È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Quantificazione simultanea di T-Cell Receptor Excision Circles (TREC) e Circles K-Eliminazione ricombinazione Excision (KRECs) per Real-time PCR
In questo articolo
Riepilogo
Here, we describe a method for simultaneous quantification of T-cell receptor excision circles (TRECs) and K-deleting recombination excision circles (KRECs). The TREC/KREC assay can be used as marker of thymic and bone marrow output.
Abstract
T-cell receptor excision circles (TRECs) and K-deleting recombination excision circles (KRECs) are circularized DNA elements formed during recombination process that creates T- and B-cell receptors. Because TRECs and KRECs are unable to replicate, they are diluted after each cell division, and therefore persist in the cell. Their quantity in peripheral blood can be considered as an estimation of thymic and bone marrow output. By combining well established and commonly used TREC assay with a modified version of KREC assay, we have developed a duplex quantitative real-time PCR that allows quantification of both newly-produced T and B lymphocytes in a single assay. The number of TRECs and KRECs are obtained using a standard curve prepared by serially diluting TREC and KREC signal joints cloned in a bacterial plasmid, together with a fragment of T-cell receptor alpha constant gene that serves as reference gene. Results are reported as number of TRECs and KRECs/106 cells or per ml of blood. The quantification of these DNA fragments have been proven useful for monitoring immune reconstitution following bone marrow transplantation in both children and adults, for improved characterization of immune deficiencies, or for better understanding of certain immunomodulating drug activity.
Introduzione
Cerchi escissione recettore T-cellulare (TREC) e cerchi ricombinazione di escissione-K (eliminazione KRECs) sono piccoli elementi di DNA della circolare che vengono asportate in una proporzione di T e cellule B, rispettivamente, durante un processo di ricombinazione DNA genomico, portando alla formazione di un repertorio altamente diversificato di T e dei recettori delle cellule B. Essi non hanno alcuna funzione, ma perché sono stabili e non possono essere replicate, vengono diluiti dopo ogni divisione cellulare, persistendo quindi solo in una delle due cellule figlie. Pertanto, i livelli nel sangue periferico può essere assunta come una stima della produzione del midollo osseo e timo.
Mentre il test TREC è stato ampiamente utilizzato nel corso degli ultimi 15 anni per valutare l'entità della produzione del timo, 1 il test KREC, che è stato sviluppato inizialmente per misurare la proliferazione delle cellule B e il suo contributo alla omeostasi delle cellule B in salute e malattia, 2 è stato solo di recente proposto come marker di osso mfreccia in uscita. 3,4 Qui, descriviamo il metodo che abbiamo sviluppato per la quantificazione simultanea di entrambi TREC e KRECs. 4
Con questo metodo combinato, la variabilità associata quantificazione DNA da PCR in tempo reale è eliminata mediante l'uso di una curva standard unico ottenuta diluendo un plasmide triplo inserto contenente frammenti di TREC, KRECs e T-costante di cella recettore alfa (TCRAC) gene in un rapporto 1: 1: 1. Questo permette una valutazione più accurata del numero di copie TREC e KREC. Inoltre, la quantificazione simultanea dei due bersagli nella stessa reazione permette la riduzione dei costi dei reagenti.
La proposta di test TREC / KREC può essere utile per misurare il grado di T e cellule B neo-produzione in bambini o adulti con grave immunodeficienza combinata (SCID), 4 immunodeficienza variabile comune, 5 malattie autoimmuni, 6-8 e l'infezione da HIV . 9 Inoltre, può essere utilizzato permonitorare il recupero immunitario dopo il trapianto di cellule staminali emopoietiche, 10 enzimatica sostitutiva, 11 e antivirale 9 o immunomodulanti terapie. 6-8, infine, perché i pazienti SCID sono rilevati sulla base del test TREC nonostante i difetti genetici sottostanti, e agammaglobulinemia pazienti possono essere identificati usando KREC quantificazione , il dosaggio TREC / KREC può essere utilizzato anche per rilevare immunodeficienze in programmi di screening neonatale. 12 In questo caso, la prova deve essere eseguita su DNA estratto da piccole macchie di sangue cancellato ed essiccato su carta da filtro, deve essere altamente sensibile e specifico per le malattie in questione, così come altamente riproducibile e conveniente.
L'introduzione di KREC quantificazione nel test dovrebbe migliorare le prestazioni di screening neonatale per la immunodeficienze, che è stato regolarmente effettuato nel alcune parti degli Stati Uniti (WI, MA, CA) dal 2008, quando Wisconsin è diventato il primo e introduzionee l'analisi del TREC nel suo programma di screening postnatale. 13
Protocollo
NOTA: Etica dichiarazione: Questo protocollo segue le linee guida della nostra istituzione, la Spedali Civili di Brescia
1. Preparazione di un "Triple-Insert" plasmidi
- Selezione e preparazione di adeguate materiale di partenza:
- Ottenere un campione contenente cellule molto che possono avere TREC e KRECs rilevabile con PCR, come il sangue periferico di un giovane soggetto sano raccolto in provette EDTA.
NOTA: In origine, si era sviluppato il metodo che utilizza timociti per TREC e cellule mononucleate da frammenti di tonsille per KRECs, ma soggetti sani di solito hanno una quantità di TREC e KRECs sufficienti per consentire la loro individuazione mediante PCR. Pertanto, sangue periferico dovrebbe essere una valida alternativa, più facile da ottenere e lavorare. Considerando che TREC, in particolare, diminuiscono con l'età negli adulti sani, sangue periferico da un giovane adulto, se non da bambini, è consigliato. - Indipendente mononuc sangue perifericocellule Lear (PBMC) di un metodo di separazione gradiente di densità standard.
- Ottenere un campione contenente cellule molto che possono avere TREC e KRECs rilevabile con PCR, come il sangue periferico di un giovane soggetto sano raccolto in provette EDTA.
- Estrazione del DNA:
- Estrarre il DNA da PBMC utilizzando un commerciale DNA Sangue Mini Kit. Seguire le istruzioni del produttore con le poche modifiche descritte nel seguito.
- Incubare i campioni a 70 ° C (invece di 56 ° C) per 10 minuti con un agitatore-incubatore a 1.400 rpm.
- Aggiungere il tampone di eluizione riscaldato a 70 ° C alla colonna, incubare per 5 min a 70 ° C ed eluire DNA per centrifugazione secondo le istruzioni del produttore. Determinare la concentrazione di DNA estratto da stima spettrofotometrica a 260/280 nm.
- Estrarre il DNA da PBMC utilizzando un commerciale DNA Sangue Mini Kit. Seguire le istruzioni del produttore con le poche modifiche descritte nel seguito.
- Preparazione di un plasmide contenente gli inserti di TREC e Krec articolazioni segnale (SJ) e gene di riferimento TCRAC:
- Amplificare il DNA estratto da PBMC (per ottenere il TREC-SJ, KREC-SJ, e frammenti TCRAC) con primer specifici per TREC, KRECs e TCRAC (vedi tabella 1).
NOTA: Al 5'-end, i primer KREC- e TCRAC specifici contengono HindIII e Spei siti di restrizione (in minuscolo nelle sequenze mostrate Tabella 1) con un numero adeguato di accompagnamento nucleotidi supplementari (indicata in corsivo); entrambe le caratteristiche non sono presenti nelle canoniche sequenze Krec e TCRAC.
- Amplificare il DNA estratto da PBMC (per ottenere il TREC-SJ, KREC-SJ, e frammenti TCRAC) con primer specifici per TREC, KRECs e TCRAC (vedi tabella 1).
| TREC | avanti | 5'-AAA GAG GGC AGC CCT CTC CAA GGC-3 ' |
| retromarcia | 5'-GGC TGA TCT TGT CTG ACA TTT GC-3 ' | |
| KRECs | avanti | 5'-CCC AAG ctt TCA GCG CCC ATT ACG TTT CT-3 ' |
| retromarcia | 5'-CCC AAG ctt GTG AGG GAC ACG CAG CC-3 ' | |
| TCRAC | avanti | 5'-tag ac G tat GAG ACC GTG ACTTGC CAG-3 ' |
| retromarcia | 5'-tag ac G CGt TGT TGT TGA AGG CGT TTG C-3 ' |
Tabella 1. Fondi utilizzati per le procedure di clonazione. Al 5'-end, in minuscolo sono mostrati nucleotidi corrispondenti ai siti di enzimi di restrizione, mentre in corsivo vengono mostrati aggiunti accompagnamento nucleotidi.
- Eseguire reazioni di PCR in un volume finale di 25 ml, consistente in 1x tampone II, 1,5 mM MgCl 2, miscela dNTP (200 pM ciascuno), primer alla concentrazione finale di 900 nM, AmpliTaq DNA Polymerase (2,5 U / 25 ml), e 100 ng di DNA.
- Utilizzare i seguenti parametri PCR: primo passo a 95 ° C per 10 minuti, seguiti da 40 cicli di 95 ° C per 30 sec; 60 ° C per 30 sec; 72 ° C per 30 sec, e infine 1 ciclo di 72 ° C per 10 min. Lunghezze di prodotto di PCR dovrebbe essere: 380 bp per TREC, 166 bp per KREC, 381 bp per TCRAC.
- Inserire il prodotto di PCR di TREC-SJ nel sito accettore TA del vettore pCR2.1-TOPO. Trasformare il DNA plasmidico in XL1-blue cellule chimicamente competenti da shock termico seguendo il protocollo fornito con le cellule. Tra le colonie che crescono a causa della resistenza ampicillina, individuare quelli che trasportano l'inserto, che appare bianco a causa della perdita di complementazione β-galattosidasi, e seminare in un piatto principale. Infine, identificare le colonie contenenti il frammento TREC mediante PCR.
- Espandere una delle colonie individuate e purificare il DNA plasmide utilizzando un kit miniprep plasmide e seguire le istruzioni del produttore. Digerire il DNA plasmide e il TCRAC prodotto amplificato con SpeI enzima di restrizione e legare il frammento TCRAC nel sito di restrizione SpeI.
- Trasformare il DNA plasmidico in cellule XL1-blue e identificare le colonie contenenti il frammento TCRAC mediante PCR. Espandere una delle colonie identificate e purificareil DNA plasmidico utilizzando il kit plasmide miniprep.
- Digerire il DNA plasmide e il prodotto KREC amplificato con HindIII e clonare il frammento KREC-SJ nel sito di restrizione HindIII. Trasformare il DNA plasmidico in cellule XL1-blue e identificare le colonie contenenti il terzo frammento da PCR.
- Verificare, mediante sequenziamento diretto, la presenza dei tre inserti e l'assenza di mutazioni. La mappa della finale triplo inserto plasmide è rappresentata in Figura 1.

Figura 1. Triple-insert plasmide mappa. Triple-insert mappa plasmide che mostra la posizione di sequenze TREC, Krec, TCRAC e siti di enzimi di restrizione. Cliccate qui per vedere una versione più grande di questa figura.
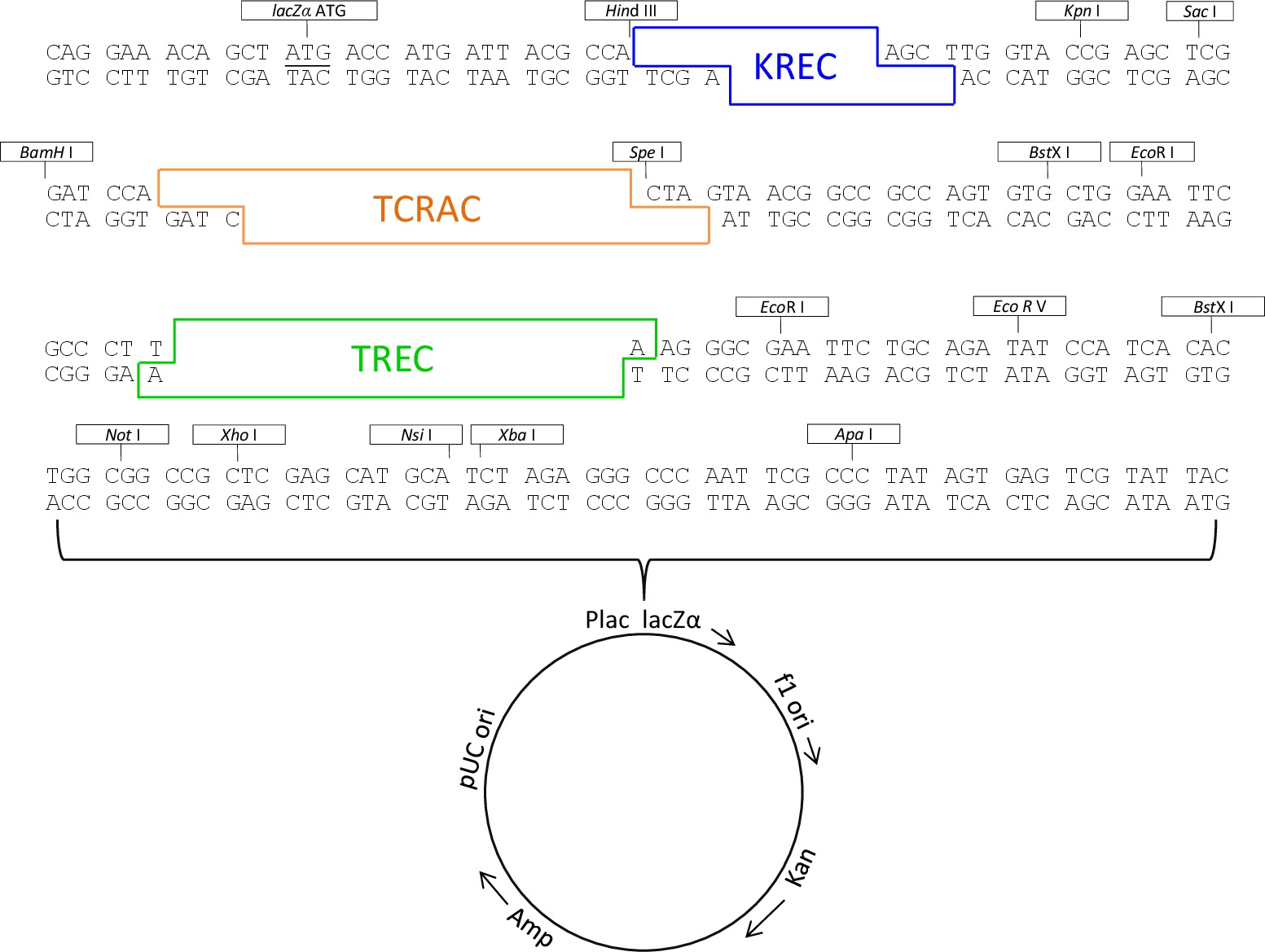{kind=link}
- Espandere una delle colonie individuate, purificare il DNA plasmidico utilizzando un kit plasmide midiprep. Determinare la concentrazione di DNA plasmidico da stima spettrofotometrica a 260/280 nm e la qualità del DNA mediante elettroforesi su gel. Conservare appropriate aliquote del plasmide a -80 ° C (ad es., 50 microlitri ciascuna).
2. Curva Preparazione standard
NOTA: Preparare tutte le diluizioni in tampone 0.1x TE in un luogo appositamente progettato per il contenimento del DNA riporto.
- Calcolare la massa dei numeri di copie plasmide di interesse (vedi tabella 2), tenendo conto che la dimensione plasmide è 4.846 bp e che la massa media per bp è 1.096 x 10 -21 g / bp, e quindi: m p = ( 4.846 bp) x (1.096 x 10 -21 g / bp) = 5,311.216 x 10 -21 g = 5,311 x 10 -18 g.
| Copia # | x 5,311 x 10 -18 g | Messa di plasmide DNA (g) |
| 1 x 10 6 | 5,311 x 10 -12 | |
| 1 x 10 5 | 5,311 x 10 -13 | |
| 1 x 10 4 | 5,311 x 10 -14 | |
| 1 x 10 3 | 5,311 x 10 -15 | |
| 1 x 10 2 | 5,311 x 10 -16 |
Tabella 2. Messa di plasmide necessario per ogni punto di diluizione curva standard.
- Calcolare le concentrazioni di DNA plasmidico dividendo masse plasmide necessari per il volume (5 mL) ad essere pipettati in ogni reazione (vedi Tabella 3).
| Messa di plasmide DNA (g) | ÷ 5 ml | Concentrazione finale di DNA plasmidico (g / ml) |
| 5,311 x 10 -12 | 1.062 x 10 -12 | |
| 5,311 x 10 -13 | 1.062 x 10 -13 | |
| 5,311 x 10 -14 | 1.062 x 10 -14 | |
| 5,311 x 10 -15 | 1.062 x 10 -15 | |
| 5,311 x 10 -16 | 1.062 x 10 -16 |
Tabella 3. Calcolo delle concentrazioni plasmide necessari per ciascun punto diluizione.
- Scongelare una aliquota del plasmide. Linearizza 2 ug del DNA plasmide con XhoI e determinare la concentrazione bStima spettrofotometrica aa 260/280 nm.
- Preparare una diluizione del DNA plasmidico linearizzato per ottenere una vantaggiosa soluzione madre di lavoro a partire da (per esempio, 100 ng / ml = 0,1 mg / mL = 1 x 10 -7 g / ml). Conservare il resto del DNA plasmidico linearizzato a -80 ° C.
- Eseguire un numero conveniente di 1:10 o 1: 100 diluizioni per portare il plasmide alla concentrazione più lavorabile di 1 x 10 -10 g / ml.
- Utilizzare la formula C 1 V 1 = C 2 V 2 per calcolare il volume di diluizione (V 2 - V 1) necessario per preparare il primo punto della curva standard della serie (1 x 10 6 copie, corrispondente a 1.062 x 10 -12 g / 5 ml; vedi tabella 4).
| Conc iniziale. (G / ml) | DNA plasmidi vol (ml) | Diluent vol (ml) | Vol finale. (Ml) | Conc finale. (G / ml) | Numero copia finale del plasmide DNA / 5μl |
| C 1 | V 1 | V -V 2 1 | V 2 | C 2 | |
| 1 x 10 -7 | 5 ml | 45 microlitri | 50 microlitri | 1 x 10 -8 | N / A |
| 1 x 10 -8 | 5 ml | 495 ml | 500 microlitri | 1 x 10 -10 | N / A |
| 1 x 10 -10 | 5 ml | 465 ml | 470 ml | 1.062 x 10 -12 | 1 x 10 6 |
| 1.062 x 10 -12 | 50 microlitri | 450 ml | 500 microlitri | 1.062 x 10 -13 | 1 x 10 5 |
| 1.062 x 10 -13 | 50 microlitri | 450 ml | 500 microlitri | 1.062 x 10 -14 | 1 x 10 4 |
| 1.062 x 10 -14 | 50 microlitri | 450 ml | 500 microlitri | 1.062 x 10 -15 | 1 x 10 3 |
| 1.062 x 10 -15 | 50 microlitri | 450 ml | 500 microlitri | 1.062 x 10- 16 | 1 x 10 2 |
Tabella 4. Calcoli diluizione.
- Procedere con una serie regolare di diluizioni 1:10 per ottenere i restanti punti della curva standard.
NOTA: Non dimenticare di vortice ogni diluizione plasmide brevemente prima di eseguire il successivo.
NOTA: La più alta diluizione del stAndard curva (10 copie / 5 ml) deve essere preparata appena prima dell'esecuzione del test, perché una piccola quantità di DNA plasmidico tale non è sufficientemente stabile per essere immagazzinato per uso futuro. - Memorizzare i punti diluizione triplo plasmid a -80 ° C in provette separate fino al momento dell'uso. Il DNA plasmidico triplo diluito è altamente stabile per almeno 6 mesi.
3. DNA Estrazione di campioni mirati
- Indipendente PBMC da sangue periferico raccolto in provette EDTA utilizzando il metodo di separazione gradiente di densità.
NOTA: In alternativa, utilizzare altro materiale di partenza adeguato (ad esempio, ordinati sottopopolazioni linfocitarie, sangue intero) come campione mirato. - Estrarre il DNA da campioni di PBMC bersaglio come riportato al punto 1.2.
4. Real-time PCR per la quantificazione di TREC e KRECs
- La preparazione del piatto e carico:
- Disporre una piastra PCR comprendente almeno due repliche per ogni campioneda analizzare: curva standard (A → F 1 → 4), controllo positivo (CTRL +; G1 → 4) e non-modello di controllo (NTC; H 1 → 4).
NOTA: Osservare uno schema tipico nella tabella 5, ma diversi formati possono essere creati. Ricordate che TREC e KRECs saranno amplificati insieme negli stessi pozzi, distinti da quelli di TCRAC.
- Disporre una piastra PCR comprendente almeno due repliche per ogni campioneda analizzare: curva standard (A → F 1 → 4), controllo positivo (CTRL +; G1 → 4) e non-modello di controllo (NTC; H 1 → 4).
| TREC + KRECs un | TCRAC b | TREC + KRECs | TCRAC | TREC + KRECs | TCRAC | |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| La | 10 6 copie | 10 6 copie | 10 6 copie | 10 6 copie | 1 | 1 | 1 | 1 | 9 | 9 | 9 | 9 |
| B | 10 5 copie | 10 5 copie | 10 5 copie | 10 5 copie | 2 | 2 | 2 | 2 | 10 | 10 | 10 | 10 |
| C | 10 4 copie | 10 4 copie | 10 4 copie | 10 4 copie | 3 | 3 | 3 | 3 | 11 | 11 | 11 | 11 |
| D | 10 3 copie | 10 3 copie | 10 3 copie | 10 3 copie | 4 | 4 | 4 | 4 | 12 | 12 | 12 | 12 |
| E | 10 2 copie | 10 2 copie | 10 2 copie | 10 2 copie | 5 | 5 | 5 | 5 | 13 | 13 | 13 | 13 |
| F | 10 copie | 10 copie | 10 copie | 10 copie | 6 | 6 | 6 | 6 | 14 | 14 | 14 | 14 |
| Sol | CTRL + | CTRL + | CTRL + | CTRL + | 7 | 7 | 7 | 7 | 15 | 15 | 15 | 15 |
| H | NTC | NTC | NTC | NTC | 8 | 8 | 8 | 8 | 16 | 16 | 16 | 16 |
Tabella 5. Esempio real-time PCR piatto.
- Calcolare il numero totale di pozzi necessari (includere 1 - 2 pozzi in più per tenere conto di eventuali errori di pipettamento) e preparare il master mix per TREC / KRECs e TCRAC in tubi separati (vedi Tabella 6).
NOTA:. I primer e sonde specifiche per TREC, KRECs e TCRAC sono menzionati nella Tabella 7 Il primer e sondare le concentrazioni finali sono 900 nm e 200 Nm rispettivamente.
- Calcolare il numero totale di pozzi necessari (includere 1 - 2 pozzi in più per tenere conto di eventuali errori di pipettamento) e preparare il master mix per TREC / KRECs e TCRAC in tubi separati (vedi Tabella 6).
| TREC / KRECs | TCRAC | ||
| H 2 O | 2 pl | H 2 O | 4.75 microlitri |
| KRECs per 20 pmol / ml | 1.125 ml | TCRAC per 20 pmol / ml | 1.125 ml |
| KRECs rev 20 pmol / ml | 1.125 ml | TCRAC rev 20 pmol / ml | 1.125 ml |
| Sonda KRECs 10 pmol / ml | 0,5 microlitri | Sonda TCRAC 10 pmol / ml | 0,5 microlitri |
| TREC-20 pmol / ml | 1.125 ml | 2x TaqMan Universal PCR Master Mix | 12,5 ml |
| TREC-rev 20 pmol / ml | 1.125 ml | ||
| Sonda TREC-10 pmol / ml | 0,5 microlitri | ||
| 2x TaqMan Universal PCR Master Mix | 12,5 ml | ||
Tabella 6. Volume di reagenti necessari per pozzetti indicati.
| TREC | avanti | 5'-CAC ATC CCT TTC AAC CAT GCT-3 ' |
| retromarcia | 5'-TGC AGG TGC CTA TGC ATC A-3 ' | |
| sonda | 5'-FAM-ACA CCT CTG GTT TTT GTA AAG GTG CCC ACT-TAMRA-3 ' | |
| KRECs | avanti | 5'-TCC CTT AGT GGC ATT ATT TGT ATC ACT-3 |
| retromarcia | 5'-AGG AGC CAG CTC TTA CCC TAG AGT-3 ' | |
| sonda | 5'-HEX-TCT GCA CGG GCA GCA GGT TGG-TAMRA-3 | |
| TCRAC | avanti | 5'-TGG CCT AAC CCT GAT CCT CTT-3 ' |
| retromarcia | 5'-GGA TTT AGA GTC TCT CAG CTG GTA CAC-3 | |
| sonda | 5'-FAM-TCC CAC AGA TAT CCA GAA CCC TGA CCC-TAMRA-3 ' |
Tabella 7. Sequenza di primer e sonde per il real-time PCR test.
- Aggiungere 20 ml di ogni mix (TREC / KRECs e TCRAC). Prima di lasciare la sala di preparazione del reagente, sigillare la piastra di reazione con una copertura adesiva ottica.
- In un acido nucleico stanza estrazione, sollevare il coperchio adesivo e aggiungere 5 ml di campione di DNA genomico (400-500 ng) ai pozzetti. Aggiungere 5 ml di DNA genomico (cioè, preparato da PBMC ottenute dal sangue del cordone ombelicale) ai pozzetti CTRL +.
NOTA: In alternativa al sangue del cordone ombelicale, utilizzare DNA dalla stessa nota, positiva campione di sangue periferico o pool di campioni, o preparare uno standard positivo "artificiale" miscelando una quantità determinata di triplo inserto plasmide con il DNA genomico da TREC- e linee cellulari KREC-negativi. - Aggiungere 5 ml di acqua ai pozzi NTC. Sigillare nuovamente la piastra di reazione con il coperchio adesivo ottico.
- Spostarsi in un luogo garantire il contenimento del plasmide DNA riporto; scongelare ogni poin diluizionet della curva standard DNA plasmidico solo prima dell'uso e preparare la diluizione più elevata (10 copie / 5 ml). Sollevare il coperchio adesivo solo da un → H 1 → 4 pozzi e aggiungere 5 ml di ogni diluizione punto di curva standard DNA plasmide. Sigillare di nuovo il coperchio adesivo.
- Controllare l'assenza di bolle sul fondo dei pozzetti, assicurando così che i reagenti sono posizionati nella parte inferiore.
- Eseguire il saggio su un sistema PCR in tempo reale. Il protocollo standard consiste prima fase a 50 ° C per 2 min, un riscaldamento iniziale a 95 ° C per 10 minuti, seguito da 45 cicli di denaturazione a 95 ° C per 15 sec, e un primer / sonda di ricottura e l'allungamento a combinata 60 ° C per 1 min.
- Salvare i dati alla fine del programma PCR.
NOTA: per evitare la contaminazione incrociata DNA, ricordate: seguire le solite buone raccomandazioni pratiche di laboratorio per PCR quantitativa per la messa in real time PCR: utilizzare aree / stanze separate per l'estrazione degli acidi nucleici,preparazione di reagenti, manipolazione plasmide, reazione di amplificazione; utilizzare set pipetta diverso; cambiare guanti / cappotti a seconda dei casi.
- Risultati e calcolo:
NOTA: Le seguenti operazioni si basano sulla nostra esperienza con il software fornito con lo strumento real-time PCR indicato nella tabella dei materiali, ma, in generale, si devono applicare ad altre piattaforme e software di analisi PCR in tempo reale. Tenete presente che, anche se si utilizza lo stesso software, di solito c'è più di un modo per eseguire i comandi necessari (ad esempio, icone, collegamenti).- Aprire il file risultato salvato nel software e fare clic sulla scheda "Risultati". Sulla parte superiore della finestra, cliccate sul menu "Analisi" e selezionare "Impostazioni di analisi". Lasciate che il software determina la soglia automaticamente selezionando "Tutte" rivelatori e "Auto Ct" (Ct ciclo = soglia. Lasciare il set software "Baseline Automatic"; per soppressore di sfondo di default.
- Fare clic sulla scheda denominata "amplificazione Plot" e, nella parte più a destra della finestra, selezionare una delle tre "rivelatori" nel menu a discesa corrispondente (ad esempio, iniziare con TREC). Appena sotto, selezionare "Manuale Ct" per impostare manualmente una soglia; andare alla parte inferiore della finestra e selezionare pozzetti corrispondenti ai punti standard diluizione curva del rivelatore scelto di mostrare i grafici di amplificazione. Per fare questo è sufficiente fare clic e trascinare il mouse sopra le corrispondenti celle della tabella.
- Per regolare la soglia manualmente, trascinare la linea di soglia rossa su e giù, e poi cliccare su "Analizza" per rianalizzare risultati. Per controllare l'impostazione della soglia influisce sui risultati, selezionare la scheda "Report", e vedere il Ct risultante dei rispettivi punti di diluizione curva standard. Quando si sposta la soglia, cercate di rispettare le seguenti raccomandazioni per ottenere un posizionamento ottimale.
- Torna alla scheda "Plot di amplificazione" e verificare che la soglia è in una regione di amplificazione esponenziale sufficientemente al di sopra del rumore di fondo, ma al di sotto della fase di plateau. Se la trama di amplificazione non è mostrata in scala logaritmica, fare clic destro sul complotto per ricordare le "Impostazioni grafico" e impostare l'impostazione post-conduzione asse Y come "log", impostare la soglia a metà strada nella parte lineare della trama, e osservare le curve di amplificazione di circa parallele tra loro.
- Fare clic sulla scheda "Report" per vedere i risultati Ct. Assicurarsi che la collocazione soglia prodotto risultati che massimizzano la precisione delle due repliche di ogni standard di punti di diluizione curva.
- Verificare che la soglia è posizionato nel punto che meglio riflette gli ordini di grandezza estremi dei punti diluizione curva standard (sensibilità ottimale). Ripetere il passaggio 4.2.2 e restanti sottofasi per i due rivelatori rimanenti (KRECs e TCRAC).
- Vai alla scheda Rapporto, trascinare il mouse di nuovo nella tabella nella parte inferiore della finestra per selezionare le celle corrispondenti ai pozzetti della curva standard per tutti i rivelatori (TREC, KRECs, TCRAC). Verificare che il Ct risultante dei tre target è molto simile agli stessi punti di diluizione (es differenza non più di 0,5 Ct). Regolare nuovamente le soglie se necessario e ricontrollare.
NOTA: Poiché il plasmide triplo inserto contiene una singola copia di ciascun target e quindi il rapporto di amplificazione dovrebbe teoricamente essere vicino a 1: 1: 1. In alcuni casi può essere necessario regolare manualmente anche la linea di base per uno o più dei rivelatori per ottenere risultati migliori. Basta ricordare la finestra "Impostazioni di analisi", selezionare il rivelatore per cui è necessario l'adeguamento, quindi impostare "Manual Baseline", ed inserire valori personalizzati per l'avvio e la Cicli finali (di solito 3 a 15). Quindi fare clic su "OK & Reanalyze" e ricontrollare i punti precedenti.
- Accettare la sessione sperimentale solo dopo aver verificato quanto segue:
- Fare clic sulla scheda "Report", andare alla parte inferiore della finestra, selezionare pozzi NTC e verificare che il corrispondente Ct è "Undet" (cioè, nessuna amplificazione è presente nei pozzi NTC).
- Selezionare la scheda "Curva Standard" e guardare a destra della finestra del grafico; selezionare rilevatori dal menu a discesa e vedere se la pendenza della curva di serie riportato compresa tra -3,55 e -3,32 (corrispondenti a PCR efficienza su 91 - 100%)
- Assicurarsi che il coefficiente di determinazione (R 2) è superiore a 0.998 (figura 2), leggendo il suo valore proprio sotto la pendenza e intercetta (nella stessa scheda "Curva Standard", dopo aver selezionato ogni rivelatore).
NOTA: Prestare attenzione ai punti di diluizione estremi perché la loro disallineamento può influenzare la pendenza più facilmente (hanno un "leve altoEffetto collera "sulla retta di regressione). In particolare, tenere presente che la diluizione plasmide massimo può degradare prima del previsto. Tuttavia, quando possibile è utile non disfarsene, perché un approccio razionale può considerare 100 come il limite di determinazione quantitativa: in altre parole, per campioni tra 10 e 100, il risultato può essere riportato come <100 o "positive , ma non quantificabili ", mentre se fuori della gamma curva (<10) può essere riportato come 0 o" non rilevabile ".
NOTA: Può essere utile per tenere un archivio dei valori Ct dei precedenti diluizioni della curva standard, al fine di calcolare un valore medio per ogni punto di diluizione da tenere come una sorta di ottimale range "di riferimento" o di controllo di qualità interno per futuri esperimenti (per esempio, calcolare media e deviazione standard per costruire carte di controllo Shewhart). Analogamente, può essere utile per tenere un archivio del passato Ct del controllo positivo.

Figura 2. Le curve standard per TREC, KRECs, e TCRAC. Appezzamenti dei punti della curva standard e la linea di log-regressione stime per TREC (A), KRECs (B), e TCRAC (C) sono costruite per verificare la conformità con l'esponenziale ideale tasso di amplificazione (slope = 3.32) che corrisponderebbe ad un'efficienza del 100%. Ct: ciclo soglia; R 2:. Coefficiente di regressione di determinazione Cliccate qui per vedere una versione più grande di questa figura.
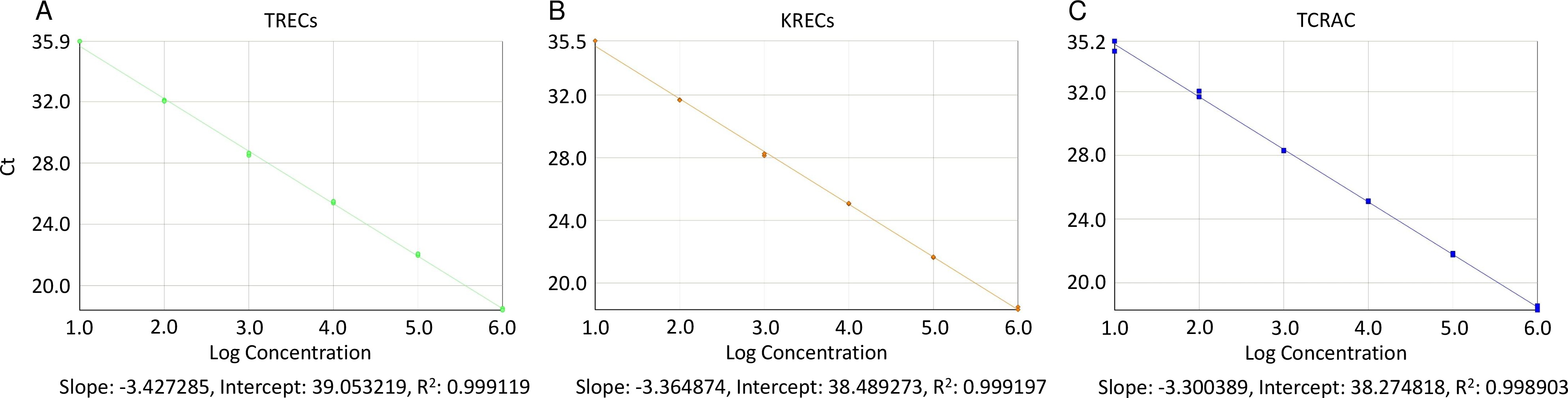{kind=link}
- Fare clic sulla scheda "Report" e verificare che il Ct del controllo positivo (che si trova nelle corrispondenti celle della tabella sotto il grafico), è coerente con i risultati delle analisi precedenti sulla same conosciuto controllo positivo.
NOTA: La scheda "Report" mostra la quantità di TREC, KRECs e TCRAC dei campioni in prova che il software ha calcolato per interpolazione dalla rispettiva curva standard, utilizzando i valori Ct ottenuti dopo la soglia e linea di base sono state correttamente impostato (un nuova analisi con una nuova soglia / basale cambierebbe questi risultati). Per vedere dove l'interpolazione avviene sulla curva standard, selezionare la scheda "Standard Curve", quindi selezionare i pozzetti desiderati, e cercare il nero "X" che appare sulla linea di regressione. Il loro valore asse y è la quantità interpolata. Se si utilizza sempre lo stesso controllo positivo, valori di riferimento adeguati e / o carte di controllo di qualità possono essere costruiti, in base ai valori precedenti determinati sul controllo positivo.
- Fare clic sulla scheda "Report" e verificare che il Ct del controllo positivo (che si trova nelle corrispondenti celle della tabella sotto il grafico), è coerente con i risultati delle analisi precedenti sulla same conosciuto controllo positivo.
- Fare clic sul menu "File", "Salva", e quindi "Export" in un file .csv per esportare le quantità di tutti i pozzetti in un tefile di xt contenente valori separati da virgola.
- Importare the.csv file in un foglio di calcolo e calcolare il numero di TREC o KRECs per 10 6 PBMC impostando la seguente formula appropriata: [(quantità di TREC o KRECs media) / (media quantità di TCRAC / 2)] x 10 6
NOTA: La quantità media di TCRAC è diviso per 2 perché in ogni cella ci sono due copie del gene TCRAC, ad esempio, uno per ciascun cromosoma.
NOTA: La consistenza del numero finale di TREC e KRECs di controllo positivo può essere considerato, ma di tenere presente che i valori non corrispondono perfettamente a causa della variabilità aggiunto dei punti di diluizione curva standard. In caso di divergenza lordo, l'esperimento potrebbe essere necessario ripetere. Ancora una volta, le carte di controllo possono essere costruiti per impostare un intervallo di riferimento appropriato. - (Opzionale, ma consigliato) Se i risultati della emocromo sono disponibili, calcolare le TREC o KRECs per ml di sangue usando la seguente formula:
TREC o KRECs a 1 x 10 6) x (+ linfociti monociti contano in 1 ml di sangue) / 10 = 6 copie / ml.
Risultati
Il dosaggio è stato effettuato in un campione rappresentativo di 87 controlli sani: 42 bambini di età compresa tra 0-17 (maschi / femmine: 25/17) e 45 adulti di età compresa tra 24-60 (maschi / femmine: 29/16). I risultati sono stati ottenuti come TREC e KRECs per 10 6 PBMC, e poi le TREC e KRECs per ml di sangue sono stati calcolati.
Il numero di TREC diminuisce con l'età a causa involuzione timica, 4 in particolare in un modo molto acuto da 0 a 3 - 4 anni. Neg...
Discussione
TREC and KREC quantification can be considered a good estimate of recent thymic and bone marrow output provided that some caveats are taken into account. Even though an absolute quantification method employing standard curve requires more reagents and more space on the real-time PCR reaction plate, it ensures highly accurate quantitative results because unknown sample quantities are interpolated from standard curves built upon known amounts of starting material. Moreover this method is better fitted to detect low amount ...
Divulgazioni
The authors have nothing to disclose.
Riconoscimenti
The authors have no acknowledgements.
Materiali
| Name | Company | Catalog Number | Comments |
| Name of Reagent/ Equipment | Company | Catalog Number | Comments/Description |
| Histopaque-1077 | Sigma Aldrich SRL | 10771-500 ML | density gradient separation method |
| QIAamp DNA Blood Mini Kit (250) | QIAGEN | 51106 | DNA extraction |
| Unmodified DNA Oligonucleotides HPSF 0.01 mmol | Eurofins MWG Operon/Carlo Erba Reagents S.r.l | Resuspend the lyophilized product to 100 picmol/µl | |
| AmpliTaq DNA Polymerase: including 10x Buffer II and 25mM MgCl2 | Applied Biosystems/Life-Technologies | N8080156 | |
| GeneAmp dNTP Blend (100 mM) | Applied Biosystems/Life-Technologies | N8080261 | |
| TOPO TA Cloning Kit for Subcloning | Invitrogen/Life-Technologies | K4500-01 | |
| XL1-Blue Subcloning Grade Competent Cells | Stratagene | 200130 | |
| PureYield Plasmid Miniprep System | Promega | A1223 | |
| SpeI 500U | New England Biolabs | R0133S | |
| HindIII-HF 10,000 U | New England Biolabs | R3104S | |
| PureYield Plasmid Midiprep System | Promega | A2492 | |
| XhoI 5,000 U | New England Biolabs | R0146S | |
| TRIS Utrapure | Sigma Aldrich SRL | T1503 | |
| EDTA | Sigma Aldrich SRL | E5134 | |
| TE buffer (1 mM TRIS and 0.1 mM EDTA) | |||
| TaqMan Universal PCR Master Mix | Applied Biosystems/Life-Technologies | 4364338 | |
| Dual labeled probes HPLC 0.01 mmol | Eurofins MWG Operon/Carlo Erba Reagents S.r.l | Resuspend the lyophilized product to 100 picmol/µl | |
| NanoDrop 2000c spectrophotometer | ThermoFisher | ||
| Applied Biosystems 2720 Thermal Cycler | Applied Biosystems/Life-Technologies | 4359659 | |
| Fast 7500 Real-Time PCR system | Applied Biosystems/Life-Technologies | ||
| SDS Sequence Detection Software 1.4 | Applied Biosystems/Life-Technologies |
Riferimenti
- Douek, D. C., et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 396 (6712), 690-695 (1998).
- Zelm, M. C., Szczepanski, T., van der Burg, M., van Dongen, J. J. M. Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. J. Exp. Med. 204 (3), 645-655 (2007).
- Fronkova, E., et al. B-cell reconstitution after allogeneic SCT impairs minimal residual disease monitoring in children with ALL. Bone Marrow Transplant. 42 (3), 187-196 (2008).
- Sottini, A., et al. Simultaneous quantification of recent thymic T-cell and bone marrow B-cell emigrants in patients with primary immunodeficiency undergone to stem cell transplantation. Clin. Immunol. 136 (2), 217-227 (2010).
- Serana, F., et al. Thymic and bone marrow output in patients with common variable immunodeficiency. J. Clin. Immunol. 31 (4), 540-549 (2011).
- Zanotti, C., et al. Opposite effects of interferon-β on new B and T cell release from production sites in sclerosis. J. Neuroimmunol. 240-241, 147-150 (2011).
- Zanotti, C., et al. Peripheral accumulation of newly produced T and B lymphocytes in natalizumab-treated multiple sclerosis patients. Clin. Immunol. 145 (1), 19-26 (2012).
- Sottini, A., et al. Pre-existing T- and B-cell defects in one progressive multifocal leukoencephalopathy patient. PLoS One. 7, e34493 (2012).
- Quiros-Roldan, E., et al. Effects of combined antiretroviral therapy on B- and T-cell release from production sites in long-term treated HIV-1+ patients. J. Transl. Med. 10, 94 (2012).
- Mensen, A., et al. Utilization of TREC and KREC quantification for the monitoring of early T- and B-cell neogenesis in adult patients after allogeneic hematopoietic stem cell transplantation. J. Transl. Med. 11, 188 (2013).
- Serana, F., et al. The different extent of B and T cell immune reconstitution after hematopoietic stem cell transplantation and enzyme replacement therapies in SCID patients with adenosine deaminase deficiency. J. Immunol. 185 (12), 7713-7722 (2010).
- Borte, S., et al. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood. 119 (11), 2552-2555 (2012).
- Baker, M. W., et al. Implementing routine testing for severe combined immunodeficiency within Wisconsin's newborn screening program. Public Health Rep. 125 (2), 88-95 (2010).
- Lorenzi, A. R., et al. Determination of thymic function directly from peripheral blood: a validated modification to an established method. J. Immunol. Meth. 339 (2), 185-194 (2008).
- De Boer, R. J., Perelson, A. S. Quantifying T lymphocyte turnover. J. Theor. Biol. 327, 45-87 (2013).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati