
Method Article
Measuring Interactions of Globular and Filamentous Proteins by Nuclear Magnetic Resonance Spectroscopy (NMR) and Microscale Thermophoresis (MST)
In This Article
Summary
Here, we present a protocol for the production and purification of proteins that are labeled with stable isotopes, and subsequent characterization of protein-protein interactions using Nuclear Magnetic Resonance (NMR) spectroscopy and MicroScale Thermophoresis (MST) experiments.
Abstract
Filamentous proteins such as vimentin provide organization within cells by providing a structural scaffold with sites that bind proteins containing plakin repeats. Here, a protocol for detecting and measuring such interactions is described using the globular plakin repeat domain of envoplakin and the helical coil of vimentin. This provides a basis for determining whether a protein binds vimentin (or similar filamentous proteins) and for measurement of the affinity of the interaction. The globular protein of interest is labeled with 15N and titrated with vimentin protein in solution. A two-dimensional NMR spectrum is acquired to detect interactions by observing changes in peak shape or chemical shifts, and to elucidate effects of solution conditions including salt levels, which influence vimentin quaternary structure. If the protein of interest binds the filamentous ligand, the binding interaction is quantified by MST using the purified proteins. The approach is a straightforward way for determining whether a protein of interest binds a filament, and for assessing how alterations, such as mutations or solution conditions, affect the interaction.
Introduction
Interactions between proteins allow the formation of molecular machines that create order within cells. The individual interactions are often weak but usually contribute to multivalent complexes that can be cooperative and dynamically regulated. Sensitive assays that provide atomic resolution and quantitative information about such complex interactions are needed to deduce mechanisms and design interventions such as drug-like molecules. NMR spectroscopy is an efficient method for obtaining such information about protein interactions, and also is used for fast screening for ligands including those that bind weakly1. The NMR methods used can be categorized into those that are protein observe or ligand observe. This manuscript uses the former approach in which a spectrum of a stable-isotope labeled protein that is comparatively small (usually under 20 kDa) is acquired and the unlabeled ligand is titrated. This allow the labeled residues involved in the interaction to be mapped in favorable cases. Once the complex forms, there are changes in the chemical environments of interacting residues that manifest themselves as changes in the chemical shift and shape of their NMR signals. The extent of such changes correlates with the degree of involvement of these groups in the interaction. Chemical shift perturbations (CSPs) can be measured by comparing a series of NMR spectra of the protein collected in the absence and presence of varying amounts of the ligand. For larger ligands or complex interactions, the change in peak shape or intensity can be measured to deduce interactions.
The most common 2D experiment used for detecting ligand interactions is the 15N-heteronuclear single quantum correlation (HSQC) experiment2. This requires that one protein be uniformly labeled with 15N, which is typically achieved by expressing them as affinity-tagged versions in E. coli bacterial cultures grown in 15N-enriched media. Binding is apparent when the HSQC spectra collected during the titration are superimposed, revealing peak changes for a subset of residues involved in the complex formation. The interaction can occur in the fast exchange regime where the free and ligand-saturated state signals collapse into one population averaged peak. Alternatively, in the case of slow exchange between the states, both signals are observed with integrals that represent their relative amounts. While NMR lineshape analysis can be used to estimate the binding affinities in some cases, methods such as MST have also proven convenient and provide cross-validation of genuine interactions.
The example provided is of two proteins found within desmosomes. They mediate junctions between cell surfaces and the cytoskeleton and mediate multivalent interactions between cell adhesion machines and intermediate filaments to maintain the integrity of skin and heart tissues and withstanding of shear forces. Diseases can result when desmosomal proteins such as desmoplakin or vimentin are compromised by mutations or autoantibodies, leading to destabilization of cell-cell junctions, and hence their interactions are of critical importance3. The structural basis of ligand binding by desmosomal proteins can be characterized by NMR spectroscopy, while the interactions can be quantified by MST. Methods herein were used to characterize the interactions between plakin repeat domains (PRDs) which often are present as tandem sets that offer basic grooves, and vimentin, an intermediate filament that interacts through an acidic surface offered by its helical bundle4. These complexes are formed at the cell membrane where they anchor for intermediate filaments of the cell cytoskeleton to desmosomes that connect to adjacent cells, thus forming a network of adhesive bonds that radiates throughout a tissue.
Protocol
1. Recombinant Protein Expression
- Expression of Envoplakin PRD (E-PRD) and Vimentin 99-249 (VimRod)
- Transform E. coli BL21(DE3) cells with the plasmid containing the desired gene. Spread the cells on agar plates containing 100 µg/mL ampicillin. Incubate the plates at 37 °C overnight.
- Pick a single colony and inoculate 20 mL of Terrific broth (TB) containing 100 µg/mL ampicillin to select for the plasmid. Grow the culture at 37 °C with shaking (180 rpm) overnight.
- Transfer the entire 20 mL culture to 1 L of TB containing 50 µg/mL ampicillin. Incubate the culture at 37 °C with shaking at 180 rpm until the OD600 = 0.6-0.8.
- Reduce the temperature to 18 °C and induce protein expression by adding isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Continue incubation at 18 °C with shaking at 160 rpm overnight to allow protein expression.
- Harvest the cells by centrifuging the culture at 8,000 x g for 15 min. Decant and discard the supernatant.
- Wash the harvested cells by resuspending the cell pellet in approximately 40 mL of phosphate buffered saline solution (PBS: 20 mM phosphate buffer, pH 7.4, 120 mM NaCl). Transfer the resuspension to a 50 mL tube. Centrifuge again at 8,000 x g for 15 min.
- Decant and discard the supernatant. Either immediately begin the purification protocol or freeze the cell pellets at -20 °C for future use.
- Expression of Isotopically Labeled Protein
- Transform E. coli BL21(DE3) cells and prepare a 20 mL starter culture as in 1.1.1-1.1.2.
- Transfer the entire 20 mL culture to 1 L of enriched TB containing an additional 4.0 g tryptone, 5.0 g NaCl, and 100 µg/mL ampicillin. Incubate the culture at 37 °C with shaking at 160 rpm until the OD600 = 1.6-1.9.
- Harvest the 1 L culture by centrifugation at 8000 x g for 15 min. Decant and discard the supernatant.
- Wash the cell pellet by gently resuspending in approximately 40 mL of PBS and transfer the resuspension to a 50 mL tube.
- Centrifuge again at 8,000 x g for 15 min. Decant and discard the supernatant.
- Resuspend the cell pellet in 20 mL of M9 minimal media (Table 1) and transfer to the remainder of the 950 mL of M9 minimal media containing 100 µg/mL ampicillin.
- Add 50 mL of filter sterilized nutrient mix (Tables 2 and 3).
- Acclimatize the culture to 18 °C for 30 min before adding IPTG to a final concentration of 1 mM.
- Incubate overnight at 18 °C with shaking at 160 rpm.
- Harvest the cells as in section 1.1.4-1.1.6.
2. Immobilized Metal Affinity Chromatography (IMAC) Purification of VimRod and E-PRD
- Purification of His6-tagged VimRod
- Resuspend the cell pellet in 5 mL/g of PBS containing a protease inhibitor cocktail lacking EDTA. Homogenize with 12 strokes in a Dounce tissue homogenizer to improve the cell lysis in the following step.
- On ice, sonicate the cell suspension at a pulse of 1 s on/1 s off, 80% amplitude for a total of 1.5 min. Repeat the sonication two additional times, swirling gently on ice between runs to prevent overheating.
- Centrifuge the sample at 75,000 x g for 45 min. Decant and filter the supernatant using a syringe filter (0.45 µm).
- Equilibrate a 5 mL IMAC column with 5 column volumes (CV) of binding buffer (20 mM HEPES, pH 7.5, 500 mM NaCl, 10 mM imidazole) at a flow rate of 1 mL/min using a fast protein liquid chromatography (FPLC) system.
- Load the filtered supernatant onto the column at a flow rate of 0.5 mL/min.
- Wash the column with 5 CV of Wash buffer (20 mM HEPES, pH 7.5, 500 mM NaCl, 50 mM imidazole) at a flow rate of 1 mL/min.
- Elute the protein with 3 CV of elution buffer (20 mM HEPES, pH 7.5, 500 mM NaCl, 350 mM imidazole) at a flow rate of 0.5 mL/min. Collect 1.5 mL fractions. If available, select the up-flow elution mode to increase the concentration of eluted protein.
- Identify the fractions from the FPLC chromatogram that contain the protein of interest by SDS-PAGE and use standard methods to measure the protein concentration5.
- Pool and concentrate the elution fractions containing the highest amounts of protein using a centrifugal ultrafiltration device (MWCO 3 kDa, 5 mL) to 2 mL. Centrifuge at 21,000 x g to remove any precipitate and pass through a 0.22 μm filter.
- Equilibrate a 120 mL size exclusion chromatography (S) column with 2 CV of S buffer (20 mM HEPES, 150 mM NaCl, pH 7.5, 0.5 mM TCEP) at a flow rate of 1 mL/min using an FPLC.
- Inject the concentrated protein from 2.1.9 onto the column and elute with 1 CV of S buffer at a flow rate of 0.5 mL/min, collecting 1 mL fractions.
- Identify the fractions containing the protein of interest as before.
- Pool the fractions containing the highest amounts protein.
- Store at 4 °C for short term use or add glycerol to 20% and store at -80 °C in small aliquots.
- Purification of E-PRD Protein with the His6 Tag Removed
- Follow the steps in 2.1.1-2.1.8 to purify His6-tagged E-PRD protein. Pool the peak fractions and determine the protein concentration.
- Add tobacco etch virus (TEV) protease (1 mg/mL) at 2 µL/mg of pooled protein. Transfer to dialysis tubing (6 kDa) and dialyze in S buffer overnight at 4 °C. This step allows cleavage of the His6 tag and removal of the imidazole that will interfere with binding to the Ni-NTA resin in the next step.
- Equilibrate 5 mL of Ni-NTA resin in a gravity column with 3 CV of S buffer. Drain excess buffer from the resin.
- Pour the cleaved E-PRD protein onto the resin and incubate for 1 hour on a rocking platform to allow the uncleaved His6-tagged E-PRD and the cleaved His6 tag to bind. The TEV protease is also His6-tagged and will to bind the resin. Collect the flow through, which contains the tag-free E-PRD. Wash the resin with 2 CV of S buffer to ensure all of the E-PRD is recovered.
- Concentrate E-PRD in the flow through to 2 mL using a centrifugal ultrafiltration device (MWCO 3 kDa, 5 mL). Centrifuge at 21 000 x g to remove any precipitate and pass through a 0.22 μm filter.
- Equilibrate a 120 mL S column with 2 CV of S buffer (20 mM HEPES, 150 mM NaCl, pH 7.5, 0.5 mM TCEP for MST or 20 mM Tris-HCl, 1 mM DTT, pH 7 for NMR) at a flow rate of 1 mL/min using an FPLC.
- Inject the concentrated E-PRD protein from 2.2.5 onto the column and elute with 1 CV of S buffer at a flow rate of 0.5 mL/min, collecting 1 mL fractions.
- Identify the fractions containing the protein of interest as before.
- Pool the fractions containing the highest amounts protein.
- Store at 4 °C for short term use or add glycerol to 20% and store at -80 °C in small aliquots.
3. NMR Methods
- NMR Sample Preparation
- Purify 15N-labeled wild-type or R1914E E-PRD protein as previously described using 20 mM Tris-HCl, 1 mM DTT, pH 7 as the S buffer for step 2.2.6. Protein stock solutions usually range from 0.3 to 1 mM with volumes of about 1 mL.
NOTE: Protein can be concentrated to >100 µM using a MWCO 3 kDa, 5 mL centrifugal ultrafiltration device to bring the concentration into a suitable range for sample preparation. - Purify a sample of unlabeled VimRod protein using 20 mM Tris-HCl, 1 mM DTT, pH 7 as the S buffer for step 2.1.10.
- In a final volume of 500 µL, add wild-type or mutant E-PRD protein to a final concentration of 100µM, deuterium oxide (D2O) to a final concentration of 10% (v/v), and DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid) to a final concentration of 20 µM. Bring the sample volume up to 500 µL using 20 mM Tris-HCl, 1 mM DTT, pH 7. A representative sample preparation is described in Table 4.
NOTE: The 0 ppm resonance of DSS is used to calibrate the 1H chemical shifts as well as for indirect referencing of the 15N chemical shifts of the protein6. D2O is used for the deuterium lock signal to keep the spectrometer operating at a constant net magnetic field. - Make up a second sample of E-PRD, D2O and DSS as in the previous step and add VimRod to a final concentration of 50 µM before bringing the volume up to 500 µL.
- Transfer the 500 µL samples to a 5 mm wide NMR tubes for the experiment.
- Purify 15N-labeled wild-type or R1914E E-PRD protein as previously described using 20 mM Tris-HCl, 1 mM DTT, pH 7 as the S buffer for step 2.2.6. Protein stock solutions usually range from 0.3 to 1 mM with volumes of about 1 mL.
- NMR Experimental Setup
- Turn on the air flow with the eject command “ej”; this will bring the sample up from the magnet. Now, place the sample within a spinner on top of the magnet by the opening and insert with the command “ij”. Wait until sample settles inside the magnet before proceeding.
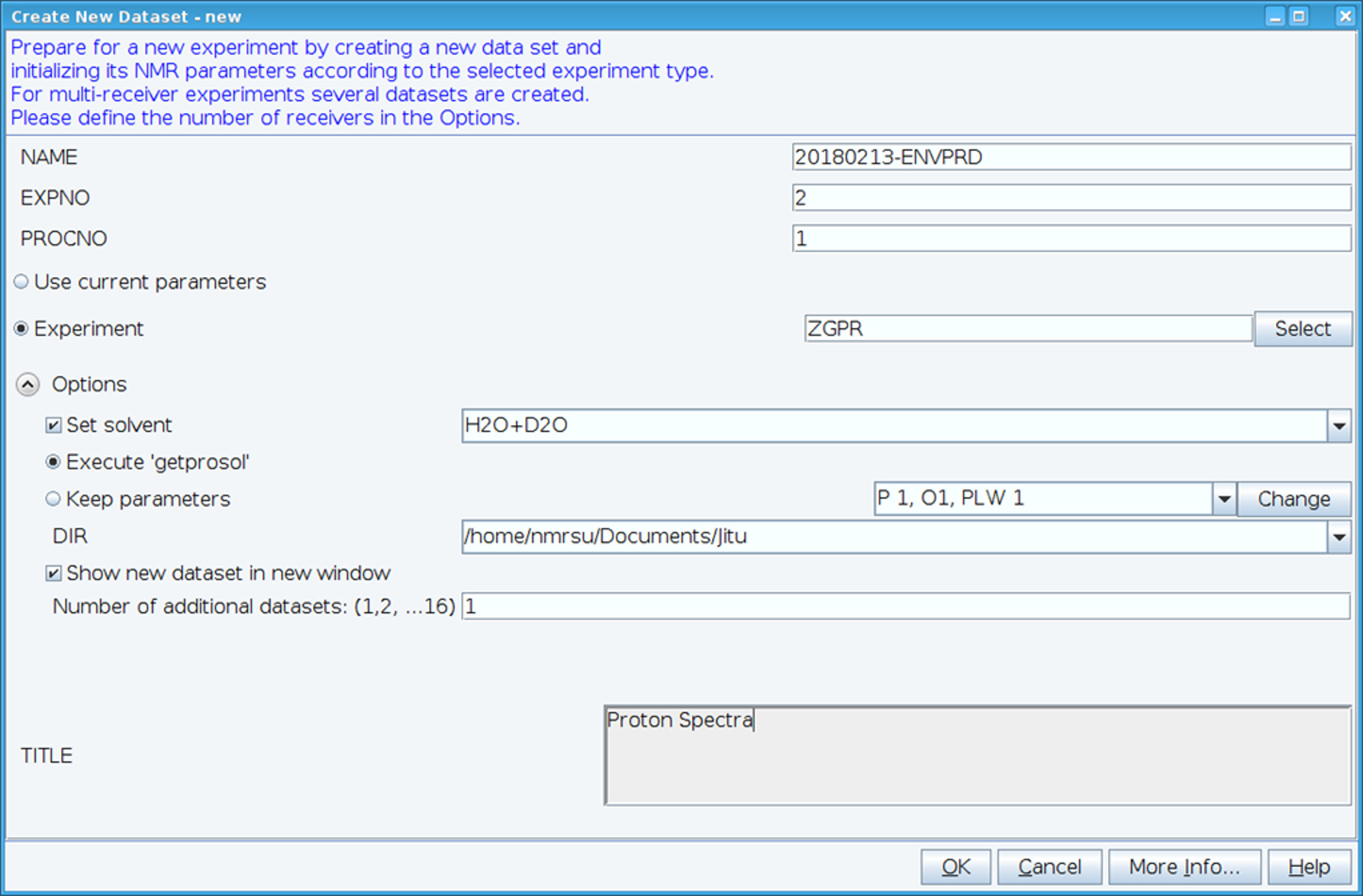
- Create a new dataset using the “edc” command and load standard 1H NMR parameters by selecting experiment “ZGPR” (Figure 1). Fill in the NAME, EXPNO (experiment number) and PROCNO (processed data folder number) fields. Select the solvent in the “Set solvent” field and click on “Execute ‘getprosol’” to read standard probehead and solvent dependent (prosol) parameters.
- Lock the sample to the deuterated solvent, i.e., D2O, using command “lock” and wait until it is finished sweeping and achieves lock.
- Correct the resonance frequency of the magnet by tuning the sample using the automatic tuning command “atma”. Monitor the wobble curve until the automatic tuning is complete.
- Shim the magnetic field using TOPSHIM (command “topshim”). Shimming is process of adjustments to magnetic field to achieve uniformity around the sample. It is good practice to store the shim values with the command “wsh” and read them using “rsh” before topshim, if using the same or similar samples.
- Adjust the receiver gain with “rga” command to achieve maximum signal to noise ratio.
- Place the center of the spectrum on the water resonance offset (o1) and set the 90 degree proton pulse (p1) at high power using “calibo1p1”.
- Collect the proton spectrum using the zero go “zg” command and process with “efp” which includes exponential multiplication (“em”), the free induction decay (FID) incorporating line broadening, “ft” fourier transformation of FID and “pk” to apply phase correction.
- Apply the automatic phase correction “apk” and the automatic baseline correction “absn” using the polynomial without integration option.
- Create a new dataset (as in 3.2.2) for the SOFAST HMBC experiment by selecting “SFHMQC3GPPH” in experiment.
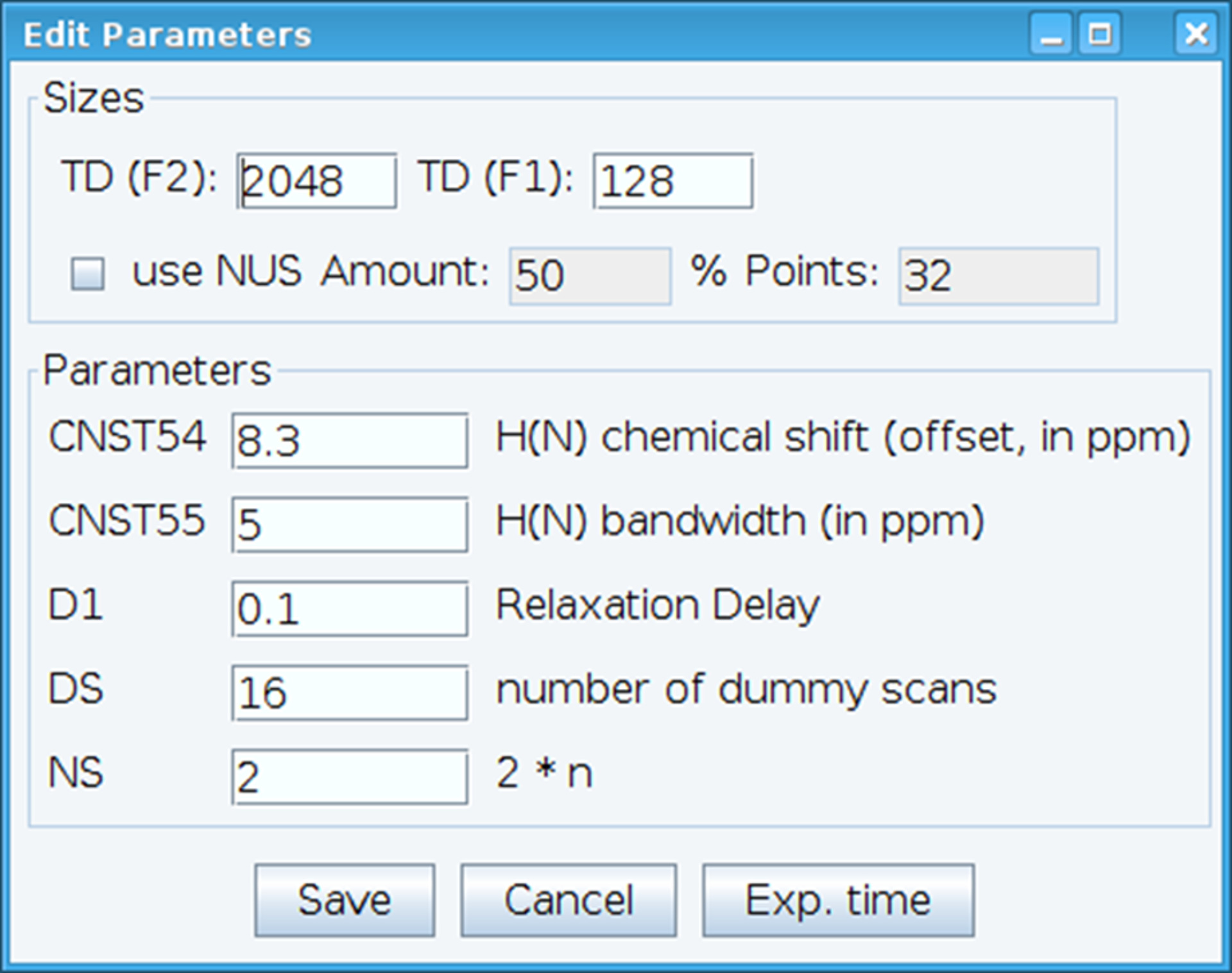
- Copy optimized P1 and O1 from proton spectrum and populate P1 dependent pulses by using command “getprosol 1H p1 plw1”, where p1 is the optimized P1 value and plw1 is the power level for P1.
- Optimize the CNST54 constant to set the offset for amide chemical shift and CNST55 to define the bandwidth in order to encompass the spectral regions of interest which allows the receiver gain to be optimized (Figure 2). To select these parameters, extract the first FID (free induction decay) from the two-dimensional spectrum and look for the observed signal to define them. In addition, vary the relaxation delay (D1), number of scans (NS), and dummy scans (DS) to obtain acceptable signal sensitivity with command “gs”, which enables go and scan to monitor data quality in real time.
- Record the spectra using Zero Go “zg”.
- NMR Data Processing
- Set the processing parameters to the size of the direct F2 (1H) and indirect F1 (15N) dimensions of the spectrum using “SI F2=2048, F1=512” with optional linear prediction in indirect dimension (Figure 3).
- Select “QSINE” as the Window function and enter a Sine bell shift (SSB) of 2 to process the two-dimensional spectrum.
- Enter the command “xfb” to process the data in both directions with window function and fourier transformation.
- Use the command “apk2d” to carry out automatic phase correction in both directions. If the automatic process does not achieve a satisfactory level of phase correction, extract FIDs with the “rser” command, calculate phase values from 1D processing, and apply them to the 2D data.
- Correct the baseline with the automatic baseline correction function “abs2” for 2D data. This applies a polynomial function between the ppm values defined in the processing parameters and will produce a 2D spectrum for further analysis.
- If planning to perform serial processing for comparison of interaction data with another molecule, store the processing parameters with the command “wpar” and recall them with “rpar”. In this way all the datasets will be processed with same parameters and variations will not be introduced due to processing differences.
- NMR Data Analysis
- Enter the command “pp” to begin the process of peak picking.
- Define the ppm range and minimum intensity/maximum number of peaks based on expected peaks (Figure 4). Click on OK and verify the results by visual inspection. If needed, re-run the process until results are satisfactory based on spectra quality.
- Generate a peaklist with the “pp” command.
NOTE: This peaklist contains data height/peak intensity information by default and can be exported to subsequent spectrums and can be read by other programs. - Observe changes in the peak intensities or movement in chemical shifts in the protein HSQC spectra that indicate interaction with another molecule. If the interacting molecule is large, expect reductions in peak intensities along with disappearance of some peaks.
- Import the peak list to the next data set by clicking on the “peaks” tab and selecting “import” with a right click in the peaks window.
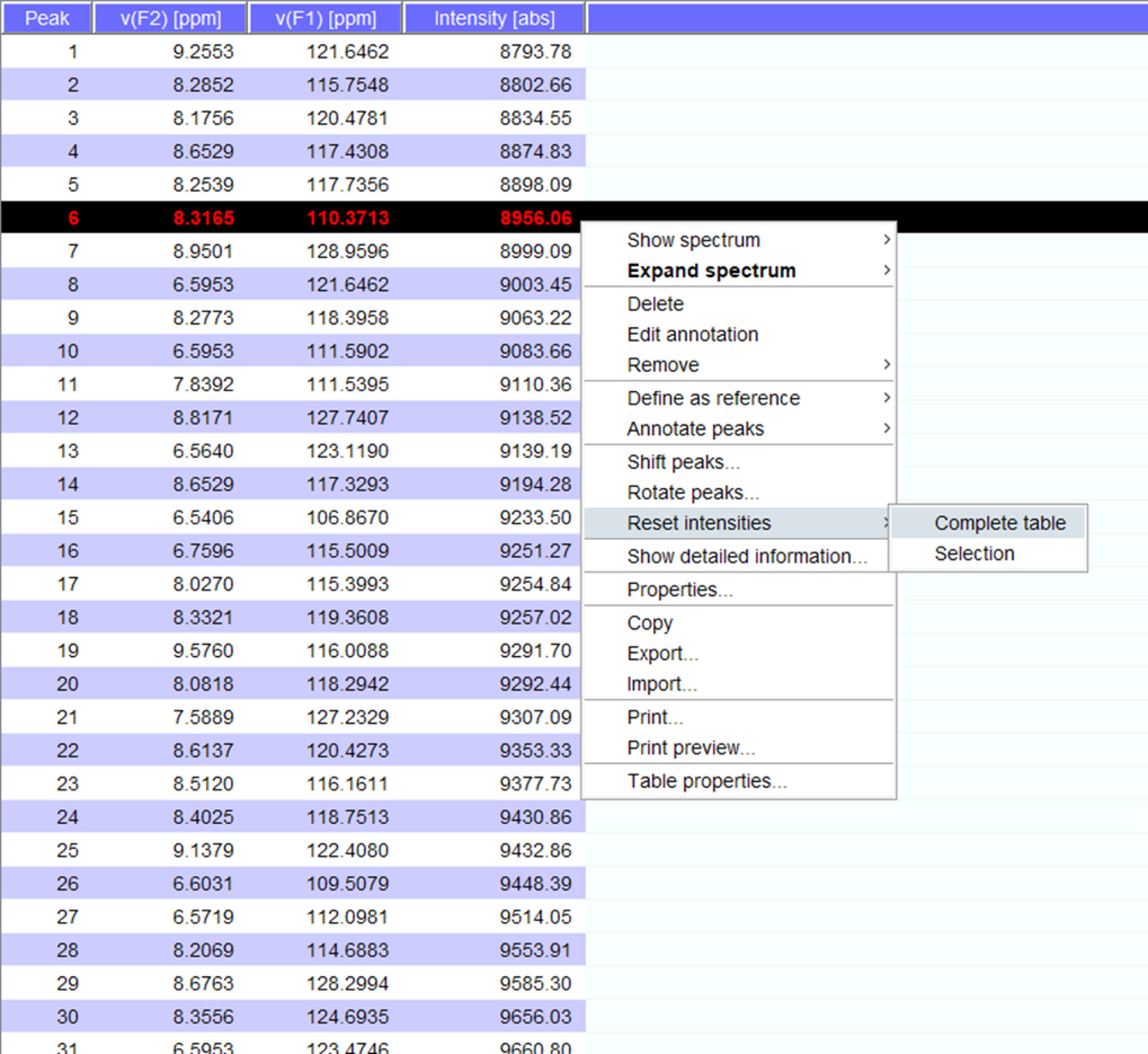
- Visualize the peaks over the spectrum and if needed shift them to new positions. Click on “reset intensities” for “complete table” to generate a peaklist for the spectrum with intensities (Figure 5). This peak list will carry over the position information from stored peak list.
- Export the peak lists from different datasets to a spreadsheet or other mathematical program for analysis by selecting the “Export” function.
- Calculate the change in peak intensities with the function “peak intensity in complex spectrum/peak intensity in protein spectrum” for each peak. Values can be converted to percentage change by multiplication of 100. Note that peak volumes are also useful although peak intensities are easier to measure for peaks that are positioned close to each other, as is usually the case for proteins with a high density of relatively broad peaks.
4. MicroScale Thermophoresis (MST)
- Preparation of the Ligand Protein E-PRD
- Exchange the ligand into an MST compatible buffer by dialyzing up to 800 μL of protein in a 3.5 kDa mini-dialysis unit suspended in 1 L of 20 mM HEPES, pH 7.5, 10 mM NaCl, stirring slowly at 4 °C overnight.
- Concentrate the ligand using a centrifugal ultrafiltration unit (3 kDa MWCO) by centrifuging at 14 000 x g for 10 min. Transfer the concentrated protein to a clean tube.
- Centrifuge the ligand at 21,000 x g for 10 min and carefully transfer the supernatant to a new tube to remove any precipitated protein. Determine the concentration of the ligand using the absorbance at 280 nm and the ligand extinction coefficient. Add 10% Tween-20 to give a final concentration of 0.015%. Tween-20 is added to the assay buffer to prevent adsorption to the capillaries. The final assay buffer that is used for the MST experiments is 20 mM HEPES, pH 7.5, 10 mM NaCl, 0.015% Tween-20.
- Preparation of Dye-labeled Target Protein VimRod
- Reconstitute the RED-tris-NTA dye by adding 50 μL of 1x PBS-T supplied with the RED-tris-NTA to give a concentration of 5 μM. Dispense 2 μL aliquots into 200 μL tubes and store at -20 °C.
- Dilute the target protein to 0.34 μM with assay buffer. Add 58 μL of target to a 2 μL aliquot of RED-tris-NTA dye and incubate for 30 min at room temperature. The final concentration of dye-labeled target is 0.33 μM. Centrifuge the labeled target at 21,000 x g for 10 min and carefully transfer the supernatant to a new tube to remove any precipitate. The RED-tris-NTA dye binds to proteins through the His6 tag and has a binding dissociation constant (KD) in the sub-nanomolar range. It is effectively 100% bound to the target protein so no further purification is required.
- Turn on the MST instrument and open the Control Software. Select the Red setting for the RED-tris-NTA dye. Turn on the temperature control to 25 °C.
- Select the Pretest to validate the labeling of the target and check for aggregation or adsorption to the cuvettes. An assessment of these parameters is automatically provided.
- Mix 17 μL of assay buffer and 3 μL of target protein and mix by pipetting. Fill two standard capillaries by dipping them in the diluted target and drawing up liquid into the center of the capillary. Place capillaries in the tray and into the instruments. Start the measurement.
- Review the results looking for a sufficient level of fluorescence and no signs of adsorption (distortion in the capillary line shape) or aggregation (distortions in the MST trace) which will be flagged in the software analysis. If results are positive move on to the ligand steps, if not a different buffer must be tried or the amount of Tween-20 may be increased to 0.05% or higher.
- Preparation of the E-PRD Ligand Two-fold Dilution Series
- Prepare a dilution series by labeling 16, 200 μL tubes from 1-16.
- Add 17 μL of the ligand at 1.17x greater concentration than the maximum ligand concentration desired to Tube1. This volume is twice the amount needed (8.5 μL) as an aliquot will then be transferred to the next tube in series. The final volume of the assay is 10 μL so 8.5 μL of 1.17x concentration of ligand will be diluted to 1x by addition of 1.5 μL of the target protein, VimRod. This maximum concentration chosen should be at least 20 times the estimated KD value.
- Add the 8.5 μL of assay buffer to Tubes2-16 using a new pipette tip for each aliquot. Reusing pipette tips can affect the accuracy (for advice on accurate pipetting see manufacturer’s instructions). Transfer 8.5 μL of ligand from Tube1 to Tube2 slowly releasing the solution into the assay buffer without generating bubbles. Mix the ligand and buffer by pipetting up and down at least 6 times, again without generating bubbles. The E-PRD in the most concentrated tube was 1.5 mM and the labeled VimRod was present at a final concentration of 50 nM.
- Transfer 8.5 μL of ligand from Tube2 to Tube3 and mix with the assay buffer. Repeat the serial dilution until all tubes have had ligand added. Discard 8.5 μL from Tube16 so that all tubes contain 8.5 μL of ligand in a series of two-fold dilutions.
- Preparation of the Binding Reaction and MST experiment
- Add 1.5 μL of the labeled target protein to each of the tubes and mix gently by pipetting up and down being careful to avoid bubbles. Incubate for 15 min.
- Select the Expert Mode for the binding assay and enter in the parameters for a serial dilution series. Make sure temperature control is set to 25 °C, the excitation power is set to 40%, and the MST power to medium. These parameters must be optimized for other binding partners being studied.
- Fill the capillaries with the binding reactions and place in the capillary tray. Load the tray into the instrument, wait for the temperature to regain 25 °C and start the measurement.
- Data Analysis
- Open the binding assay file in the Affinity analysis software. Review the capillary scans; Fluorescence levels should not vary more than 10% from the average, no aggregation or adsorption should be detected as described in section 3.6.
- Select the MST analysis on the right panel and drag the assay data into the analysis set. If there are multiple runs of the same target-ligand binding assay under identical conditions they can be merged by dropping in the same set. Alternatively, each run can be dropped into analysis independently.
- Move to the Dose Response Fit tab to graph the data. Choose the KD model if a single binding site is expected. The Hill model is also an option for multiple binding sites with cooperative behavior. Outlier points that did not pass quality control can be removed from the fit at this stage. Merge sets with multiple assays will be averaged and errors calculated as the standard deviation.
- Open the final tab and compare results between all plots on a single graph. Retrieve fitting results for each curve from the table that is generated. Export the data or fitted curves to other presentation or analysis software if desired.
Results
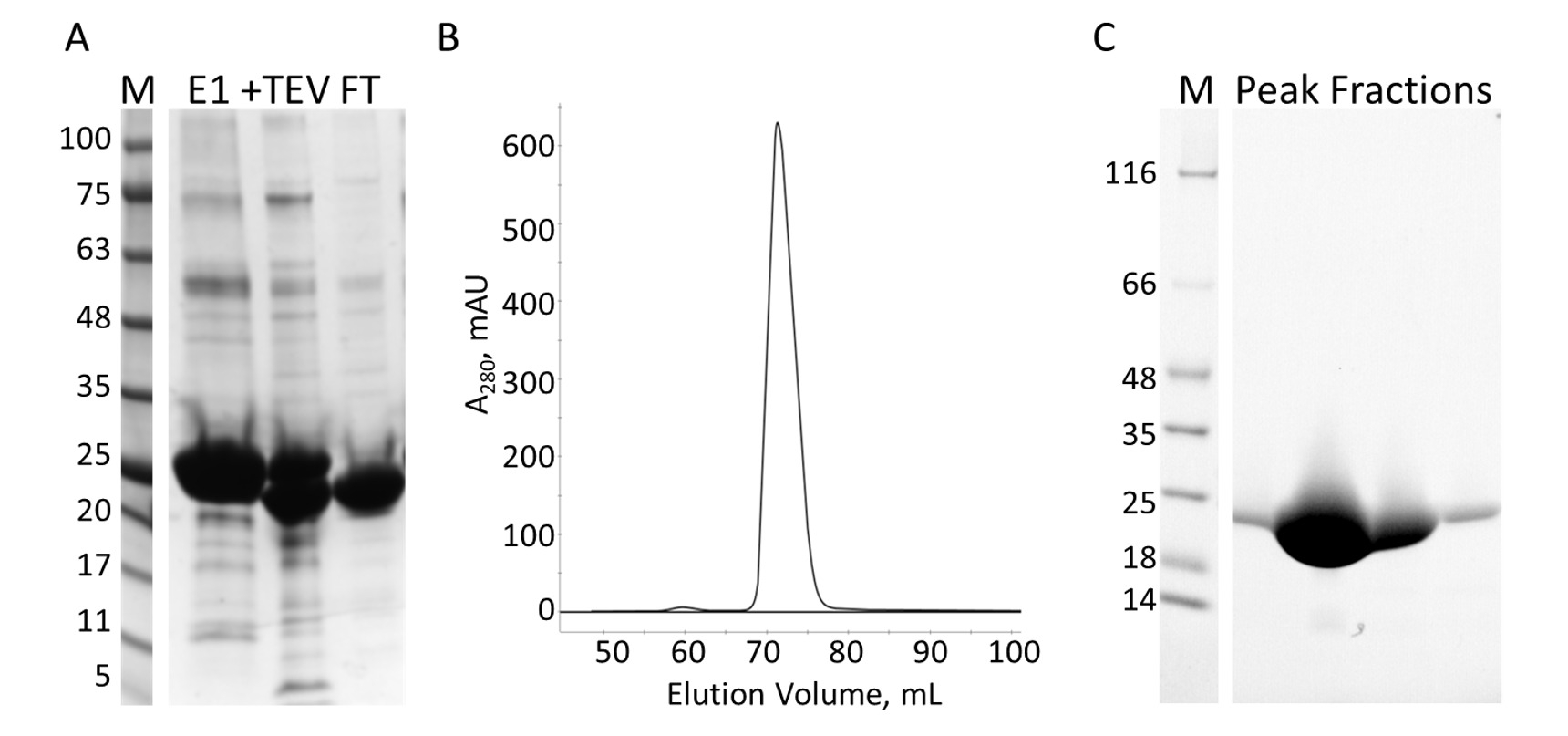
The E-PRD domain (residues 1822-2014 cloned into pProEX-HTC) of the human envoplakin gene and the VimRod domain (residues 99-249 cloned into pET21a) of human vimentin4 were expressed with His6 tags and purified. Figure 6 and Figure 7 demonstrate the levels of purity of VimRod (18.8 kDa) and E-PRD (21.8 kDa) obtained from this method of protein purification. The removal of the His6 tag from the E-PRD construct is essential for the MST experiments as the VimRod protein is labeled using a His6 tag binding dye and any E-PRD retaining its His6 tag may compete for binding of the dye. The second IMAC column after cleavage of the tag with TEV protease removes the TEV protease, the cleaved tag and any uncleaved His6-E-PRD that remained. The final polishing step of the purification is size exclusion chromatography. Despite both proteins being of a similar size, the VimRod elutes from the column at a 51 mL while the E-PRD elution peak is centered at 72 mL where a protein monomer of this size would be expected. The apparent increase in size of VimRod is likely due to the its characteristics as a filamentous long rod shaped protein as analytically ultracentrifuge experiments demonstrated that VimRod was monomeric4. Lower yields of protein are obtained from the cultures grown in M9 than those from rich broth due to a lower amount of cells being produced in the minimal media. The initial growth of larger starter cultures for M9 preparations in TB allows improvement of cell yields while maintaining the extent of 15N labeling necessary for the NMR experiments.
The 15N-1H HSQCs were acquired for the wild type and R1914E mutant of E-PRD in presence or absence of VimRod (Figure 8A-8D). The spectrum of E-PRD in Figure 8A shows the expected number of well resolved peaks, indicative of a properly folded protein. In the presence of VimRod (Figure 8B) the spectrum shows extensive line broadening and peak disappearance, corresponding to binding between the E-PRD and VimRod. This binding is lost by mutation of R1914E as evidenced by comparison of Figure 8C and 8D. Little change is observed in the spectrum upon addition of VimRod to the R1914E mutant indicating a lack of binding between this mutant E-PRD and VimRod. The E-PRD peak intensities in the presence/absence of VimRod were compared and plotted as the relative peak intensities in Figure 8E, which indicates the range of peak broadening in the E-PRD complex. The R1914E mutant of E-PRD (not shown) retained about 97% of peaks at 20% or higher peak intensities in presence of VimRod compared to about 20% for the wild type (Figure 8E). This represents a loss of function point mutant, with additional mutants having intermediate effects also having been studied4.
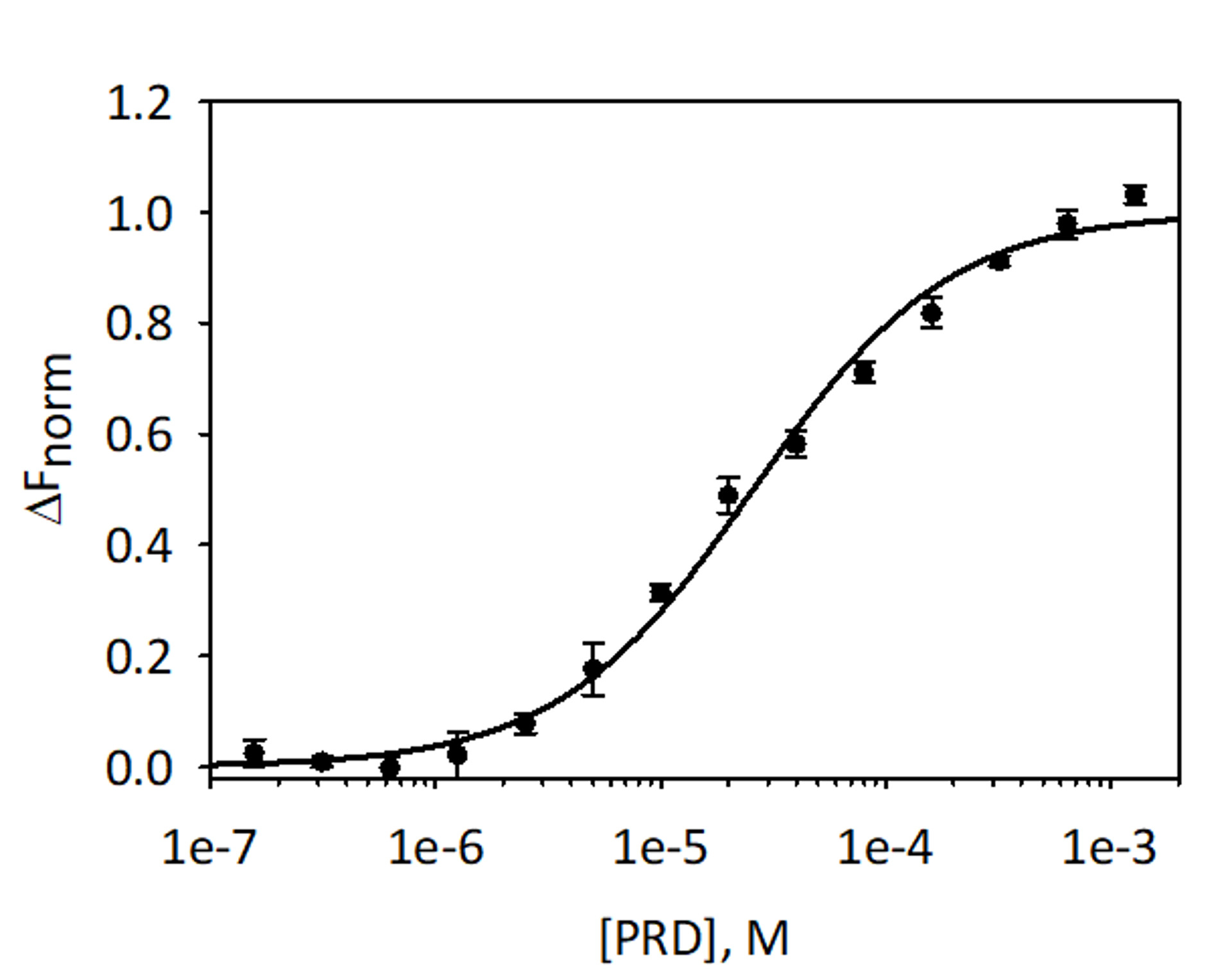
To validate and quantitate the binding of VimRod and E-PRD MST analysis using His6-VimRod labeled with fluorescent RED-tris-NTA dye as the target mixed with decreasing concentrations of the ligand E-PRD from 1.28 mM to 39.1 nM were performed. Three binding titrations were carried out and the results are averaged and shown in Figure 9. The data were fit with a standard model of one-site ligand binding and gave a KD of 25.7 ± 2.1 μM. Evaluation of the binding between VimRod and E-PRD by surface plasmon resonance gave a similar KD value of 19.1 ± 1.3 μM4.

Figure 1: Screen Capture of the Setup of the NMR Experiment. The window shown is used to set up a standard experiment to collect a HSQC dataset. Experiment parameters are read in adjacent to Experiment. The ZGPR experiment shown is chosen as an initial experiment to load the standard and solvent dependent proton parameters. The Title window is used to input experimental details for record keeping purposes. To collect the HSQC spectrum the ZGPR experiment is replaced with SFHMQC3GPPH. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Adjusting of NMR Experimental Parameters. The window shown is used for entering the basic parameters for the NMR pulse sequence in order to optimize the signal. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: NMR Data Processing. Parameters used for processing each of the two dimensions of the NMR spectrum are shown, with arrows indicating those that are typically adjusted. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Parameters for NMR Peak Picking. The parameters used for picking NMR peaks in the processed NMR spectrum are shown with typical values. Adjust the ppm range, intensity and number of peaks to optimize the spectra. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Representative Peaklist with Intensities. Each peak that is picked in the NMR spectrum is given a number, and its 1H and 15N chemical shifts and signal intensity are displayed. This peaklist can then be used to compare spectra obtained in the presence/absence of an interacting partner. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Purification of His6-tagged VimRod by IMAC and S. A. The chromatogram for the elution from the IMAC column shows one major peak of VimRod. B. The chromatogram for the elution from the S column shows one major peak. C. SDS-PAGE of fractions collected over the course of purification: MW standards with the MW indicated in kDa to the left of the gel (M), cell lysate (1), IMAC flow-through (2), wash (3), pooled elution (E1), pooled S elution (E2). Bands visible at higher molecular weights in lanes E1 and E2 are oligomers of pure VimRod as confirmed by western blot (data not shown). Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Purification of E-PRD by IMAC and S. A. SDS-PAGE of the IMAC purification showing the molecular weight standards with the MW indicated in kDa to the left of the gel (M) and the eluate from the first IMAC column (E1), the TEV cleavage products (+TEV), and the flow through from the second IMAC column (FT). B. The chromatograph from the S column shows one major peak. C. SDS-PAGE of the molecular weight standards (M) and the fractions from S peak. Please click here to view a larger version of this figure.
{kind=link}

Figure 8: HSQC Spectra of Wild-type and R1914E Mutant of E-PRD in the Presence and Absence of VimRod. The HSQC spectra show wild-type E-PRD (100 µM) in 20 mM Tris-HCl, 150 mM NaCl, 1 mM DTT, pH 7 in the absence (A) or presence of 50 µM VimRod (B). Panels C and D are the HSQC spectra of the R1914E mutant (100µM) in the absence or presence of 50 µM VimRod, respectively. In panel E the relative 1H-15N peak intensities of the E-PRD with or without VimRod binding are shown as a function of the peak number, which is arbitrarily assigned and not based on sequence position. These values can be used to define a significance cutoff for peak intensity reduction upon addition of a ligand. If assignments are available, the significant values can often be seen to map to a binding area. Please click here to view a larger version of this figure.
{kind=link}

Figure 9: Binding of E-PRD to VimRod. E-PRD was diluted in a series of two-fold dilutions from 1.28 mM to 39.1 nM and incubated with labeled VimRod before performing MST analysis. Data from three independent assays were combined. The data were fit to a KD model giving a KD of 25.7 μM with a KD confidence of ± 2.1 μM. Please click here to view a larger version of this figure.
{kind=link}
| Reagent | Quantity |
| Sodium phosphate, dibasic (anhydrous) | 6.0 g |
| Potassium phosphate, monobasic (anhydrous) | 3.0 g |
| Sodium Chloride | 0.5 g |
| H2O | Up to 950 mL |
Table 1. M9 media for Isotopic Labeling.
| Reagent | Quantity |
| 15NH4Cl | 1.0 g |
| Glucose (or 13C-glucose) | 2.0 g |
| 1 M MgSO4 | 2 mL |
| 50 mM CaCl2 | 4 mL |
| 20 mg/mL Thiamine | 1.0 mL |
| 3 mM FeCl3 | 400 µL |
| Metal Mix (Table 3) | 500 µL |
| H2O | Up to 50 mL |
Table 2. Nutrient Mix for Supplementation of M9 media.
| Reagent | Quantity |
| 4 mM ZnSO4 | 323 mg |
| 1 mM MnSO4 | 75.5 mg |
| 4.7 mM H3BO3 | 145 mg |
| 0.7 mM CuSO4 | 55.9 mg |
| H2O | Up to 500 mL |
Table 3. Metal Mix Supplement for Enriching the MT Nutrient Mix.
| Sample | 1 mM E-PRD in buffer A1 (µL) | 1mM VimRod in buffer A (µL) | Buffer A (µL) | 200 µM DSS in D2O (µL) | Buffer B2 (µL) | Total Volume (µL) |
| E-PRD alone | 50 | 0 | 50 | 50 | 350 | 500 |
| E-PRD + VimRod | 50 | 50 | 0 | 50 | 350 | 500 |
| 1Buffer A: 20 mM Tris-HCl, 1 mM DTT, pH 7 | ||||||
| 2Buffer B: 23 mM Tris-HCl, 1.14 mM DTT, pH 7 | ||||||
Table 4. NMR Sample Preparation.
Discussion
The 2D 15N-resolved NMR experiment is one of the most widely used methods to show how two molecules interact. It is the most information-rich method that allows both partners' signals to be continuously monitored throughout a titration experiment in solution state. Although typically qualitative in the case of large complexes, the method can also be used in favorable cases to measure binding affinities where NMR signals can be tracked in high resolution spectra. Where assignments can be conveniently made, such as in the case of many proteins under 20 kDa in size, the binding sites can also be mapped. Complementary assays such as MST provide quantitative information about interactions in solution, and require less protein in unlabeled states. Comparison of mutant binding data is useful for providing controls to ensure that interactions evidenced by NMR line broadening are genuine and not artifacts of, for example, aggregation or viscosity changes.
Protein Expression
Streamlining the expression process reduces the amount of labor intensive protein production. Part of this optimization process involves identification of an appropriate strain of E. coli for the recombinant expression of protein. Strain preference depends on elements including the nature of the vector in use and, more specifically, the ultimate stability of the recombinant protein being expressed7. The risk of degradation of the heterologous protein by endogenous E. coli proteases can be reduced by use of protease deficient E. coli such as the BL21 strain. For genes containing rare codons, a strain such as BL21-CodonPlus (DE3)RIPL may be preferred. This strain combines the protease deficient nature of the BL21 strain with additional endogenous copies of rare codon tRNAs for arginine, isoleucine, proline, and leucine. Alternatively, rare codons that can compromise overexpression may be avoided by ordering a codon-optimized construct from a commercial source. Many strains of E. coli are available for recombinant gene expression, each optimized for circumvention of a particular problem during expression7. In the case of this study, the standard protease deficient strain BL21(DE3) produced adequate quantities of soluble protein for subsequent purification and analysis.
Protein Purification
The purification protocol for a given protein is often unique in the sense that each protein remains stable and soluble under different conditions such as temperature, salt concentration, or pH. The overall effectiveness of purification through affinity chromatography is also sensitive to the concentration of eluting species such as imidazole at various steps during the purification process. In this work, critical buffer conditions for IMAC were pH for the E-PRD, and imidazole concentration for the VimRod. A pH of 7.5 was required to avoid precipitation of the E-PRD following initial elution from the IMAC column. For the IMAC purification of VimRod, increasing the concentration of imidazole from 30 to 50 mM during the column wash step was found to have a substantial improvement in the purity of the final elution fractions. For the elution step, increasing the concentration of imidazole from 250 to 350 mM also was found to improve the yield of the final elution. Initial attempts to elute protein using 250 mM imidazole led to incomplete elution of VimRod as revealed by a final 1 M imidazole strip of the column (data not shown). Increasing the imidazole concentration to 350 mM for the elution was sufficient to recover all of the protein bound to the column. S can serve a dual purpose because it acts as a polishing step for protein purification while simultaneously performing buffer exchange. Buffer exchange is a critical step for subsequent binding analysis since it removes the imidazole used to elute His6-tagged protein. It also serves as an opportunity to change conditions such as salt concentration or pH, which may impact the efficacy of certain downstream techniques or assays. Protein thermal shift (PTS) can be used to identify optimal buffers for downstream assays, especially for those requiring stable protein for prolonged periods of time at room temperature8,9.
Binding Analysis
Protein that is freshly prepared is critical for accurate binding assays, although frozen protein can also be used as long as the results are compared. Filamentous proteins such as vimentin multimerize in a salt and pH dependent fashion, and hence the solution condition need to be optimized and the oligomeric state estimated by a method such as SEC10,11, dynamic light scattering12 or analytical ultracentrifugation13,14,15. NMR spectroscopy is well suited for measuring ligand interactions of small proteins at atomic resolution. However, when a protein interacts with larger molecule, slower tumbling ensues, and this results in loss of signals, which can confirm binding although it does not necessarily allow mapping of binding sites, which would also require assignment of at least backbone resonances. In this scenario, NMR experiments do not allow identification of the interaction site. Hence site directed mutagenesis is applied to identify the critical residues needed for the binding. Such mutants therefore do not exhibit signal loss. In this protocol, a mutant form with a substitution at position 1914 retains the peak intensities in presence of VimRod and therefore confirms disruption of the interaction of E-PRD and VimRod. Assignment of the backbone and sidechain resonances would add value to this approach, particularly as the structure for the free E-PRD has been solved by X-ray crystallography4. Future applications of NMR include characterization of complex interactions between larger molecules and will benefit from ultra high field magnets and the use of other observable groups such as 13C-labeled and trifluoro methyl groups as reporters.
MST has a number of advantages for studying binding interactions16. The binding partners are free in solution and not immobilized. Analysis of the quality of the samples is built into the software with quality control reporting of aggregation, adsorption to the capillaries or insufficient fluorescent labeling of the target molecule. Small amounts of the target are typically used, the concentration of the labeled target is usually between 20-50 nM in a 10-20 μL volume/reaction. This protocol uses very small reaction volumes (10 μL) to maximize the concentration of ligand that can be achieved in the titrations allowing weak binding interactions to be characterized. This necessitates accurate pipetting and care being taken to avoid introducing bubbles while still thoroughly mixing. Adequate mixing is critical for accurate, consistent fluorescence measurements along the set of serial dilutions. The amount of Tween-20 in the MST experiments was reduced from a standard 0.05% to 0.015% to lower the tendency to create bubbles and improve mixing.
The RED-Tris-NTA dye provides a quick, easy and convenient way to fluorescently label any protein that has a His tag. The labeling is effectively complete in only 30 minutes and is very tight so that no dye removal procedure is necessary. No modifications are made to amino acid residues in the protein that might alter the ligand binding properties. A caveat is that only the protein to be labeled should have a His6 tag. This required the cleavage of the tag from the ligand protein, E-PRD, and the removal of the tag and uncleaved E-PRD with a second IMAC column step. If possible, the ligand protein should be prepared without the use of a His tag. Alternatively, proteins may be covalently labeled with a fluorophore through amine coupling to lysine residues or thiol coupling to cysteine residues. However, care must be taken when using such systems since the covalent attachment of a fluorophore may affect electrostatic or polar binding interactions relying on lysine or cysteine residues. The quantification of binding affinity between the VimRod and E-PRD by MST was unusually sensitive to salt concentration. This problem was mitigated by initially dialyzing both the target and ligand into the same batch of assay buffer. Nonetheless, saturation of the MST binding curve could not be achieved when performing the MST assay in the presence of 150 mM NaCl due to the complex behavior of VimRod. Reliable, complete data was obtained once the concentration of NaCl was lowered to 10 mM allowing accurate calculation of the KD. Hence, careful optimization of solution conditions and comparison with complementary assays are recommended to achieve robust results. Furthermore, MST may be used to quantify the salt dependence for a given interaction, quantify stoichiometric properties of protein interactions, monitor protein folding, and probe into enzyme kinetics17.
Disclosures
The authors disclose no conflicts of interest.
Acknowledgements
This project has been supported by NSERC RGPIN-2018-04994, Campus Alberta Innovation Program (RCP-12-002C) and Alberta Prion Research Institute / Alberta Innovates Bio Solutions (201600018), awarded to M.O and Genome Canada and Canada Foundation for Innovation grants awarded to The Metabolomics Innovation Centre (TMIC) and NANUC.
Materials
| Name | Company | Catalog Number | Comments |
| Monolith NT.115, includes control and analysis software | NanoTemper Technologies | MO-G008 | Instrument for microscal thermophoresis |
| His-Tag Labeling Kit RED-Tris-NTA | NanoTemper Technologies | MO-L008 | RED-tris-NTA dye for MST |
| Standard capillaries | NanoTemper Technologies | MO-K022 | capillaries for MST |
| HisTrap HP, 5 mL | GE Healthcare | 17524801 | IMAC column |
| HisPur Ni-NTA resin, 100 mL | Thermo Fisher Scientific | 88222 | IMAC resin |
| HiLoad 16/600 Superdex 75 pg | GE Healthcare | 28989333 | SEC column |
| TEV protease | Sigma Aldrich | T4455 | cleavage of his tag from E-PRD |
| BL21(DE3) Competent E. coli | New England Biolabs | C2527H | cells for protein expression |
| cOmplete Protease Inhibitor Cocktail Tablets EDTA-Free | Sigma Aldrich | 11873580001 | protease inhibitors for protein purification by IMAC |
| TCEP, Tris(2-carboxyethyl)phosphine hydrochloride | Sigma Aldrich | C4706 | reducing agent for protein purification |
| Reagents (HEPES, NaCl, etc) | Sigma Aldrich | various | Preparation of media and buffers |
| Ammonium chloride (15N, 99%) | Cambridge Isotope Laboratories | NLM-467 | isotope labelling for NMR |
| D2O, Deuterium Oxide (D, 99.8%) | Cambridge Isotope Laboratories | DLM-2259 | NMR sample preparation |
| DSS, Sodium 2,2-dimethyl-2-silapentane-5-sulfonate-D6 (D,98%) | Cambridge Isotope Laboratories | DLM-8206 | reference for NMR |
| Precision 5 mm NMR Tubes, 7” long | SJM/Deuterotubes | BOROECO-5-7 | NMR tubes |
| NMR spectrometer (14.1 Tesla) | Bruker | acquisition of NMR data | |
| TCI 5mm z-PFG cryogenic probe | Bruker | acquisition of NMR data | |
| Name | Company | Catalog Number | Comments |
| Software | |||
| Bruker TopSpin 4.0.1 | Bruker | processing of NMR data | |
| MO.Control | NanoTemper Technologies | included with Monlith NT.115 | |
| MO.Affinity Analysis | NanoTemper Technologies | included with Monlith NT.115 |
References
- Vinogradova, O., Qin, J. NMR as a unique tool in assessment and complex determination of weak protein-protein interactions. Topics in Current Chemistry. 326, 35-45 (2012).
- McKercher, M. A., Wuttke, D. S. NMR Chemical Shift Mapping of SH2 Peptide Interactions. Methods in Molecular Biology. 1555, 269-290 (2017).
- Al-Jassar, C., Bikker, H., Overduin, M., Chidgey, M. Mechanistic Basis of Desmosome-Targeted Diseases. Journal of Molecular Biology. 425 (21), 4006-4022 (2013).
- Fogl, C., et al. Mechanism of intermediate filament recognition by plakin repeat domains revealed by envoplakin targeting of vimentin. Nature Communications. 7, (2016).
- Walker, J. M. . The Protein Protocols Handbook. , (2002).
- Wishart, D. S., et al. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR. 6, 135-140 (1995).
- Casali, N. Escherichia coli Host Strains. E. coli Plasmid Vectors. , 27-48 (2003).
- Ericsson, U. B., Hallberg, B. M., DeTitta, G. T., Dekker, N., Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Analytical Biochemistry. 357 (2), 289-298 (2006).
- Kozak, S., et al. Optimization of protein samples for NMR using thermal shift assays. Journal of Biomolecular Nmr. 64, 281-289 (2016).
- Burgess, R. R. A brief practical review of size exclusion chromatography: Rules of thumb, limitations, and troubleshooting. Protein Expression and Purification. , (2018).
- Kunji, E. R. S., Harding, M., Butler, P. J. G., Akamine, P. Determination of the molecular mass and dimensions of membrane proteins by size exclusion chromatography. Methods. 46 (2), 62-72 (2008).
- Stetefeld, J., McKenna, S. A., Patel, T. R. Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophysical Reviews. 8 (4), 409-427 (2016).
- Patel, T. R., Winzor, D. J., Scott, D. J. Analytical ultracentrifugation: A versatile tool for the characterisation of macromolecular complexes in solution. Methods. 95, 55-61 (2016).
- Pearson, J. Z., Cole, J. L., et al. Chapter One - Next-Generation AUC Adds a Spectral Dimension: Development of Multiwavelength Detectors for the Analytical Ultracentrifuge. Methods in Enzymology. 562, 1-26 (2015).
- Gorbet, G. E., Pearson, J. Z., Demeler, A. K., Cölfen, H., Demeler, B., Cole, J. L. Chapter Two - Next-Generation AUC: Analysis of Multiwavelength Analytical Ultracentrifugation Data. Methods in Enzymology. 562, 27-47 (2015).
- Wienken, C. J., Baaske, P., Rothbauer, U., Braun, D., Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nature Communications. 1, 100 (2010).
- Jerabek-Willemsen, M., et al. MicroScale Thermophoresis: Interaction analysis and beyond. Journal of Molecular Structure. 1077, 101-113 (2014).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved