
Method Article
Separating Bacteria by Capsule Amount Using a Discontinuous Density Gradient
In This Article
Summary
We demonstrate the use of discontinuous density gradients to separate bacterial populations based on capsule production. This method is used to compare capsule amount between cultures, isolate mutants with a specific capsule phenotype, or to identify capsule regulators. Described here is the optimization and running of the assay.
Abstract
Capsule is a key virulence factor in many bacterial species, mediating immune evasion and resistance to various physical stresses. While many methods are available to quantify and compare capsule production between different strains or mutants, there is no widely used method for sorting bacteria based on how much capsule they produce. We have developed a method to separate bacteria by capsule amount, using a discontinuous density gradient. This method is used to compare capsule amounts semi-quantitatively between cultures, to isolate mutants with altered capsule production, and to purify capsulated bacteria from complex samples. This method can also be coupled with transposon-insertion sequencing to identify genes involved in capsule regulation. Here, the method is demonstrated in detail, including how to optimize the gradient conditions for a new bacterial species or strain, and how to construct and run the density gradient.
Introduction
Many bacterial species produce a polysaccharide capsule, which protects the bacterial cell from various physical stresses and from recognition and killing by the immune system. In Klebsiella pneumoniae, capsule production is an absolute requirement for infection1,2. K. pneumoniae capsule mediates resistance to antimicrobial peptides, resistance to complement-mediated killing, prevention of phagocytosis, and suppression of the innate immune response3. Excess capsule production is associated with increased virulence and community-acquired (rather than nosocomial) infections4.
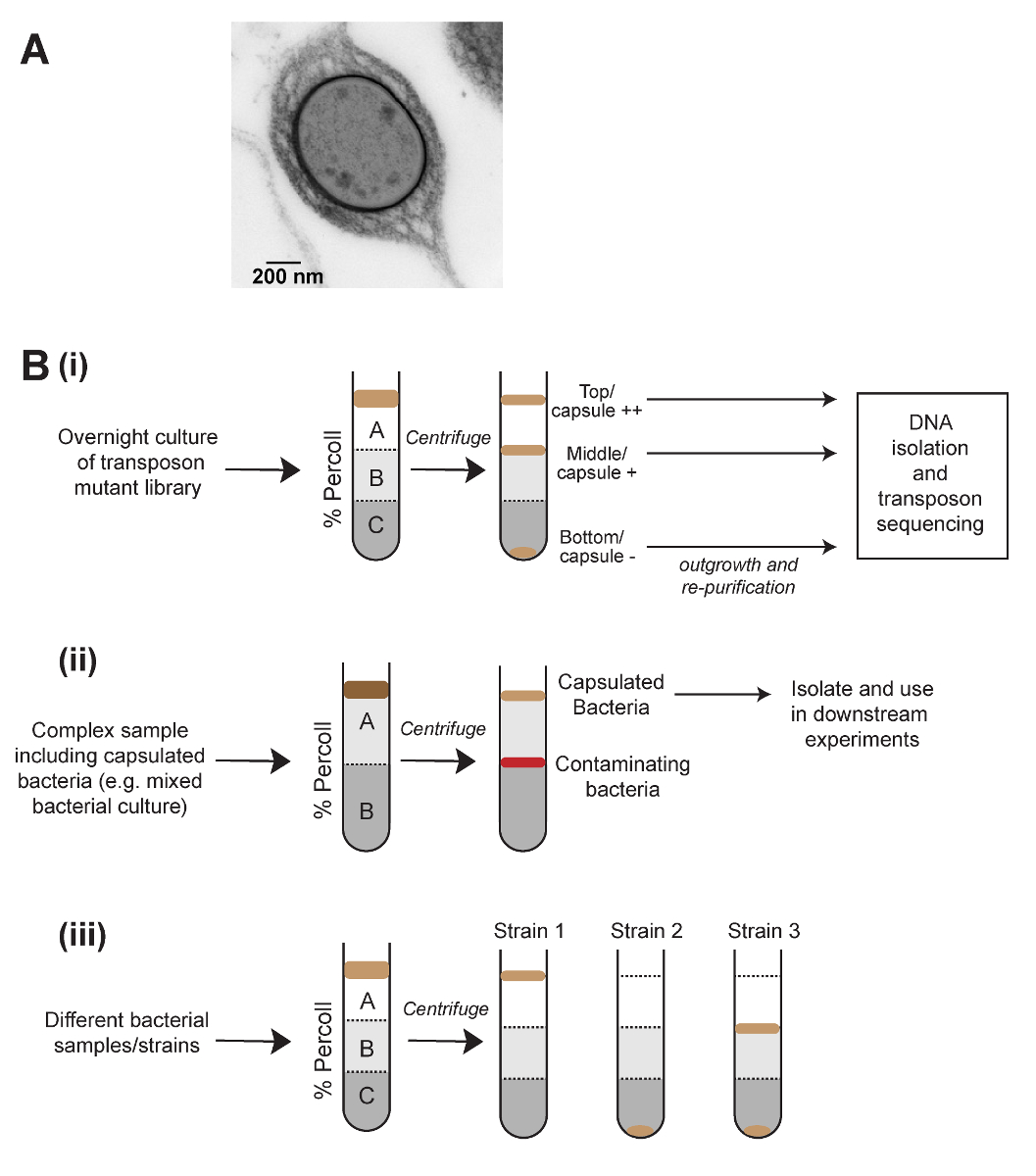
A range of quantitative and qualitative tests are available to investigate capsule production. For Klebsiella species, these include the string test5, in which a toothpick touched to a colony is pulled upwards and the length of the string produced measured, and the mucoviscosity assay6, which involves the slow centrifugation of a culture followed by measuring the optical density of the supernatant. These methods are simple and quick, but lack sensitivity when used on classical Klebsiella strains rather than capsule overproducing strains. Another method of capsule quantification is the uronic acid assay, which is technically challenging and requires the use of concentrated sulfuric acid1. Finally, capsule is visible directly by microscopy (Figure 1A). Of these methods, only microscopy allows the user to observe different capsulation states within a single population, and none of these methods enables the physical separation of capsulated and non-capsulated bacteria.
Density-based separations by gradient centrifugation are routinely used in cell biology to purify different eukaryotic cell types7, but are rarely used in microbiological research. The mucoviscosity assay for Klebsiella is based on the observation that highly capsulated bacteria take more time to pellet by centrifugation, and we reasoned that this may be due to reduced overall density of capsulated cells. The method shown here was developed to separate K. pneumoniae populations physically by capsule amount, using density gradient centrifugation (Figure 1). This method was applied successfully to Streptococcus pneumoniae, indicating that it is applicable to other bacterial species. Density-gradient separation of a saturated transposon mutant library coupled with transposon-insertion sequencing (density-TraDISort) has been used to identify genes involved in the capsule production and regulation8. Similarly, this method was used in conjunction with random-prime polymerase chain reaction (PCR) of individual colonies to isolate non-capsulated K. pneumoniae mutants. This method can also be used for rapid comparisons of capsule production between different populations and conditions, or to purify capsulated bacteria from complex samples (Figure 1B). Finally, there is the option to assay other phenotypes that affect density, such as cell size or aggregation.
This manuscript demonstrates how to optimize the procedure for a new bacterial species or strain and demonstrates the construction and running of a discontinuous density gradient to separate hyper-capsulated, capsulated and non-capsulated bacteria.
Protocol
NOTE: Ensure that any risk assessments applicable to the bacterial strains are adhered to when culturing and handling samples. Be aware that setting up too many gradients at one time can lead to musculoskeletal disorders due to the pressure on joints from the slow pipetting involved. Plan work and take precautions to avoid injury.
1. Preparation of Bacterial Strains or Mutant Libraries
- Streak out the strains to be tested on appropriate agar plates. These are stock plates for the experiment.
- Incubate the plates overnight at the desired temperature to achieve single colonies. For this experiment, culture K. pneumoniae (NTUH-K2044 and ATCC43816 strains) on Luria broth (LB) agar at 37 °C, and S. pneumoniae (23F wild type and 23F Δcps) on blood agar in a humidified candle jar at 37 °C.
- Pick a single colony from a stock plate (step 1.1) to inoculate 10 mL of appropriate broth using a sterile loop or cocktail stick. For screening of random mutant libraries, inoculate the broth with 10 µL of the random mutant library stock (TraDIS Library).
- Incubate K. pneumoniae strains in low salt LB media at 37 °C with shaking, and S. pneumoniae strains in brain heart infusion (BHI) media, statically at 37 °C.
- Transfer the overnight culture to a 15 mL tube and centrifuge in a bench-top centrifuge for 10 min at 3,200 x g in swing out buckets with 15 mL tube inserts and aerosol tight lids.
- Discard the supernatant and resuspend the pellet in 2 mL of 1x phosphate-buffered saline (PBS).
NOTE: Dispose of supernatant via the appropriate liquid biological waste route in the laboratory. The purpose of the centrifugation and resuspension steps (1.2.2 and 1.2.3) is to concentrate the bacteria for easy visualization on the gradient. Bacterial cultures can be loaded directly onto the gradient if preferred. Heavily capsulated strains may not form a tight pellet. If this occurs, remove as much supernatant as possible without removing any bacterial cells, add 1x PBS to a final volume of 5 mL and resuspend the pellet. Continue the protocol with step 1.2.5. - For non-mucoid strains, go to step 2.
- Centrifuge the tubes as described in step 1.2.2.
- Discard the supernatant and resuspend the pellet in 2 mL of 1x PBS. Cells are now ready to use.
NOTE: The density of the bacterial cell suspension is not critical but needs to be sufficient for visualization in the gradient. A minimum OD600 (optical density at 600 nm) of 4 is suggested.
2. Preparation of Gradient Dilutions and Mini-gradient Test
- Prepare gradient dilutions.
NOTE: Exact concentrations of density gradient medium (e.g., Percoll) needed in the density gradients to achieve good separation will differ depending on the bacterial strain and growth conditions used. Mini-gradient tests are performed first, to identify the concentrations that will give the best separation. These comprise 500 µL of a single dilution in a 2 mL tube. If bacteria will be extracted from the gradient and subcultured for downstream applications, these steps should be performed under aseptic conditions.- Combine density gradient medium with 1x PBS to make the density gradient dilutions. Make dilutions of 20%, 30%, 40%, 50%, 60%, 70%, and 80% (e.g., 2 mL of density gradient medium plus 8 mL of 1x PBS = 10 mL of 20% density gradient medium).
- Aliquot 500 µL of the 20% gradient dilution into a 2 mL tube for each strain to be tested (e.g., 4 strains = 4 tubes containing 500 µL of 20% gradient dilution).
- Repeat step 2.1.2 for the rest of the gradient dilutions.
- Apply bacteria to the gradient.
- Apply 100 µL of bacterial cells prepared in steps 1.2.3 and 1.2.6 to the top of each gradient dilution following the steps below.
- Take up 100 µL of cells using a 200 µL pipette and place the pipette tip on the side of the tube just below the meniscus of the mini-gradient.
- Aspirate the bacterial cells onto the gradient extremely slowly so that they form a layer on the top of the gradient, without any mixing of the interface.
- Repeat steps 2.2.1.1–2.2.1.2 for all strains to be tested.
- Using a fixed angle rotor with an aerosol tight lid, centrifuge the prepared tubes in a microcentrifuge for 10 min at 8,000 x g.
- After centrifugation, transfer the tubes to a rack to visualize the minimum gradient dilution required to retain cells just above the gradient layer following centrifugation.
- If the results are not clear, repeat the mini-gradient test with increments of 5% density gradient medium dilutions above and below the concentrations defined in step 2.1.1 (e.g., 25% and 35% dilutions should be tested if the result at 30% is ambiguous).
NOTE: See Figure 2A for typical results at a single density gradient medium dilution. - Use the results from steps 2.2.3–2.2.4 to determine the ideal gradient dilutions to use to separate cells in larger-scale discontinuous gradients.
- Apply 100 µL of bacterial cells prepared in steps 1.2.3 and 1.2.6 to the top of each gradient dilution following the steps below.
3. Preparation of Cells for the Main Experiment
- Prepare fresh overnight cultures by inoculating 10 mL of appropriate broth as described in step 1.2.
- Incubate the cultures overnight in appropriate conditions as described in step 1.2.1.
- Pellet the overnight culture as described in step 1.2.2.
- Wash the cells as described in step 1.2.3.
- Discard the supernatant and resuspend the pellet in 1 mL of 1x PBS. If the strain is mucoid and does not pellet easily, resuspend the pellet in up to 2 mL of PBS or residual media. Cells are now ready to use.
4. Preparation of Discontinuous Density Gradients
NOTE: An alternative method of gradient preparation from bottom (most concentrated) to top (least concentrated) using a pipette is described in step 7.
- Pipette 1 mL of the most dilute density gradient dilution into a 5 mL polypropylene round bottom tube to form the top, most dilute, layer of the gradient.
NOTE: Subsequent layers of more concentrated gradient dilutions will be added below this layer using a needle, so as not to disrupt the layers. A minimum of two and maximum of three gradient layers of 1 mL each are recommended for robust separation and easy visualization of bacteria.- Using a 1 mL disposable syringe with a 1.5 inch needle attached, take 1 mL of the next most concentrated density gradient medium dilution into the syringe. Avoid taking up any air, as bubbles can disrupt the gradient layers.
- Place the needle end at the bottom of the tube containing the first layer. Aspirate the syringe contents very slowly to avoid mixing the interface.
NOTE: The interface of the two dilutions will rise as the more concentrated gradient dilution is added. Observe the interface by holding it up to light or an outside window. If no distinct interface is observed, discard the gradient and start again.- Remove the needle from the gradient very gently so as not to disturb the interface between the different gradient dilutions. Place the tube in a suitable rack to keep it upright.
- If using three different dilutions, repeat steps 4.1.1–4.1.2.1 so that the densest dilution is at the bottom. If the gradient has been constructed successfully, there will be three distinct layers with no mixing at the interfaces.
5. Adding Prepared Cells to Gradients and Separation by Centrifugation
- Add 600 µL of prepared cells from step 3.3 to the top of the gradient in the tube very slowly and without mixing the interface, as described in step 2.2.
- Place the tubes in tube adapters and weigh the combined adapters and tubes to ensure they are balanced.
- Place the tube adapters in a fixed angle rotor within a bench-top centrifuge. Centrifuge for 30 min at 3,000 x g.
- After centrifugation, carefully remove the tubes, place them in a suitable rack, and photograph the results as a record.
- Recover the bacterial fractions for uronic acid quantification or DNA extraction by following step 6.
- If downstream applications are not required, dispose of the samples/gradients via the appropriate liquid biological waste route in the laboratory.
NOTE: It is important to validate the separation for new species/strains of bacteria. Individual fractions from the gradient should be examined by microscopy, by uronic acid assay (step 8), or another suitable quantitative assay to confirm the capsule phenotype of each fraction. If the aim is to separate based on aggregation or cell size, independent appropriate assays should be used.
- If downstream applications are not required, dispose of the samples/gradients via the appropriate liquid biological waste route in the laboratory.
6. Recovering Sample Fractions and Optional Outgrowth Step
- Recover the fractions from a gradient by removing and discarding any liquid from the top dilution if no bacterial fraction is present within that layer.
- To remove the top fraction, use a P200 pipette and gently pass the pipette tip through the gradient to the fraction, and take up the fraction. Place the fraction into a 1.5 mL tube and label appropriately.
- To recover further fractions, still using the P200 pipette, insert the tip gently through the gradient to the fraction and recover the fraction to a fresh 1.5 mL tube. Remove excess gradient as the fractions lower down the gradient are accessed.
- If DNA extraction from a fraction containing very low cell numbers is required (e.g., for density-TraDISort), subculture the fraction by following the protocol from step 6.2 for optional outgrowth to obtain more cells.
NOTE: There will be a low level of carryover from cells at the top of the gradient into lower fractions, as fractions are removed from top to bottom. Minimize this by working carefully so as not to mix the gradient, by using a needle to remove lower fractions, and by performing re-purification of very low abundance samples where carryover may affect results.
- Transfer cells from the low-abundance fraction recovered in steps 6.1.1–6.1.2 into 5 mL of appropriate liquid media. Place in an incubator and grow for 2 h at 37 °C.
- After 2 h of growth or when the sample has reached an OD600 of 1, transfer the culture to a 15 mL tube. Centrifuge the tube for 10 min at 3,200 x g in a centrifuge with swing out buckets and aerosol tight lids. Discard the supernatant
- Resuspend the cell pellet in 1 mL of 1x PBS.
- Prepare a fresh single-concentration density gradient in a 5 mL polypropylene tube. Use the gradient concentration from just above the location of the fraction in the original gradient (e.g., for purification of the ΔslyA mutant shown in Figure 2D, 15% density gradient medium would be used).
NOTE: This re-purification step is optional.- Apply the cells to the top of the gradient and centrifuge as described in steps 2.2 and 5.1.
- Remove the tubes from the centrifuge and place in an appropriate rack. Photograph the gradient and recover the fraction for DNA extraction as described in 6.1–6.1.2.
NOTE: The sample is now ready for DNA extraction or other downstream applications.
7. Alternative Method for Gradient Preparation from Bottom (Most Concentrated) to Top (Least Concentrated) Using a Pipette
- Use the gradient dilutions determined in step 2.2.3. Using a 1,000 µL pipette, add 1 mL of the densest gradient dilution to a 5 mL tube.
- Using a 200 µL pipette, add 200 µL of the next dense gradient dilution very slowly to the top of the layer in the tube, so as not to mix the interface. Add another 200 µL and then the final 600 µL. Adding multiple smaller volumes gives more control over pipetting speed and prevents mixing of the interface. If the interface mixes, discard and start again.
- Repeat step 7.1.1 with the third, least dense gradient concentration if three dilutions are being used. The tube should now have gradients of either 2 mL or 3 mL depending on the results from step 2.2.3.
- Go to step 5 of the protocol to add cells and continue the experiment.
8. Measurement of Capsule Amount by Uronic Acid Assay
- Adjust the OD600 of each recovered fraction to 4.0 by dilution with PBS. Proceed with quantification of uronic acids as previously published1.
Results
Representative results are shown in Figure 2. The exact result to expect will depend on the bacterial species, the set-up of the density gradients, and whether the user is examining a single strain or a pool of mutants. Most strains will migrate to a single location within a gradient, as shown in Figure 2A and 2D. Applying the method to a bacterial mutant library will give rise to a major band above the gradient, a less dense band distributed through the uppermost layer of the gradient, and a minor acapsular fraction at the bottom (Figure 2B). These fractions differ in capsule amount as shown by an assay for uronic acids (Figure 2B). Transposon insertion sequencing of individual fractions results in clear localization of specific mutants within different gradient fractions, as shown for the capsule biosynthesis locus of K. pneumoniae ATCC43816 (Figure 2C). Representative results for pure cultures of K. pneumoniae NTUH-K2044 and S. pneumoniae, and different capsule biosynthesis or regulatory mutants, are shown in Figure 2D.

Figure 1: Schematic of the density centrifugation method to separate bacteria based on capsule, and its applications. (A) An electron microscopy image of a capsulated Klebsiella pneumoniae cell. The capsule is visible as a dense layer on the outside of the cell. (B) Applications of density centrifugation to the study of capsulated bacteria. (Bi) Density separation can be used to generate high-, low-, and no-capsule fractions of a transposon mutant library and followed by transposon insertion sequencing to define genes that influence capsule production. (Bii) Purification of capsulated bacteria from a complex sample. (Biii) Use of density-based separation for rapid comparisons of capsule amount between samples. This method also allows the visualization of heterogenous capsule production in bacterial populations, as shown in (Biii). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative results. (A) Example of the output from mini-gradient tests. Two different K. pneumoniae strains were centrifuged on 1 mL of 15% density gradient medium. The hypermucoviscous NTUH-K2044 strain is retained above the density gradient medium layer, while ATCC43816 (which makes less capsule) migrates to the bottom of the layer. (Bi) Use of a density gradient to separate a transposon mutant library into three fractions. Note that the bottom fraction contains a low proportion of mutants and is not visible on this picture. (Bii) Validation of different capsule amounts in the top, middle, and bottom fractions using an assay for uronic acids. Cells from the top, middle, and outgrown bottom fractions were isolated and resuspended in PBS to an OD600 of 4, then capsule polysaccharides extracted and uronic acids measured1. (C) Example density-TraDISort results. Mutation locations identified are shown by blue lines above the chromosome diagram. Mutants lacking capsule can be identified as those that are present in the input library but are depleted in the top fraction while being enriched in the bottom fraction, as shown here for the capsule biosynthesis locus8. (D) An example of using density gradient centrifugation to compare capsule amount between wild type and mutant strains of K. pneumoniae NTUH-K2044 and S. pneumoniae 23F. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Capsule is an important virulence factor in many bacterial species including K. pneumoniae3, Streptococcus pneumoniae9, Acinetobacter10, and Neisseria11 species. Although various methods exist for quantification and visualization of bacterial capsules, at present there is no widely used method to physically separate capsulated and non-capsulated cells. In this article, we have demonstrated a robust method for capsule-based separation of bacterial populations, with multiple potential applications in conjunction with different upstream or downstream protocols.
The presence of a surface capsule can reduce bacterial cell density, which allows separation by density gradient centrifugation (Figure 2D). We have validated this method in K. pneumoniae NTUH-K204412 and ATCC4381613 as well as in Streptococcus pneumoniae 23F14 and its Δcps mutant15. This method uses Percoll16 as the main constituent of the density gradient, which is a suspension of coated colloidal silica particles that has low viscosity and no toxicity towards bacteria - in principle, other substances meeting these criteria could be used to establish the density gradient.
It can be challenging to ensure that layers of different density do not mix when constructing density gradients, and if mixing does occur, the separation method will not give clean results. We have included two alternative methods for pouring the gradients, using a needle or a pipette — both are effective, and which method to use is simply a matter of preference. For all steps that involve pipetting a substance (either a bacterial suspension, or a more dilute gradient layer) above a gradient layer, pipetting multiple aliquots of smaller volumes can make it easier to achieve a sharp interface without any mixing of layers.
A limitation of this protocol is that its performance with other bacterial species cannot be guaranteed. Therefore, it is critical when examining a new bacterial species or strain to validate the density-based separation using an additional, independent capsule quantification method. Visualizing the bacteria present in each fraction by microscopy with appropriate capsule stains is a reliable method for which detailed protocols are available17. Alternatively, capsules containing uronic acids (such as those of Escherichia coli and K. pneumoniae) can be quantified by a specific assay as shown in Figure 2B1. The centrifugation-based mucoviscosity test is not suitable as an independent validation method, as this assay also depends on the density of the bacterial cells.
Another limitation of this method is that capsule production is very sensitive to culture conditions, and even small changes to growth medium, temperature, or aeration may affect the results of this assay. To minimize this issue, researchers can use a defined growth medium or a batch-consistent complex medium, keep all other growth parameters identical between experiments, and include appropriate control strains to enable the interpretation of unexpected results. Some bacterial capsules are fragile and can shear away from the cell when cultures are pipetted. To avoid the shearing of capsules, cultures should be centrifuged and resuspended no more than twice during preparation for loading on the gradient. If loss of capsule during concentration of the cultures remains problematic, bacterial cultures can be applied to a density gradient directly, with a larger volume of bacterial suspension added if necessary for visualization.
Future applications of this method are to apply it to other bacterial species, and to use this separation in conjunction with different upstream and downstream technologies. In addition to density-TraDISort8, we suggest that density gradient separation of capsulated bacteria could be used for isolation of mutants with altered capsule, for purification of capsulated cells from mixed cultures or complex samples, and for rapid profiling of capsule production in multiple strains. Finally, this technology could be used to examine other bacterial phenotypes such as aggregation.
Disclosures
The authors have no financial interests to disclose.
Acknowledgements
We thank Jin-Town Wang and Susannah Salter for supplying strains, and members of the Parkhill group for helpful discussions. This work was funded by the Wellcome Sanger Institute (Wellcome grant 206194), and by a Sir Henry Wellcome postdoctoral fellowship to F.L.S. (grant 106063/A/14/Z). M.J.D. is supported by a Wellcome Sanger Institute PhD Studentship.
Materials
| Name | Company | Catalog Number | Comments |
| Percoll | GE Healthcare | 17-0891-01 | |
| Centrifuge 5810R with Rotor A-4-81 and 500 mL buckets | Eppendorf | 5810 718.007 | |
| Adapters for 15 mL tubes | Eppendorf | 5810 722.004 | |
| Fixed andgle rotor F-34-6-38 | Eppendorf | 5804 727.002 | |
| 2.6 to 7 mL tube adapter | Eppendorf | 5804 739.000 | |
| Centrifuge 5424 including Rotor FA-45-24-11 | Eppendorf | 5424 000.460 | |
| 2 mL tubes | Eppendorf | 0030 120.094 | |
| 1.5 mL tubes | Eppendorf | 0030 120.086 | |
| 5 mL polypropylene round bottom tube | Falcon | 352063 | |
| 1 mL disposable syringe Luer slip | Becton Dickinson | 300013 | |
| AGANI Needle 21G Green x 1.5" | Terumo | AN 2138R1 | |
| P1000 pipette and tips | |||
| P200 pipette and tips |
References
- Favre-Bonté, S., Licht, T. R., Forestier, C., Krogfelt, K. A. Klebsiella pneumoniae capsule expression is necessary for colonization of large intestines of streptomycin-treated mice. Infection and Immunity. 67 (11), 6152-6156 (1999).
- Bachman, M. A., et al. Genome-wide identification of Klebsiella pneumoniae fitness genes during lung infection. mBio. 6 (3), 1-9 (2015).
- Paczosa, M. K., Mecsas, J. Klebsiella pneumoniae: Going on the offense with a strong defense. Microbiology and Molecular Biology Reviews : MMBR. 80 (3), 629-661 (2016).
- Shon, A. S., Bajwa, R. P. S., Russo, T. A. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae. Virulence. 4 (2), 107-118 (2014).
- Fang, C. -. T., Chuang, Y. -. P., Shun, C. -. T., Chang, S. -. C., Wang, J. -. T. A novel virulence gene in Klebsiella pneumoniae. strains causing primary liver abscess and septic metastatic complications. The Journal of Experimental Medicine. 199 (5), 697-705 (2004).
- Lai, Y. -. C., Peng, H. -. L., Chang, H. -. Y. RmpA2, an activator of capsule biosynthesis in Klebsiella pneumoniae CG43, regulates K2 cps gene expression at the transcriptional level. Journal of Bacteriology. 185 (3), 788-800 (2003).
- Menck, K., et al. Isolation of human monocytes by double gradient centrifugation and their differentiation to macrophages in Teflon-coated cell culture bags. Journal of Visualized Experiments. (91), 1-10 (2014).
- Dorman, M. J., Feltwell, T., Goulding, D. A., Parkhill, J., Short, F. L. . The capsule regulatory network of Klebsiella pneumoniae defined by density-TraDISort. , (2018).
- Geno, K. A., et al. Pneumococcal capsules and their types: Past, present, and future. Clinical Microbiology Reviews. 28 (3), 871-899 (2015).
- Weber, B. S., Harding, C. M., Feldman, M. F. Pathogenic Acinetobacter: From the cell surface to infinity and beyond. Journal of Bacteriology. 1986 (6), 880-887 (2016).
- Mubaiwa, T. D., Semchenko, E. A., Hartley-Tassell, L. E., Day, C. J., Jennings, M. P., Seib, K. L. The sweet side of the pathogenic Neisseria.: The role of glycan interactions in colonisation and disease. Pathogens and Disease. 75 (5), 1-9 (2017).
- Wu, K. -. M. M., et al. Genome sequencing and comparative analysis of Klebsiellapneumoniae. NTUH-K2044, a strain causing liver abscess and meningitis. Journal of Bacteriology. 191 (14), 4492-4501 (2009).
- Broberg, C. A., Wu, W., Cavalcoli, J. D., Miller, V. L., Bachman, M. A. Complete genome sequence of Klebsiellapneumoniae. Strain ATCC 43816 KPPR1, a rifampin-resistant mutant commonly used in animal, genetic, and molecular biology studies. Genome Announcements. 2 (5), (2014).
- Croucher, N. J., et al. Role of conjugative elements in the evolution of the multidrug-resistant pandemic clone Streptococcuspneumoniae Spain23F ST81. Journal of Bacteriology. 191 (5), 1480-1489 (2009).
- Croucher, N. J., et al. Selective and genetic constraints on Pneumococcal serotype switching. PLoS Genetics. 11 (3), 1-21 (2015).
- Pertoft, H., et al. Density gradients prepared from colloidal silica particles coated by polyvinylpyrrolidone (Percoll). Analytical Biochemistry. 88, 271-282 (1978).
- Breakwell, D. P., Moyes, R. B., Reynolds, J. Differential staining of bacteria: Capsule stain. Current Protocols in Microbiology. 15 (1), 1-4 (2009).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved