
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Identificação de codificação e Classes de RNA não-codificante expressa em suínos, sangue total
Neste Artigo
Resumo
Aqui, apresentamos um protocolo otimizado para o processamento de codificação (mRNA) e não-codificantes (ncRNA) globina reduzido RNA-seq bibliotecas de uma amostra da unidade de sangue total.
Resumo
O advento da inovadora e cada vez mais poderosas técnicas de sequenciamento próxima geração abriu novos caminhos para a capacidade de examinar a expressão de gene subjacente relacionada a processos biológicos de interesse. Essas inovações não só permitem que os pesquisadores a observar a expressão das sequências de mRNA que codificam genes essa função celular do efeito, mas também as moléculas de RNA (ncRNA) não-codificantes que permanecem untranslated, mas ainda têm funções reguladoras. Embora os pesquisadores têm a capacidade de observar a expressão tanto do mRNA e ncRNA, tem sido habitual para um estudo para se concentrar em um ou outro. No entanto, quando estudos estão interessados na expressão tanto do mRNA e ncRNA, muitas vezes eles usam amostras separadas para examinar ou codificação ou não-codificantes RNAs devido à diferença de preparações de biblioteca. Isso pode levar à necessidade de mais amostras que pode aumentar o tempo, consumíveis e estresse animal. Além disso, pode causar pesquisadores decidir para preparar amostras para análise de apenas um, geralmente o mRNA, limitando o número de questões biológicas que podem ser investigados. No entanto, ncRNAs abrangem várias classes com funções reguladoras aquela expressão de RNAm de efeito. Porque ncRNA são importantes para processos biológicos fundamentais e desordem destes processos em durante a infecção, eles podem, portanto, possam ser alvos para terapêutica. Este manuscrito demonstra um protocolo modificado para a geração do mRNA e não-codificantes do RNA expressão bibliotecas, incluindo o RNA viral, de uma única amostra de sangue total. Otimização do presente protocolo, melhorou a pureza do RNA, aumentou a ligadura para recuperação de RNAs metilados e omitido a seleção de tamanho, para permitir a captura de mais espécies de RNA.
Introdução
Sequenciamento de próxima geração (NGS) emergiu como uma poderosa ferramenta para a investigação das alterações que ocorrem no nível genômico de organismos biológicos. Preparação da amostra para métodos NGS pode variar dependendo do organismo, tipo de tecido, e mais importante as perguntas os pesquisadores fazem questão de endereço. Muitos estudos se voltaram para NGS como um meio de estudar as diferenças na expressão do gene entre Estados como indivíduos sãos e doentes,1,2,3,4. O sequenciamento tomar lugar numa base de genoma inteiro e permite que um pesquisador capturar a maioria, se não todos, a informação genômica para um marcador genético específico cada vez apontam.
Os marcadores mais comuns de expressão observadas são os RNAs mensageiro (mRNAs). Os procedimentos mais utilizados para preparar as bibliotecas para RNA-seq são otimizados para a recuperação das moléculas de mRNA através do uso de uma série de purificações, fragmentações e ligadura5,6. No entanto, a decisão sobre como um protocolo é para ser realizado depende muito do tipo de amostra e as questões sendo colocadas sobre amostra disse. Na maioria dos casos, o RNA total é extraído; ainda, nem todas as moléculas de RNA são de interesse e em casos tais como estudos de expressão de RNAm excessivamente abundantes espécies de RNA, como RNA ribossómico (rRNA) precisam ser removidos para aumentar o número de transcrições detectáveis associado com os mRNAs. O método mais popular e amplamente utilizado para a remoção das moléculas de RNA ribossomal abundante é a redução do polyadenylated RNA transcrições referidas como polyA depleção7. Essa abordagem funciona bem para a análise da expressão do mRNA como não afeta as transcrições do mRNA. No entanto, em estudos que estão interessados em RNAs virais ou não-codificantes, polyA depleção também remove essas moléculas.
Muitos estudos optar por focar a preparação de biblioteca de sequência de RNA para examinar qualquer expressão de RNAm (codificação) ou uma determinada classe de RNA não-codificante de pequena ou grande. Apesar de existirem outros procedimentos8 como a nossa que permitem a preparação da amostra dupla, muitos estudos preparem bibliotecas de amostras separadas para estudos separados quando disponível. Para um estudo como o nosso, isso normalmente exigiria várias amostras de sangue, aumentando o tempo, consumíveis e estresse animal. O objetivo do nosso estudo era ser capaz de usar todo o sangue de animais para identificar e quantificar as diferentes classes de ambos mRNA e RNA não-codificante expressas entre saudável e altamente patogénica reprodutiva suína e o vírus da síndrome respiratória (HP-PRRSV) desafiou os porcos9,10 , apesar de ter apenas uma unidade de sangue total de amostra (2,5 mL) de cada porco. Para isso, precisávamos para otimizar a extração típica e protocolos de criação de biblioteca para gerar os dados apropriados para permitir a análise de mRNA e não-codificantes do RNA (ncRNA) expressão11 de uma única amostra.
Isto levou a uma necessidade de um protocolo que permitiu mRNA e a análise de RNA não-codificante porque os kits de padrão disponíveis e métodos para criação de biblioteca e extração de RNA-destinavam-se principalmente para mRNA e usam uma depleção de poli-A etapa12. Esta etapa que tornaram impossível recuperar de RNA não-codificante ou transcrições virais da amostra. Portanto, um método otimizado era necessário que permitido para extração de RNA total sem esgotamento de polyA amostra. O método apresentado neste manuscrito foi otimizado para permitir o uso de sangue como um tipo de amostra e construir bibliotecas de sequenciamento para mRNA e ncRNAs de tamanhos pequenos e grandes. O método foi otimizado para permitir a análise de todos os RNAs detectáveis não-codificantes, bem como reter RNAs virais para posterior investigação13. Ao todo, nosso protocolo de preparação de biblioteca otimizado permite para a investigação de várias moléculas de RNA de uma amostra da unidade de sangue total.
O objetivo geral por trás o uso desse método foi desenvolver um processo que permitiu para a coleção de não-codificantes do RNA e mRNA de uma amostra de sangue total. Isto nos permite ter mRNA, ncRNA e RNA viral para cada animal em nosso estudo, originário de uma única amostra9. Isto, em última análise, permite a descoberta científica mais sem custos adicionais de animais e dá uma imagem mais completa da expressão de cada amostra individual. O método descrito permite o exame dos reguladores da expressão gênica, bem como permitindo a conclusão dos estudos correlativos comparando ambos mRNA e expressão de RNA não-codificante usando uma amostra da unidade de sangue total. Nosso estudo utilizou este protocolo para examinar as alterações na expressão gênica e possíveis epigenéticos reguladores em Viralmente infectado comerciais de suínos machos 9 - semana de idade.
Protocolo
Protocolos de animais foram aprovados pelo Comitê de uso e cuidado do Animal centro de doença Animal nacional (USDA-ARS-NADC).
1. coleta de sangue de porco amostras
- Colete amostras de sangue em tubos de RNA. Colete ~2.5 mL ou mais, se a maior coleção de tubos está disponíveis.
2. processamento de sangue de porco amostras
- Centrifuga os tubos de sangue em 5.020 x g durante 10 minutos à temperatura ambiente (15-25 ° C). Se amostras congelada de processamento incubam o tubo em temperatura ambiente por um mínimo de 2 h antes da centrifugação.
- Remover o sobrenadante e adicionar 8 mL de água livre de RNase a pelota. Fechar e vortex a pelota até que é visivelmente dissolvido. Centrifugar tubos de amostra em 5.020 x g durante 10 minutos à temperatura ambiente para recuperar a pelota. Descartar o sobrenadante todos e preservar a pelota.
3. orgânica extração de RNA Total e RNA pequeno (miRNA Kit de isolamento)
- Começa a extração de RNA total por pipetagem 300 μL de tampão de Lise vinculação ao pellet da etapa 2.2.
- Vórtice e transferir a mistura para um novo rotulagem tubo de centrífuga de 1,5 mL. Adicione 30 µ l de aditivo homogenate do kit. O tubo de vórtice e lugar no gelo por 10 min.
- Retire o tubo e adicionar 300 µ l do fenol-ácido: reagente de clorofórmio do kit. Vórtice do tubo para misturar. Centrifugar a 10.000 x g durante 5 min à temperatura ambiente.
- Remova cuidadosamente a fase aquosa (300-350 µ l) para um tubo de fresco. Anote o volume para o próximo passo.
4. procedimento de isolamento de RNA total
- Com base na quantidade de recuperação aquosa (300-350 µ l) adicione 1,25 x volume, de etanol 100% (~ 375 µ l) a fase aquosa. Misture a amostra com uma pipeta.
- Set-up nova coleção tubos contendo um cartucho de filtro para cada amostra. Pipeta ~ 675 µ l da mistura etanol/lisado para o refil do filtro.
Nota: Não adicione > 700 µ l para o refil do filtro ao mesmo tempo. Para volumes superiores aplicar em sucessão. - Centrifugue brevemente (~ 15-20 s) a 10.000 x g para passar líquido através do filtro. Não gire mais do que isso.
- O fluxo através de descartar e, se necessário, repita a centrifugação com a restante mistura de lisado (etanol), até que tudo foi aplicado. Manter o mesmo filtro cartucho e coleção do tubo para o próximo passo.
- Adicione 700 µ l de solução de lavagem 1 do kit para o refil do filtro e centrifugue brevemente (~ 10 s) para puxar através do filtro. Descartar o fluxo através de e manter o mesmo filtro cartucho e coleção do tubo.
- Adicione 500 µ l de solução de lavagem 2/3. Centrífuga para desenhar o líquido através do cartucho do filtro. Descarte a fluir. Repita a etapa de lavagem.
- Inthe mesmo tubo, girar o refil do filtro, um adicional de 60 s para remover qualquer líquido residual do filtro. Transfira o refil do filtro para um tubo de coleção fresca.
- Adicione 100 µ l de água livre de nuclease pré-aquecido (95 ° C) para o centro do refil do filtro. Rotação para aproximadamente 20-30 s para a velocidade máxima de centrifugação do tabletop.
Nota: RNA é contido o eluato e pode agora ser mais processado ou armazenado a-20 ° C ou abaixo. Enriquecimento para RNAs pequeno não foi realizado.
5. globina redução (baseada em um protocolo otimizado para amostras de sangue de suínos total)14,15
Nota: Globina redução é realizada para que as bibliotecas não estão superpovoadas com lê mapeamento de genes de globina, o que reduziria o número de leituras disponíveis para mapear para outros genes de maior interesse14,15 .
- Hibridação com globina redução oligos
- Desnature o RNA (6 µ g de RNA no volume máximo de 7 µ l de amostra) Pipetar a amostra extraída em um tubo de parede fina livre de nuclease reação de 0,2 mL e colocando em um termociclador a 70 ° C por 2 min. É a chave para tubos de gelo imediatamente após a primeira etapa do denature de óptima qualidade de RNA. Nenhum tratamento de DNase é necessária.
- Enquanto cool tubos preparar um 400 µ l de mistura de oligo redução globina de 10x: 100 µ l cada de dois HBA oligos (5'-GATCTCCGAGGCTCCAGCTTAACGGT-3' e 5'-TCAACGATCAGGAGGTCAGGGTGCAA-3') a 30 µM, dois HBB oligos (5'-AGGGGAACTTAGTGGTACTTGTGGGC-3', e 5'-GGTTCAGAGGAAAAAGGGCTCCTCCT-3') em 120 µM por reação, produzindo uma concentração final de 7,5 µM HBA oligos e 30 µM HBB oligos. Preparar o tampão de hibridização de x oligo 10: 100 mM Tris-HCL, pH 7,6; 200 mM KCl.
- Preparar a mistura de hibridização: 6 µ g de amostra de RNA (máximo 7 µ l de volume), 2 µ l da 400 µ l de mistura de oligo de redução de globina (concentração final 2 X), 1 µ l de tampão de hibridização 10 x oligo de 10x (concentração final 1 x). Adicione água nuclease livre até um volume final de 10 µ l.
- Definir thermocycler a 70 ° C por 5 min. Cool imediatamente a 4 ° C e proceder à digestão de RNase H.
- Digestão de RNase H
- Diluir 10 x RNase H (10 U / µ l) de 1x RNase H com tampão de RNase H 1x.
Nota: O RNase buffer vem em 10x e precisará ser diluído com 1 x buffer de RNase H 1 x antes de usar. - Preparar a mistura de reação de RNase H combinando: 2 µ l de RNase tampão, 1 µ l de inibidor de RNase em 2 µ l de RNase H de 1x e 5 µ l de água nuclease livre para um volume total de 10 µ l de 10x.
- Misture bem as amostras de hibridação de globina redução com 10 µ l da mistura de reação de RNase H e digerir a 37 ° C durante 10 min. esfriar a 4 ° C.
- Parar a digestão por adição de 1,0 µ l de EDTA 0,5 M para cada amostra e proceda imediatamente para a etapa de limpeza.
- Diluir 10 x RNase H (10 U / µ l) de 1x RNase H com tampão de RNase H 1x.
- RNase H-tratada Total RNA limpeza.
- Purificar o RNase H tratados RNA usando um kit de limpeza de purificação de eluição baseados em sílica-membrana de acordo com as instruções do fabricante. Premix amortecedores: para o tampão de lavagem suave, adicionar 44 mL de etanol a 100%.
- Não transfira a amostra para um tubo novo. Adicione 80 µ l de água livre de RNase e 350 µ l de tampão de Lise. Adicione 250 µ l de etanol 100% para o RNA diluído e misture bem por pipetagem.
Nota: Não centrifugar. Proceda de imediato à etapa 5.3.3. - Agora transfira a 700 µ l da amostra a um cartucho de filtro de eluição colocado em um tubo de coleta de 2 mL para coletar o fluxo através de. Centrifugar por 15 s a ≥ 8.000 x g. Descarte o escoamento.
- Repita isto colocando o cartucho de filtro de eluição para um novo tubo de coleta de 2 mL. Adicionar 500 µ l de buffer de lavagem suave para o refil do filtro e centrifugar por 15 s a ≥ 8.000 x g a fim de lavar o filtro de membrana de cartucho. Descarte o escoamento. Reutilize o tubo de coleta na etapa 5.3.5.
- Agora, usando o mesmo tubo de amostra, adicionar outro 500 µ l de etanol 80% para o refil do filtro. Desta vez centrifugar o tubo para 2 min a ≥ 8.000 x g. Recolher a coluna de rotação de eluição para a próxima etapa e descartar o escoamento e coleta de tubo.
- Colocar o refil do filtro de eluição em um novo tubo de coleta de 2 mL da última etapa. Deixe a tampa Abra no refil do filtro e centrifugar a toda velocidade por 5 min secar a membrana de coluna de rotação e evitar etanol transitar. Descarte o tubo de escoamento e a coleção.
Nota: Para evitar danos ângulo as tampas para apontar no sentido oposto do rotor. - Leve o cartucho de filtro seco e coloque em um novo tubo de coleta de 1,5 mL. Adicione 14 µ l de água livre de RNase para filtro de membrana de cartucho, certificando-se de adicionar a água livre de RNase diretamente para o centro. Centrífuga para 60 s a toda a velocidade para Eluir o RNA.
- Avalie a qualidade da globina-redução de amostras de RNA (etapa 6). Proceda à preparação da amostra de mRNA (passo 7) e o pequeno RNA biblioteca preparação (passo 8)16,17.
Nota: Amostras do RNA globina-reduzido agora podem ser armazenadas a-20 ° C, porém o armazenamento a-80 ° C é recomendado para preservação a longo prazo.
6. avaliação do RNA
- Quantificar a concentração de RNA usando um espectrofotômetro. Examine a relação do comprimento de onda entre 260 e 280 nm. Rácios de ~ 2 ou mais são considerados puros para o RNA e valores mais baixos indicam alguma contaminação. O aparelho utiliza esta relação para determinar a concentração de RNA como ng / µ l.
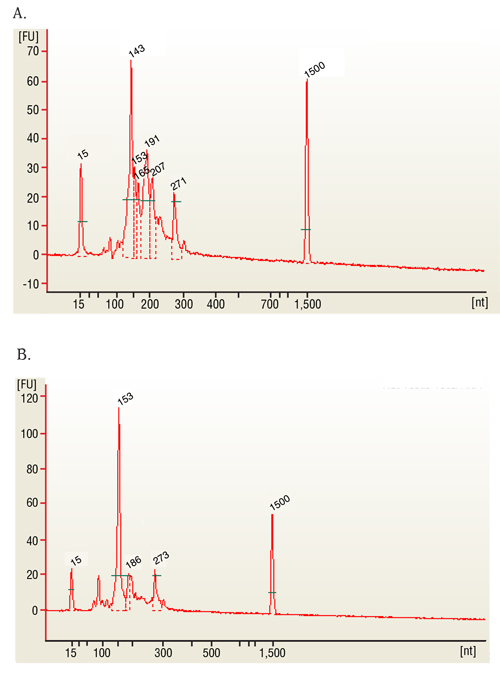
- Avaliar a qualidade do RNA usando 1 µ l (100 ng) da amostra em um chip apropriado. Produto final deve ser um RIN ~ 2 ou superior e um pico em ~ 280 nt para mRNA único ler bibliotecas; pequenos picos de bibliotecas de RNA em 143 correspondem os miRNAs.
7. preparação de amostras de RNA Total de mRNA e bibliotecas ncRNA longo de presos. 16
- Nota: Levar tampão de eluição e rRNA grânulos de remoção à temperatura. Pre-etiqueta 0,2 ml paredes finas da polimerase (placas também podem ser usadas). Etapas de protocolo baseadas em instruções de16 do fabricante.
- Comece com 4 µ l de RNA reduzida da globina. Adicione 6 µ l de água livre de nuclease, 5 µ l de tampão de ligação de rRNA e 5 µ l do mix de remoção rRNA por tubo (1st ). Pipete para misturar e re-cap. Coloque em thermocycler com aquecida-tampa do 100 ° C por 5 min em 68° c. Retire e deixe em temperatura ambiente por 60 s.
- Resuspenda grânulos de remoção rRNA pelo vórtice; Adicione 35 µ l de grânulos para novo tubo de PCR (2nd) e pipetar amostra dos tubos de 1st em grânulos nos 2 tubosnd . Incube o tubo do PCR de 2nd em temperatura ambiente por 3 min. Coloque sobre o suporte magnético para 7 min.
Nota: Misture bem cada amostra pipetando para esgotamento de rRNA ideal. Aumento/redução do tubo no suporte pode ajudar a acelerar o processo de separação. - Transferência o sobrenadante do tubo 2nd PCR para correspondência 3rd tubo PCR e lugar para o magnético defende um mínimo de 60 s. Repeat, a transferência para um novo tubo de PCR somente se as contas não são movidas para os lados do tubo.
- Grânulos de mistura amostra purificação pelo vórtice; Adicione 99 µ l de cada tubo PCR 3rd . Permita a sentar-se à temperatura ambiente por 15 min. lugar em suporte magnético para 5 min. Certifique-se de grânulos move-se para os lados. Pipeta para descartar o sobrenadante.
- Mantenha 3 tubo derd , definido em suporte magnético. Adicione 200 µ l de etanol 70%, enquanto tomando cuidado para não empurram os grânulos. Deixe para descansar pelo menos 30 s e pipeta para descartar o sobrenadante. Repita a etapa.
- Permitir que a amostra secar à temperatura ambiente por 15 min no stand magnético. Centrifugue o tampão de eluição kit para 5 s a 600 x g. Retire o tubo e adicionar 11 µ l de tampão de eluição, misturando cuidadosamente. Incube durante 2 min no banco, em seguida, pelo menos de 5 min a depor magnético à temperatura ambiente.
- Transferência de 8,5 µ l do sobrenadante da 3rd para uma nova (4th) tubo do PCR. Adicione 8,5 µ l do mix elevado Elute/Prime/fragmento do kit. Misture bem. Tampa e coloque em um thermocycler com pré-aquecido tampa durante 8 min a 94 ° C, manter a 4 ° C. Remover e centrifugar brevemente.
- Sintetizar o cDNA da primeira costa
- Permitir que a primeira vertente mistura de síntese do kit para a temperatura e centrifugar a 600 × g por 5 µ l de s. transferir 50 de transcriptase reversa na mistura de síntese primeira vertente. Transcriptase reversa pode ser adicionado a primeira vertente síntese na proporção de 1:9.
- Pipetar 8 µ l do transcriptase reversa combinado/primeira vertente mistura de síntese no 4º tubo com a amostra. Cap e centrifugar 600 x g durante 5 s. lugar em thermocycler com tampa pré-aquecido conjunto a 100 ° C. Executado por: 10 min a 25 ° C, 15 min a 42 ° C, 15 min a 70 ° C, então descansar a 4 ° C. Volume final é ~ 25 µ l por bem. Vá imediatamente para a próxima etapa.
- Sintetizar a segunda vertente do cDNA
- Levar o reagente final de reparação (ERR) e a segunda vertente Mix (SSM) à temperatura e centrifugar a 600 x g por 5 s. ERR Mix com buffer de ressuspensão em 01:50 diluição. Destampe 4th tubo e adicionar 5 µ l de ERR diluído e 20 µ l de SSM para cada um; Pipeta para misturar bem. O volume final ~ 50 µ l ds cDNA.
- Cap e incubar em thermocycler durante 1 hora a 16 ° C. Quando o ciclo está completo, retire as tampas e permitir para vir à temperatura ambiente no topo do banco.
- etapa de limpeza do cDNA DS
- Comece misturando os grânulos paramagnética de fase sólida reversível imobilização (SPRI) pelo vórtice. Transferir 90 µ l dos grânulos para a amostra no tubo 4th(cDNA ds) e misture. Volume final é 140 µ l. Allow tubos para sentar-se à temperatura ambiente por 15 min. incubam em suporte magnético para ~ 5 min. Remova ~ 135 µ l do sobrenadante de cada poço. Lá deve 5 µ l em cada poço.
- Deixar o tubo sobre o suporte magnético e lave adicionando-se 200 µ l de etanol 80%. Deixe para descansar por 30 s em seguida, remover e descartar o sobrenadante. Repita a etapa de lavagem. Deixe os tubos no stand magnético por 15 min deixar para secar.
- Centrifugue o buffer de ressuspensão, a 600 x g durante 5 s após a vinda à temperatura ambiente. Retire os tubos do carrinho. 17.5 µ l de tampão de ressuspensão de transferência para o tubo e misture. Deixe os tubos incubar na parte superior do banco para 2 min, em seguida, mover para carrinho magnético por 5 min.
- Pipete 15 µ l do sobrenadante contendo as amostras de cDNA de ds para novos tubos de conjunto da (5th) 0,2 mL paredes finas. Mover para a próxima etapa prontamente ou selar e armazenar a-15 ° C a-25 ° C, por não mais de 7 dias.
- Adenilato 3' extremidades.
- Pipetar 2,5 µ l de tampão de ressuspensão de temperatura para o tubo de amostra, em seguida, adicione 12,5 µ l da mistura de A-Tailing descongelada. Misture completamente por pipetagem.
Nota: Não A-tailing controle usado. - Cap e incubá-los em um thermocycler com tampa previamente aquecida de 100 ° C. Execute a 37 ° C por 30 min, em seguida a 70 ° C, por 5 min com tampa aquecida. Permitir que o thermocycler descansar a 4 ° C.
- Pipetar 2,5 µ l de tampão de ressuspensão de temperatura para o tubo de amostra, em seguida, adicione 12,5 µ l da mistura de A-Tailing descongelada. Misture completamente por pipetagem.
- Ligate adaptadores de índice
- Levar os tubos adaptador de RNA e a mistura de Buffer de ligadura parar à temperatura. Para cada um, centrifugar a 600 x g durante 5 s. mistura de ligadura deixe no congelador até que esteja pronto para uso. Pool de arranjo para indexação de amostra deve ser conhecida.
- Adicione 2,5 µ l de tampão de ressuspensão e 2,5 µ l de mistura de ligadura para cada tubo de amostra. Agora Pipete em 2,5 µ l do adaptador de RNA apropriado em cada tubo de amostra. Seleção do adaptador deve ser realizada por instruções do fabricante para o kit escolhido.
- Fechar e por centrifugação por 1 min a 280 x g. Incubar em um termociclador durante 10 minutos a 30 ° C. Adicione 5 µ l de Buffer de ligadura parar a amostra para deter a reação e misture bem.
- Comece limpar pela mistura de grânulos paramagnéticos de SPRI pelo vórtice para 60 s. µ l 42 de transferência de contas para cada tubo. Thoroughlythen Mix incube por 15 min na bancada.
- Mover os tubos para suporte magnético e deixe até vira líquida claro (~ 5 min). Uma vez claro, remover e descartar 79,5 µ l do sobrenadante. Deixar os tubos no stand magnético e lave adicionando-se 200 µ l de etanol 80%. Deixe para descansar pelo menos 30 s em seguida, remover e descartar o sobrenadante. Repita a etapa de lavagem. Deixe na cabeceira magnética para adicional ~ 15 min permitir a secagem. Não perturbem grânulos durante as etapas de lavagem.
- Adicionar 52,5 µ l de tampão de ressuspensão para o tubo de amostra e misture até que os grânulos são completamente re-suspensos. Incube por 2 min em cima do banco. Tubo de amostra movimento volta ao carrinho magnético até o líquido é claro (~ 5 min).
- Deixar na cabeceira e suavemente, pipete 50 µ l do sobrenadante para novos (6th) tubos. Adicione 50 µ l de grânulos SPRI vortexed. Permita a incubar na bancada por 15 min.
- Mover para carrinho magnético e deixar a placa ficar até vira líquida claro (~ 5 min). Sobrenadante de descarte 95 µ l dos deixando 5 µ l em cada tubo.
- Deixar os tubos no suporte magnético e lave adicionando-se 200 µ l de etanol 80%. Permitir que o tubo descansar por 30 s, em seguida, remover e descartar o sobrenadante. Repita a lavagem. Seque os tubos magnéticos depor por 15 min.
- Adicionar em 22,5 µ l de tampão de ressuspensão e misture até grânulos estão suspensos. Incubar em banco por 2 min, em seguida, mover para carrinho magnético até vira líquido claro (~ 5 min). Pipete 20 µ l de amostra para novos tubos de PCR (7th).
- Levar os componentes do kit necessários à temperatura. Transferir 5 µ l de mistura de primeira demão do PCR e 25 µ l de mistura PCR mestre do kit de amostra (7th PCR tubo) contendo amostras indexadas. Misture o tubo de amostra e cap.
- Coloque os tubos no termociclador com tampa pré-aquecido. Executar programa: 98 ° C por 30 s e, em seguida, 15 ciclos de 98 ° C por 10 s, 60 ° C por 30 s, 72 ° C por 30 s, 72 ° C por 5 min. espera a 4 ° C.
- Limpe as amostras pela adição de 50 µ l de grânulos SPRI suficientemente misturados ao tubo da amostra e pipetar para combinar. Permitir que a reação a incubar na parte superior da bancada para 15 min.
- Mover o tubo para um suporte magnético e incubar até vira líquida claro (~ 5 min). Sobrenadante de descarte 95 µ l dos deixando 5 µ l em cada tubo.
- Deixar o tubo no suporte magnético e lave adicionando-se 200 µ l de etanol 80%. Deixe para descansar pelo menos 30 s e depois remover e descartar o sobrenadante. Repita essa etapa de lavagem. Deixe na cabeceira magnética por 15 min deixar para secar.
- Adicionar em 32,5 µ l de tampão de ressuspensão e misture até grânulos são completamente re-suspensos. Incubar em banco por 2 min, em seguida, mover para carrinho magnético até vira líquido claro (~ 5 min). Pipete 30 µ l da amostra de cada tubo para um novo tubo PCR.
- Avalie a qualidade e quantidade de DNA, repetindo as etapas 6.1 e 6.2 com o chip apropriado. Seguir em frente ao pool (passo 9).
8. pequeno RNA biblioteca de preparação para o sncRNAs. 17
Nota: Etapas de protocolo dependendo instruções17 do fabricante.
- Preparar pequeno RNA biblioteca com ~ 220 ng - ~1.1 µ g / µ l de RNA reduzida globina amostras (4 µ l).
- Ligadura de adaptador
- Ligam o 3'-adaptador misturando completamente 1 µ l de adaptador, 2 µ l de água livre de nuclease e 4 µ l da amostra de RNA reduzida da globina. Incubar a 70 ° C por 2 min e coloque imediatamente no gelo.
- Adicionar 10 µ l de buffer de ligadura de 2x, e 3 µ l do mix de enzima de ligadura, misturar e incubar a 16 ° C por 18 h.
Nota: O aumento tempo de incubação da amostra de 18 horas a uma temperatura mais baixa de 16 ° C é recomendado para estudos interessados em espécies de RNA metilados, como piwi RNAs. O tempo de incubação permite maior eficiência de ligadura devido à aula de modificações durante a fase de ligadura adaptador 3'. - Adicione 1 µ l de primer de transcrição reversa em 4,5 µ l de água livre de nuclease à mistura de ligadura para evitar a formação de adaptador-dímero de excesso 3' adaptador.
- Incubar em um termociclador pré-aquecido programado por 5 min a 75 ° C, 15 min a 37 ° C, 15 min a 25 ° C e segurar a 4° C.
- Desnaturar 1 µ l de pre-diluído 5'-adaptador por amostra em um termociclador a 70 ° C por 2 min e coloque imediatamente no gelo.
- Adicione 1 µ l de desnaturado 5'-adaptador, 1 µ l de tampão de ligadura 10 x 5' e 2,5 µ l de enzima de ligadura 5'. Misturar e incubar a 25 ° C, durante 1 h.
- amplificação e a síntese do cDNA
- Misture bem por pipetagem µ l 30 de RNA 5 ' / 3 '-adaptador-ligados, 8 µ l de buffer de primeiro-costa, 1 µ l de inibidor de RNase e 1 µ l de transcriptase reversa. Incubar a mistura a 50 ° C por 60 min. proceder imediatamente a amplificação por PCR ou calor inativar a reação a 70 ° C por 15 min e armazenar a-20 ° C.
- PCR amplificar a 40 µ l de reação Transcriptase reversa pela adição de 50 µ l de mistura mestre PCR, 2,5 µ l de primer de RNA PCR, adicione 2,5 µ l de designada RNA PCR Primer índice de amostra e nuclease água livre para um volume total de 100 µ l. misturar por pipetagem. Executar o PCR no termociclador usando as seguintes condições de ciclismo: desnaturação inicial a 94 ° C por 30 s; 11 ciclos de 94 ° C por 15 s, recozendo a 62 ° C, durante 30 s e extensão a 70 ° C por 15 s; seguido por uma extensão final a 70 ° C por 5 min; as amostras podem ser mantidas a 4 ° C no cycler.
- Nota: Índices são adicionados para efeito de pool de amostra.
- Amostra limpa-up
- Do kit de limpeza de DNA adicionar 500 µ l de tampão de ligação (5 M Gu-HCl, 30% de isopropanol) para a 100 µ l de amostra amplificada de PCR para permitir a ligação eficiente para a membrana de rotação-coluna.
Nota: Se o buffer de vinculação tem indicador de pH incluído Verifique a cor da mistura é amarela. - Pipeta mistura de 600 µ l de amostra e tampão de ligação em um cartucho de filtro dentro de um tubo de coleta de 2 mL, rotação para 30-60 s a 17.900 x g em uma centrífuga de mesa. Descarte-se de passagem.
- Lavar o cartucho de filtro no tubo de coleta mesmo com 0,75 mL de tampão de lavagem do kit fornecido (10 mM Tris-HCl pH 7,5, 80% de etanol) girando para 30-60 s a 17.900 x g em uma centrífuga de mesa e descartando o fluxo-através de. Girar o refil do filtro seco para um adicional de 60 s.
- Coloque o cartucho de filtro em um tubo de coleta limpa 1,5 mL. Adicionar 30 µ l de tampão de eluição (10 mM Tris-HCl pH 8,5), deixe a coluna repousar durante 60 s e em seguida girar por 60 s a 17.900 x g em uma centrífuga do tabletop.
- Do kit de limpeza de DNA adicionar 500 µ l de tampão de ligação (5 M Gu-HCl, 30% de isopropanol) para a 100 µ l de amostra amplificada de PCR para permitir a ligação eficiente para a membrana de rotação-coluna.
- Avalie a qualidade de amostra, repetindo os passos 6.1 e 6.2 com o chip de DNA apropriado. Mover para o pool de step (passo 9).
9. amostra pool para sequenciamento
- Código de barras piscina e QC'ed amostras pela criação e rotulagem novos tubos (ou um prato PCR bem 96) para conter o mRNA ou ncRNA amostras. Transferência 13 µ l de cada biblioteca de 10nM de código de barras para tubos correspondentes (ou poços da placa de novo) por instruções16,17 de fabricante. Apresentar em pool de amostras para sequenciamento, certifique-se de escolher comprimentos de leitura semelhantes se as amostras estão a ser executado no mesmo chip.
Resultados
Em nosso estudo, as amostras representativas são a globina e amostras de sangue de todo ribo-esgotada. O resultado representativo do protocolo consiste em uma amostra de biblioteca empobrecido de globina com um número de integridade do RNA (RIN) acima de proporções de concentração de nm 260/280 igual ou superior a 2 (Figura 1b e 1C) e 7 (Figura 1um). Validação dos resultados da amostra fo...
Discussão
O primeiro passo crítico no protocolo que foi otimizado incluiu as etapas de depleção de globina adicionado, que tornou possível obter leituras de qualidade de amostras de sangue total. Uma das maiores limitações sobre o uso de sangue total em estudos de sequenciamento são o elevado número de leituras na amostra que irão mapear para moléculas de globina e reduzir as leituras que poderiam mapear para outras moléculas de interesse18. Portanto, na otimização do protocolo para o nosso tip...
Divulgações
Os autores não têm nada para divulgar.
Menção de nomes comerciais ou produtos comerciais neste artigo é exclusivamente com o propósito de fornecer informações específicas e não implica em recomendação ou endosso pelo departamento E.U. da agricultura. USDA é um provedor de igualdade de oportunidades e o empregador.
Agradecimentos
Este trabalho foi apoiado principalmente pela pelo USDA NIFA AFRI 2013-67015-21236 e em parte pelo USDA NIFA AFRI 2015-67015-23216. Este estudo foi suportado em parte por uma nomeação para o agrícola pesquisa serviço programa de participação de pesquisa administrada pelo Instituto de Oak Ridge para ciência e educação (ORISE) através de um acordo interinstitucional entre o departamento de energia ( DOE) e o departamento de agricultura dos EUA. ORISE é gerido pela Oak Ridge Associated universidades sob contrato DOE não. DE-AC05-06OR2310.
Gostaríamos de agradecer o Dr. Kay Faaberg para os clones HP-PRRSV infecciosos, Dr. Susan Brockmeier por sua ajuda com animais envolvidos no experimento e Sue Ohlendorf para secretariado em preparação do manuscrito.
Materiais
| Name | Company | Catalog Number | Comments |
| PAXgene Tubes | PreAnalytix | 762165 | |
| Molecular Biology Grade Water | ThermoFisher | 10977-015 | |
| mirVana miRNA Isolation Kit | ThermoFisher | AM1560 | |
| Rneasy MinElute Clean Up Kit | QIAGEN | 74204 | |
| 100% Ethanol | Decon Labs, Inc. | 2716 | |
| 0.2 mL thin-walled tubes | ThermoFisher | 98010540 | |
| 1.5 mL RNase/DNase - free tubes | Any supplier | ||
| Veriti 96-well Thermocycler | ThermoFisher | 4375786R | |
| Globin Reduction Oligo (α 1) | Any supplier | Sequence GAT CTC CGA GGC TCC AGC TTA ACG GT | |
| Globin Reduction Oligo (α 2) | Any supplier | Sequence TCA ACG ATC AGG AGG TCA GGG TGC AA | |
| Globin Reduction Oligo (β 1) | Any supplier | Sequence AGG GGA ACT TAG TGG TAC TTG TGG GT | |
| Globin Reduction Oligo (β 2) | Any supplier | Sequence GGT TCA GAG GAA AAA GGG CTC CTC CT | |
| 10X Oligo Hybridization Buffer | |||
| -Tris-HCl, pH 7.6 | Fisher Scientific | BP1757-100 | |
| -KCl | Millipore Sigma | 60142-100ML-F | |
| 10X RNase H Buffer | |||
| -Tris-HCl, pH 7.6 | Fisher Scientific | BP1757-100 | |
| -DTT | ThermoFisher | Y00147 | |
| -MgCl2 | Promega | A351B | |
| -Molecular Biology Grade Water | ThermoFisher | 10977-015 | |
| RNase H | ThermoFisher | AM2292 | |
| SUPERase-IN | ThermoFisher | AM2694 | Rnase inhibitor |
| EDTA | Millipore Sigma | E7889 | |
| Microcentrifuge | Any supplier | ||
| 2100 Electrophoresis BioAnalyzer Instrument | Agilent Technologies | G2938C | |
| Agilent RNA 6000 Nano Kit | Agilent Technologies | 5067-1511 | |
| Agilent High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 | |
| TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero | Illumina | RS-122-2201 | mRNA kit; Human/Mouse/Rat Set A (48 samples, 12 indexes) |
| TruSeq Stranded Total RNA Sample Preparation Guide | Illumina | Available on-line | |
| RNAClean XP Beads | BeckmanCoulter | A63987 | |
| AMPure XP Beads | BeckmanCoulter | A63880 | |
| MicroAmp Optical 8-tube Strip | ThermoFisher | N8010580 | 0.2 ml thin-walled tubes |
| MicroAmp Optical 8-tube Strip Cap | ThermoFisher | N801-0535 | |
| RNase/DNase - free reagent reservoirs | Any supplier | ||
| SuperScript II Reverse Transcriptase | ThermoFisher | 18064-014 | |
| MicroAmp Optical 96 well plates | ThermoFisher | N8010560 | These were used in place of .3mL plates as needed |
| MicroAmp Optical adhesive film | ThermoFisher | 4311971 | |
| NEBNext Multiplex Small RNA Library Prep Set for Illumina® (Set 1) | New England Biolabs | E73005 | small RNA kit |
| NEBNext Multiplex Small RNA Library Prep Set for Illumina® (Set 2) | New England Biolabs | E75805 | small RNA kit |
| QIAQuick PCR Purification Kit | QIAGEN | 28104 | |
| 96S Super Magnet Plate | ALPAQUA | A001322 |
Referências
- Finotello, F., Di Camillo, B. Measuring differential gene expression with RNA-seq: challenges and strategies for data analysis. Briefings in Functional Genomics. 14 (2), 130-142 (2015).
- Coble, D. J., et al. RNA-seq analysis of broiler liver transcriptome reveals novel responses to high ambient temperature. BMC Genomics. 15, 1084 (2014).
- Koltes, J. E., et al. Identification of a putative quantitative trait nucleotide in guanylate binding protein 5 for host response to PRRS virus infection. BMC Genomics. 16, 412 (2015).
- Miller, L. C., et al. Comparative analysis of signature genes in PRRSV-infected porcine monocyte-derived cells to different stimuli. PLoS One. 12 (7), 0181256 (2017).
- Head, S. R., et al. Library construction for next-generation sequencing: overviews and challenges. Biotechniques. 56 (2), 61-64 (2014).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10 (1), 57-63 (2009).
- Dominic, O. N., Heike, G., Martin, S. Ribosomal RNA Depletion for Efficient Use of RNA-Seq Capacity. Current Protocols in Molecular Biology. 103 (1), 11-14 (2013).
- Fleming, D. S., Miller, L. C. Identification of small non-coding RNA classes expressed in swine whole blood during HP-PRRSV infection. Virology. 517, 56-61 (2018).
- Fleming, D. S., Miller, L. C. Small non-coding RNA expression status in animals faced with highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV). Proceedings of the World Congress on Genetics Applied to Livestock Production. (11), (2018).
- Bivens Nathan, J., Zhou, M. RNA-Seq Library Construction Methods for Transcriptome Analysis. Current Protocols in Plant Biology. 1 (1), 197-215 (2016).
- Cui, P., et al. A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics. 96 (5), 259-265 (2010).
- Guo, Y., et al. RNAseq by Total RNA Library Identifies Additional RNAs Compared to Poly(A) RNA Library. BioMed Research International. 2015, 9 (2015).
- Choi, I., et al. Increasing gene discovery and coverage using RNA-seq of globin RNA reduced porcine blood samples. BMC Genomics. 15, 954 (2014).
- . TruSeq® Stranded Total RNA Sample Preparation Guide. Illumina. , (2018).
- . New England Biolabs Inc. Protocol for use with NEBNext Multiplex Small RNA Library Prep Kit for Illumina (Index Primers 1-48) (E7560) Available from: https://www.neb.com/~/media/Catalog/All-Protocols/758F75A29CDE4D03954E1EF75E78EA3D/Content/manualE7560_Figure_1.jpg (2018)
- Shin, H., et al. Variation in RNA-Seq transcriptome profiles of peripheral whole blood from healthy individuals with and without globin depletion. PLoS One. 9 (3), 91041 (2014).
- Costa, V., Angelini, C., De Feis, I., Ciccodicola, A. Uncovering the complexity of transcriptomes with RNA-Seq. Journal of Biomedicine and Biotechnology. 2010, 853916 (2010).
- Martens-Uzunova, E. S., Olvedy, M., Jenster, G. Beyond microRNA--novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer Letters. 340 (2), 201-211 (2013).
{kind=link}
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados