîte enrobée d’agarose (réalisée à l’étape 1.5.4) remplie de 10 mL de milieu L-15. Assurez-vous que la parabole et le milieu L-15 sont préchauffés à 28 °C dans un incubateur pendant 30 minutes avant de transférer les embryons.
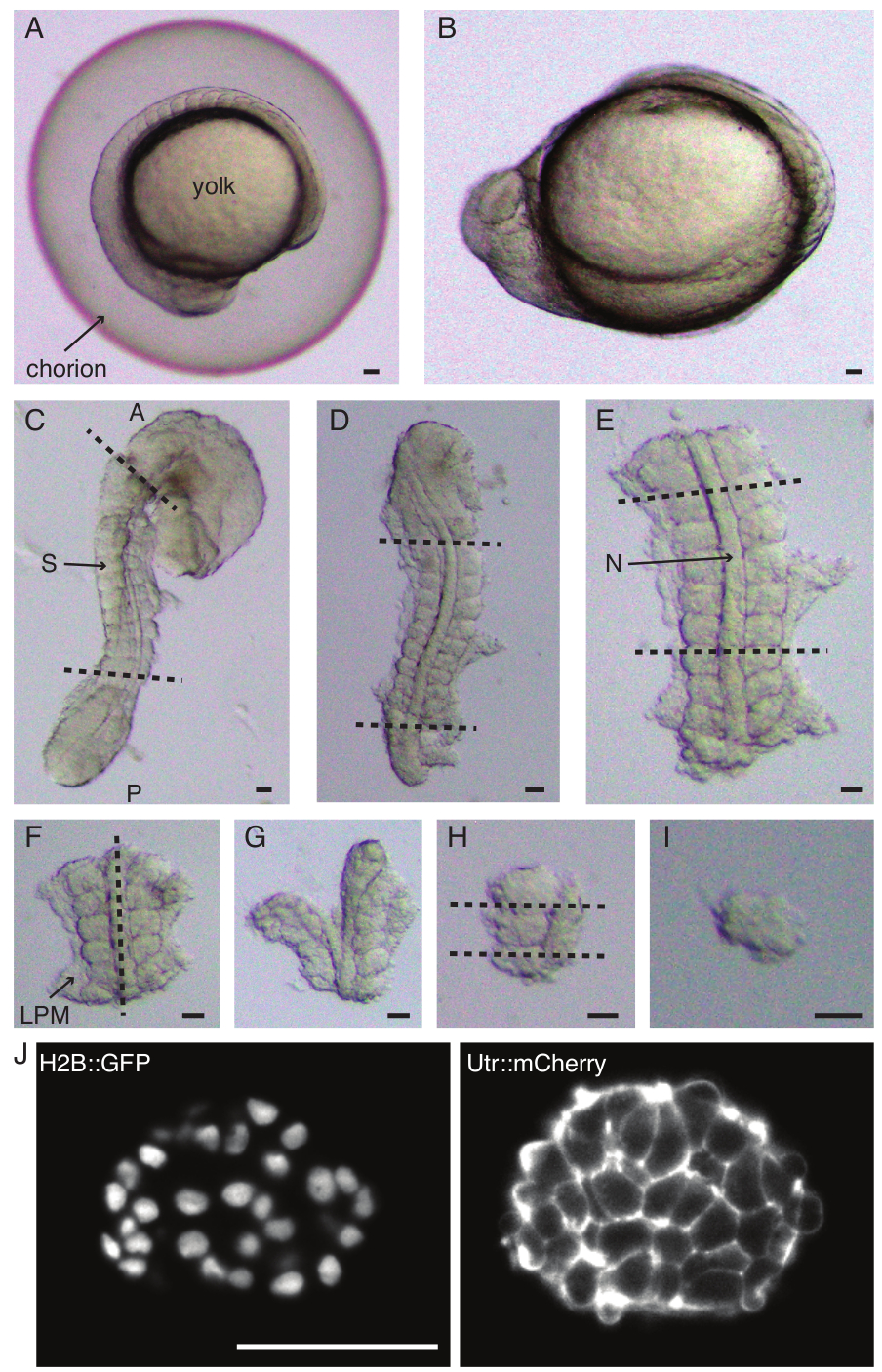
- Déchorionnez soigneusement (Figure 1 A, B) un embryon à l’aide d’une paire de pinces. Tenez le chorion avec l’une des pinces et utilisez la deuxième pince pour pincer et tirer sur le chorion près de l’endroit où la première pince est positionnée.
- Répétez cette étape une fois de plus pour ouvrir largement le chorion, après quoi l’embryon peut être doucement poussé vers l’extérieur. Assurez-vous que l’embryon n’est pas endommagé ou comprimé pendant la délivrion.
- Orientez l’embryon sur sa face latérale et tenez la peau entre la tête et la queue de l’embryon avec l’une des fines pinces. Utilisez la deuxième pince fine pour pincer la peau près de l’endroit où la première pince est encore positionnée.
- Éloignez les deux pinces l’une de l’autre, tout en les tenant à la peau, créant ainsi une grande ouverture. Relâchez l’une des pinces de la peau et déplacez-la près de l’endroit où la deuxième pince est tenue et répétez le processus d’élimination de la peau jusqu’à ce que la peau soit retirée dans les régions formant des somites.
REMARQUE : Il est essentiel d’enlever complètement la peau entourant les somites qui seront explantés, sinon il devient difficile de faire des dissections plus fines dans les dernières étapes du protocole. - À l’aide d’un micro-couteau, grattez le jaune (Figure 1C - explant avec la peau enlevée et le jaune gratté) et faites une série d’incisions comme suit :
- Trancher l’embryon avec la première coupe antérieure au premier somite morphologique de l’embryon et la seconde coupe postérieure au somite le plus récemment formé (figure 1D). Trancher une fois de plus en arrière et en avant des somites d’intérêt (Figure 1E).
- À l’aide du micro-couteau, grattez le jaune restant dans les régions explantées de l’embryon. Essayez d’enlever autant de jaune que possible pour réduire le risque que les explants se collent au micro-couteau. Si cela se produit, utilisez un outil à cils pour ramener l’explant dans la solution tout en gardant le micro-couteau immergé dans le milieu.
- Positionnez l’explant dorsalement et faites une tranche le long de l’axe antéropostérieur à travers la notochorde, près des somites d’un côté (Figure 1F,G).
- Prenez l’explant sans la notocorde et coupez à travers le mésoderme de la plaque latérale (LPM) près des somites.
REMARQUE : Cette étape peut être remplacée par l’étape précédente, c’est-à-dire qu’elle fonctionne tout aussi bien pour couper le LPM en premier, suivi de trancher la notochorde.
- Choisissez le somite à explanter et faites deux tranches respectivement en arrière et en avant du somite (Figure 1H).
- Enfin, faites pivoter le somite de 90° à l’aide de l’outil œil-cil pour visualiser la plaque neurale toujours attachée au somite. Faites une tranche entre le somite et la plaque neurale, libérant ainsi un seul somite (Figure 1I).
REMARQUE : La plaque neurale peut également être retirée immédiatement après l’étape 2.6.4, c’est-à-dire après l’élimination de la notocorde et du LPM et avant de disséquer un somite des somites voisins.
3. Imagerie d’explants monosomites
REMARQUE : Ici, la procédure d’imagerie des explants monosomites à l’aide d’un microscope à feuillet de lumière à vue unique est décrite. Comme alternative, les somites uniques explantés peuvent également être imagés dans un microscope confocal, qui est plus largement disponible, ou même dans un microscope à grand champ si l’on s’intéresse exclusivement à la morphologie globale des explants.
- Préparez une chambre d’imagerie comme suit :
- Remplissez une seringue de 1 mL avec une formulation de caoutchouc de silicone. Coupez une pointe de pipette de 200 μL à environ 1 cm de la bouche et insérez la seringue dans la pointe de la pipette.
- Enveloppez manuellement le support de membrane à l’aide d’une bande de membrane d’éthylène-propylène fluoré (FEP) prédécoupée (Figure 2A-C) et fixez la chambre d’imagerie sur le support de membrane, qui s’emboîtent parfaitement les uns dans les autres (Figure 2D).
- Ajoutez une formulation de caoutchouc de silicone le long de l’intersection de la chambre et de la membrane et séchez-la pendant la nuit, ce qui permet à la membrane de coller à la chambre. Retirez le support de membrane 1 h avant l’expérience d’explantation.
- Chauffer une aliquote contenant 0,5 mL d’agarose à bas point de fusion à 2 % faite avec du milieu L-15 à 75 °C pendant 10 min. Ajoutez 160 μL d’agarose à bas point de fusion chauffé au centre de la chambre et placez un moule d’imagerie dans l’agarose (figures 2E, F) et maintenez-le debout jusqu’à ce que l’agarose se solidifie.
- Ajoutez encore 160 μL d’agarose à bas point de fusion chauffé aux deux extrémités du moule et transférez l’ensemble de l’unité à 4 °C pendant 30 min.
- Ajoutez 600 μL de fluide L-15 dans la chambre et retirez le moule de la chambre à l’aide d’une pince. Attendez que la chambre avec le milieu L-15 revienne à température ambiante.
- Transférez le somite explanté à l’aide d’une pipette en verre poli par pipetage dans la chambre d’imagerie et positionnez l’explant au centre de la chambre à l’aide d’un outil à cils.
- Alignez les lasers d’imagerie conformément aux instructions du fabricant et transférez soigneusement la chambre d’imagerie dans le microscope équipé d’une chambre d’incubation pour maintenir les explants à 28 °C.
- Effectuez un deuxième tour d’alignement laser avec les régions d’intérêt de la raie transgénique, puis configurez l’empilement z, l’intervalle de temps et la durée souhaités pour l’imagerie en accéléré. Pour cette expérience, acquérez 70 tranches z avec un espacement de 2 μm et un intervalle de trame de 2 min.
- Effectuez une imagerie par feuille de lumière bicolore (Figure 1J) des explants avec des lasers de 561 nm (puissance laser de 10 %, exposition de 100 ms) et de 488 nm (puissance laser de 10 %, exposition de 100 ms). Dans ce protocole, le signal a été collecté avec un objectif NA 25x/1.1 et à travers des filtres passe-bande 561/25 nm et 525/50-25 respectivement sur une caméra sCMOS.
Résultats
Les explants permettent de quantifier les changements de forme à grande échelle en 3D. Pour illustrer cela, nous avons explanté la somite quatre (N = 3) à partir d’embryons précoces de poisson-zèbre obtenus à partir d’un croisement entre des lignées hétérozygotes Utr ::mCherry (Tg(actb2 :mCherry-Hsa.UTRN) ; e119Tg) et H2B ::GFP (Tg(h2az2a :h2az2a-GFP) ; kca6Tg). L’utrophine est une protéine liant l’actine, et la ligne Utr ::mCherry montre la distribution des structures d’actine filamenteuses. Ce transgène a été utilisé ici comme marqueur pour les contours cellulaires. H2B ::GFP est une histone marquée par fluorescence et marque par conséquent la distribution de la chromatine, fournissant une description efficace de l’emplacement et de la forme nucléaires, ainsi que des figures mitotiques qui mettent en évidence la division cellulaire.
Nous avons évalué le succès du protocole d’explantation lors de l’imagerie. Nous avons observé que chez un somite endommagé lors de la dissection, soit l’intégrité du tissu du somite est compromise, avec de nombreuses cellules se dissociant et extrudant du somite explanté, et/ou de nombreuses cellules mourant, ce qui peut être noté par la présence de noyaux fragmentés dans le canal nucléaire. Les explants réussis sont restés sains pendant 4 à 6 heures, après quoi des changements dans l’intégrité du somite ont été observés, les cellules se dissociant de l’explant et mourant.
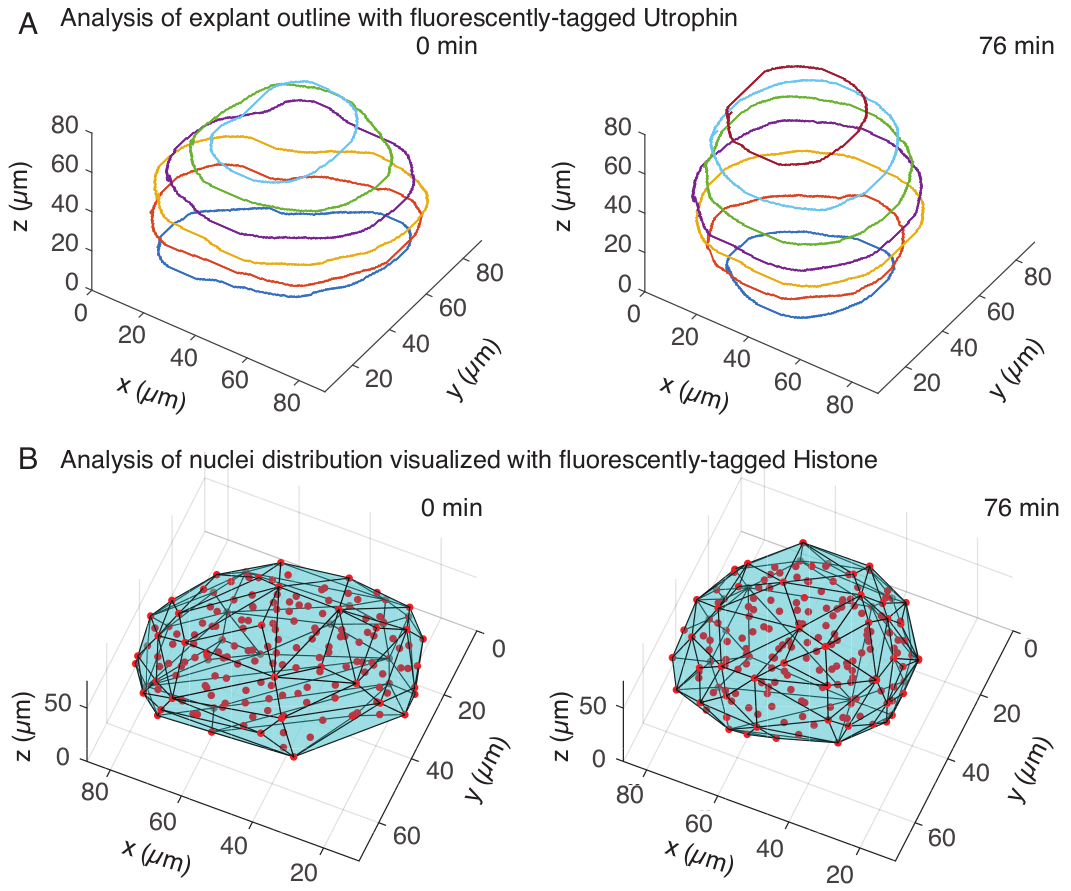
Dans le canal utrophine, nous avons effectué une segmentation manuelle des explants dans MATLAB (R2018b) sur des tranches z espacées tous les 10 μm. Nous avons développé un algorithme de segmentation personnalisé dans MATLAB, qui peut être téléchargé gratuitement (https://github.com/sundar07/SomSeg). Dans l’algorithme, le fichier, le cadre et la tranche z à segmenter peuvent être définis, suivis d’une invite de l’utilisateur, ce qui permet de dessiner manuellement un contour autour de la région d’intérêt. Cela a été répété pour plusieurs coupes z et tracé, ce qui a montré l’arrondi des explants au fil du temps en 3D (Figure 3A). De plus, des informations complémentaires sur la forme des tissus à l’aide du canal nucléaire ont également été obtenues. Pour cela, nous avons utilisé Mastodon (version 1.0.0-beta-19, https://github.com/mastodon-sc/mastodon), un plugin FIJI15 , pour obtenir les positions des centroïdes des noyaux grâce à la détection ponctuelle. Nous avons d’abord converti les fichiers tif du microscope au format xml/hdf5 dans FIJI, après quoi un nouveau projet a été ouvert à l’aide du plugin Mastodon. Dans le plugin, nous avons choisi l’option de détection ponctuelle, où nous avons défini une région d’intérêt qui couvrait l’explant et utilisé la différence de détecteur gaussien d’un diamètre de 5 μm et d’un facteur de qualité de 25 pour la détection des taches. Nous avons ensuite transféré les positions centroïdes des noyaux dans MATLAB et utilisé une fonction intégrée (convhull) pour obtenir une enveloppe convexe (Figure 3B), qui a caractérisé la géométrie de l’explant. De même, on a vu l’arrondi des explants dans le temps (figure 3B). Les données des noyaux permettent en outre de quantifier les mouvements cellulaires en suivant en 3D l’évolution du nombre de cellules au fil du temps. D’autre part, le canal utrophine permet de quantifier les changements de forme cellulaire à mesure que les explants s’arrondissent. Ensemble, ces paramètres sont précieux pour caractériser les propriétés intrinsèques des matériaux des somites, ce qui aide à développer des descriptions physiques efficaces des changements de forme à l’échelle des tissus.
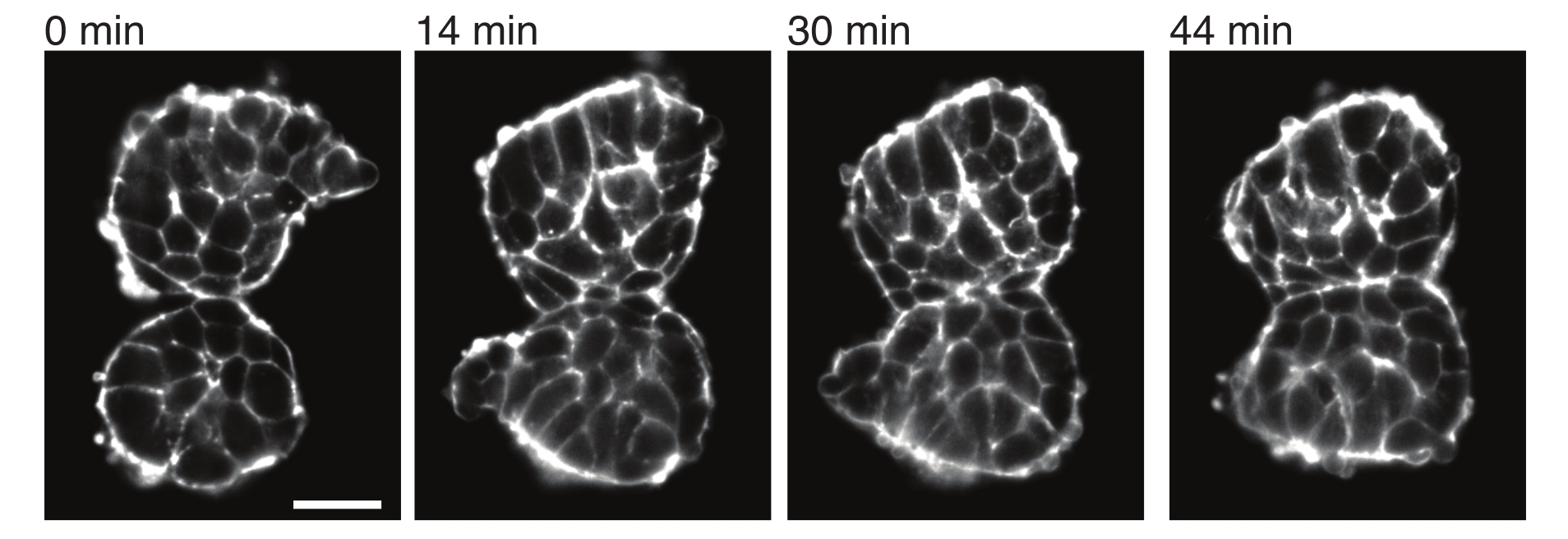
Les explants permettent également de caractériser les contraintes de contact avec les tissus voisins de manière quantitative. Pour illustrer cela, nous avons isolé manuellement deux somites et les avons placés à proximité immédiate et observé leur dynamique (N = 2). Pour cette expérience, l’orientation des somites par rapport aux axes corporels in vivo n’a pas été suivie. Il est intéressant de noter qu’au fil du temps, les somites explantés ont adhéré les uns aux autres le long d’une surface, tandis que les surfaces libres, c’est-à-dire les régions éloignées du site de contact, se sont arrondies vers le haut (Figure 4). Cela suggère que les forces d’adhérence surmontent les contraintes générées par la tension superficielle aux sites de contact dans les explants. Cela peut être caractérisé en suivant les changements de forme décrits ci-dessus et en quantifiant les angles de contact entre les deux tissus au fil du temps en 3D. Ainsi, les explants fournissent un système attrayant pour quantifier les forces concurrentes en action qui conduisent à des formes tissulaires spécifiques, dont les implications peuvent ensuite être explorées in vivo.

Figure 1 : Préparation d’explants mono-somite. L’embryon de poisson-zèbre (A) est d’abord déchorionné (B), suivi de l’ablation de la peau et du vitellus (C). La région de l’embryon contenant le somite est ensuite sélectionnée en enlevant le reste des tissus (D, E). Les régions autour du somite d’intérêt sont ensuite retirées en série (F-H) pour finalement isoler un seul somite (I), suivie d’une imagerie en accéléré à l’aide d’un microscope à feuillet de lumière (section en Z centrale du somite illustrée ici) (J). Abréviations : S = somite ; N = notochorde ; LPM = mésoderme de la plaque latérale ; A = antérieur ; P = postérieur. Les lignes pointillées indiquent les positions de coupe. Barre d’échelle = 50 μm dans tous les panneaux. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Assemblage d’une chambre d’imagerie à feuillet léger. (A-D) Une bande de membrane FEP (dimensions en (B)) est enroulée autour du support de membrane, fixée sur la chambre d’imagerie et la membrane est collée à la chambre d’imagerie. (E-F) Le lendemain, le support de membrane est retiré et un moule d’imagerie (dimensions en (E)) est placé au centre de la chambre d’imagerie dans une agarose à bas point de fusion. L’ensemble de l’unité est maintenu à 4 °C pendant 30 min, après quoi le moule est retiré et la chambre avec l’auge est utilisée pour l’imagerie des explants. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Analyse 3D de formes de tissus. (A) Les contours des cellules ont été visualisés avec de l’utrophine marquée par fluorescence (Utr ::mCherry) et les noyaux ont été visualisés avec des histones marquées par fluorescence (H2B ::GFP). Les contours de l’explant ont été segmentés manuellement à plusieurs profondeurs à l’aide de MATLAB et présentés ici. (B) Des noyaux (rouges) ont été détectés dans le même explant à l’aide de Mastodon, un plugin FIJI, et soumis à une enveloppe convexe (cyan), qui informe sur la géométrie de l’explant. Notez que l’arrondi des explants est évident dans les deux analyses. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Les somites explantés adhèrent les uns aux autres. Deux somites isolés d’embryons et placés manuellement à proximité ont tendance à adhérer dans le temps (N = 2). Plusieurs coupes z avec un intervalle d’image de 2 min ont été acquises à l’aide d’un microscope à feuillet de lumière et des sections centrales de somites à partir de points temporels sélectionnés sont montrées ici. Les contours des cellules ont été visualisés avec de l’utrophine marquée par fluorescence (Utr ::mCherry). Barre d’échelle = 25 μm. Veuillez cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Discussion
Le domaine de la somitogenèse a été dominé par des études sur le rôle de l’horloge de segmentation dans la définition de la longueur des segments au cours du développement embryonnaire. Cependant, il est tout aussi important de considérer le rôle de la mécanique tissulaire dans la détermination de la morphologie finale des somites. Le protocole décrit ici permet d’explanter des somites uniques, dont les propriétés physiques intrinsèques peuvent être étudiées isolément de l’embryon. Cependant, la préparation manuelle limite le nombre de somites qui sont généralement préparés à quatre à six par séance d’imagerie. Étant donné que le protocole implique des dissections fines et soigneuses de plusieurs tissus dans les embryons sans l’utilisation d’enzymes pour faciliter l’élimination des tissus, il peut falloir quelques semaines de pratique pour maîtriser le processus de dissection. Entre nos mains, les étapes critiques du protocole impliquent l’élimination minutieuse de la peau et du jaune des explants, ce qui empêche les explants à différentes étapes du protocole de coller aux outils utilisés, ce qui permet à son tour des dissections plus faciles d’éliminer en série les tissus entourant un somite.
Le protocole de dissection peut être arrêté à des étapes intermédiaires pour obtenir des somites attachés à un seul des tissus environnants ou pour obtenir des explants de groupes de somites. Il s’agit d’une méthode puissante pour démonter en série les tissus et étudier l’impact des tissus voisins sur les changements de forme des somites ou des tissus voisins. D’autre part, en utilisant ce protocole, on peut laisser des tissus explantés individuels adhérer et s’auto-organiser mécaniquement, comme le démontre le placement de deux somites explantés à proximité.
Les explants préparés selon cette méthode ne nécessitent aucun ingrédient ajouté dans le tampon ni de contraintes pour assurer la survie pendant des périodes allant jusqu’à plusieurs heures. Cependant, on peut imaginer les cultiver dans des hydrogels et suivre leur dynamique en présence de contraintes externes, ce qui pourrait servir de modèle pour les stress de contact rencontrés par les somites in vivo. De plus, les explants permettent de sonder directement les propriétés de leurs matériaux par microscopie à force atomique, aspiration à pipette ou à l’aide d’outils micro-robotiques16. Enfin, nous pensons que cette méthode peut être facilement adaptée à la culture et à l’étude d’autres tissus développementaux à des stades similaires tels que la notochorde, la plaque neurale et le mésoderme de la plaque latérale.
Déclarations de divulgation
Les auteurs ne déclarent aucun intérêt concurrent.
Remerciements
Nous remercions les membres du laboratoire Oates pour leurs commentaires sur le protocole et l’installation piscicole de l’École polytechnique fédérale de Lausanne (EPFL). En particulier, nous remercions Laurel Ann Rohde pour ses précieux conseils sur l’élimination de la peau et du jaune dans le protocole de dissection ; Arianne Bercowsky Rama pour la construction d’un pipeline efficace pour le traitement d’ensembles de données de feuille de lumière via Mastodon ; Jean-Yves Tinevez pour la création du logiciel open source Mastodon ; Marko Popović pour des conseils sur l’analyse des données ; Chloé Jollivet, Guillaume Valentin et Florian Lang pour leur soutien important dans la pisciculture ; Petr Strnad et Andrea Boni pour la construction du microscope à feuillet de lumière et pour les conseils sur l’imagerie à feuillet de lumière. Ce travail a été soutenu par l’EPFL et S.R.N. a été soutenu par une bourse postdoctorale du Long-Term Human Frontier Science Program (LT000078/2016).
matériels
| Name | Company | Catalog Number | Comments |
| Agarose | Sigma | 9012-36-6 | For coating bottom of petri dishes |
| Agarose, low gelling temperature | Sigma | 39346-81-1 | For preparing Viventis imaging chamber |
| Camera | Andor | Andor Zyla 4.2 Plus | For image acquisition in the light-sheet microscope |
| Detection objective | Nikon | Nikon CFI75 Apo LWD 25x/1.1 NA | For imaging explants |
| FEP membrane strip | Lohmann Technologies UK Ltd | Dupont FEP Fluorocarbon film, 200A | For preparing Viventis imaging chamber |
| Fine forceps | Dumont | Dumont 5SF 11252-00 | For removal of skin of embryos |
| Forceps | Dumont | Dumont 55 | For dechorionating embryos |
| Leibovitz's L-15 medium | Gibco | 21083-027 | Explant culture medium |
| Light-sheet microscope | Viventis | LS1 live | For imaging explants |
| Micro knife | Fine Science Tools | 10318-14 | For making incisions in embryos |
| Silicone rubber formulation | Wacker Chemie AG | Silpuran 4200 | For preparing Viventis imaging chamber |
Références
- Oates, A. C., Morelli, L. G., Ares, S. Patterning embryos with oscillations: structure, function and dynamics of the vertebrate segmentation clock. Development. 139 (4), 625-639 (2012).
- Pourquié, O. Segmentation of the vertebrate spine: From clock to scoliosis. Cell. 145 (5), 650-663 (2011).
- Naganathan, S. R., Oates, A. C. Patterning and mechanics of somite boundaries in zebrafish embryos. Seminars in Cell & Developmental Biology. 107, 170-178 (2020).
- Hubaud, A., Regev, I., Mahadevan, L., Pourquié, O. Excitable Dynamics and Yap-Dependent Mechanical Cues Drive the Segmentation Clock. Cell. 171 (3), 668-682 (2017).
- Dias, A. S., de Almeida, I., Belmonte, J. M., Glazier, J. A., Stern, C. D. Somites without a clock. Science. 343 (6172), 791-795 (2014).
- Nelemans, B. K. A., Schmitz, M., Tahir, H., Merks, R. M., Smit, T. H. Somite Division and New Boundary Formation by Mechanical Strain. iScience. 23 (4), 100976 (2020).
- Grima, R., Schnell, S. Can tissue surface tension drive somite formation. Developmental Biology. 307 (2), 248-257 (2007).
- Naganathan, S. R., Popovic, M., Oates, A. C. Left–right symmetry of zebrafish embryos requires somite surface tension. Nature. 605, 516-521 (2022).
- Campàs, O. A toolbox to explore the mechanics of living embryonic tissues. Seminars in Cell & Developmental Biology. 55, 119-130 (2016).
- Langenberg, T., Brand, M., Cooper, M. S. Imaging brain development and organogenesis in zebrafish using immobilized embryonic explants. Developmental Dynamics: An Official Publication of The American Association of Anatomists. 228 (3), 464-474 (2003).
- Henry, C. A., Poage, C. T., McCarthy, M. B., Campos-Ortega, J., Cooper, M. S. Regionally autonomous segmentation within zebrafish presomitic mesoderm. Zebrafish. 2 (1), 7-18 (2005).
- Picker, A., Roellig, D., Pourquié, O., Oates, A. C., Brand, M. Tissue micromanipulation in zebrafish embryos. Methods in Molecular Biology. 546, 153-172 (2009).
- Manning, A. J., Kimelman, D. Tbx16 and Msgn1 are required to establish directional cell migration of zebrafish mesodermal progenitors. Developmental Biology. 406 (2), 172-185 (2015).
- Simsek, M. F., Özbudak, E. M. A 3-D Tail Explant Culture to Study Vertebrate Segmentation in Zebrafish. Journal of Visualized Experiments:JoVE. (172), e61981 (2021).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Özkale, B., et al. Modular soft robotic microdevices for dexterous biomanipulation. Lab on a Chip. 19 (5), 778-788 (2019).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.