
Sign In
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
Identification of Coding and Non-coding RNA Classes Expressed in Swine Whole Blood
In This Article
Summary
Here, we present a protocol optimized for the processing of coding (mRNA) and non-coding (ncRNA) globin reduced RNA-seq libraries from a single whole blood sample.
Abstract
The advent of innovative and increasingly powerful next generation sequencing techniques has opened new avenues into the ability to examine the underlying gene expression related to biological processes of interest. These innovations not only allow researchers to observe expression from the mRNA sequences that code for genes that effect cellular function, but also the non-coding RNA (ncRNA) molecules that remain untranslated, but still have regulatory functions. Although researchers have the ability to observe both mRNA and ncRNA expression, it has been customary for a study to focus on one or the other. However, when studies are interested in both mRNA and ncRNA expression, many times they use separate samples to examine either coding or non-coding RNAs due to the difference in library preparations. This can lead to the need for more samples which can increase time, consumables, and animal stress. Additionally, it may cause researchers to decide to prepare samples for only one analysis, usually the mRNA, limiting the number of biological questions that can be investigated. However, ncRNAs span multiple classes with regulatory roles that effect mRNA expression. Because ncRNA are important to fundamental biologic processes and disorder of these processes in during infection, they may, therefore, make attractive targets for therapeutics. This manuscript demonstrates a modified protocol for the generation mRNA and non-coding RNA expression libraries, including viral RNA, from a single sample of whole blood. Optimization of this protocol, improved RNA purity, increased ligation for recovery of methylated RNAs, and omitted size selection, to allow capture of more RNA species.
Introduction
Next generation sequencing (NGS) has emerged as a powerful tool for the investigation of the changes that occur at the genomic level of biological organisms. Sample preparation for NGS methods can be varied depending on the organism, tissue type, and more importantly the questions the researchers are keen to address. Many studies have turned to NGS as a means of studying the differences in gene expression between states such as healthy and sick individuals1,2,3,4. The sequencing take place on a whole genome basis and allows a researcher to capture the most, if not all, of the genomic information for a particular genetic marker at a time point.
The most common markers of expression observed are the messenger RNAs (mRNAs). The most used procedures for prepping libraries for RNA-seq are optimized for the recovery of mRNA molecules through the use of a series of purifications, fragmentations, and ligations5,6. However, the decision on how a protocol is to be performed relies heavily on the sample type and the questions being posed about said sample. In most cases total RNA is extracted; yet, not all RNA molecules are of interest and in cases such as mRNA expression studies overly abundant RNA species, like ribosomal RNAs (rRNA) need to be removed to increase the number of detectable transcripts associated with the mRNAs. The most popular and widely used method for removing the abundant rRNA molecules is the reduction of polyadenylated RNA transcripts referred to as polyA depletion7. This approach works well for the analysis of mRNA expression as it does not affect the mRNA transcripts. However, in studies that are interested in non-coding or viral RNAs, polyA depletion also removes these molecules.
Many studies choose to focus on the RNA sequence library preparation to examine either mRNA expression (coding) or a particular class of small or large non-coding RNA. Although there are other procedures8 like ours that allow for the dual sample preparation, many studies prepare libraries from separate samples for separate studies when available. For a study like ours, this would normally require multiple blood samples increasing time, consumables, and animal stress. The goal of our study was to be able to use whole blood from animals to identify and quantify the different classes of both mRNA and non-coding RNA expressed between healthy and highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV) challenged pigs9,10 despite having only a single whole blood sample (2.5 mL) from each pig. In order to do this, we needed to optimize the typical extraction and library creation protocols to generate the proper data to allow for analysis of both mRNA and non-coding RNA (ncRNA) expression11 from a single sample.
This prompted a need for a protocol that allowed for mRNA and non-coding RNA analysis because the available standard kits and methods for RNA-extraction and library creation were intended chiefly for mRNA and use a poly-A depletion step12. This step would have made it impossible to recover non-coding RNA or viral transcripts from the sample. Therefore, an optimized method was needed that allowed for total RNA extraction without sample polyA depletion. The method presented in this manuscript has been optimized to allow for the use of whole blood as a sample type and to build sequencing libraries for both mRNA and ncRNAs of small and large sizes. The method has been optimized to allow for the analysis of all detectable non-coding RNAs as well as retain viral RNAs for later investigation13. In all, our optimized library preparation protocol allows for the investigation of multiple RNA molecules from a single whole blood sample.
The overall goal behind the use of this method was to develop a process that allowed for the collection of both non-coding RNA and mRNA from one sample of whole blood. This allows us to have mRNA, ncRNA, and viral RNA for each animal in our study sourced from a single sample9. This, ultimately, allows for more scientific discovery without additional animal costs and gives a more complete picture of the expression of each individual sample. The described method allows for the examination of the regulators of gene expression as well as allowing for completion of correlative studies comparing both mRNA and non-coding RNA expression using a single whole blood sample. Our study used this protocol to examine the changes in gene expression and possible epigenetic regulators in virally infected 9-week old male commercial pigs.
Protocol
Animal protocols were approved by the National Animal Disease Center (USDA-ARS-NADC) Animal Care and Use Committee.
1. Collection of Swine Blood Samples
- Collect blood samples into RNA tubes. Collect ~2.5 mL or more if larger collection tubes are available.
2. Processing of Swine Blood Samples
- Centrifuge the blood tubes at 5,020 x g for 10 min at room temperature (15-25 °C). If processing frozen samples incubate tube at room temperature for a minimum of 2 h prior to centrifugation.
- Remove supernatant and add 8 mL of RNase-free water to the pellet.Close and vortex the pellet until it is visibly dissolved. Centrifuge sample tubes at 5,020 x g for 10 min at room temperature to recover the pellet. Discard all supernatant and preserve the pellet.
3. Organic Extraction for Total RNA and Small RNA (miRNA Isolation Kit)
- Begin total RNA extraction by pipetting 300 µL of lysis binding buffer to the pellet from the step 2.2.
- Vortex and transfer the mixture to a new labelled 1.5 mL centrifuge tube. Add 30 µL of homogenate additive from the kit. Vortex the tube and place on the ice for 10 min.
- Remove the tube and add 300 µL of the acid-phenol: chloroform reagent from the kit. Vortex the tube to mix. Centrifuge at 10,000 x g for 5 min at room temperature.
- Carefully remove aqueous phase (300-350 µL) to a fresh tube. Note the volume for the next step.
4. Total RNA Isolation Procedure
- Based on the amount of aqueous recovery (300-350 µL) add 1.25x volume of 100% (~375 µL) ethanol to aqueous phase. Mix the sample using a pipette.
- Set-up new collection tubes containing a filter cartridge for each sample. Pipette ~ 675 µL of the lysate/ethanol mixture onto the filter cartridge.
NOTE: Do not add > 700 µL to filter cartridge at one time. For larger volumes apply in succession. - Centrifuge briefly (~15-20 s) at 10,000 x g to pass liquid through the filter. Do not spin harder than this.
- Discard the flow through and, if necessary, repeat the centrifugation with the remaining lysate/ethanol mixture until it has all been applied. Retain the same filter cartridge and collection tube for the next step.
- Add 700 µL of Wash Solution 1 from the kit to the filter cartridge and centrifuge briefly (~10 s) to pull through the filter. Discard the flow through and retain the same filter cartridge and collection tube.
- Add 500 µL of Wash Solution 2/3. Centrifuge to draw the liquid through the filter cartridge. Discard the flow through. Repeat wash step.
- Inthe same tube, spin the filter cartridge an additional 60 s to remove any residual liquid from filter. Transfer the filter cartridge to a fresh collection tube.
- Add 100 µL of pre-heated (95 °C) nuclease-free water to the center of the filter cartridge. Spin for approximately 20-30 s at the tabletop centrifuge max speed.
NOTE: RNA is contained in the eluate and can now be further processed or stored at -20 °C or below. Enrichment for small RNAs was not performed.
5. Globin Reduction (based on a protocol optimized for porcine whole blood samples)14,15
NOTE: Globin reduction is performed so that libraries are not overpopulated with reads mapping to globin genes, which would lower the number of reads available to map to other genes of greater interest14,15 .
- Hybridization with globin reduction oligos
- Denature the RNA (6 µg RNA sample in maximum 7 µL volume) by pipetting the extracted sample into a 0.2 mL thin-walled nuclease-free reaction tube and placing in a thermal cycler at 70 °C for 2 min. It is key to ice tubes immediately after the first denature step for optimal RNA quality. No DNase treatment is needed.
- While tubes cool prepare a 400 µL of 10x globin reduction oligo mix: 100 µL each of two HBA oligos (5'-GATCTCCGAGGCTCCAGCTTAACGGT-3', and 5'-TCAACGATCAGGAGGTCAGGGTGCAA-3') at 30 µM, two HBB oligos (5'- AGGGGAACTTAGTGGTACTTGTGGGC-3', and 5'- GGTTCAGAGGAAAAAGGGCTCCTCCT-3') at 120 µM per reaction, yielding a final concentration of 7.5 µM HBA oligos and 30 µM HBB oligos. Prepare 10x oligo hybridization buffer: 100 mM Tris-HCL, pH 7.6; 200 mM KCl.
- Prepare the hybridization mix: 6 µg of RNA sample (maximum 7 µL volume), 2 µL of the 400 µL of 10x globin reduction oligo mix (final concentration 2X), 1 µL of 10x oligo hybridization buffer (final concentration 1x). Add nuclease-free water to a final volume of 10 µL.
- Set thermocycler at 70 °C for 5 min. Cool immediately to 4 °C and proceed to RNase H digestion.
- RNase H digestion
- Dilute 10x RNase H (10 U/ µL) to 1x RNase H with 1x RNase H buffer.
NOTE: The RNase buffer comes at 10x and will need to be diluted with 1x RNase H buffer to 1x prior to use. - Prepare RNase H reaction mix by combining: 2 µL of 10x RNase buffer, 1 µL of RNase inhibitor in 2 µL of 1x RNase H, and 5 µL of nuclease-free water to a total volume 10 µL.
- Mix thoroughly the Globin Reduction hybridization samples with 10 µL of the RNase H reaction mix and digest at 37 °C for 10 min. Cool to 4 °C.
- Stop digestion by addition of 1.0 µL of 0.5 M EDTA to each sample and proceed immediately to the cleanup step.
- Dilute 10x RNase H (10 U/ µL) to 1x RNase H with 1x RNase H buffer.
- RNase H-Treated Total RNA Cleanup.
- Purify the RNase H treated RNA using a silica-membrane-based elution purification cleanup kit according to manufacturer's instructions. Premix buffers: For the mild washing buffer, add 44 mL of 100% ethanol.
- Do not transfer the sample to a new tube. Add 80 µL of RNase-free water and 350 µL of lysis buffer. Add 250 µL of 100% ethanol to the diluted RNA and mix well by pipetting.
NOTE: Do not centrifuge. Proceed immediately to step 5.3.3. - Now transfer the 700 µL sample to an elution filter cartridge placed in a 2 mL collection tube to collect the flow through. Centrifuge for 15 s at ≥ 8,000 x g. Discard the flow-through.
- Repeat this by placing the elution filter cartridge into a new 2 mL collection tube. Add 500 µL of the mild washing buffer to the filter cartridge and centrifuge for 15 s at ≥ 8,000 x g in order to wash the filter cartridge membrane. Discard the flow-through. Reuse the collection tube in step 5.3.5.
- Now using the same sample tube, add another 500 µL of 80% ethanol to the filter cartridge. This time centrifuge the tube for 2 min at ≥ 8,000 x g. Collect the elution spin column for the next step and discard both the flow-through and collection tube.
- Put the elution filter cartridge from the last step into a new 2 mL collection tube. Leave the lid open on filter cartridge, and centrifuge at full speed for 5 min to dry the spin column membrane and prevent ethanol carry over. Discard the flow-through and collection tube.
NOTE: To avoid damage angle the lids to point in a direction opposite to that of the rotor. - Take the dried filter cartridge and place into a new 1.5 mL collection tube. Add 14 µL of RNase-free water to filter cartridge membrane making sure to add the RNase-free water directly to the center. Centrifuge for 60 s at full speed to elute the RNA.
- Assess quality of globin-reduced RNA samples (step 6). Proceed to mRNA sample preparation (step 7) and small RNA library preparation (step 8)16,17.
NOTE: Globin-reduced RNA samples can now be stored at -20 °C, however storage at -80 °C is recommended for long-term preservation.
6. Assessment of RNA
- Quantify RNA concentration using a spectrophotometer. Examine the wavelength ratio of 260 and 280 nm. Ratios of ~2 or greater are considered pure for RNA and lower values indicate some contamination. The instrument uses this ratio to determine RNA concentration as ng/µL.
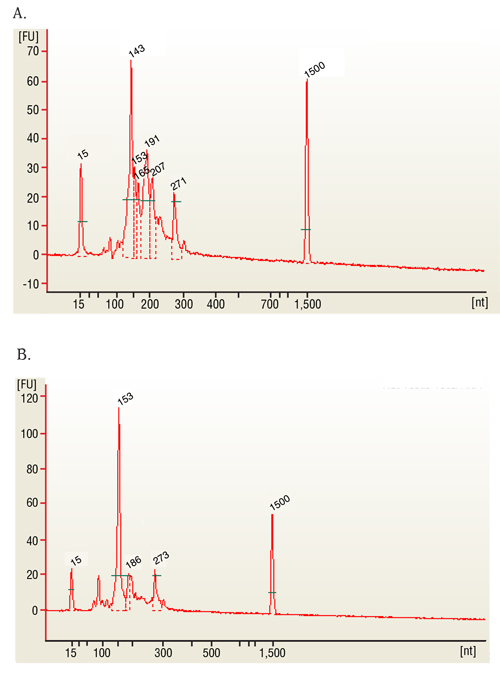
- Assess RNA quality by using 1 µL (100 ng) of sample on an appropriate chip. Final product should be a RIN of ~2 or higher and a peak at ~280 nt for mRNA single read libraries; small RNA libraries peaks at 143 correspond to miRNAs.
7. Stranded Total RNA Sample Preparation for the mRNA and long ncRNA libraries.16
- NOTE: Bring elution buffer and rRNA removal beads to room temperature. Pre-label 0.2ml thin-walled PCR tubes (plates may also be used). Protocol steps based on manufacturer's instructions16.
- Start with 4 µL of globin-reduced RNA. Add 6 µL of nuclease-free water, 5 µL of rRNA binding buffer, and 5 µL of rRNA removal mix per tube (1st tube). Pipette to mix and re-cap. Place in thermocycler with 100 °C heated-lid for 5 min at 68° C. Remove and leave at room temperature for 60 s.
- Resuspend rRNA removal beads by vortex; add 35 µL of beads to new (2nd) PCR tube and pipette sample from the 1st tubes onto beads in the 2nd tubes . Incubate 2nd PCR tube at room temperature for 3 min . Place on the magnetic stand for 7 min.
NOTE: Mix each sample thoroughly by pipetting for optimal rRNA depletion. Raising/Lowering tube in the stand may help expedite separation process. - Transfer the supernatant from the 2nd PCR tube to matching 3rd PCR tube and place on to the magnetic stand for a minimum of 60 s. Repeat the transfer into a new PCR tube only if the beads are not moved to the tube sides.
- Mix Sample Purification beads by vortex; add 99 µL to each 3rd PCR tube. Allow to sit at room temperature for 15 min. Place on magnetic stand for 5 min. Ensure beads moves to sides. Pipette to discard the supernatant.
- Keep 3rd tube set on magnetic stand. Add 200 µL of 70% ethanol whilst being careful not to jostle the beads. Allow to sit for at least 30 s and pipette to discard the supernatant. Repeat step.
- Allow sample to dry at room temperature for 15 min on the magnetic stand. Centrifuge the kit elution buffer for 5 s at 600 x g. Remove tube and add 11 µL of elution buffer mixing thoroughly. Incubate for 2 min on the bench then at least 5 min on the magnetic stand at room temperature.
- Transfer 8.5 µL of supernatant from the 3rd to a new (4th) PCR tube. Add 8.5 µL of Elute/Prime/Fragment High mix from the kit. Mix well. Cap and place in a thermocycler with pre-heated lid for 8 min at 94 °C, hold at 4 °C. Remove and centrifuge briefly.
- Synthesize First Strand cDNA
- Allow first strand synthesis mix from kit to come to room temperature and centrifuge at 600 × g for 5 s. Transfer 50 µL of reverse transcriptase into the first strand synthesis mix. Reverse transcriptase can be added to first strand synthesis at a ratio of 1:9.
- Pipette 8 µL of the combined reverse transcriptase/first strand synthesis mix into the 4th tube with sample. Cap and centrifuge 600 x g for 5 s. Place into thermocycler with pre-heated lid set at 100 °C. Run for: 10 min at 25 °C, 15 min at 42 °C, 15 min at 70 °C, then rest at 4 °C. Final volume is ~25 µL per well. Move immediately to next step.
- Synthesize Second Strand cDNA
- Bring End Repair Reagent (ERR) and Second Strand Mix (SSM) to room temperature, and centrifuge at 600 x g for 5 s. Mix ERR with resuspension buffer at 1:50 dilution. Uncap 4th tube and add 5 µL of diluted ERR and 20 µL of SSM to each; Pipette to mix well. Final volume ~50 µL ds cDNA.
- Cap and incubate in thermocycler for 1 hour at 16 °C. When the cycle is complete, remove caps and allow to come to room temperature on bench top.
- ds cDNA clean-up step
- Begin by mixing the Solid Phase Reversible Immobilization (SPRI) paramagnetic beads by vortex. Transfer 90 µL of the beads to the sample in the 4thtube (ds cDNA) and mix. Final volume is 140 µL. Allow tubes to sit at room temperature for 15 min. incubate on magnetic stand for ~5 min . Remove ~135 µL of supernatant from each well. There should 5 µL left in each well.
- Leave the tube on the magnetic stand and wash by adding 200 µL of 80% ethanol. Allow to sit for 30 s then remove and discard supernatant. Repeat wash step. Leave tubes on the magnetic stand for 15 min to allow to dry.
- Centrifuge the resuspension buffer at 600 x g for 5 s after coming to room temperature. Remove tubes from stand. Transfer 17.5 µL of resuspension buffer to the tube and mix. Let tubes incubate on the bench top for 2 min then move to magnetic stand for 5 min.
- Pipette 15 µL of the supernatant containing the ds cDNA samples to new set of (5th) 0.2 mL thin-walled tubes. Move to next step promptly or seal and store at -15 °C to -25 °C for no more than 7 days.
- Adenylate 3' Ends.
- Pipette 2.5 µL of room temperature resuspension buffer into the sample tube then add 12.5 µL of thawed A-Tailing mix. Mix completely by pipetting.
NOTE: No A-tailing control used. - Cap and incubate in a thermocycler with 100 °C pre-heated lid. Run at 37 °C for 30 min, then at 70 °C for 5 min with heated lid. Allow the thermocycler to rest at 4 °C.
- Pipette 2.5 µL of room temperature resuspension buffer into the sample tube then add 12.5 µL of thawed A-Tailing mix. Mix completely by pipetting.
- Ligate index adaptors
- Bring the RNA Adaptor tubes and the Stop Ligation Buffer mix to room temperature. For each, centrifuge at 600 x g for 5 s. Leave ligation mix in freezer until ready for use. Sample pooling arrangement for indexing should be known.
- Add 2.5 µL of resuspension buffer and 2.5 µL of ligation mix to each sample tube. Now pipette in 2.5 µL of the proper RNA Adaptor into each sample tube. Adaptor selection should be performed by manufacturer's instructions for the chosen kit.
- Recap and mix by centrifugation for 1 min at 280 x g. incubate in a thermal cycler for 10 min at 30 °C. Add 5 µL of Stop Ligation Buffer to sample to halt the reaction and mix well.
- Begin clean-up by mixing SPRI paramagnetic beads by vortex for 60 s. Transfer 42 µL of beads to each tube. Mix thoroughlythen incubate for 15 min on bench top.
- Move tubes to magnetic stand and leave until liquid turns clear (~5 min). Once clear remove and discard 79.5 µL of supernatant. Leave tubes on the magnetic stand and wash by adding 200 µL of 80% ethanol. Allow to sit for at least 30 s then remove and discard supernatant. Repeat wash step. Leave on magnetic stand for additional ~15 min to allow for drying. Do not disrupt beads during wash steps.
- Add 52.5 µL of resuspension buffer to the sample tube and mix until the beads are completely re-suspended. Incubate for 2 min on bench top. Move sample tube back to magnetic stand until liquid is clear (~5 min).
- Leave on stand and gently pipette 50 µL of supernatant to new (6th) tubes. Add 50 µL of vortexed SPRI beads. Allow to incubate on bench top for 15 min.
- Move to magnetic stand and let plate stay until liquid turns clear (~5 min). Discard 95 µL of supernatant leaving 5 µL in each tube.
- Leave tubes on magnetic stand and wash by adding 200 µL of 80% ethanol. Allow tube to sit for 30 s then remove and discard supernatant. Repeat wash. Let tubes dry on magnetic stand for 15 min.
- Add in 22.5 µL of resuspension buffer and mix until beads are suspended. Incubate on bench for 2 min then move to magnetic stand until liquid turns clear (~5 min). Pipette 20 µL of sample into new PCR tubes (7th).
- Bring needed kit components to room temperature. Transfer 5 µL of PCR primer mix and 25 µL of PCR master mix from kit to sample (7th PCR tube) containing indexed samples. Mix sample and cap tube.
- Place tubes into thermal cycler with pre-heated lid. Run program: 98 °C for 30 s, then 15 cycles of 98 °C for 10 s, 60 °C for 30 s, 72 °C for 30 s, 72 °C for 5 min. Hold at 4 °C.
- Clean up samples by adding 50 µL of sufficiently mixed SPRI beads to sample tube and pipette to combine. Allow the reaction to incubate on the bench top for 15 min.
- Move the tube to a magnetic stand and incubate until liquid turns clear (~5 min). Discard 95 µL of supernatant leaving 5 µL in each tube.
- Leave tube on magnetic stand and wash by adding 200 µL of 80% ethanol. Allow to sit for at least 30 s and then remove and discard the supernatant. Repeat this wash step. Leave on magnetic stand for 15 min to allow to dry.
- Add in 32.5 µL of resuspension buffer and mix until beads are completely re-suspended. Incubate on bench for 2 min then move to magnetic stand until liquid turns clear (~5 min). Pipette 30 µL of sample from each tube into a new PCR tube.
- Assess DNA quantity and quality by repeating steps 6.1 and 6.2 with the appropriate chip. Move on to pooling step (step 9).
8. Small RNA Library Preparation for the sncRNAs.17
NOTE: Protocol steps based on manufacturer's instructions17.
- Start small RNA library prep with ~220 ng - ~1.1 µg/ µL of globin-reduced RNA samples (4 µL).
- Adaptor ligation
- Ligate the 3'-adaptor by mixing thoroughly 1 µL of adaptor, 2 µL of nuclease-free water, and 4 µL of globin-reduced RNA sample. Incubate at 70 °C for 2 min and then immediately place on ice.
- Add 10 µL of 2x ligation buffer, and 3 µL of ligation enzyme mix, mix and incubate 16 °C for 18 h.
NOTE: Increase sample incubation time to 18 hours at a lower temperature of 16 °C is recommended for studies interested in methylated RNA species, such as piwi RNAs. The longer incubation time allows for greater ligation efficiency due to the class of modifications during the 3' adaptor ligation phase. - Add 1 µL of reverse transcription primer in 4.5 µL of nuclease-free water to the ligation mixture to prevent adaptor-dimer formation of excess 3' adaptor.
- Incubate in a preheated thermal cycler programmed for 5 min at 75 °C, 15 min at 37 °C, 15 min at 25 °C, and hold at 4°C.
- Denature 1 µL of pre-diluted 5'-adaptor per sample in a thermal cycler at 70 °C for 2 min and then immediately place on ice.
- Add 1 µL of denatured 5'-adaptor, 1 µL of 10x 5' ligation buffer and 2.5 µL of 5' ligation enzyme. Mix and incubate 25 °C for 1 h.
- cDNA synthesis and amplification
- Mix thoroughly by pipetting 30 µL of 5′/3′-adapter-ligated RNA, 8 µL of first-strand buffer, 1 µL of RNase inhibitor and 1 µL of reverse transcriptase. Incubate the mixture at 50 °C for 60 min. Proceed immediately to PCR amplification or heat inactivate the reaction at 70 °C for 15 min and store at -20 °C.
- PCR amplify the 40 µL of Reverse Transcriptase reaction by adding 50 µL of PCR master mix, 2.5 µL of RNA PCR primer, add 2.5 µL of designated RNA PCR Primer Index to sample, and nuclease-free water to a total volume of 100 µL. Mix by pipetting. Run PCR in the thermal cycler using the following cycling conditions: initial denaturation at 94 °C for 30 s; 11 cycles of 94 °C for 15 s, annealing at 62 °C for 30 s, and extension at 70 °C for 15 s; followed by a final extension at 70 °C for 5 min; samples can be held at 4 °C in the cycler.
- NOTE: Indexes are added for purpose of sample pooling.
- Sample clean-up
- From the DNA cleanup kit add 500 µL of binding buffer (5 M Gu-HCl, 30% isopropanol) to the 100 µL of PCR amplified sample to enable efficient binding to the spin-column membrane.
NOTE: If the binding buffer has pH indicator included check that the color of the mixture is yellow. - Pipette 600 µL mixture of sample and binding buffer into a filter cartridge inside a 2 mL collection tube, spin for 30-60 s at 17,900 x g in a tabletop centrifuge. Discard flow-through.
- Wash the filter cartridge into the same collection tube with 0.75 mL of kit supplied wash buffer (10 mM Tris-HCl pH 7.5, 80% ethanol) by spinning for 30-60 s at 17,900 x g in a tabletop centrifuge and discarding the flow -through. Spin the filter cartridge dry for an additional 60 s.
- Place the filter cartridge in a clean 1.5 mL collection tube. Add 30 µL of elution buffer (10 mM Tris-HCl pH 8.5), let the column stand for 60 s, and then spin for 60 s at 17,900 x g in a tabletop centrifuge.
- From the DNA cleanup kit add 500 µL of binding buffer (5 M Gu-HCl, 30% isopropanol) to the 100 µL of PCR amplified sample to enable efficient binding to the spin-column membrane.
- Assess sample quality by repeating steps 6.1 and 6.2 with the appropriate DNA chip. Move to pooling step (step 9).
9. Sample pooling for sequencing
- Pool barcoded and QC'ed samples by setting up and labeling new tubes (or a 96 well PCR plate) to contain either the mRNA or ncRNA samples. Transfer 13 µL of each barcoded 10nM library to corresponding tubes (or wells in the new plate) per manufacturer's instructions16,17. Submit pooled samples for sequencing, be certain to choose similar read lengths if samples are to be run on the same chip.
Results
The representative samples in our study are the globin and ribo-depleted whole blood samples. The representative outcome of the protocol consists of a globin depleted library sample with an RNA integrity number (RIN) above 7 (Figure 1a) and 260/280 nm concentration ratios at or above 2 (Figure 1b and 1c). Validation of the sample outcome was performed using spectrophotometer to give the final con...
Discussion
The first critical step in the protocol that made it optimized included the added globin depletion steps, which made it possible to get quality reads from whole blood samples. One of the largest limitations on using whole blood in sequencing studies are the high numbers of reads in the sample that will map to globin molecules and reduce the reads that could map to other molecules of interest18. Therefore, in optimizing the protocol for our sample type, we needed to incorporate a globin depletion s...
Disclosures
The authors have nothing to disclose.
Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Acknowledgements
This work was mainly supported by the by USDA NIFA AFRI 2013-67015-21236, and in part by USDA NIFA AFRI 2015-67015-23216. This study was supported in part by an appointment to the Agricultural Research Service Research Participation Program administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the US Department of Energy (DOE) and the US Department of Agriculture. ORISE is managed by Oak Ridge Associated Universities under DOE contract no. DE-AC05-06OR2310.
We would like to thank Dr. Kay Faaberg for the HP-PRRSV infectious clones, Dr. Susan Brockmeier for her help with animals involved in the experiment, and Sue Ohlendorf for secretarial assistance in preparation of the manuscript.
Materials
| Name | Company | Catalog Number | Comments |
| PAXgene Tubes | PreAnalytix | 762165 | |
| Molecular Biology Grade Water | ThermoFisher | 10977-015 | |
| mirVana miRNA Isolation Kit | ThermoFisher | AM1560 | |
| Rneasy MinElute Clean Up Kit | QIAGEN | 74204 | |
| 100% Ethanol | Decon Labs, Inc. | 2716 | |
| 0.2 mL thin-walled tubes | ThermoFisher | 98010540 | |
| 1.5 mL RNase/DNase - free tubes | Any supplier | ||
| Veriti 96-well Thermocycler | ThermoFisher | 4375786R | |
| Globin Reduction Oligo (α 1) | Any supplier | Sequence GAT CTC CGA GGC TCC AGC TTA ACG GT | |
| Globin Reduction Oligo (α 2) | Any supplier | Sequence TCA ACG ATC AGG AGG TCA GGG TGC AA | |
| Globin Reduction Oligo (β 1) | Any supplier | Sequence AGG GGA ACT TAG TGG TAC TTG TGG GT | |
| Globin Reduction Oligo (β 2) | Any supplier | Sequence GGT TCA GAG GAA AAA GGG CTC CTC CT | |
| 10X Oligo Hybridization Buffer | |||
| -Tris-HCl, pH 7.6 | Fisher Scientific | BP1757-100 | |
| -KCl | Millipore Sigma | 60142-100ML-F | |
| 10X RNase H Buffer | |||
| -Tris-HCl, pH 7.6 | Fisher Scientific | BP1757-100 | |
| -DTT | ThermoFisher | Y00147 | |
| -MgCl2 | Promega | A351B | |
| -Molecular Biology Grade Water | ThermoFisher | 10977-015 | |
| RNase H | ThermoFisher | AM2292 | |
| SUPERase-IN | ThermoFisher | AM2694 | Rnase inhibitor |
| EDTA | Millipore Sigma | E7889 | |
| Microcentrifuge | Any supplier | ||
| 2100 Electrophoresis BioAnalyzer Instrument | Agilent Technologies | G2938C | |
| Agilent RNA 6000 Nano Kit | Agilent Technologies | 5067-1511 | |
| Agilent High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 | |
| TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero | Illumina | RS-122-2201 | mRNA kit; Human/Mouse/Rat Set A (48 samples, 12 indexes) |
| TruSeq Stranded Total RNA Sample Preparation Guide | Illumina | Available on-line | |
| RNAClean XP Beads | BeckmanCoulter | A63987 | |
| AMPure XP Beads | BeckmanCoulter | A63880 | |
| MicroAmp Optical 8-tube Strip | ThermoFisher | N8010580 | 0.2 ml thin-walled tubes |
| MicroAmp Optical 8-tube Strip Cap | ThermoFisher | N801-0535 | |
| RNase/DNase - free reagent reservoirs | Any supplier | ||
| SuperScript II Reverse Transcriptase | ThermoFisher | 18064-014 | |
| MicroAmp Optical 96 well plates | ThermoFisher | N8010560 | These were used in place of .3mL plates as needed |
| MicroAmp Optical adhesive film | ThermoFisher | 4311971 | |
| NEBNext Multiplex Small RNA Library Prep Set for Illumina® (Set 1) | New England Biolabs | E73005 | small RNA kit |
| NEBNext Multiplex Small RNA Library Prep Set for Illumina® (Set 2) | New England Biolabs | E75805 | small RNA kit |
| QIAQuick PCR Purification Kit | QIAGEN | 28104 | |
| 96S Super Magnet Plate | ALPAQUA | A001322 |
References
- Finotello, F., Di Camillo, B. Measuring differential gene expression with RNA-seq: challenges and strategies for data analysis. Briefings in Functional Genomics. 14 (2), 130-142 (2015).
- Coble, D. J., et al. RNA-seq analysis of broiler liver transcriptome reveals novel responses to high ambient temperature. BMC Genomics. 15, 1084 (2014).
- Koltes, J. E., et al. Identification of a putative quantitative trait nucleotide in guanylate binding protein 5 for host response to PRRS virus infection. BMC Genomics. 16, 412 (2015).
- Miller, L. C., et al. Comparative analysis of signature genes in PRRSV-infected porcine monocyte-derived cells to different stimuli. PLoS One. 12 (7), 0181256 (2017).
- Head, S. R., et al. Library construction for next-generation sequencing: overviews and challenges. Biotechniques. 56 (2), 61-64 (2014).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10 (1), 57-63 (2009).
- Dominic, O. N., Heike, G., Martin, S. Ribosomal RNA Depletion for Efficient Use of RNA-Seq Capacity. Current Protocols in Molecular Biology. 103 (1), 11-14 (2013).
- Fleming, D. S., Miller, L. C. Identification of small non-coding RNA classes expressed in swine whole blood during HP-PRRSV infection. Virology. 517, 56-61 (2018).
- Fleming, D. S., Miller, L. C. Small non-coding RNA expression status in animals faced with highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV). Proceedings of the World Congress on Genetics Applied to Livestock Production. (11), (2018).
- Bivens Nathan, J., Zhou, M. RNA-Seq Library Construction Methods for Transcriptome Analysis. Current Protocols in Plant Biology. 1 (1), 197-215 (2016).
- Cui, P., et al. A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics. 96 (5), 259-265 (2010).
- Guo, Y., et al. RNAseq by Total RNA Library Identifies Additional RNAs Compared to Poly(A) RNA Library. BioMed Research International. 2015, 9 (2015).
- Choi, I., et al. Increasing gene discovery and coverage using RNA-seq of globin RNA reduced porcine blood samples. BMC Genomics. 15, 954 (2014).
- . TruSeq® Stranded Total RNA Sample Preparation Guide. Illumina. , (2018).
- . New England Biolabs Inc. Protocol for use with NEBNext Multiplex Small RNA Library Prep Kit for Illumina (Index Primers 1-48) (E7560) Available from: https://www.neb.com/~/media/Catalog/All-Protocols/758F75A29CDE4D03954E1EF75E78EA3D/Content/manualE7560_Figure_1.jpg (2018)
- Shin, H., et al. Variation in RNA-Seq transcriptome profiles of peripheral whole blood from healthy individuals with and without globin depletion. PLoS One. 9 (3), 91041 (2014).
- Costa, V., Angelini, C., De Feis, I., Ciccodicola, A. Uncovering the complexity of transcriptomes with RNA-Seq. Journal of Biomedicine and Biotechnology. 2010, 853916 (2010).
- Martens-Uzunova, E. S., Olvedy, M., Jenster, G. Beyond microRNA--novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer Letters. 340 (2), 201-211 (2013).
{kind=link}
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ISSN 2689-3649
Copyright © 2025 MyJoVE Corporation. All rights reserved
We use cookies to enhance your experience on our website.
By continuing to use our website or clicking “Continue”, you are agreeing to accept our cookies.