
Method Article
クライオ電子断層図におけるアクチンネットワークのセグメンテーションのための効果的な戦略としての簡略化された体積モデル
要約
ここでは、簡略化された体積モデルをノイズが多く複雑な断層撮影3Dボリュームに配置するためのプロトコルを紹介します。これにより、アクチンフィラメント密度の迅速なセグメンテーション、系統的なフィラメントの曲げや毛束フィラメントのギャップの検出、および距離などの体積モデル特性の簡便な定量化が可能になります。
要約
関心のある特徴を抽出するための効率的な方法は、クライオ電子断層撮影法の解釈における最大の課題の1つです。さまざまな自動化アプローチが提案されており、その多くは、関心のある特徴を簡単に検出でき、互いに明確に分離されている高コントラストのデータセットに適しています。当社の内耳立体繊毛クライオ電子断層撮影データセットは、頻繁に交差する六角形に詰め込まれたアクチンフィラメントの密集した配列によって特徴付けられます。これらの特徴により、自動セグメンテーションは非常に困難であり、クライオ電子断層撮影の高ノイズ環境や高密度に詰め込まれた特徴の複雑性によってさらに悪化します。アクチンバンドルの構成に関する予備知識を使用して、非常に単純化されたボールアンドスティックアクチンモデルのレイヤーを配置して、最初に密度マップへの全体的な適合を取得し、次にモデルの地域的および局所的な調整を行いました。ボリュームモデルの構築により、高い複雑さを処理できるだけでなく、アクチンバンドルに関する正確な測定と統計も提供できることを示します。ボリュームモデルは、アクチン-アクチンクロスコネクターの場合など、ローカルセグメンテーションのアンカーポイントとしても機能します。ボリュームモデルの構築は、特にコンピューターベースの自動フィッティングアプローチによってさらに強化される場合、従来の自動セグメンテーションアプローチが成功しない場合の強力な代替手段になる可能性があります。
概要
クライオ電子線トモグラフィーは、突入凍結4または高圧凍結超急速ガラス化5のいずれかを使用して、細胞小器官全体または細胞および組織の一部を、ほぼ天然の状態1,2,3でナノメートルの分解能で視覚化することを可能にする。凍結保存された未染色の凍結水和サンプルでは、限られた電子線量しか許容できないため、断層撮影の3Dデータは非常にノイズが多くなります。このノイズは、非線形異方性拡散8、両側フィルタリング9、および再帰的中央値フィルタリング10を含む、さまざまなノイズフィルタリングアルゴリズム6,7によって大幅に低減できることがよくあります。
さらに、顕微鏡ステージの傾斜制限により情報のくさびが欠落し、高傾斜角で試料の厚さが増加するという事実により、異方性分解能の3D再構成が可能になります。これは、Z方向の解像度が低いため、密度が3次元で汚れていることを意味します。その結果、高分子の形状は歪んで見えます(つまり、3次元で明確に定義されておらず、細長くなっています)。
断層撮影データの解釈における最大の課題は、セグメンテーション11とも呼ばれる関連特徴の自動抽出です。十分なユニークな形状の特徴と低ノイズにより、複雑な3Dボリュームの高分子マシンは、テンプレートマッチング12,13,14によって識別できます。ただし、テンプレートマッチングの成功は、断層撮影の解像度、適切な検索モデル、および特徴量のサイズと形状の特性に依存します。対象の特徴が十分に離れており、繰り返されるモチーフ(大型の高分子機械など)を容易に識別できる場合、トモグラムサブボリュームを組み合わせてS/N比を高め、個々の粒子形状の歪みを平均化することができます。テンプレートマッチングによる凍結水和Dictyostelium discoideum細胞の細いエッジの電子断層図におけるアクチンフィラメントネットワークの自動セグメンテーションが報告されています15。
しかし、対象の特徴が近接して配置されている場合、データ解像度の異方性により、Z方向(電子ビームの方向)のマップ密度がにじみ出ることになり、近接した高分子マシンや超分子複合体の密度エンベロープが明らかに融合することがあります。このような場合、セグメンテーションのための自動化されたアプローチ、例えば、流域16、境界セグメンテーション17、または様々な機械学習ベースの分類アプローチ18、19は、関心のある特徴を認識することができないか、または関心のある物体の周囲に正しい境界を確立することができないかもしれない。多くの場合、非常に大きなピースがいくつかあるか、セグメント化が進んだボリュームが非常に多いため、関心のあるフィーチャが完全であると認識されるまで、多くの小さなピースをマージするために多くの努力が必要になります。このようなセグメンテーション結果の手動キュレーションは、非常に手間がかかる可能性があり、対象の構造が短いリンカー を介して 相互接続された間隔の狭いフィラメントの配列である場合、完全に失敗する可能性さえあります。この巨大な糸状構造のネットワークでは、自分の向きを決めるのが難しい場合があります。これは、解像度の異方性により、密度が互いに混ざり合っているように見え、自動化されたセグメンテーションアプローチとインタラクティブな手動セグメンテーションアプローチの両方にとって手ごわい課題となるためです。その結果、小さな領域を目視で検査するだけで、フィラメント間を簡単に「ジャンプ」できます。
幸いなことに、内耳有毛細胞立体繊毛のアクチン束の場合、全体的なアクチン束の組織化とアクチンフィラメントの方向性についての知識があります20,21。アクチン束は、直径6〜8nmの数百の六角形に密集したアクチンフィラメントからなり、これらは互いに約12〜13nm間隔で配置されています22。
これにより、アクチンフィラメントを表現するための簡略化されたボールアンドスティックモデルに基づくセグメンテーションに対して、かなり異なるアプローチを取ることができました。この戦略では、理想的なフィラメントモデルの規則的な配列をクライオ電子線トモグラフィー密度マップのスラブに同時に配置して、アクチンバンドルの3Dモデルをレイヤーごとに構築しました。個々のフィラメントモデルまたはフィラメントモデルのグループに密度マップに厳密に一致するように局所的な調整を行う前に、モデルが密度マップに全体的に適合していることを確認しました。フィラメントモデルの位置でマップ密度値を自動的に色分けすることで、アクチンバンドルの見かけのギャップを簡単に検出することができました。体積モデルを使用すると、アクチンフィラメント間の距離などの体積特性を定量的に分析できるだけでなく、全体的な3Dフィラメントネットワーク構成を簡単に表示できます。
さらに、モデルは、個々のフィラメントモデル(の一部)を選択できるため、アクチン-アクチンリンカーなどの追加機能のセグメンテーションのための固定構造としても機能し、その周囲に適切な半径マップ密度ゾーンを生成して検査およびさらなるセグメンテーションを行うことができます。
私たちの体積モデルベースのセグメンテーションアプローチは、ギャップやフィラメント間の相互接続を含む可能性のある大規模なフィラメント構造ネットワークに特に役立つと考えています。セグメンテーションアルゴリズムは局所的に動作する傾向がありますが、人間の脳はより広い領域を考慮に入れるため、複雑でノイズの多い環境でもフィラメント構造を認識することに関してはコンピューターよりも優れています。
プロトコル
このプロトコルは、サウスイースト大学の人間研究倫理委員会のガイドラインに従っています。
1. 体積モデル構築のためのクライオ電子線トモグラフィーデータソース
注:立体モデルの構築に使用された立体繊毛クライオ電子断層撮影再構成は、以前に発表されており22,23、Metlagel et al.22に記載されているように取得されました。
立体繊毛モデリング用の UCSF Chimera Python スクリプトは 、Supplementary File 1、Supplementary Coding File 1、Supplementary Coding File 2、Supplementary Coding File 3、Supplementary Coding File 4、および Supplementary Coding File 5 で提供されています。
- 簡単に説明すると、マウス卵形嚢感覚上皮の頂端表面から電子顕微鏡(EM)グリッドのレーシーカーボン支持膜に立体繊毛を吸い取ります。次に、300 kV、公称焦点ぼけ3.5〜4.5 μmで操作する極低温透過型電子顕微鏡(cryo-TEM)で、0.47〜0.59 nmピクセルサイズのCMOS統合モードカメラを使用して、超高速プランジ凍結ガラス固化および単軸クライオ電子断層撮影データ収集を行います( 材料表を参照)。
- 単軸データ収集の一般的な線量は、通常、80-100電子/Å2です。ソフトウェアパッケージIMOD24を使用して、加重後方投影またはSIRT法25,26のいずれかにより、断層撮影3Dボリュームを再構築します。
- Priism27 の再帰的中央値または両側フィルタリング、または IMOD の非線形異方性拡散オプションを使用して、断層像のノイズを除去します。
注:インタラクティブな視覚化、体積モデルの構築、および定量分析に使用された主要なソフトウェアパッケージは、UCSF Chimeraソフトウェア28,29でした。使用されているすべてのソフトウェアパッケージは、資料の表に記載されています。
2. 体積モデル構築のためのクライオ電子線トモグラフィーデータ作成
- 断層撮影の回転
注:この手順の目的は、アクチンコアが3つの軸(xとz)のうちの2つに位置合わせされている「最適な角度」を見つけることです。これにより、モデルを3Dに配置する際に、1つの軸だけを気にする必要があります。次の手順は、IMODソフトウェアパッケージ内で実行されます。- 3dmod(IMOD)を開き、3D画像スタックファイルを.mrcファイル形式で開きます。3D スタックが 16 ビット グレースケール モードであり、画像ファイル ヘッダーに正しい X、Y、Z 寸法が表示されていることを確認します。必要に応じて、コマンドプロンプトで alterheader -d (x_pixelspacing)、(y_pixelspacing)、(z_pixelspacing) inputfilename.mrc コマンドを使用して修正します。 3D スタックが TIFF ファイル形式の場合は、コマンド プロンプトで tif2mrc inputfilename.tif outputfilename.mrc コマンドを使用して .mrc ファイルを作成します。
- 断層撮影を目視検査し、「スライサー」を開き(バックスラッシュ「\」を押して)、アクチンフィラメント平面とZ平面の位置合わせに最適なX、Y、Z寸法の回転角度を見つけます。 X回転、Y回転、Z回転バーを操作して、アクチンフィラメントがX平面とZ平面に整列する最適な角度を見つけます。トモグラムの表示の平均厚さ(厚さ:Img)を変更すると、Z平面の密度の理想的な平均化量を見つけるのに役立ち、コントラストが向上することに注意してください。キーボードの+キーと-キーをそれぞれ使用して、トモグラムをズームインおよびズームアウトします。
- 回転の理想的な角度が特定されたら、IMODコマンド rotatevol -a(Z回転角度)、(Y回転角度)、(X回転角度)-s(x幅)、(y-高さ)、(z深さ)inputfilename.mrcを コマンドプロンプトに入力して、断層撮影を回転させます。ローテーション時にマップの一部が途切れないように、ローテーションに対応するためにマップに十分なスペースを確保してください。
注: rotatevol コマンドの使用方法の詳細については、ヘルプ メニューのオプションを使用するか、次の URL (https://bio3d.colorado.edu/imod/doc/man/rotatevol.html) を参照してください。このコマンド (rotatevol) は、IMOD ウィンドウに現在表示されているマップを変更せず、密度マップが回転する新しいマップ ファイルを作成します。 - スライサー ウィンドウを使用して、マップの 2 つの対向する角の X、Y、Z 座標を特定し、 trimvol コマンドを使用してステレオシリウムを含むトリミング領域を特定します。
- IMODコマンドの Trimvol を使用して、コマンドプロンプトに trimvol -x (x-coordiante 1),( x-coordiante 2) -y (y-coordiante 1),( y-coordiante 2) -z (z-coordiante 1),( z-coordiante 2) inputfilename.mrc outputfilename.mrc と入力し て、以前に取得した座標でマップをトリミングします。現在、マップははるかに小さくなっているため、この時点からはより簡単かつ迅速に作業できます。
注: trimvol コマンドの使用方法の詳細については、ヘルプ メニューのオプションを使用するか、次の URL (https://bio3d.colorado.edu/imod/doc/man/trimvol.html) を参照してください。これらの手順により、回転およびトリミングされた .mrc ファイルが作成されます。
- 断層撮影フィルタリング
注:この手順では、ノイズリダクションのためにIMODが提供する非線形異方性拡散(NAD)フィルターを使用します。初期値として、IMODの非線形異方性拡散フィルタリングのヘルプページで推奨されているもの(次のURL(https://bio3d.colorado.edu/imod/doc/NADexample.html)を参照)を使用し、初期値として使用しました。- コマンドプロンプトで 「etomo 」と入力して、IMODのetomoグラフィックユーザーインターフェイスを開始します。
- etomoメニューから Nonlinear Anisotropic Diffusion オプションを選択し、マップファイルを選択します(ボリュームを選択)。
- 目視検査で判断される最良のフィルタリング結果を提供するK値と反復回数を見つけるには、NADフィルターを小さなテストボリュームに適用して、指定されたNADフィルターテストボリュームを抽出します。3dmod インターフェースの上部にある Rubberband Tool をクリックし、左クリック ドラッグしてテスト ボリュームを選択し、 Hi ボタンと Lo ボタンをクリックして Z スライスをテスト ボリュームの境界に指定します (3dmod からテスト ボリューム範囲を取得)。 「Extract Test Volume」をクリックして、テストボリュームを抽出します。
- 0.1、1、5、10、15、25、30、50、75 などのさまざまな K 値 ([テスト ボリュームの K 値の検索] > K 値のリスト) を使用して、テスト ボリュームをフィルター処理します。指定した K 値ごとに NAD フィルタリングを実行します (異なる K 値での実行>異なる K 値のテスト結果の表示)。スライサー ウィンドウを使用して NAD フィルターのパフォーマンスを評価し、ボリューム全体のフィルター処理に使用する K 値を選択します。
- ステップ 2.2.4 で特定した K 値を使用して、2、5、8、11、15、21 など、さまざまなイテレーション数をテストします (テスト ボリュームのイテレーション数の検索>イテレーションのリスト)。さまざまな反復の NAD フィルターのパフォーマンスを評価するには、[ 異なる反復テスト結果の表示 ] をクリックするか、スライサー ツールを使用します。異なる反復回数の値を選択します。
- 特定された K 値と反復回数 (Filter Full Volume >K value and Iterations) を使用し、ボリューム全体をフィルタリングします (Filter Full Volume)。新しくフィルタリングされたボリュームは、ロードされたフルボリュームが配置されているのと同じディレクトリに表示され、.mrcファイルには.nadファイル拡張子が添付されます。
注:これらの手順では、NADフィルタでフィルタリングされた.mrcファイルが作成され、ノイズが少なくなるため、信号対雑音比が向上します。
3.ボリュームモデルの構築
- 3Dモデル配置準備
注:この手順の目的は、UCSF Chimeraプログラムを使用して、回転およびノイズ除去された密度マップに3Dボリュームモデルを配置するためのマップを準備および分析することです。- スライサーウィンドウを使用して、断面図(30スライス/28.4nmのスラブ)を取得し、アクチンフィラメントモデルが配置される密度スラブの中心のZ座標を特定します。フィラメント密度の中心をクリックし、IMODメインウィンドウのZ座標を確認します。
- UCSF Chimera での将来のモデル配置のために特定された座標をメモしておいてください。
注:このステップで示したZ座標は、ステップ4で使用され、既製のアクチンフィラメントモデルを正しいZ高さに簡単に見つけて配置し、モデルの配置を高速化します。
- 3Dモデルの配置
注:この手順の目的は、UCSF Chimeraを使用して、準備した密度マップに3Dボリュームモデルを配置することです。- UCSF Chimera で、フィルタリングされた回転マップを開きます ([ファイルを開く] >)
- マップのパラメーターが正しく設定されているかどうかを確認するには、ボリューム ビューアーで [フィーチャ > 平面] と [フィーチャ > 座標 ] を選択して、フィーチャと座標のインターフェイスを開きます。 ボクセル サイズ(Voxel Size )を確認し、コマンド プロンプト(IMOD)の ヘッダー コマンドを使用して、正しいボクセル間隔が設定されているかどうかを確認します。そうでない場合は、 ボクセル サイズ(Voxel Size )インタフェースを正しいボクセル間隔に修正します。マップを中央に配置して、カメラを表示ウィンドウの中心 (Origin Index > Center) に設定します。
- カメラ制御ウィンドウを開きます(ツール>表示制御>カメラ)。次に、カメラの表示を正投影ビュー(投影>正投影)に設定します
- 事前に作成されたPythonスクリプトモデル(File > Open > ActinFilamentPlane.py TASK )をロードし、マップ内のアクチンフィラメント平面と同じ数のアクチンフィラメント平面スクリプトを開きます。
- ロードしたモデルを再配置するには、 移動マウスモード (ツール>移動]>移動マウスモード)をオンにします。移動マウス オプションを有効にします (移動マウス モードの設定>選択移動)。 Ctrl + ドラッグ を使用して、セッションに存在する任意のモデルを複数選択し、右クリック ( または「環境設定」>「マウス」で指定されている他のキー) して、モデルを目的の方向に移動します。
注: モデルは、特定の厚さ (ActinFilamentPlane.py スクリプトの半径 ) と特定の間隔 (ActinFilamentPlane.py スクリプトのLattice_Spacing ) のボールとスティックを使用して作成されています。 - 関心のあるモデルだけを視覚化するには、モデルパネルを開きます([ツール]>[一般コントロール]>[モデルパネル])。[S (表示)]の下のチェックボックスをクリックして、特定のモデルの表示を有効または無効にします。
- コマンドラインパネルをアクティブにします([ツール]>[一般コントロール]>コマンドライン)。
- 各アクチン フィラメント平面モデルを適切な Z 高さに配置した後、モデルの余分な部分を選択し、UCSF Chimera メイン ビュー ウィンドウの下部にあるコマンド ラインに 「del sel (選択を削除)」と入力して、各アクチン フィラメント平面モデル内の余分なアクチン フィラメントを削除します。
- 一度に1つのアクチンフィラメント平面のみを視覚化するには、 モデルパネル ウィンドウに移動し、他のすべてのアクチンフィラメントモデルを選択して非表示にします(Ctrl + Click > Hide)。
- 対象の単一アクチンフィラメント平面モデルのすぐ近くにあるマップ密度の部分を可視化するには、モデルを選択し([モデルパネル]>選択)、ボリュームビューアでゾーンコントロールパネルを開きます([ボリュームビューア]>[フィーチャ]>ゾーン)。 ゾーン半径 を 100 Å に設定し、[ ゾーン ] をクリックして、選択したモデルから半径 100 Å (= 10 nm) でマップをゾーニングします。Chimera の寸法はオングストローム (1 Å = 0.1 nm) で設定されていることに注意してください。
- 必要に応じて、 移動マウス モードをオンにして、密度マップに合うようにモデルを調整します。適切な位置にないモデルを複数選択して、モデルをゾーン密度マップ内に適切に配置するように移動します。
- ActinFilamentPlane.py のプログラミングのバグを解決するには、FixingMarkerID.py スクリプトを実行して、"atoms" (ボール アンド スティック モデルのボール) の欠落している MarkerID を修正します。モデル全体を選択し、コマンドラインにrunscript FixingMarkerID.py と入力します(スクリプトは、C:\ directory \ Script.py などの正しいディレクトリで指定する必要があります)。
- 密度マップでアクチンフィラメントが湾曲していることが示されている場合は、目的のモデルを選択して runscript dividelinks.py 番号を入力して、曲率に対応するために直線のアクチンフィラメントを複数の部分に分割してください。追加のマーカー(コマンドの番号で指定)は、アクチンモデルの内側に互いに等距離に配置され、モデルを曲げることができる「ジョイント」を作成します。
- モーションマウスモードを使用して、ゾーン化されたマップに沿って追加のマーカー「ジョイント」を移動することにより、曲率を反映するようにジョイントを調整します。必要に応じて、dividelinks.py スクリプトを使用してモデルにマーカーを追加します。
- すべてのアクチン フィラメントがアクチン密度内に正しく配置されていることを確認するには、UCSF Chimera ドロップダウン メニューの [Tools] > [Viewing Control (表示制御)] > [Camera (カメラ)] をクリックしてクリッピング バーを使用し、[ Side View (サイド ビュー )] を選択してカメラの 2 つのクリッピング バー (近距離および遠距離のクリッピング平面) を確認します。左クリックして 2 本の黄色のバーを狭いスリットにドラッグし、 マウスの中央ボタン を使用してクリッピング部分をドラッグして、モデルを一度に 1 ビットずつ検査します。
注:これで、stereocilium actinコアモデルが完成しました。
- メンブレンセグメンテーション
注:この手順の目的は、立体繊毛膜の表面モデルを作成することです。- サーフェスのセグメンテーションは、 ボリュームトレーサー ツール(ボリュームデータツール>ボリュームトレーサー>ツール)を使用します。
- Volume TracerウィンドウでMouseをクリックし、Place Markers on Data PlanesオプションとLink new marker to selected markerオプションのみをオンにします。これにより、マーカーを同じZ面に正確に配置できます(単一のZ面が表示されている場合)。また、Volume TracerウィンドウのPlace Markers Using the Middle Mouse Buttonもチェックします。
- 新しいマーカーセットを作成します(ファイル>新しいマーカーセット)。マーカーセット 1 を作成し、[ ボリューム トレーサー ] ウィンドウで選択する必要があります。
- ボリュームビューアを使用して、メイン表示ウィンドウに単一のZ平面を表示します(軸からZ面>1)。
- マウスの中央ボタンを使用して、表示されているメンブレンの一方の端からもう一方の端までメンブレンをトレースします。すべてのマーカーは、一方の端からもう一方の端へのリンクで接続する必要があります。
- 最後に配置したマーカーの選択を解除するには、選択可能なアイテムが 1 つもない背景で Ctrl + クリック します。これにより、次に配置されるマーカーが、別のZ平面の以前の膜トレースではなく、新しい平面のリンクに接続されるようになります。
- 可視膜の一方の端からもう一方の端まで、同じ方向に10 nmごとに膜トレーシングを繰り返します。
- メンブレンの多数の平行バンドがセグメント化されたら、Volume Tracerウィンドウに移動し、Features > Surfacesをクリックします。これにより、Volume Tracerウィンドウのサーフェスインターフェースが有効になります。Surfacesインターフェースの横のCreateをクリックし、メンブレンセグメンテーションのバンド間にサーフェスを作成します。これにより、サーフェス モデルの薄いフラップが作成され、間隔が空けられたメンブレン トレースを埋めます。
注:これで、ステレオ繊毛膜表面モデルの作成は完了です。
- 架橋剤モデリング
注:このステップの目的は、アクチンフィラメント間の架橋剤をモデル化することです。- 架橋剤は、 ボリュームトレーサー ツールを使用して配置することもできます。
- Volume Tracerウィンドウで、左クリックしてPlace Markers on SurfacesとLink new marker to selected markerのみにチェックを入れます。次に、ボリュームトレーサーウィンドウでマウスの中央ボタンを使用してマーカーを配置します。
- 対象の 1 つのアクチン フィラメント平面モデルの周囲の密度は、zone 関数を使用してのみ可視化します。ゾーン 半径 を 100 Å に設定すると、選択したモデルから半径 100 Å 以内のマップが表示されます。
- クロスリンカーの可視密度全体にマーカーを配置する には、マウスの中央ボタンをクリックします。2 つのマーカーを配置したら (つまり 1 つの架橋剤がモデル化されます)、リンクが次の架橋剤モデルに連続して接続されないように、2 番目のマーカーの選択を解除してください。
- すべてのアクチンフィラメント平面に対して上記の手順を繰り返し、架橋剤が配置される可能性のある3つの主要な方向すべてに対して上記のプロセス全体を繰り返します。アクチンフィラメント平面を対角線方向にゾーニングする場合は、アクチンフィラメントモデルを個別に手動で選択します。
注:アクチン架橋剤の各モデルについて、推定架橋剤が接続するアクチンフィラメント上の位置を記録できます。この情報は、原則として、サブトモグラム平均化を使用したフォローアップ分析のために抽出できますが、これはこの研究の範囲を超えています。
4. 3Dモデルの定量解析
- ギャップ分析
注:このステップの目的は、アクチンフィラメントのギャップを検出することです。- 選択した項目を分析できます。色などのパラメータを指定するには、選択項目の 検査 ツール (Action > Inspect) を使用します。
- 選択した項目の色を指定するには、UCSF Chimera で選択可能な項目を選択し、[Inspect Selection] ウィンドウの [Color] セクションの横にある色を変更します。
- マーカーとリンクのサイズを指定するには、UCSF Chimera で任意のマーカーとリンクを選択し、[選択項目の検査] ウィンドウの半径セクション (単位がオングストロームにある) の横にある値を変更します。
- Values at Atom Positions機能(Tools > Volume Data > Values at Atom Positions)を利用して、密度ギャップを自動検出し、ギャップ解析を行います。
- 値を読み取るマップを選択するには、[Values at Atom Positions] ウィンドウの [Volume Data] セクションの横にある目的のマップを選択します。すべてのマーカーにパラメータが割り当てられるモデルを選択するには、Moleculeセクションの横で目的のモデルを選択します。
- Values at Atom Positions ウィンドウの下部にある Histogram ボタンを使用すると、Render/Select by Attribute という新しいウィンドウにヒストグラムが作成されます。ヒストグラムをCtrl +クリックすると、カットオフポイントが追加または削除され、さまざまな色を選択できます。[不透明を維持] をオフにして、シーンに透明色を適用します。ギャップを表す特定のしきい値を下回る密度のマーカーは、色を付けるだけでなく選択することもできるため、ギャップ領域内のマーカーの数をカウントできます。選択するには、[Select (選択)] > Attribute > value_mapnameをクリックします。
- アクチン間距離の決定
- アクチン間距離を測定するには、ボリュームトレーサーウィンドウを使用してマウスをクリックし、データプレーン上にマーカーを配置するのみをオンにし、選択したマーカーに新しいマーカーをリンクするチェックを外して、単一のXZ平面(UCSF ChimeraではY平面と呼ばれます)にマーカーを配置します。これにより、マーカーを同じY面に正確に配置できます(単一のZ面が表示されている場合)。また、Volume TracerウィンドウでPlace Markers Using Middle Mouse Buttonにチェックを入れ、アクチン間距離測定のために対象のターゲットY平面にマーカーを配置します。
- 単一のY平面のアクチン位置が正常にマークされたら、すべてのマーカーを選択し、コマンドfindclash #model-spec test self overlapCutoff -200 hbondAllowance 0 log true Linewidth 10 pbアクチンフィラメント平面の下部にあるコマンドラインで黒色を入力します。これにより、200 Å (overlapCutoff -200) の距離内にあるすべてのマーカー間干渉が検出され、マーカー間に 10 Å (線幅 10) の太さの黒い線 (pbColor black) が作成されます。
- 指定された200 Åの範囲内に重なり合うマーカーがあるため、最も近い隣接するマーカー間のリンクではない冗長な測定値を削除します。これを行うには、UCSF Chimera の Web サイトで提供されている RemoveCross.py スクリプトを使用して、隣接するマーカーとの最短リンクではない重複するリンクをすべて削除します。
- 残っているボンドの距離を測定するには、すべての擬似ボンド (findclash によって作成されたリンク) を選択し、runscript pblengths.py と入力して python スクリプト pblengths.py を使用します。返信ログ(「ツール」>「ユーティリティ」>「返信ログ」)の測定値をご覧ください。次に、取得した値を返信ログからコピーし、スプレッドシートまたはその他の適切なツールに転送してヒストグラムを作成します。
結果
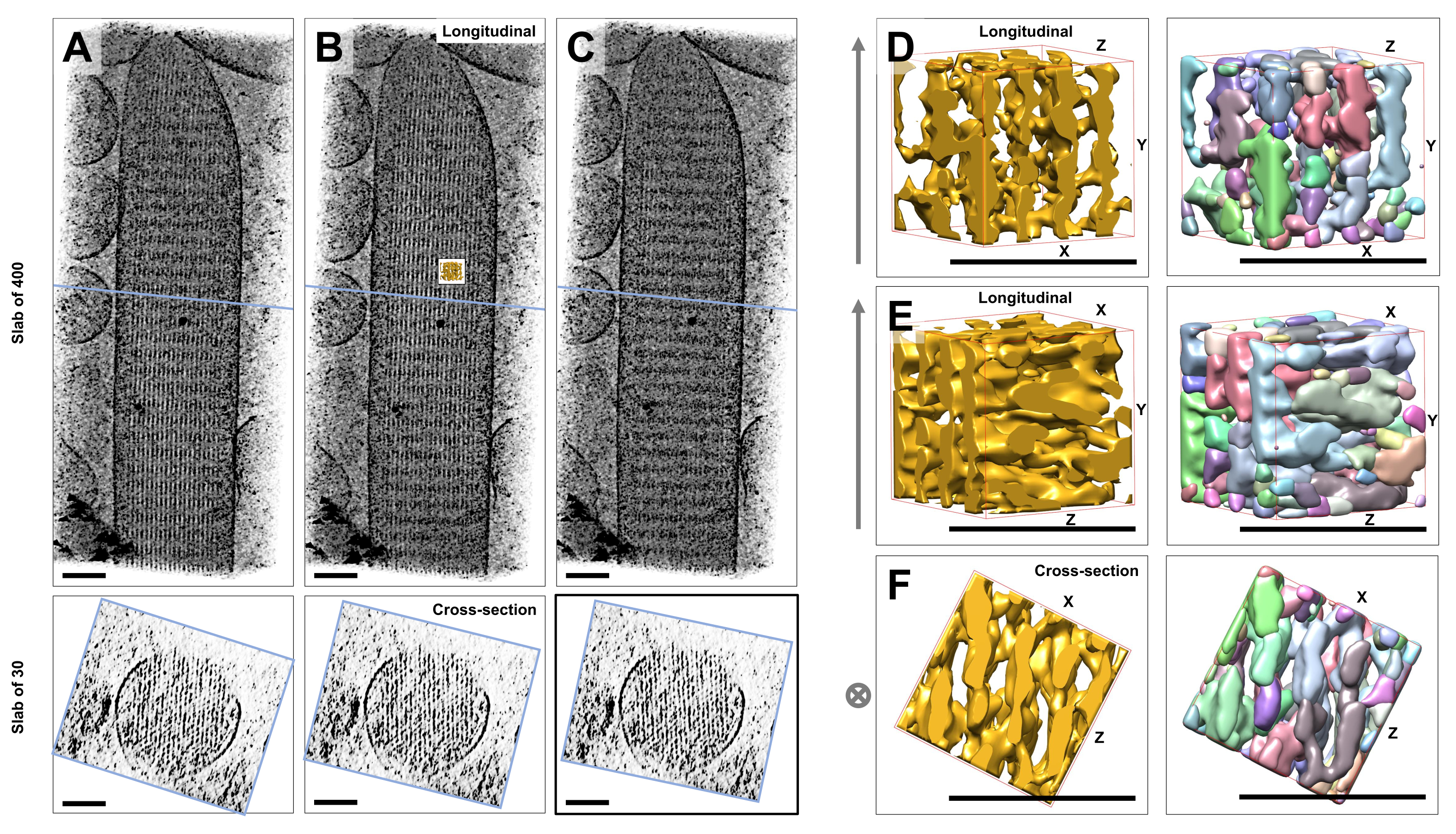
ガラス質の氷に埋め込まれた未染色の凍結水和した個々の立体繊毛のクライオ電子線トモグラフィーを使用して、クロスコネクタータンパク質23によって連結された、六角形に配置されたアクチンフィラメントを持つアクチン束の密度マップを取得しました。個々のボクセルの寸法は0.947nmでした。IMODスライサープログラムでトモグラム全体(400スライス/379 nm)のボリュームレンダリングを目視検査したところ、縦方向の図(XY平面;図1A-C、トップパネル)、および断面図(XZ平面;図1A-C、下部パネル)。400スライス/379nmのフィラメントネットワークを通る投影ビューは、元の再構築ボリュームをX軸を中心に-6°、Y軸を中心に-13.5°、Z軸を中心に5°回転させると最も鮮明になることがわかりました。この角度では、すべてのフィラメントが互いに重なり合っているため、断面図から理解できるように、コントラストが最大になります(図1B)。単一の断面スライスではアクチンフィラメントを明確に区別するのに十分なシグナルがないため、断面図で六角形のパターンがはっきりと示す30スライス/28.4nmのスラブをボリュームレンダリングすることを選択しました。図1C(上部パネル)の青い線は、下部パネルの対応する30スライス/28.4nm断面スラブの中央の位置を示しています。
この最適な視野角からわずか±2°のわずかな偏差で、アクチンフィラメントネットワークの知覚される順序が大幅に減少しました(図1A、C)、これは断層撮影の3Dボリュームで失われやすいことを示しています。
流域セグメンテーションなどの自動セグメンテーションアプローチを使用する際の課題を説明するために、UCSF Chimera ソフトウェアパッケージ (Tools > Volume Data > Segger > Segment) に実装されているように、流域セグメンテーションに小さなサブボリューム (金で表示) を選択しました。立体繊毛マップ全体に対するサブボリュームの位置は、図1Bの小さな挿入図で示されています。
図1D-Fは、選択したサブボリュームをさまざまな向きで示し、図1D、Eは縦方向の視線方向、図1Fは断面の視線方向を示しています。図1D-Fの左側の矢印は、アクチンフィラメントの方向を示しています。
図1D-F(右パネル)は、流域セグメンテーションの結果を示しています。サブボリュームはオブジェクトの ID によって色分けされ、色はさまざまなオブジェクトにランダムに割り当てられます。異なる色は異なるオブジェクトの同一性を示すため、図1D-Fから、フィラメントのマップ密度は両方ともフィラメント軸に沿って断片化されているのに対し、隣接するフィラメントを接続するマップ密度には同じ色、つまりオブジェクトの同一性が与えられていることが明らかになります。言い換えれば、流域セグメンテーションアルゴリズムは、アクチンフィラメントの密度マップを長期間追跡することができず、代わりに隣接するフィラメントからの接続密度につながったのです。選択を手動でキュレーションすることは可能ですが(たとえば、オブジェクトを削除またはマージする)、このアプローチはかなり手間がかかり、時間がかかります。
私たちのボリュームモデル構築戦略が機能するために絶対に必要というわけではありませんが、アクチンフィラメントネットワークの軸がY軸に、アクチンフィラメントのモデル平面がトモグラムのX-Y平面に揃うように、3Dマップの向きを変更(回転)するのに役立ちました。この向きを立体繊毛断層撮影ディスプレイの標準向きと呼んでいます。
そこで、アクチンフィラメントが全体的に規則的な組織(六角形のパッキング)を示し、規則的な間隔と定義された全体的な束の向きを示すという事実を利用して、画像セグメンテーションの異なる戦略を模索することにしました。私たちの戦略は、アクチンバンドルのモデルの全体的な適合度をフィラメントの配列として見つけ、その後、実験密度マップに適合するようにモデル位置を地域的および局所的に調整することでした。全体的なモデルを最初に配置することで、ローカルマップのあいまいさを克服し、フィラメントの曲げなど、モデルの元の組織からの逸脱の地域的傾向を検出できます。
モデルを配置するために、アクチンフィラメントの単層の厚さに対応する標準配向の密度(10スライス/9.47nm)のスラブを表示し、それに等間隔の直線的なアクチンフィラメントモデルの層を取り付けました。もちろん、これはアクチンフィラメントを単純化しすぎたものであり、各フィラメントはらせん対称性を持つアクチンモノマーの線形配列で構成されています。図2A-Cは、Z高さの異なる3つの代表的な層を示しており、赤色のロッドはアクチンフィラメントを表しています。上面パネルは~30スライス/厚さ28.4nmの断面で、19本のロッドからなる個々のアクチンモデル層がZ高さに配置された位置を示しており、下面パネルは縦方向の向きを示しています(斜視図では示されていますが)。図 2D は、断面図 (上部パネル) と縦方向の透視図 (下部パネル) の両方で、完全な簡略化されたモデルを示しています。断面の向きにより、フィラメントを自信を持って配置することができました。ここでは、トモグラムの主軸と一致するようにボリューム全体の向きを変更するという当初の動きが、モデルの標準的な視線方向も主軸と平行になることを意味するため、役に立ったことが証明されました。しかし、厳密に言えば、私たちのアプローチは、トモグラムの向きを変えなくてもうまくいったでしょうし、モデルを密度に配置するだけでも、より困難になっていたでしょう。
密度マップの個々のスラブを注意深く調べたところ、完全にまっすぐなアクチンモデルが、立体繊毛の近位端から遠位端(つまり先端に向かって)移動する観察された密度マップに適合しないことに気づきました(図3A-C)。立体繊毛の先端付近では、フィラメントのマップ密度が13 nm以上ずれていました(アクチン-アクチン間隔)ため、立体繊毛密度マップの近位部分から遠位部分に移動しながらモデルを調整することでこれを補正することができ、アクチンモデルに小さいながらも識別可能な緩やかな曲率を導入しました。図3Dは、アクチンフィラメントのマップ密度の単一のスラブと、密度マップに適合する体積モデルを示しています。直線モデル(赤)と曲線モデル(黄色)の比較を図3Eに示します。この曲率は、配置されたモデルと密度マップのスラブをX軸を中心に80°傾けることで最もよく理解でき、アクチンフィラメントの方向に沿った透視図が可能になります(図3D、E)。
先端付近のアクチンモデルの位置がアクチンフィラメントの間隔とほぼ同じ距離だけずれているという2つのモデルの偏差は、私たちが行った方法で進めなかった場合、多くの混乱を引き起こした可能性があります。アクチンフィラメントモデルの層のこの「全体的」な位置決めとそれに続く「領域的」な調整により、縦方向または断面図ではほとんど目立たないこの曲率を検出することができました。ただし、 図 3E に示すように、2 つのモデルを重ね合わせると、微妙な違いが明らかになります。
このアプローチを複数の層で繰り返すと、断面の向きで見たときに、立体繊毛の最上部と下部のデータの不確実性によってのみ制限される完全な3Dモデルを取得できます(図3F)。この密度の欠如は、(単軸)断層撮影データ収集のくさびの欠落とそれに対応するデータ解像度の異方性によって引き起こされ、その影響は、立体繊毛膜の明確に定義されたマップ密度の欠如によって示されます。
3Dモデルを作成したら、その位置のマップ密度値に従って、ボリュームモデルの各位置を色分けしました。基礎となる弱いマップ密度を持つモデルの領域は赤色で表示され、マップ密度信号が強いモデルの領域は黄色で色付けされました(図4A)。このような数十ナノメートルにも及ぶ赤色の領域は、アクチンフィラメント構造のギャップであり、その程度から、クライオ電子顕微鏡マップの高ノイズ環境で頻繁に遭遇する密度変動に起因するとは考えられません。ノイズは、個々のボクセルまたはボクセルの小さなグループに影響を与える傾向がありますが、フィラメント密度が欠落している数百のボクセルで構成されるボリュームの原因になる可能性は低いです。むしろ、このようなギャップは、立体繊毛アクチンメッシュワークの実際の特徴である可能性が高く、アクチン代謝回転の部位を構成する可能性があります。 図 4A には、水色と濃色で示されている 2 つの異なるマップ密度値があります。私たちの体積モデル構築アプローチは、密度の弱い領域でのモデルの自動色分けと組み合わせることで、アクチンフィラメントモデルにおけるそのようなギャップの分布を検出して視覚化するための高速で便利な方法であることに明示的に注意する必要があります。
図 4B に示すように、密度が比較的弱い位置の体積モデルの一部は、図 4A で得られた結果に基づいて簡単に隠すことができます。これにより、より断片化されたモデルが得られ、ステレオ繊毛のアクチンモデルをよりリアルに描写できる可能性があります。アクチンフィラメントを小さく伸ばすという代替案は、図1を説明する際に説明した問題のために、非常に労働集約的であり、完全に失敗する可能性があります。
さらに、体積モデルでは、アクチンフィラメントモデルのモデルポイント位置の間に接続部(赤で表示)をクロスコネクトの両側に配置するだけで、クロスコネクタを簡単にモデル化できます(図4C)。私たちの単純化されたアプローチでは、各交差結合タンパク質の正確な同一性について仮定する必要はなく、そのためにはより高い分解能や高度な標識アプローチが必要になります。そうではなく、私たちが判断する必要があるのは、隣接するアクチンフィラメントを橋渡しする密度が存在するかどうかです。ある場合は、1つのフィラメントから隣接するフィラメントに短い接続を配置できます。 図4Dでは、5つのアクチンフィラメントとそのクロスコネクターのモデルが示されており、アクチンフィラメント軸に沿ったクロスコネクターの分布を示しています。
アクチンバンドルの体積モデルを構築するもう1つの利点は、隣接するアクチンフィラメント間の間隔を迅速に決定できることです(図4E-H)。図4E,Fは、マップ密度の六角形格子にフィットしたモデルがない場合とした場合の密度マップの断面図を示しています。図 4G は、最も近い隣接するボール間の接続を持つモデルを示しています。UCSF Chimeraでは、最近傍中心の距離を自動的に計算でき、その結果を距離分布としてプロットできます(図4H)。2 つの追加データセットのモデル構築を補足図 1 と補足図 2 に示します。

図1:有毛細胞立体繊毛断層撮影の分水界セグメンテーションが直面する課題 (A-C) XY平面の断層撮影3Dマップ(上部パネル)とXZ平面(下部パネル)の断面図(30スライス/28.4 nm)による縦方向の投影(400スライス/379 nm)。(A)最適な方向からY軸に沿って-2°回転した断層撮影マップ。(B)X軸、Y軸、Z軸の回転角度(X = -6°、Y = -13.5°、Z = 5°)の調整によって決定され、密度マップの高度な秩序を明らかにする最適な方向の断層撮影マップは、高秩序のアクチンフィラメントネットワークを示唆しています。(C)最適な方向からY軸に沿って+2°回転した断層撮影マップ。最適な視線方向から Y 軸を中心にわずか 2° 回転すると、密度マップの知覚される規則性が大幅に損なわれます。底面のパネルは、断面方向で見たときにアクチンフィラメントアレイの規則性を示しています。A-Cの青い線は、断面スラブの位置を示しています。(D-F)流域セグメンテーションの前(左のパネル)と後(右のパネル)の3つの異なる方向から見た50 nm x 50 nm x 50 nmの立方体。流域セグメンテーションでは連続的なアクチンフィラメント密度を検出できないのに対し、隣接するアクチンフィラメントとその交差接続は同じオブジェクト同一性を共有していることから、流域セグメンテーションはトモグラムセグメンテーションに適したアプローチではないことが示唆されています。パネルD-Fでは、Chimeraの密度マップがマップスタイル「Surface」として表示されています。(A-C)スケールバー= 100nm。(D-F)スケールバー = 50 nm。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
補足図1:2つの追加の立体繊毛データセットの最初のモデル構築。(A-C)立体繊毛密度マップの ~10 スライス/厚さ 9.47 nm の縦スラブの小さな領域を、メッシュ モード表示を使用して青色で示します。最初に配置したモデルは赤で、修正されたモデルは黄色で示されます。(A)マップ密度のみ。(B)密度マップに配置された初期モデル。(C)修正されたモデルを密度マップに配置しました。(D-E)(D) なしと (E) の補正モデルを立体繊毛密度マップの ~10 スライス/厚さ 9.47 nm の縦スラブに適合させたより大きな立体繊毛領域。(F-G)立体繊毛断層撮影領域全体が表示されます。(F)地図のみ。(G)修正されたモデルでマッピングします。(H) 初期モデルと修正モデルの重ね合わせ。スケールバー = 100 nm このファイルをダウンロードするには、ここをクリックしてください。
補足図2:2つの追加の立体繊毛データセットのうちの2番目のモデル構築。(A-C)立体繊毛密度マップの ~10 スライス/厚さ 9.47 nm の縦スラブの小さな領域を、メッシュ モード表示を使用して青色で示します。最初に配置されたモデルは赤で表示され、修正されたモデルは黄色で示されます。(A)マップ密度のみ。(B)密度マップに配置された初期モデル。(C)修正されたモデルを密度マップに配置しました。(D-E)(D) なしと (E) の補正モデルを立体繊毛密度マップの ~10 スライス/厚さ 9.47 nm の縦スラブに適合させたより大きな立体繊毛領域。(F-G)立体繊毛断層撮影領域全体が表示されます。(F)地図のみ。(G)修正されたモデルでマッピングします。(H)初期モデルと修正モデルの重ね合わせ。スケールバー= 100nm。このファイルをダウンロードするには、ここをクリックしてください。
補足ファイル1:立体繊毛モデリング用のUCSF Chimera Pythonスクリプト。このファイルをダウンロードするには、ここをクリックしてください。
補足コーディングファイル1:pblengths.py。このファイルをダウンロードするには、ここをクリックしてください。
補足コーディングファイル2:RemoveCross.py。このファイルをダウンロードするには、ここをクリックしてください。
補足コーディングファイル3:ActinFilamentPlane.py。このファイルをダウンロードするには、ここをクリックしてください。
補足コーディングファイル4:dividelinks.py。このファイルをダウンロードするには、ここをクリックしてください。
補足コーディングファイル5:FixingMarkerID.py。このファイルをダウンロードするには、ここをクリックしてください。
ディスカッション
私たちは、分水界セグメンテーションなどのセグメンテーションの自動化アプローチが、有毛細胞立体繊毛クライオ電子断層撮影の高ノイズで高複雑な環境では失敗する可能性があることを示しました。この糸状ネットワークのどの部分がアクチンフィラメントを表し、何が局所環境レベルで架橋を構成しているのかを区別することは、小さな断層撮影サブボリュームを検査するだけでは、せいぜい難しいように思われます。この研究で使用されたモデル構築アプローチは、アクチンバンドルの大規模次数に関する事前知識の恩恵を受けており、アクチンフィラメントの配向と架橋剤密度に関する期待を発展させるのに役立ちます。おそらくさらに重要なことは、人間の脳は局所的な密度分布を超えたより大きなコンテキストを考慮することでパターンを簡単に見つけることができるのに対し、コンピューターアルゴリズムはアルゴリズムによって考慮される比較的小さな領域に対してのみ機能するということです。したがって、より大きなトレンドは容易に考慮に入れることはできません。モデルを密度の層にグローバルにフィットさせることで、一度に単一のアクチンフィラメントの小さな部分のモデルを作成しようとするときに発生する可能性のある混乱を回避しました。もちろん、このようなグローバルフィッティングは、長い距離に広がる順序を前提としています。しかし、アクチンフィラメントが予想外の小さいながらも大幅に緩やかに曲がることがわかったため、全体近似は初期近似にすぎず、密度マップに適合させるためにモデルの局所的な調整が必要でした。初期モデルが良いスタート地点だったので、高い自信を持って調整を行うことができました。私たちのアプローチの大きな利点の 1 つは、定義された密度ゾーンのみを表示できるため、風景の複雑さを軽減することができたことです。さらに、マップ密度スラブをフィラメントモデルの軸に沿って表示することで、単に小さなサブボリュームを表示するだけでは見逃しがちな予期しない曲率を特定するのに役立ちました。また、初期モデルを配置することで、アクチンフィラメントの各層の全体像とモデル調整のための詳細ビューを交互に表示し、ズームインとズームアウトを素早く行うことができました。
このプロトコルの重要なステップには、目視検査後のマップの回転、モデルの作成と密度マップへの配置、フィラメントモデルの小さなセグメントへの分割が含まれていました。その後、セグメントの原子位置を密度マップに合うように空間的に調整したり、ギャップを検出するために色分けしたりできます。
アクチンモデル構築のこのアプローチは、10-30スライス/9.47-28.4nmの平均密度スラブの断面図を使用して、一連の「原子」(つまり、ボールアンドスティックモデルのボール)をフィラメント密度に配置し、それを結合(つまり、ボールアンドスティックモデルのスティック)で接続することによって変更することもできます。我々は、ここで詳細に述べたプロトコルからの修正であるこのアプローチを、有毛細胞立体繊毛23のテーパ領域における体積モデル構築に使用した。さらに、ここで説明したように、当社の体積モデル構築アプローチは、メンブレンのセグメンテーションとモデル構築にも適しています。
ボリュームモデルの構築は、糸状の特徴を示す任意の密度マップに適用できますが、ここで説明した手法は、等間隔のフィラメントの配列がある場合に最も効率的であり、ボリュームモデルのグローバルフィットを取得できます。また、糸状の特徴が徐々に方向性を変えるかどうかにも依存します。糸状構造に突然のねじれや急激な曲がりがある場合、私たちのアプローチはセグメンテーションに特に役立たない可能性があります。
その間、私たちの共同研究者は、ここで手動セグメンテーション30,31に使用されているのと同様の概念に従って、自動フィラメントトレースのための自動アプローチを開発しました。今後、最善のアプローチは、手動で識別し、最初のスパースモデル(わずか数個のボール)を出発点として密度に配置し、その後、探索とフィッティングのアルゴリズムでフィラメントのトレースを完了するというハイブリッドになるかもしれません。
要約すると、インタラクティブな手動モデルポイント配置は、その後の自動ローカルフィッティングおよびフィラメントトレース機能によってさらに強化される可能性があり、電子断層撮影の細胞内ボリュームの視覚化と定量分析のためのかなり有望なアプローチです。これは、パターン認識には人間の脳の力を、モデルの最適化にはコンピュータサイエンスの力を利用しているためです。
開示事項
著者は、競合する金銭的利益またはその他の利益相反がないことを宣言します。
謝辞
サンプル調製における役割について、Peter Barr-Gillespie博士と彼のチーム、および断層撮影データ収集におけるAuer研究室とDr. Dorit Hanein研究室の元メンバーに感謝します。また、UCSF Resource for Biocomputing, Visualization, and Informatics (RBVI) の Tom Goddard 氏にも、さまざまな UCSF Chimera スクリプトを提供していただいたことに感謝いたします。
資料
| Name | Company | Catalog Number | Comments |
| Chimera | RBVI | Version 1.16 https://www.cgl.ucsf.edu/chimera/download.html | |
| Chimera | RBVI | Version 1.16 https://www.cgl.ucsf.edu/chimera/cgi-bin/secure/chimera-get.py?file=win64/chimera-1.16-win64.exe | |
| Chimera | RBVI | Version 1.16 https://www.cgl.ucsf.edu/chimera/cgi-bin/secure/chimera-get.py?file=mac64/chimera-1.16-mac64.dmg | |
| Chimera | RBVI | Version 1.16 https://www.cgl.ucsf.edu/chimera/cgi-bin/secure/chimera-get.py?file=linux_x86_64/chimera-1.16-linux_x86_64.bin | |
| Excel | Microsoft | Version 2211 https://www.office.com/?auth=1 | |
| Falcon II | Thermofisher | https://www.thermofisher.com/de/de/home/electron-microscopy/products/accessories-em/falcon-detector.html | |
| IMOD | University of Colorado | Version 4.11.1 https://bio3d.colorado.edu/imod/download.html | |
| PC Desktop | Intel | Windows 10, ver. 22H2 | |
| PC Laptop | Gigabyte | Windows 10, ver. 22H2 | |
| Powerpoint | Microsoft | Version 2211 https://www.office.com/?auth=1 | |
| Titan Krios Electron Microscope | Thermofisher | https://www.thermofisher.com/de/de/home/electron-microscopy/products/transmission-electron-microscopes/krios-g4-cryo-tem.html | |
| Word | Microsoft | Version 2211 https://www.office.com/?auth=1 |
参考文献
- Downing, K. H., Sui, H., Auer, M. Electron tomography: A 3D view of the subcellular world. Analytical Chemistry. 79 (21), 7949-7957 (2007).
- Koning, R. I., Koster, A. J. Cellular nanoimaging by cryo electron tomography. Methods in Molecular Biology. 950, 227-251 (2013).
- Asano, S., Engel, B. D., Baumeister, W. In situ cryo-electron tomography: A post-reductionist approach to structural biology. Journal of Molecular Biology. 428 (2), 332-343 (2016).
- Serwas, D., Davies, K. M. Getting started with in situ cryo-electron tomography. Methods in Molecular Biology. 2215, 3-23 (2021).
- McDonald, K. L., Auer, M. High-pressure freezing, cellular tomography, and structural cell biology. Biotechniques. 41 (2), 137-143 (2006).
- Narasimha, R., et al. Evaluation of denoising algorithms for biological electron tomography. Journal of Structural Biology. 164 (1), 7-17 (2008).
- Frangakis, A. S. It's noisy out there! A review of denoising techniques in cryo-electron tomography. Journal of Structural Biology. 213 (4), 107804(2021).
- Frangakis, A. S., Hegerl, R. Noise reduction in electron tomographic reconstructions using nonlinear anisotropic diffusion. Journal of Structural Biology. 135 (3), 239-250 (2001).
- Jiang, W., Baker, M. L., Wu, Q., Bajaj, C., Chiu, W. Applications of a bilateral denoising filter in biological electron microscopy. Journal of Structural Biology. 144 (1-2), 114-122 (2003).
- vander Heide, P., Xu, X. -P., Marsh, B. J., Hanein, D., Volkmann, N. Efficient automatic noise reduction of electron tomographic reconstructions based on iterative median filtering. Journal of Structural Biology. 158 (2), 196-204 (2007).
- Volkmann, N. Methods for segmentation and interpretation of electron tomographic reconstructions. Methods in Enzymology. 483, 31-46 (2010).
- Böhm, J., et al. Toward detecting and identifying macromolecules in a cellular context: template matching applied to electron tomograms. Proceedings of the National Academy of Sciences. 97 (26), 14245-14250 (2000).
- Frangakis, A. S., et al. Identification of macromolecular complexes in cryoelectron tomograms of phantom cells. Proceedings of the National Academy of Sciences. 99 (22), 14153-14158 (2002).
- Lebbink, M. N., et al. Template matching as a tool for annotation of tomograms of stained biological structures. Journal of Structural Biology. 158 (3), 327-335 (2007).
- Rigort, A., et al. Automated segmentation of electron tomograms for a quantitative description of actin filament networks. Journal of Structural Biology. 177 (1), 135-144 (2012).
- Volkmann, N. A novel three-dimensional variant of the watershed transform for segmentation of electron density maps. Journal of Structural Biology. 138 (1-2), 123-129 (2002).
- Bajaj, C., Yu, Z., Auer, M. Volumetric feature extraction and visualization of tomographic molecular imaging. Journal of Structural Biology. 144 (1-2), 132-143 (2003).
- Yu, Z., Frangakis, A. S. Classification of electron sub-tomograms with neural networks and its application to template-matching. Journal of Structural Biology. 174 (3), 494-504 (2011).
- Moebel, E., et al. Deep learning improves macromolecule identification in 3D cellular cryo-electron tomograms. Nature Methods. 18 (11), 1386-1394 (2021).
- Tilney, L. G., Derosier, D. J., Mulroy, M. J. The organization of actin filaments in the stereocilia of cochlear hair cells. The Journal of Cell Biology. 86 (1), 244-259 (1980).
- Tilney, L. G., Tilney, M. S., DeRosier, D. J. Actin filaments, stereocilia, and hair cells: how cells count and measure. Annual Review of Cell Biology. 8, 257-274 (1992).
- Metlagel, Z., et al. Electron cryo-tomography of vestibular hair-cell stereocilia. Journal of Structural Biology. 206 (2), 149-155 (2019).
- Song, J., et al. A cryo-tomography-based volumetric model of the actin core of mouse vestibular hair cell stereocilia lacking plastin 1. Journal of Structural Biology. 210 (1), 107461(2020).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology. 116 (1), 71-76 (1996).
- Agulleiro, J. I., Fernandez, J. J. Fast tomographic reconstruction on multicore computers. Bioinformatics. 27 (4), 582-583 (2011).
- Fernandez, J. J. Computational methods for electron tomography. Micron. 43 (10), 1010-1030 (2012).
- Chen, H., Clyborne, W. K., Sedat, J. W., Agard, D. A. Priism: an integrated system for display and analysis of 3-D microscope images. Biomedical Image Processing and Three-Dimensional Microscopy. 1660, 784-790 (1992).
- Pettersen, E. F., et al. UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Goddard, T. D., Huang, C. C., Ferrin, T. E. Visualizing density maps with UCSF Chimera. Journal of Structural Biology. 157 (1), 281-287 (2007).
- Sazzed, S., et al. Tracing actin filament bundles in three-dimensional electron tomography density maps of hair cell stereocilia. Molecules. 23 (4), 882(2018).
- Sazzed, S., Scheible, P., He, P., Wriggers, J. Spaghetti tracer: A framework for tracing semiregular filamentous densities in 3D tomograms. Biomolecules. 12 (8), 1022(2022).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved