Method Article
Probing Nicotinic Acetylcholine Receptor Function in Mouse Brain Slices via Laser Flash Photolysis of Photoactivatable Nicotine
In This Article
Summary
This article presents a method for studying nicotinic acetylcholine receptors (nAChRs) in mouse brain slices by nicotine uncaging. When coupled with simultaneous patch clamp recording and 2-photon laser scanning microscopy, nicotine uncaging connects nicotinic receptor function with cellular morphology, providing a deeper understanding of cholinergic neurobiology.
Abstract
Acetylcholine (ACh) acts through receptors to modulate a variety of neuronal processes, but it has been challenging to link ACh receptor function with subcellular location within cells where this function is carried out. To study the subcellular location of nicotinic ACh receptors (nAChRs) in native brain tissue, an optical method was developed for precise release of nicotine at discrete locations near neuronal membranes during electrophysiological recordings. Patch-clamped neurons in brain slices are filled with dye to visualize their morphology during 2-photon laser scanning microscopy, and nicotine uncaging is executed with a light flash by focusing a 405 nm laser beam near one or more cellular membranes. Cellular current deflections are measured, and a high-resolution three-dimensional (3D) image of the recorded neuron is made to allow reconciliation of nAChR responses with cellular morphology. This method allows for detailed analysis of nAChR functional distribution in complex tissue preparations, promising to enhance the understanding of cholinergic neurotransmission.
Introduction
Cholinergic signaling modulates numerous brain processes, including attentional control, volitional movement, and reward1,2. Drugs that enhance acetylcholine (ACh) transmission are used to treat cognitive impairment associated with Alzheimer's disease, implying an important role for cholinergic systems in cognition3. An improved understanding of cholinergic receptors and circuitry in healthy and diseased states could lead to better therapeutic approaches for several neurological diseases/disorders.
Nicotinic ACh receptors (nAChRs) are a family of ligand-gated ion channels that flux cations in response to endogenous ACh or exogenous nicotine from tobacco products. Given the fact that they were among the very first neurotransmitter receptors to be described4, nAChR pharmacology and location in muscle fibers is well-understood for muscle-type receptors. By contrast, comparatively little is known about the pharmacology and subcellular distribution of native nAChRs in the brain. This gap in knowledge was recently addressed by developing a novel chemical probe that allows for spatially restricted and rapid activation of nAChRs in brain tissue during cellular imaging and electrophysiological recording5. Here, the key methodological steps involved in this approach are described, with the overall goal of enhancing the ability to connect nAChR function with neuronal structure.
Photoactivatable nicotine (PA-Nic; chemical name: 1-[7-[bis(carboxymethyl)- amino]coumarin-4-yl]methyl-nicotine) can be photolyzed with ~405 nm laser flashes to efficiently release nicotine5,6. Prior to uncaging, PA-Nic is stable in solution and exhibits no untoward pharmacological or photochemical features5. After photolysis, released nicotine predictably activates nAChRs and the uncaging byproducts are pharmacologically inert5. A continuous-wave laser is used as the photolysis light source with an output power >1 mW measured at the sample. When localized, targeted photo-stimulation is combined with the ability to locate cellular membranes with 2-photon laser scanning microscopy (2PLSM), and the two key advantages of this approach are fully realized: photolysis speed and spatial precision.
In most respects, photolysis of PA-Nic is superior to other methods of delivering nAChR ligands to receptors within brain slices. Such approaches include bath application7 and local drug delivery via a puffer pipette8. Whereas the former approach tends to over-emphasize the long-term effects of the applied drug, the latter approach can suffer from variability in response kinetics between trials and across cells. Neither of these alternative approaches can adequately distinguish receptor activities in different cellular locations from the same neuron. Optogenetically-activated release of ACh has been used for investigation of native nAChRs9,10,11, but it has not proven useful for mapping subcellular nAChR locations. Furthermore, most studies utilizing this approach have relied on a ChR2-expressing bacterial artificial chromosome transgenic mouse with abnormal cholinergic transmission12,13,14,15,16,17.
PA-Nic photolysis is not the only optical approach for studying cholinergic receptors. A caged carbachol was used to functionally map ACh receptor activities in cultured cells18 and brain slices19, but was not commercially available for comparative studies during the development of PA-Nic. A ruthenium bis(bipyridine)-nicotine complex (RuBi-nicotine) was reported to allow for nicotine uncaging20, but commercial preparations of RuBi-nicotine proved inferior to PA-Nic in a head-to-head comparison study5. It may be useful to repeat such comparative experiments with non-commercial, highly purified RuBi-nicotine, as its visible absorption could compliment PA-Nic's features for cholinergic studies. Finally, nAChRs have also been optically manipulated using a combination of photo-switchable ligands and genetically-modified receptors21. This approach is complementary to PA-Nic photolysis in brain tissue, with the ability/requirement of genetic targeting of the modified nAChR seen as both an advantage and a drawback.
Several key requirements of this approach should be noted. First, an appropriate visualization method is needed to accurately locate the neuronal membrane. Imaging with conventional epi-fluorescence microscopy may be sufficient when studying cultured cells, but for recording from neurons in brain slices or other thick tissue preparations, 2PLSM or confocal microscopy is a requirement. Second, a suitable method is needed to position the photolysis laser beam. This approach utilizes a dual-galvanometer scan head with two independent x-y mirrors for raster scanning of the imaging beam and point photoactivation using the uncaging laser beam22,23,24. Other, more limited solutions are possible, such as (1) a single-galvanometer scan head that alternatively raster scans the imaging beam and the uncaging beam, or (2) simply directing the uncaging beam to the center of the field of view such that the cell is brought to this position for flash photolysis. Third, a system is required for simultaneous electrophysiological recording if one wishes to collect physiological signals during experiments. The above requirements can be met with a suitable all-optical imaging technique, as recently described5. Below, a detailed protocol is included that describes the key steps of this approach.
Protocol
Work pertaining to brain slice preparation was reviewed and approved by the Northwestern University Animal Care and Use Committee (protocol #IS00003604).
CAUTION: Lasers used for point photo-stimulation are visible Class IIIb lasers which have the potential to cause harm to the eyes. 2PLSM requires a near-infrared (NIR) Class IV laser (>500 mW), which has the potential to cause serious harm to the eyes and even burns in other tissues. Proper laser beam containment, system interlocks, plus engineered and administrative controls are required to ensure safe operation of laser-based equipment. Always seek out local laser safety personnel when working with lasers.
1. Calibration and Verification of the Uncaging Laser(s)
- Quantify laser power to be delivered to the sample.
- Turn on the 405 nm laser (100 mW maximum power with 5 V control signal) and let the laser system warm up for about 10 min.
NOTE: The laser is still shuttered (with 0 V drive) and there is no output power until the laser is sent a control voltage to modulate the output power. - Place a power meter in the tissue sample plane or in place of the condenser lens. Manually center the meter relative to the optical path/objective lens.
- Set the meter to the correct wavelength range (400-1100 nm). Zero the meter by depressing the appropriate button.
- Using software controls, select 100 (out of max 1000; 1000 = 5 V) for the 405 nm laser power, which sets the laser to 10% of full power. If desired, the laser control voltage can also be fed into the PrairieView system via VoltageRecord to provide a digital record of the command signal timing and power level.
- Record the reading from the power meter.
- Select 150 (15% of max 1000) for the 405 nm laser power output and record the reading from the power meter. Repeat this for the following output powers to collect a laser power curve: 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, and 1000.
- Turn on the 405 nm laser (100 mW maximum power with 5 V control signal) and let the laser system warm up for about 10 min.
- Calibrate the uncaging laser galvanometers. Execute the following steps whenever there is a change to the optical components of the system, whenever there is a concern about accurate spot positioning, or regularly every month.
- Install the 60x water-dipping microscope objective lens that will be used in photostimulation and imaging experiments. In the acquisition/imaging software, select the 60x objective lens and an optical zoom setting of 1 (see step 3.1.4.2.).
- Mark a filled circle onto a clean glass microscopy slide with a red permanent marker. Place the slide on the microscope stage, with marker toward the objective.
- Pre-focus the microscope on the red marker region with a 4x or 10x objective. Add 1-2 mL of water to the top of the red marker dot/spot and then switch to the 60x objective and submerge the objective into the water. Focus the objective lens on the thin red marker region.
- Switch to 2-photon laser scanning. For most systems, move turret to position #1, move the trinocular prism out of the light path, move the scan head mirror to front, and set the laser wavelength to ~900 nm. Select the 512 x 512 pixel box option for the image acquisition parameters, which is the default pixel element for the stimulation mirror calibration routine.
- Start system scanning with an imaging laser power greater than minimum and fine tune the objective focus onto the thin red marker fluorescence layer. Choose a field in the fluorescence field clear of debris and evenly coated with marker.
NOTE: The setup in this protocol uses PrairieView 5.4 acquisition/imaging software. - Open the Uncaging Galvo Calibration function within the Tools – Calibration/Alignment menu of the software. Walk through the burn spots tutorial for the spatial calibration of the second galvanometer mirror pair.
- Within the burn spots tutorial, choose the 405 nm laser, select a laser stimulation power of 400 and a stimulation duration of 20 ms. This should yield small (~1-5 µm diameter) holes in the red marker.
NOTE: Settings such as ~2-4 mW and 1-10 ms are typically used, but the settings are determined by the sample. PA-Nic photo-stimulation power settings are likely to be far lower than the power required during calibration to ablate a visible hole in the red marker slide. This calibration routine is useful for locating photostimulation spots but should not be used to infer absolute photostimulation volumes during physiological responses. - Select Update to stimulate and refresh the image after the center spot burn and move the round red indicator to the actual spot location. Do this for the center spot, the right center spot, the lower center spot, and finally for a grid of nine spots (all corners and edges plus the center of image).
NOTE: The center, right, and lower corrected spot locations determine the stimulation galvanometer voltages to define the true center and X and Y scaling factors to match the stimulation mirror pair to the imaging mirror pair. The software will scale, and update, all subsequent MarkPoints experiments performed at zoom settings different from the spatial calibration zoom.
- Within the burn spots tutorial, choose the 405 nm laser, select a laser stimulation power of 400 and a stimulation duration of 20 ms. This should yield small (~1-5 µm diameter) holes in the red marker.
- Test the calibration by opening the MarkPoints window and manually activating the stimulation parameters in defined spot(s) in a new area of the sample. Ensure that the correct, latest calibration file is loaded into the MarkPoints window. Activate the MarkPoints/Group or MarkPoints Series feature at a defined spot(s) or utilize the Live/Ablation feature to right/left click the mouse anywhere on the image during live scanning to apply a test pulse and to verify correct calibration.
NOTE: The laser burn spot should now be perfectly centered on the MarkPoints indicator. - Monitor and record the activation of the laser pulse power and temporal duration by tapping off the drive voltage into the VoltageRecord program (see step 1.1.4). Similarly, record the position of each stimulation spot using a scaled voltage signal from the feedback signals derived from the pair of photo-stimulation galvanometer mirrors.
2. Preparation of Photoactivatable Nicotine (PA-Nic)
- Retrieve an aliquot of lyophilized photoactivatable drug from storage.
NOTE: The following protocol is specific for PA-Nic; adjust as needed for other photoactivatable drugs. Although PA-Nic demonstrates exceptional stability5, take reasonable precautions to protect it from exposure to bright light during preparation and/or experiments. This can be accomplished by simply working in low light; restricting to red filtered light is not necessary. - Perform local application of PA-Nic.
- Pull a glass micropipette with an opening diameter of 20-40 µm with a programmable pipette puller.
- Filter ~1 mL of recording solution with a 0.22 µm filter. Resuspend a quantity of PA-Nic in filtered recording solution to yield a final concentration of 2 mM. For example, dissolve a 100 nmol lyophilized aliquot in 50 µL of filtered recording solution.
NOTE: A suggested recording solution composition can be found in recent publications5,6 employing PA-Nic photolysis. - Back-fill the local application pipette with 50 µL of 2 mM PA-Nic.
- Secure the local application pipette into a pipette holder mounted on a micromanipulator. Connect the pipette holder via appropriate tubing to a pressure ejection system capable of sustained low-pressure application (1-2 psi).
- Using the micromanipulator, maneuver the local application pipette into the extracellular recording solution and position the pipette tip slightly above the mouse brain tissue located ~50 μm from the cell of interest. Consult a previous report for a detailed protocol of mouse brain slice preparation and patch clamp recordings8.
- Check the application parameters by briefly applying pressure (1-2 psi). There should be minimal to no displacement of the cell of interest. If significant movement does occur, reposition the local application pipette further away (in the lateral and/or axial direction) from the cell of interest.
- After achieving stable whole cell patch clamp (details for which are included in a prior publication8), turn on low pressure (1-2 p.s.i.) application using the appropriate manual switch on the pressure ejection device. Saturate the tissue surrounding the cell with PA-Nic for 1-2 min before proceeding to the next step.
- Perform bath application (superfusion) of PA-Nic to the brain slice.
- Dissolve a quantity of PA-Nic in a volume of recording solution appropriate for continuous recirculation to yield a final concentration of 100 μM. For example, dissolve a 1 μmol aliquot into 10 mL of recording solution using a standard 15 mL tube.
- Begin recirculation of the PA-Nic solution at a rate of 1.5-2 mL/min by opening the appropriate flow control in the perfusion system. Recirculation occurs for the duration of the recording. To conserve valuable drug, minimize the recirculation volume by using tubing with a minimal inner diameter, and/or by shortening the overall length of tubing used in the perfusion system.
NOTE: By taking these steps, the volume for bath recirculation can be reduced to 5 mL of PA-Nic solution. PA-Nic solutions can often be used for two consecutive recording days within the same week if stored protected from light at 4 °C. - During recirculation, continuously bubble the solution with carbogen (5% O2, 95% CO2) and maintain the bath temperature at 32 °C.
- Retain the brain slice in recording solution while working with PA-Nic in low light conditions.
3. Imaging Neurons with 2-Photon Laser Scanning Microscopy
- Perform live visualization of the cell.
- Identify/visualize a medial habenula (MHb) neuron using transmitted light or infra-red differential interference contrast (IR/DIC) optics and a video camera and establish a stable whole cell patch clamp recording. Refer to a previous protocol for details on patch clamp recordings from neurons in acutely-prepared mouse brain slices8.
- After establishing the high-resistance (>1 GΩ) cell-attached configuration, but before break-in, switch the set-up and software to laser scanning mode.
- After break-in, use laser scanning to verify that an imaging dye (diluted to a final concentration of 100-200 µM into a standard intracellular pipette solution described previously8) is passively (by diffusion) filling the neuron. Allow the dye (e.g., Alexa Fluor 488 in green photomultiplier tube [PMT] channel, PMT 2; or Alexa Fluor 568 or 594 in red PMT channel, PMT 1) to fill the cellular compartments for at least 20-30 min before attempting experiments that require visualization of any cellular compartments outside of the soma.
NOTE: Distal compartments (dendritic structures, spines, axons, etc.) may require 30-40 min to fill completely25. - Use the software Live Scan function to visualize the neuron and subcellular compartment of interest. Choose imaging parameters that allow for accurate live visualization of neuronal features. Manipulate various settings to affect or alter the display visualization (contrast), resolution, signal-to-noise (S/N) ratio, and image frame acquisition time:
- Look-up-table (LUT). Open the LUT window using the appropriate icon on the side of any image window. Once open, adjust the LUT floor (min) and ceiling (max) setting of the specific image channel to enhance visualization of signal contrast shown on the screen. Lower the maximum value to ~1000 (out of 4096, 12-bit detection), which will help to pull out the dimmer signals when first searching for cells, signal, and structure.
NOTE: These settings only affect the displayed signal, not the detected/recorded values. Human eyes can typically only make out contrast to ~50 grey levels26. - Optical zoom. Use software controls to select 1X optical zoom and use panning controls to locate the desired area in the tissue. This zoom setting yields the largest square, scanned field of view, and sends the largest voltages/scan angles, to the galvanometer mirrors.
NOTE: The default configuration is for a 12 mm x 12 mm field of view inside the scan head which translates to 12 mm divided by objective magnification inside the sample. Therefore, a 60x objective lens yields scanned images of 200 µm per side at 1x optical zoom. Higher optical zoom values scan less area. 2x optical zoom is often the most useful setting for visualizing whole neurons. 4x can be useful for visualizing subcellular aspects of neurons. - Number of pixels. To complement 1x optical zoom, select 1024 x 1024 pixels per line using software controls. Set the number of pixels per line in the captured and displayed image to not lose details possible from the objective lens. Use the following practical pixel values for a 60x/1.0 numerical aperture (NA) objective: 1024 x 1024 for zoom 1, 512 x 512 for zoom 2, and 256 x 256 for zoom 4. The end pixel size (~0.17 µm; 12 mm/magnification/zoom/pixels) should be half, or less, of the lateral resolution defined by the objective lens.
NOTE: The image resolution is only defined by the laser wavelength and the objective NA (0.4 µm resolution from two-photon excited [2PE] with 920 nm and a 1.0 NA objective)27. The criteria for the full excitation NA, as listed on the lens, is that the 1/e2 intensity of the laser beam diameter matches (or “fills”) the entrance pupil (2 x tube lens focal length x NA / magnification) of the objective lens. The tube lens in the system described here has a focal length of 180 mm. - Pixel dwell time. Use software controls to select 4 µs for the pixel dwell time, a useful default value.
NOTE: Changing the pixel dwell time does not change the mean signal detected; it only affects the intra-pixel averaging and these changes can be visualized through the image quality via the S/N. The image pixel dwell time is always a multiple of 0.4 µs units, and for larger dwell times the 12-bit-limited intensity value of each image pixel is the average of the 0.4 µs samples. Since the S/N ratio improves as the square root of the number of samples per pixel dwell time (4 µs equals ten samples, or 3.16-fold improvement in S/N), the improvement in image quality reaches diminishing returns for values much larger than 12 µs. - Scan Rotation and Region of Interest (ROI). Set the image angle to 0° rotation using software controls (no action may be needed, as 0° rotation is the default setting for most imaging systems). If the sample is placed in an “upside-down” orientation, select 180° rotation to “flip” the image.
NOTE: Rotating the image, at any given angle, can provide a better fit for the entire area of interest of a filled cell. Rotation can also yield a clearer basis for aligning structural changes and for performing subsequent analysis. Selecting a region of interest within the scanned image at a given zoom (step 3.1.4.2) setting retains the native pixel number (step 3.1.4.3), but the restriction in total number of area and pixels can dramatically increase the frame rate, providing improved temporal resolution of signal changes. - Frame Averaging. Select a starting frame average setting of 2 frames using software controls.
NOTE: The final image contrast (S/N) is defined by the total photons collected/detected within the signal pixels of the image. Averaging of multiple image frames can improve the S/N ratio, provided the sample does not move or is not bleached during imaging. The signal of interest remains the same value during frame averaging while the noise in the image is reduced by the square root of the number of frames averaged. Small structures in fluorescence images often require inter-pixel averaging, combining pixels within the image (often called ROIs), and/or frame averaging. Frame averaging increases scanning time by the number of images one chooses to average.
- Look-up-table (LUT). Open the LUT window using the appropriate icon on the side of any image window. Once open, adjust the LUT floor (min) and ceiling (max) setting of the specific image channel to enhance visualization of signal contrast shown on the screen. Lower the maximum value to ~1000 (out of 4096, 12-bit detection), which will help to pull out the dimmer signals when first searching for cells, signal, and structure.
- Use the Pan Control, Scan Rotation, and Optical Zoom tools to orient the sample location while scanning. If motor stage manipulation is necessary to position the sample, avoid large step sizes to the X, Y, and Z axis to prevent objective/condenser collisions, vibrations, or exposure of the laser beam to reflective surfaces.
- Collect a Z-stack. Using the Z-series tool, select a start and stop position that contains the cell of interest. Select a step size (1 µm) and then consecutively image the neuron in every Z-plane that contains the cell.
NOTE: Z-stack acquisition settings will vary between neuron type and filling dye. Optimal parameters for Z-stack acquisition should be determined independent of parameters used for live imaging. Z-stack acquisition can be performed before and/or after optopharmacology experiments. If possible, perform Z-stack acquisition after optopharmacology to avoid any cellular damage induced from 2PLSM and to allow for optimal dye filling of small cellular compartments.
4. Laser Flash Photolysis During Electrophysiological Recordings
NOTE: Applying 405 nm or 473 nm laser powers of ≥ 1 mW produces phosphorescence inside the glass of objective and condenser lenses. This generated light is directly related to the laser illumination power; the emission is present in the green and red spectral windows and has excited-state lifetimes in the ms range. This background stimulation artifact is seen in all lenses tested and in water-dipping objective lenses from all major manufacturers of objective lenses. Condenser lenses produce much higher phosphorescence than objective lenses. This “signal” motivates the use of mechanical shuttering for protection of the sensitive gallium arsenide phosphide (GaAsP) PMT cathodes during photo-stimulation events. Using a normally closed mechanical shutter (closed when not actively scanning) represents the best solution for protection of cooled GaAsP PMTs.
- For an hourglass-type photostimulation beam geometry, remove any focusing lenses in the light path optics that would otherwise narrow/constrict the laser beam as it enters the objective entrance pupil.
- Using MarkPoints, select the single spot setting.
NOTE: Other photo-stimulation settings (multiple spots, a grid of spots, spiral scanning) are possible within MarkPoints. Single spot is the simplest. Experimental goals and biological differences may necessitate a different setting. - Update the image using the Live Scan option to briefly image and locate the subcellular area of interest. Periodically update the image to identify any potential small drifts in focus.
- Use software controls to increase the optical zoom (i.e., select a higher optical zoom setting than the current one), if necessary, to visualize small structures (i.e., spines or distal dendrites).
- Place the MarkPoints single spot crosshairs immediately adjacent (~0.5 μm) to the cell membrane. Do not place the photo-stimulation spot directly over a cellular feature, as this could lead to photodamage.
- Set the parameters for photo-stimulation using software controls in MarkPoints. Apply the starting guidelines as follows: 1-50 ms duration, 1-4 mW laser power, and ≥1 trial.
- Select Run MarkPoints to initiate the MarkPoints protocol and observe electrophysiology data acquisition in real time.
- Repeat steps 4.2-4.7 several times to evaluate consistency and stability, or lack thereof, of the response amplitude and kinetics.
Results
For photolysis stimulation, the exposure dose (intensity and time), exposure location, and beam geometry are key variables. The system described in this article is capable of two different photostimulation beams, adjustable via moving a lens in/out of the photostimulation light path before the beam enters the galvanometer system. Without this lens, the photostimulation beam fills the entrance pupil of the 60x/1.0 NA water-dipping [60x WD] objective, producing a near-diffraction-limited, sub-µm spot at the focal plane inside the sample. This is associated with photostimulation light with an hourglass shape, extending above and below the focal spot symmetric with the optical axis. With the lens inserted into the path, photostimulation laser light is focused into the entrance pupil of the objective lens and then exits as a pencil-like beam. This beam, which is expected to be ~10 µm in diameter for a 60x objective, extends uniformly/vertically throughout the sample. In this mode, the light intensity at any given location within the stimulation spot will be ~1% of the near-diffraction-limited small spot intensity. Thus, higher laser powers are typically required when using ~10 µm spot stimulation. For all experiments reported in this article, an hourglass-type photostimulation beam was used.
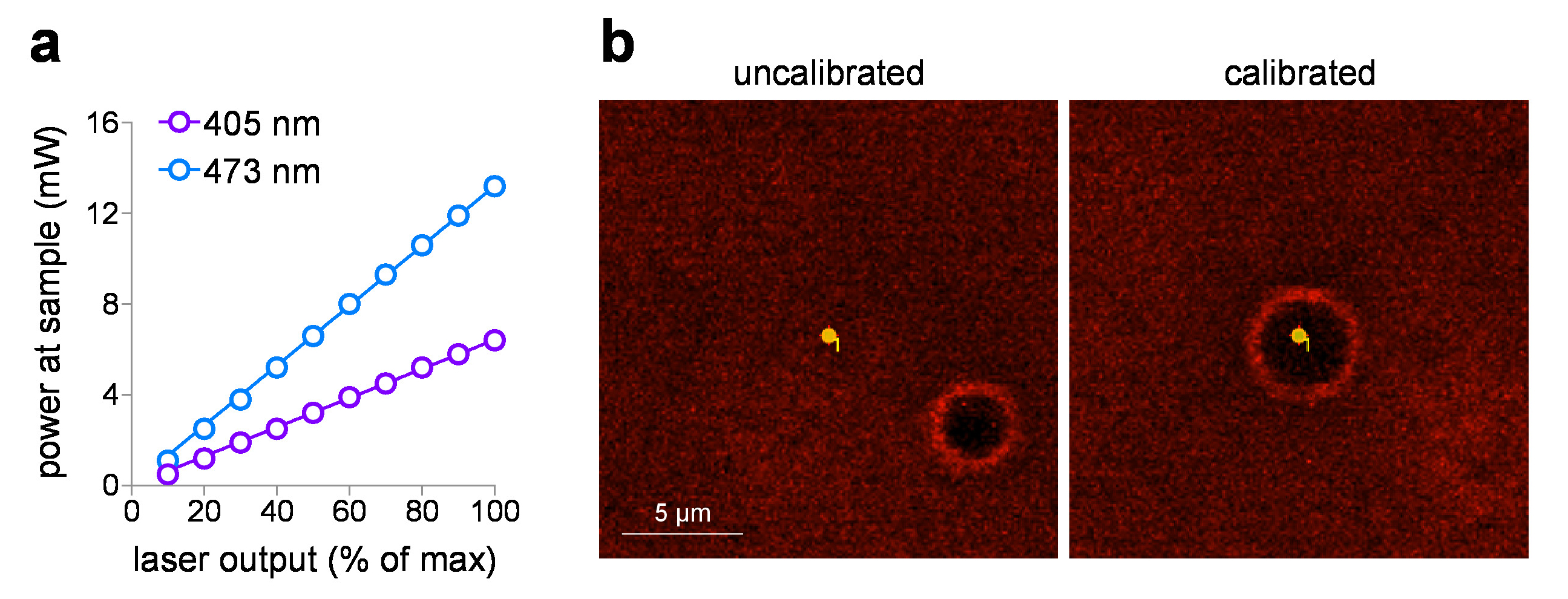
The delivered sample power can be plotted against the input voltage setting, after measuring the laser power at the sample using a power meter. These studies use a 60x WD objective with a working distance of 2 mm, but it is not submerged in water for power measurements to avoid potential damage to the detector element. When objectives with listed NA > 0.95 are measured in air (without immersion fluid), there can be total internal reflection losses at the lens front face element due to the lower index (air). In this case, for a more accurate sample power measurement (to correct for total internal reflection losses), increase the measured power by the 1.0 NA objective (1.0/0.95)2 measured in air. Figure 1a shows a typical input/output plot for 405 nm and 473 nm visible lasers that are incorporated into the laser launch system in this study. These laser systems are ideal for photo-stimulation exposure dose control for the following reasons: (1) they are pre-calibrated to provide directly linear power output relative to input voltage (0-5 V), (2) they provide a silent shutter operation (with no laser output), and (3) they have rapid, sub-ms intensity pulse duration control (0.1 ms response). When using spot photo-stimulation with a laser/galvo system, routine calibration of MarkPoints spots is an essential task. Figure 1b (left panel) demonstrates a system that is out of calibration (desired point to photo-stimulate does not result in accurate stimulation of that point, as indicated by burn-hole location), with a return to accurate positioning of the spot after calibration (Figure 1b, right panel).
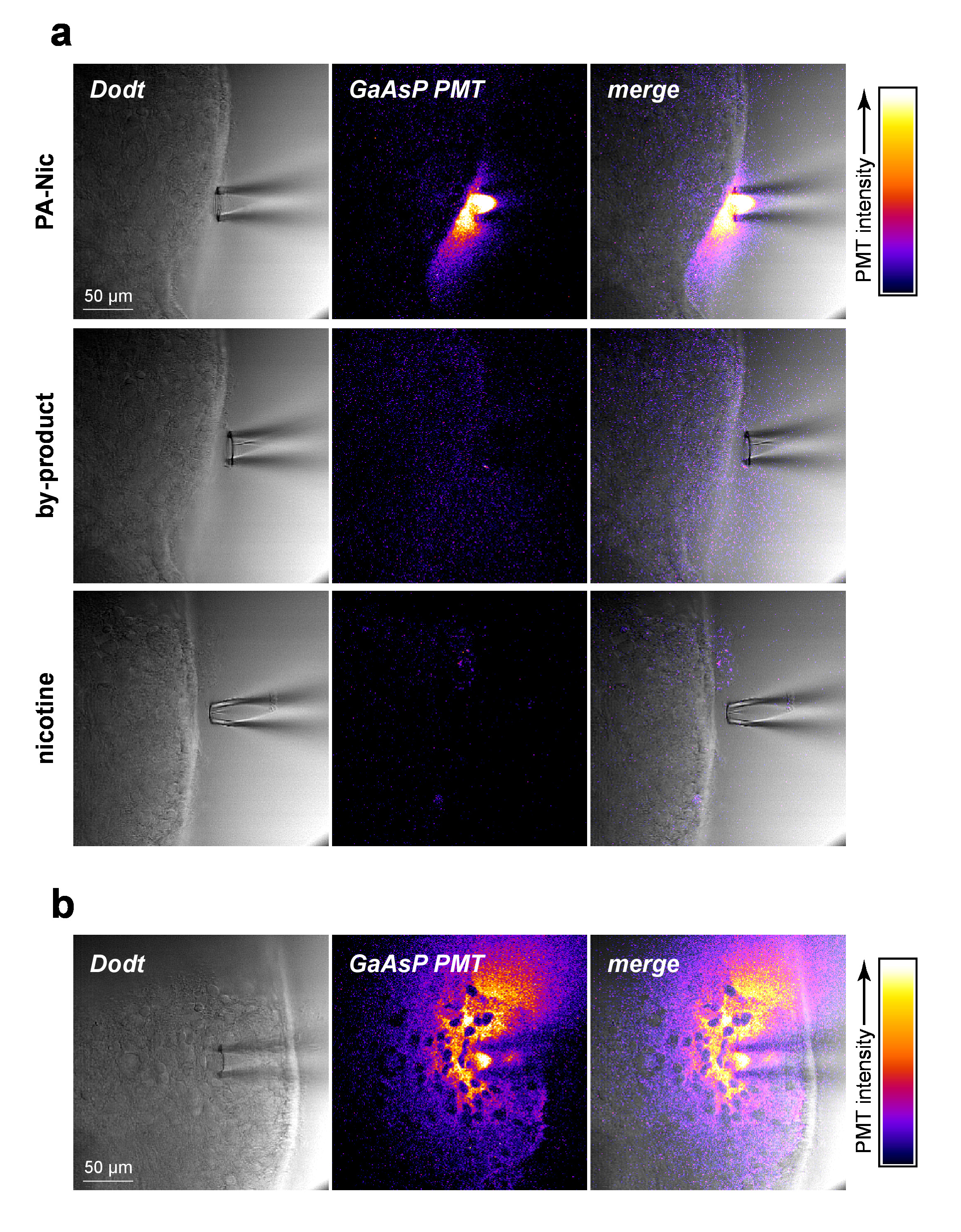
PA-Nic is modestly fluorescent (emission peak at ~510 nm), exhibiting effective excitation between 350-450 nm (1-photon excitation) or 700-900 nm (2-photon excitation)5. To visualize PA-Nic during local application, PA-Nic (1 mM) was applied near brain tissue followed by simultaneous imaging of (1) brain tissue optical sectioned transmission context via Dodt contrast and (2) the emitted fluorescence from excitation (900 nm) of PA-Nic. PA-Nic 2PE fluorescence was easily detectable during pressure ejection from a local application pipette (Figure 2a). Nicotine and a monoalkylcoumarin, 7-carboxymethylamino-4-methyl coumarin, are the main photochemical products of the PA-Nic photolysis reaction5. Using the same imaging settings/parameters that were used for PA-Nic imaging, the tissue was imaged during delivery of either nicotine (1 mM) or 7-carboxymethylamino-4-methyl coumarin (1 mM). No fluorescent signal was detected (Figure 2a, middle and lower panels), demonstrating the specificity of the PA-Nic results. Finally, PA-Nic was applied within brain tissue and PA-Nic fluorescence emission was imaged (Figure 2b). This approach confirms that PA-Nic is present within 100-200 µm of the local application pipette. Together, these data confirm that PA-Nic is effectively delivered to brain tissue via local application.
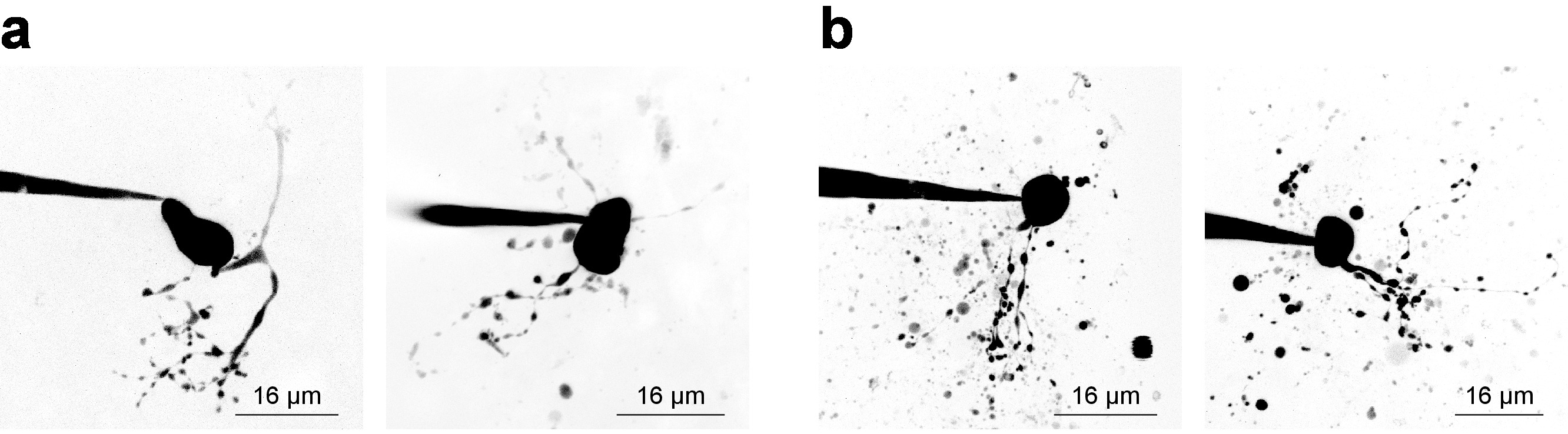
Electrophysiology recordings with simultaneous 2PLSM for visualization of cellular structures requires the investigator to balance considerations for both components of the experiment, and often a narrow time window (~20 min) is available for valid sample data acquisition from a patched cell. Without considering cellular visualization, it is best practice to begin recording as soon as possible after break-in because recording stability tends to decline with time. However, when imaging is a requirement, electrophysiological considerations must allow adequate time for fluorescence concentration increases in small/remote structures. This is exemplified by examining a dye concentration filling curve28, which is sometimes useful to derive when imaging a new cell type. Figure 3 shows several example neurons imaged as Z-stacks via 2PLSM and collapsed into a maximum intensity projection for presentation purposes. Figure 3a shows high-quality images where neuronal morphology appears to be complete, noise is minimized, and debris does not interfere with interpretation of cellular morphology. Figure 3b shows images of lower quality, owing to a lower signal-to-background ratio and substantial debris. This debris often appears as spherical pockets of intense fluorescence, arising from ejection of imaging dye from the patch pipette while approaching the cell. Notably, inclusion of 100 µM PA-Nic in the bath (when performing bath application) tends to reduce the signal-to-background ratio and leads to sub-optimal image contrast. Alexa Fluor 568 or 594 is often quite useful in local application experiments as a finder dye or as a normalizing 2PE reference/normalization signal. An effective wavelength for two-photon excitation of these dyes is ~780 nm27, which allows simultaneous visualization of PA-Nic and identification of cellular compartments. This wavelength, however, does not fully avoid two-photon photolysis of PA-Nic5. Alexa Fluor 488 is advantageous in PA-Nic bath-application experiments; when excited with a suitable wavelength ≥900 nm, two-photon photolysis of PA-Nic5 can be avoided while still maintaining suitable visualization of cellular compartments.
Figure 4 shows example data for localized PA-Nic laser flash photolysis at MHb neurons in brain slices. Figure 4a (upper panels) shows an example of a "reference" image, which is a screen-capture of the last 2PLSM image taken before the MarkPoints protocol was run, overlaid with the photo-stimulation spot location. Figure 4a (lower image panels) shows a zoomed view of the photo-stimulation spot overlaid on the cellular morphology. The corresponding time-correlated electrophysiological response to PA-Nic photolysis is shown in the lower panels of Figure 4a. Previous work demonstrated that these currents are sensitive to nAChR antagonists5. Figure 4b shows representative data from different cells where single spot photolysis was performed at an interval of either 1 s or 10 s. Whereas a 10 s interval allowed sufficient recovery time for the baseline holding current, a shorter 1 s interval led to a gradual increase in the holding current as the protocol proceeded. The increasing current suggests that nicotine did not have enough time to diffuse away from the system with the 1 Hz interval29. Such temporal response analyses must be performed de novo on any new cell type being studied, as the neuropharmacology of nAChRs may differ between cell types.

Figure 1: Photostimulation laser calibration. (a) Photo-stimulation laser power output. Power at the sample plane (through a 60x/1.0 NA water-dipping objective) was measured for 405 nm and 473 nm photo-stimulation lasers at the indicated output setting. (b) Photo-stimulation laser calibration. Screen capture images show the spatial relationship between the intended photo-stimulation spot and the corresponding location where photo-stimulation occurred (burn-hole) before (left) and after (right) running calibration in MarkPoints. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: PA-Nic local application. (a) Detection of PA-Nic from a local application pipette. 1 mM PA-Nic, photolysis by-product, or nicotine were dissolved in ACSF, loaded into a local application pipette, and dispensed onto brain tissue during 2PLSM (900 nm excitation) imaging using the same imaging settings for each drug. Laser scanning Dodt contrast transmission image shows the tissue/pipette whereas a GaAsP cathode PMT was used to capture fluorescence emission. (b) PA-Nic (1 mM) was perfused into brain tissue and imaged via 2PLSM as in (a) to show the lateral spread of PA-Nic using its intrinsic fluorescence. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Acquisition of 2-photon laser scanning microscopy images. (a) Optimal 2PLSM Z-stacks. Two examples of 2PLSM Z-stack maximum intensity projections are shown for MHb neurons with well-resolved dendrites and little to no interfering debris. (b) Sub-optimal 2PLSM Z-stacks. Two examples of 2PLSM Z-stack maximum intensity projections are shown for MHb neurons surrounded by debris (dye expelled from the pipette during cell approach). Such images are more difficult to interpret than images like those shown in (a). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Laser flash photolysis of PA-Nic. (a) MarkPoints reference images and inward currents evoked by PA-Nic photolysis. For one MHb neuron, raw reference images are shown for MarkPoints photo-stimulation trials at a single (indicated) cellular location. Note that for some photo-stimulation locations (the right-most image in this series), the dendritic structure is in focus but the soma and proximal dendrite are not. Below each reference image, the nicotine uncaging-evoked inward current is plotted. (b) Inter-stimulus intervals for PA-Nic photolysis. Exemplar recordings are shown for MHb neurons where nicotine was repeatedly uncaged at the same perisomatic location with an inter-stimulus interval of 1 s or 10 s. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The choice of PA-Nic application/delivery method is the most critical step in this localized photo-stimulation technique. The two methods, bath application and local perfusion, each offer distinct advantages and limitations. The choice is largely impacted by the nAChR functional expression level in the cell type of interest. It is often preferable to use bath application when functional expression levels are high, as bath application allows for a uniform probe concentration surrounding the recorded cell, facilitating data interpretation. Bath application also eliminates the need for a second perfusion pipette in the tissue, making the entire process easier. However, bath application of expensive compounds costs more per experiment.
Commonly, troubleshooting involves trying to understand why no nAChR activation is seen following flash photolysis. When working with a cell type that has not been previously studied with PA-Nic, the investigator should perform local puff-application of ACh or nicotine to determine whether sufficient receptors are functionally expressed5. To validate that the system is capable of detecting photolysis responses, control experiments should be done in medial habenula neurons that express large quantities of receptor30. In this brain area, PA-Nic bath application is possible, which is preferable for validation experiments. Only after performing these validation experiments should one move on to an unstudied cell type. If the experimental system has been validated and responses remain very small or undetectable, it may be warranted to increase the concentration of PA-Nic, increase the flash intensity or pulse duration, add a nAChR positive allosteric modulator to enhance nAChR activity6, or some combination of these.
Occasionally, uncaging responses are too large, with significant nAChR activation resulting in indirect voltage gated Na+ channel activation and unclamped inward currents due to poor space clamp. These artifacts, which completely obscure nAChR inward currents and make data interpretation impossible, can be eliminated by inclusion of QX-314 (2 mM) in the recording pipette. They may also be eliminated by reducing the concentration of PA-Nic or by reducing the flash intensity or pulse duration. In all visible light photo-stimulation experiments, care must be exercised when selecting stimulation sites to avoid unintended stimulation/photolysis above or below the desired focal plane. Additionally and when applicable, the laser power must always be titrated to reproduce physiological responses. It is especially important to be aware of z-axis photostimulation when working with caged ligands, as ligands that are activated above/below the focal spot may still diffuse and interact with the biological system (i.e., receptors) under study.
PA-Nic laser flash photolysis may not be appropriate for all investigators, as several limitations exist. The first is the relatively high cost of a suitable set-up. When working with intact brain slices, uncaging near small-diameter structures like dendrites requires a sophisticated visualization system such as a 2-photon microscope. Aside from the high cost of a Ti:sapphire, tunable IR pulsed laser for performing 2-photon microscopy, a dual-galvanometer system capable of independently positioning two laser beams further increases the system cost. Total system costs can be reduced by using a home-built system if the investigator has sufficient expertise and time to construct, troubleshoot, and maintain such a system. A second limitation often involves low nAChR functional expression, which can be partially mitigated by taking steps as mentioned above, but this may not guarantee success. Typically, if one cannot measure ligand-activated currents following puff-application of agonists, PA-Nic flash photolysis under voltage clamp may not yield acceptable results. A third limitation involves the intrinsic fluorescence of PA-Nic. PA-Nic absorbs ~405 nm light and emits in a similar range as green fluorescent protein (GFP) or Alexa 4885. When PA-Nic concentrations exceed ~1 mM, this fluorescence property can make it challenging to simultaneously visualize neuronal structures. To mitigate this, it is critical to be able to easily control the flow of PA-Nic from the perfusion pipette. Periodically, the PA-Nic flow was stopped to allow fluorescent molecules to diffuse away. This permitted re-imaging of the neuron for checking the spot position of the uncaging beam. A fourth potential limitation to mention involves the use of 405 nm light for photolysis. Shorter wavelengths such as 405 nm are more prone to scattering in complex tissue such as a brain slice. Thus, at a given flash intensity and duration, uncaging response amplitudes and decay kinetics may be differentially affected by the depth of the uncaging focus within the slice. Conclusions about biological aspects of nAChRs should take this important caveat into account.
This localized laser flash photolysis technique has recently been used to uncover new details about nAChR neurobiology. For example, chronic nicotine exposure enhances perisomatic and dendritic nAChR function in medial habenula neurons5. It was also used to help demonstrate, for the first time, that ventral tegmental area glutamate neurons express functional nAChRs in their perisomatic and dendritic cellular compartments6. There are many potential future uses of this technique, and the approach could be applied to other key neuron types that are known to express nAChRs, such as cortical pyramidal neurons31 or interneurons in cerebral cortex32, striatum33, and hippocampus19. This technique could also be combined with pharmacology and/or nAChR gene editing34 to localize specific receptor subtypes to different neuronal compartments. The approach may be easily adapted to other coumarin-caged compounds, including, but not limited to, those developed in parallel with PA-Nic5. Finally, PA-Nic flash photolysis may one day be used in an awake/behaving animal to study nicotine's action in novel behavioral pharmacology paradigms.
Disclosures
D.L.W. serves as a paid consultant for Bruker Nano Fluorescence Microscopy.
Acknowledgements
The authors thank laboratory members of the following Northwestern principal investigators: Ryan Drenan, D. James Surmeier, Yevgenia Kozorovitskiy, and Anis Contractor. This work was supported by the US National Institutes of Health (NIH) (grants DA035942 and DA040626 to R.M.D.), the PhRMA Foundation (fellowship to M.C.A.), and HHMI.
Materials
| Name | Company | Catalog Number | Comments |
| Instruments, Consumables, and Miscellaneous Chemicals | |||
| Multiclamp 700B | Molecular Devices Corp. | Patch clamp amplifier | |
| Pneumatic Picopump | World Precision Instruments | PV820 | |
| Micropipette puller | Sutter Instrument Co | P-97 | |
| Temperature Controller | Warner Instruments | TC-324C | |
| Vibrating blade microtome | Leica Biosystems | VT1200S | |
| Ultrafree-MC Centrifugal Filter | MilliporeSigma | UFC30GV0S | internal solution filter |
| Borosilicate Glass Capillaries | World Precision Instruments | 1B150F-4 | patch and local application pipette |
| (-)-Nicotine hydrogen tartrate salt | Glentham | GL9693 | nicotine salt |
| 7-carboxymethylamino-4-methyl coumarin | Janelia Research Campus | PA-Nic by-product | |
| 1-[7-[bis(carboxymethyl)- amino]coumarin-4-yl]methyl-nicotine | Janelia Research Campus | PA-Nic | |
| Euthasol (Pentobarbital Sodium and Phenytoin Sodium) | Virbac | ANADA #200-071 | |
| Alexa FluorTM 488 Hydrazide | ThermoFisher | A10436 | green fill dye |
| Alexa FluorTM 568 Hydrazide | ThermoFisher | A10437 | red fill dye |
| 6-carboxy-AF594 (Alexa Fluor 594) | Janelia Research Campus | red fill dye | |
| QX 314 chloride | Tocris | 2313 | voltage-gated sodium channel blocker |
| Power Meter | ThorLabs | S120C | |
| Name | Company | Catalog Number | Comments |
| Chemicals for Solutions | |||
| N-Methyl-D-glucamine | Sigma | M2004 | |
| Potassium chloride | Sigma | P3911 | |
| Sodium phosphate monobasic monohydrate | Sigma | S9638 | |
| Sodium bicarbonate | Sigma | S6014 | |
| HEPES | Sigma | H3375 | |
| D-(+)-Glucose | Sigma | G5767 | |
| (+)-Sodium L-ascorbate | Sigma | A4034 | |
| Thiourea | Sigma | T8656 | |
| Sodium pyruvate | Sigma | P2256 | |
| Magnesium sulfate heptahydrate | Sigma | 230391 | |
| Calcium chloride dihydrate | Sigma | 223506 | |
| Sodium chloride | Sigma | S9625 | |
| Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid | Sigma | E3889 | |
| Adenosine 5′-triphosphate magnesium salt | Sigma | A9187 | |
| Guanosine 5′-triphosphate sodium salt hydrate | Sigma | G8877 | |
| Name | Company | Catalog Number | Comments |
| Components of 2-Photon Microscope | |||
| Ultima Laser Scanner for Olympus BX51 Microscope | Bruker Nano, Inc. | imaging software and galvos | |
| Imaging X-Y galvanometers | Cambridge Technology | ||
| Mai Tai HP1040 | Spectra-Physics | Tuneable IR laser | |
| Pockels cell M350-80-02-BK with M302RM Driver | Conoptics, Inc. | for IR laser attenuation | |
| Integrating Sphere Photodiode Power Sensor | Thorlabs, Inc | laser power pick-off photodiode | |
| Uncaging X-Y galvanometers | Cambridge Technology | ||
| Helios 2-Line Laser Launch | Bruker Nano, Inc. | uncaging laser components | |
| OBIS LX/LS 405 nm (100 mW) | Coherent, Inc. | ||
| OBIS LX/LS 473 nm (75 mW) | Coherent, Inc. | ||
| Point-Photoactivation / Fiber Input Module for Limo Sidecar - Uncaging | Bruker Nano, Inc. | ||
| Name | Company | Catalog Number | Comments |
| Upright Microscope | Olympus | BX51WIF | Upright microscope chasis |
| Objective: Olympus M Plan FL 10x; NA 0.3 WD 11 mm | Olympus | 10x objective | |
| Objective: Olympus M Plan Fluorite 60x/1.0 WD=2mm NIR | Olympus | 60x water-dipping objective | |
| X-Cite 110, four-LED LLG coupled epi-fluorescence light source | Excelitas Technologies | LED Light Source | |
| Epi-Fluorescence Filter: ET-GFP (FITC/CY2) for Epi-Turret | Chroma Technologies | LED Filter for blue light excitation | |
| Epi-Fluorescence Filter: ET-DsRed (TRITC/CY3) for Epi-Turret | Chroma Technologies | LED Filter for green light excitation | |
| B&W CCD camera; Watec, 0.5in B/W CCD | Watec Co., LTD. | CCD camera for patch clamp recording | |
| Name | Company | Catalog Number | Comments |
| External Detectors - Dual Reflected Emission - Olympus Upright (Multi-Alkali, GaAsP) | Bruker Nano, Inc. | ||
| Multi-alkali side-on PMT | Hamamatsu | R3896 | red channel PMT |
| 595/50m | Chroma Technologies | red channel emission filter | |
| 565lpxr | Chroma Technologies | dichroic beam splitter | |
| GaAsP end-on PMT | Hamamatsu | 7422PA-40 | green channel PMT |
| 525/70m | Chroma Technologies | green channel emission filter | |
| High-Speed Shutter for Hamamatsu H7422 PMT | Vincent Associates / Bruker | 517329 | PMT shutter mount |
| Name | Company | Catalog Number | Comments |
| Dodt Gradient Contrast Transmission Detection Module | Bruker Nano, Inc. | ||
| Multi-alkali side-on PMT | Hamamatsu | R3896 | Dodt PMT |
References
- Zhang, C., et al. Cholinergic tone in ventral tegmental area: Functional organization and behavioral implications. Neurochemistry International. 114, 127-133 (2018).
- Sarter, M., Parikh, V., Howe, W. M. Phasic acetylcholine release and the volume transmission hypothesis: time to move on. Nature Reviews Neuroscience. 10 (5), 383-390 (2009).
- Coyle, J. T., Price, D. L., DeLong, M. R. Alzheimer's disease: a disorder of cortical cholinergic innervation. Science. 219 (4589), 1184-1190 (1983).
- Katz, B., Thesleff, S. A study of the desensitization produced by acetylcholine at the motor end-plate. Journal of Physiology. 138 (1), 63-80 (1957).
- Banala, S., et al. Photoactivatable drugs for nicotinic optopharmacology. Nature Methods. 15 (5), 347-350 (2018).
- Yan, Y., et al. Nicotinic Cholinergic Receptors in VTA Glutamate Neurons Modulate Excitatory Transmission. Cell Reports. 23 (8), 2236-2244 (2018).
- Engle, S. E., Shih, P. Y., McIntosh, J. M., Drenan, R. M. α4α6β2* nicotinic acetylcholine receptor activation on ventral tegmental area dopamine neurons is sufficient to stimulate a depolarizing conductance and enhance surface AMPA receptor function. Molecular Pharmacology. 84 (3), 393-406 (2013).
- Engle, S. E., Broderick, H. J., Drenan, R. M. Local application of drugs to study nicotinic acetylcholine receptor function in mouse brain slices. Journal of Visualized Experiments. (68), (2012).
- Ren, J., et al. Habenula "cholinergic" neurons co-release glutamate and acetylcholine and activate postsynaptic neurons via distinct transmission modes. Neuron. 69 (3), 445-452 (2011).
- Koppensteiner, P., Melani, R., Ninan, I. A Cooperative Mechanism Involving Ca(2+)-Permeable AMPA Receptors and Retrograde Activation of GABAB Receptors in Interpeduncular Nucleus Plasticity. Cell Reports. 20 (5), 1111-1122 (2017).
- Zhang, J., et al. Presynaptic Excitation via GABAB Receptors in Habenula Cholinergic Neurons Regulates Fear Memory Expression. Cell. 166 (3), 716-728 (2016).
- Chen, E., et al. Altered Baseline and Nicotine-Mediated Behavioral and Cholinergic Profiles in ChAT-Cre Mouse Lines. The Journal of Neuroscience. 38 (9), 2177-2188 (2018).
- Nagy, P. M., Aubert, I. Overexpression of the vesicular acetylcholine transporter increased acetylcholine release in the hippocampus. Neuroscience. 218, 1-11 (2012).
- Ting, J. T., Feng, G. Recombineering strategies for developing next generation BAC transgenic tools for optogenetics and beyond. Frontiers in Behavioral Neuroscience. 8, 111 (2014).
- Crittenden, J. R., Lacey, C. J., Lee, T., Bowden, H. A., Graybiel, A. M. Severe drug-induced repetitive behaviors and striatal overexpression of VAChT in ChAT-ChR2-EYFP BAC transgenic mice. Frontiers in Neural Circuits. 8, 57 (2014).
- Kolisnyk, B., et al. ChAT-ChR2-EYFP mice have enhanced motor endurance but show deficits in attention and several additional cognitive domains. The Journal of Neuroscience. 33 (25), 10427-10438 (2013).
- Nagy, P. M., Aubert, I. Overexpression of the vesicular acetylcholine transporter enhances dendritic complexity of adult-born hippocampal neurons and improves acquisition of spatial memory during aging. Neurobiology of Aging. 36 (5), 1881-1889 (2015).
- Denk, W. Two-photon scanning photochemical microscopy: mapping ligand-gated ion channel distributions. Proceedings of the National Academy of Sciences of the United States of America. 91 (14), 6629-6633 (1994).
- Khiroug, L., Giniatullin, R., Klein, R. C., Fayuk, D., Yakel, J. L. Functional mapping and Ca2+ regulation of nicotinic acetylcholine receptor channels in rat hippocampal CA1 neurons. The Journal of Neuroscience. 23 (27), 9024-9031 (2003).
- Filevich, O., Salierno, M., Etchenique, R. A caged nicotine with nanosecond range kinetics and visible light sensitivity. Journal of Inorganic Biochemistry. 104 (12), 1248-1251 (2010).
- Tochitsky, I., et al. Optochemical control of genetically engineered neuronal nicotinic acetylcholine receptors. Nature Chemistry. 4 (2), 105-111 (2012).
- Wokosin, D. L., Squirrell, J. M., Eliceiri, K. W., White, J. G. Optical workstation with concurrent, independent multiphoton imaging and experimental laser microbeam capabilities. Review of Scientific Instruments. 74 (1), 193-201 (2003).
- Plotkin, J. L., Day, M., Surmeier, D. J. Synaptically driven state transitions in distal dendrites of striatal spiny neurons. Nature Neuroscience. 14 (7), 881-888 (2011).
- Galtieri, D. J., Estep, C. M., Wokosin, D. L., Traynelis, S., Surmeier, D. J. Pedunculopontine glutamatergic neurons control spike patterning in substantia nigra dopaminergic neurons. Elife. 6, (2017).
- Yasuda, R., et al. Imaging calcium concentration dynamics in small neuronal compartments. Science STKE. (219), pl5 (2004).
- Inoue, S., Spring, K. . Video microscopy: The fundamentals. , 163-186 (1997).
- Zipfel, W. R., Williams, R. M., Webb, W. W. Nonlinear magic: multiphoton microscopy in the biosciences. Nature Biotechnology. 21 (11), 1369-1377 (2003).
- Maravall, M., Mainen, Z. F., Sabatini, B. L., Svoboda, K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophysical Journal. 78 (5), 2655-2667 (2000).
- Wathey, J. C., Nass, M. M., Lester, H. A. Numerical reconstruction of the quantal event at nicotinic synapses. Biophysical Journal. 27 (1), 145-164 (1979).
- Shih, P. Y., et al. Differential expression and function of nicotinic acetylcholine receptors in subdivisions of medial habenula. The Journal of Neuroscience. 34 (29), 9789-9802 (2014).
- Verhoog, M. B., et al. Layer-specific cholinergic control of human and mouse cortical synaptic plasticity. Nature Communications. 7, 12826 (2016).
- Koukouli, F., et al. Nicotine reverses hypofrontality in animal models of addiction and schizophrenia. Nature Medicine. 23 (3), 347-354 (2017).
- Xiao, C., et al. Chronic nicotine selectively enhances α4β2* nicotinic acetylcholine receptors in the nigrostriatal dopamine pathway. The Journal of Neuroscience. 29 (40), 12428-12439 (2009).
- Peng, C., et al. Gene Editing Vectors for Studying Nicotinic Acetylcholine Receptors in Cholinergic Transmission. European Journal of Neuroscience. , (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved