
Method Article
Extremely Rapid and Specific Metabolic Labelling of RNA In Vivo with 4-Thiouracil (Ers4tU)
In This Article
Summary
The use of thiolated uracil to sensitively and specifically purify newly transcribed RNA from the yeast Saccharomyces cerevisiae.
Abstract
The nucleotide analogue, 4-thiouracil (4tU), is readily taken up by cells and incorporated into RNA as it is transcribed in vivo, allowing isolation of the RNA produced during a brief period of labelling. This is done by attaching a biotin moiety to the incorporated thio group and affinity purifying, using streptavidin coated beads. Achieving a good yield of pure, newly synthesized RNA that is free of pre-existing RNA makes shorter labelling times possible and permits increased temporal resolution in kinetic studies. This is a protocol for very specific, high yield purification of newly synthesized RNA. The protocol presented here describes how RNA is extracted from the yeast Saccharomyces cerevisiae. However, the protocol for purification of thiolated RNA from total RNA should be effective using RNA from any organism once it has been extracted from the cells. The purified RNA is suitable for analysis by many widely used techniques, such as reverse transcriptase-qPCR, RNA-seq and SLAM-seq. The specificity, sensitivity and flexibility of this technique allow unparalleled insights into RNA metabolism.
Introduction
RNA has a dynamic nature; soon after it is produced much RNA is rapidly processed and degraded. Currently, most studies of RNA metabolism analyze the total cellular RNA, which is mostly fully processed and at steady state level. This level depends on the balance between the rates of transcription, post-transcriptional maturation and degradation. Analysis of the processes that lead to the steady state equilibrium requires specialized techniques to capture very short-lived RNA species.
Metabolic labelling of RNA with nucleotide analogues such as 4-thiouracil (4tU) or 4-thiouridine (4sU) (see Duffy et al.1 for an excellent review), offers the ability to isolate thio-labelled nascent RNAs and their processing intermediates. However, published protocols involve labelling times of several minutes2,3, which is slow relative to the rate of production of many transcripts. It takes in the order of one minute to transcribe the average yeast gene, so labelling yeast RNA for less than one minute can be considered extremely short. The extremely rapid and specific 4 thiouracil protocol (ers4tU) maximizes the signal to noise ratio by maximizing 4tU incorporation and minimizing the recovery of unlabeled, pre-existing RNA making very short labelling times possible4.
The thio-modified base must be imported into the cells rapidly and in sufficient quantity to efficiently label the newly synthesized RNA (nsRNA). To promote this, cells are grown in uracil-free medium, and expression of an appropriate permease helps to boost 4tU or 4sU uptake (see Table 1 for a list of plasmids that carry suitable permease genes and Supplementary Figure 1). 4tU's solubility in sodium hydroxide avoids the need for toxic organic solvents required by other nucleotide analogues. Unfortunately, growing cultures for long periods with thio-modified nucleosides at concentrations greater than 50 µM has been observed to disrupt ribosomes5. However, the concentration (10 µM) used here, and the extremely short labelling times, minimize deleterious effects5 (Figure 1a), while still yielding sufficient RNA for analysis.
This technique can be combined with rapid and specific auxin-mediated depletion of a target protein6,7 (Figure 2), referred to as the "β-est AID 4U" protocol, in which β-estradiol regulated expression of the auxin inducible degron (AID) system is combined with 4tU labelling. With the β-est AID 4U approach, a target protein can be depleted and the effect on RNA metabolism closely monitored (Figure 2). The timing is critical; it is advisable to view the accompanying video and pay close attention to Figure 2 and its animated form (see Supplementary Figure 2).
Processing and degradation of RNA must be stopped extremely rapidly for accurate time resolution. This is achieved using methanol at low temperature, which fixes the cell contents very rapidly and degrades the cell membrane while preserving the nucleic acid content8. The RNA extraction should be efficient and not damage the RNA. Mechanical lysis is effective in the absence of chaotropic agents (often these contain thio groups, so should be avoided). Lithium chloride precipitation of RNA is preferred, as tRNAs are less efficiently precipitated. tRNAs are rapidly transcribed and naturally thiolated9, so removing tRNAs reduces competition for the biotinylation reagent. If small, highly structured RNAs are of interest, alcohol-based RNA precipitation methods are recommended.
To recover the thiolated RNA, biotin is covalently attached via the thio groups incorporated into the RNA with 4tU. The use of modified biotin, which attaches via a cleavable disulfide bond (e.g., HPDP-biotin (N-[6-(Biotinamido)hexyl]-3´-(2´-pyridyldithio)propionamide, ) or MTS-biotin (Methane thiosulfonate)) is recommended as it permits release of the RNA by addition of a reducing agent. The biotinylated RNA is affinity purified on streptavidin coupled to magnetic beads. This protocol is similar to others listed previously10 but has been intensively optimized to reduce background.
There are two types of thiol-labelling experiment that can be performed, continuous and discontinuous labelling. Each has its own advantages. In continuous labelling the 4tU is added to the culture and samples taken at regular intervals. This type of experiment shows how the RNA is processed and how levels change over time. Examples include comparison of mutant with wild-type experiments and a pulse-chase experiment. The experiments shown in Figure 3b,c are of this type. For discontinuous labelling a change is induced into the system and the RNA monitored. Once the change has been induced the culture must be split into several sub-cultures, and at specific times, each one is then thio-labelled for a brief period. One example is β-est AID 4U shown in Figure 27. This type of experiment is particularly useful for monitoring the effect of a metabolic change on RNA processing (see Figure 3d).
A graphical representation of a thio-labelling experiment is presented in Figure 4 and Figure 5, and a spreadsheet that greatly simplifies the performance of the protocol is available (see 4tU experiment template.xlsx). As well as this the Supplementary Information contains an extensive troubleshooting guide. For the β-est AID 4U protocol that integrates 4tU labelling with the auxin depletion protocol, see Figure 2 and Supplementary Figure 2. See Barrass et al.7 for the detailed AID depletion protocol.
Protocol
1. Growth and thio-labelling
NOTE: Time for completion of this section of the protocol is highly variable, depending on cell growth rate. Allow 1 h to prepare the solutions and equipment prior to thio-labelling and 30 min post-labelling to process samples.
- Ensure the S. cerevisiae strain contains a plasmid encoding a permease (Table 1) to boost 4tU import into the cell.
NOTE: Without an importer, labelling for less than 2 min is unlikely to be successful11 (see Supplementary Figure 1). 4tU incorporation is more efficient if growth is in medium without uracil, so the strain must be URA3+; several of the plasmids in Table 1 carry URA3 as marker. If this protocol is to be combined with β-est AID depletion7, additional strain modifications are required. - Prepare YMM uracil-free medium by adding 6.9 g of yeast nitrogen base without amino acids, 20 g of glucose, and 1.92 g of SCSM single drop-out−ura (Table of Materials) to 1 L of water. Autoclave or filter sterilize the growth medium before use.
NOTE: Filter sterilization is preferred as peptide/sugar complexes produced by autoclaving co-precipitate with the cells in the methanol used in sample collection. - Grow yeast in YMM uracil-free medium to an optical density at 600 nm (OD600) of 0.6−0.8. Ensure the culture is in log phase growth and has been for at least two doublings. Growth at 30 °C is normally recommended, but other temperatures may be used, for example, for temperature-sensitive strains.
NOTE: Depending on the strain, growth conditions and RNA yield, approximately 30 mL sample volume will be needed. This amount will be assumed throughout the protocol. 30 mL of culture is the most that will fit into a 50 mL centrifuge tube with 20 mL of methanol, so is a convenient volume to start optimization. Consider using more sample volume for early time points to increase RNA recovery, up to 2000 mL has been used for slower growing cells at really short labelling times (<1 min). - Chill about 50 mL of H2O on ice. For each sample, add 200 µL of zirconia beads to a 2 mL screw-cap tube and chill on ice. Also put 20 mL of methanol (CAUTION), into 50 mL centrifuge tubes, and place on dry ice (CAUTION). The methanol should be 1/3 to 2/3 the volume of the sample.
CAUTION: Methanol is toxic by inhalation, contact and consumption. Dispense large volumes in a fume hood, and wear two pairs of gloves, as methanol can penetrate nitrile laboratory gloves. Methanol is highly flammable, keep away from all sources of ignition.
NOTE: As dry ice can cause cold burns on contact and produces asphixiant gas, use gloves when handling and use in a well-ventilated space.
NOTE: Adding the beads at this point is easier than after the sample has been added as the tube is dry and when spinning down the cell pellet the beads are also spun clear of the tube thread saving some time. Additionally, this allows the beads to cool before the sample is added. - If an S. pombe spike is to be added to the culture (rather than later), thaw an aliquot of thiolated S. pombe cells on ice and vortex thoroughly, at least 30 s, then add to the culture. If prepared according to the instructions below, one S. pombe aliquot is sufficient for 400 mL of culture (enough for twelve 30 mL samples plus a little to allow for errors in handling). If more or less culture is used, adjust the volume of S. pombe added to the culture.
- Grow 1 L of S. pombe culture to OD600 to 0.8 exactly as described in the protocol for S. cerevisiae.
- Thio-label as step 1.7, but for 10 min.
- Fix all of the culture using 400 mL of methanol on dry ice, essentially as described in step 1.9.
- Pellet the cells by centrifugation at 3000 x g for 3 min.
- Discard the supernatant and resuspend the cell pellet in 3.3 mL of H2O.
- Split into aliquots of 80 µL each. Store at -80 °C.
- Use all of one aliquot for 400 mL culture or 10 µL per 30 mL sample.
NOTE: Reduce the volume of spike to 1/10 if performing RNAseq. Do not reuse aliquots; discard any unused spike. This spike is useful to normalize and compare results across time points and experiments.
- For discontinuous labelling, induce the required metabolic perturbation (e.g., growth conditions, gene induction or depletion such as β-est AID7 (Figure 2 and Supplementary Figure 2), then split the culture. Ensure all flasks and media are at the required temperature and, if possible, aerate the medium before adding the culture.
- Add 4tU to the culture to a concentration of 10 µmM and mix vigorously (1/10,000 of the culture volume of 100 mM 4tU dissolved in 1 M NaOH). Thiol-label for 15 s to 5 min.
NOTE: Thirty seconds is a good starting point. Thio-labelling for less than 20 s gives more variable results due to difficulties manipulating the culture under time pressure. However, labelling for more than 1 min reduces the temporal resolution of the technique. - If a chase experiment is to be performed; allow thio-labelling for 20−30 s then chase by adding 1/200 culture volume of 1 M uridine (not thiolated), to a final concentration of 5 mM.
NOTE: Uridine is preferable to uracil for the chase, as uridine is more water soluble allowing a smaller volume to be added to the culture and so there is less disturbance to the growth of the cells. - Take samples of culture at regular intervals (at least 15 s), to the end of the time course. Sampling intervals shorter than this are difficult to perform reliably. Add the sample to the methanol on dry ice prepared in step 1.4. For convenience, add 30 mL of culture to a 50 mL tube containing 20 mL of methanol.
NOTE: Carbon dioxide dissolves in the methanol when cold; this comes out of solution on addition of the sample and foams vigorously upon mixing―resulting in sample loss. To avoid this, chill the methanol to <-70 °C in a tightly sealed tube until close to the time it is needed, then transfer to dry ice. - Seal the tube and mix thoroughly by shaking. Place the samples on ice. Check that none of the samples have frozen; if so, gently warm in the hand, inverting constantly. This is best done in the hand as the sample’s temperature can be assessed, it should always feel cold. Place on ice. This is not a pause point; once all the sample is fluid proceed to the next step.
- Spin at 3000 x g for 2 min (at 4 °C if possible) to pellet the cells. Pour off the liquid and resuspend the pellet in at least 1 mL of ice-cold water by gently pipetting up and down.
NOTE: The residual methanol in the sample pellet aids resuspension. - Transfer to 2 mL screw cap tubes as prepared in step 1.4. Spin briefly (e.g., 10 s total time) at >13,000 x g to re-pellet the cells, place back on ice and remove liquid.
NOTE: The cell pellet can be stored at -70 to -80 °C for several months.
2. Preparation of total RNA
NOTE: The time for completion is 90 min.
- Use diethyl pyrocarbonate (DEPC)-treated solutions to protect the RNA from degradation. Aliquot the solutions using filter pipette tips and wear gloves at all times.
- To a solution add 1/1000 volume of DEPC and mix by vigorous shaking.
- Leave at room temperature (RT) for 24 h, then autoclave.
- Solutions with amine groups (such as tris) cannot be DEPC treated. Aliquot the powder and store specially for RNA work. Use previously DEPC treated H2O to make the solution.
NOTE: As the thio-group on the RNA is photoactivatable, minimize exposure to UV light from this point on. Storage should be in the dark and incubation is best done in a PCR machine with a lid.
- If an S. pombe spike is to be added to the cell pellet rather than the culture (do not do both), add it now. Thaw an aliquot of thiolated S. pombe cells on ice and vortex thoroughly, at least 30 s, before adding to the pellet.
NOTE: If prepared according to steps 1.5.1−1.5.7, 10 µL of S. pombe aliquot is required for one pellet derived from 30 mL of culture. - Before putting on the cap, spin very briefly for 1−2 s to ensure that no zirconia beads are trapped between the cap and the tube, which can cause sample and phenol to leak from the tube.
- Resuspend the cells in 400 µL of acetate EDTA (AE) buffer (50 mM sodium acetate pH 5.3, 10 mM EDTA pH 8.0), by vortexing vigorously. Add 40 µL of 10% (w/v) sodium dodecyl sulfate (SDS). Do not vortex, as SDS will foam.
- If the β-est AID 4U protocol is to be used, take 40 µL of the cell suspension for protein analysis7. Add 40 µL of AE to make the volume back up to 400 µL.
- Add 800 µL of phenol (CAUTION) at low pH and vortex for 10 s.
CAUTION: Phenol is toxic and corrosive by inhalation and contact. Always perform procedures involving phenol in a fume hood and wear two pairs of gloves. - Lyse the cells in a homogenizer (e.g., Table of Materials) for three 2-min bursts at the lowest power setting. Leave the samples on ice for 2 min between pulses of homogenization.
NOTE: Optimize the conditions if using other homogenizers. Insufficient shaking will result in poor yields, whereas excessive shaking results in apparent higher yield, as determined by absorbance at 260 nm (A260), but the RNA may be degraded. A homogenizer is preferred, but hot phenol RNA purification12 can be used. - Place the lysed sample on dry ice for 5 min, until it solidifies, this reduces genomic DNA carry over into the RNA. Do not freeze for too long as the sample will not thaw. Spin 5 min in microfuge at >13,000 x g at RT; do not be tempted to do this at 4 °C, as the sample/phenol mix will remain solid throughout the spin if performed at low temperature.
NOTE: If the sample is still frozen at the end of the spin, re-spin for another 5 min until the sample has completely thawed. - Phenol/chloroform extract then chloroform extract with an equal volume (approximately 600 µL) of phenol:chloroform 5:1 then chloroform (CAUTION). Transfer the top phase to another tube containing phenol:chloroform 5:1 or chloroform. Vortex, then spin for 5 min in a microfuge at RT. Then transfer the top phase to a new 1.5 mL tube.
CAUTION: Chloroform is toxic by inhalation and contact. Always perform procedures involving chloroform in a fume hood and wear two pairs of gloves. - Add a third to half volume (approximately 300 µL) of 10 M LiCl, and mix to precipitate the RNA. The sample should go cloudy immediately but leave for at least 10 min on ice or at 4 °C (do not store below -20 °C as it will freeze), or until the precipitate flocculates.
- Spin for 5 min at >13,000 x g in a microfuge. Remove the fluid, briefly re-spin and remove the dregs. Wash pellet with 300−500 µL of 70% ethanol, spin briefly and remove remaining ethanol.
NOTE: During these washes keep the pellet on the same side of the tube as the first spin, this way the pellet will not move and break; if it breaks some of the RNA could be lost accidently.
NOTE: Do not dry the pellet; as long as most of the fluid has been removed it will not interfere with subsequent steps. The RNA can also be stored at this stage at -20 °C for a few months or -70 to -80 °C for long-term storage. - Re-dissolve the RNA pellet in 90 µL of TE pH 7.0 (10 mM Tris HCl pH 7.0, 1 mM EDTA pH 8.0) by heating at 65 °C with shaking as the RNA pellet can be difficult to re-dissolve. This must be for no more than 5 min as RNA degrades at higher temperatures. Check for full RNA solubilization and then transfer to a 0.2 mL tube. Pipette the sample up and down; there should be no "lumps", and the fluid should rise and fall smoothly in the tip. This solution is viscous so the final pipetting motion should be slow.
NOTE: The RNA can be stored at -20 °C in the dark at this stage; this can also be beneficial to RNA solubility.
3. Biotinylation
NOTE: The time for completion is 60 min. The following steps are conveniently done in a strip of tubes with integral caps as they have less tendency to open on vortexing than strips with separate caps.
- Biotinylate by adding 10 µL (1/10 final volume) of a 5 mM HPDP-biotin solution (MTS-biotin can be used in exactly the same way as HPDP-biotin), to the RNA and mix thoroughly. Preheat the RNA for no more than a few seconds at 65 °C before adding the biotin. Incubate at 65 °C for 15 min to a maximum of 30 min in the dark.
NOTE: This heating is required as some HPDP batches precipitate at RT in the RNA sample. A PCR block with a heated lid is ideal for this. - Prepare a small resin volume, size exclusion column (Table of Materials) to exclude the unincorporated biotin. Remove the bottom tag of the column and loosen the cap, place in a 2 mL centrifuge tube. Spin at 1500 x g for 1 min to flush out the buffer Add 0.3 mL of TE gently to the top of the column and spin again. Repeat the wash and spin twice more for a total of 3 washes. Finally transfer the washed column to a fresh 1.5 mL tube.
- Once the sample incubation (step 3.1) is complete, add the sample to the top of the column. Spin at 1500 x g for 2 min. The biotinylated RNA sample is now in the bottom of the tube.
NOTE: A 1 min spin is not sufficient to elute the entire sample. - Add a third to half volume (approximately 40 µL) of 10 M LiCl, mix to re-precipitate the RNA as step 2.10. The sample should go cloudy immediately but leave for at least 5 min on ice or at 4 °C or until the precipitate flocculates; do not store below -20 °C as it will freeze. Centrifuge the sample for 5 min at >13,000 x g in a microfuge.
- Wash with 80% ethanol, ≤1 h rotating. Follow the procedure in step 2.11 to remove as much of the fluid as possible.
NOTE: As HPDP-biotin is very soluble in 80% ethanol, this is an additional purification step. - Repeat the 80% ethanol wash to remove as much un-incorporated biotin as possible.
NOTE: The RNA can also be stored at this stage at -20 °C in the dark.
4. Purification of the newly synthesized RNA
NOTE: The time for completion is 2 h.
- Re-dissolve the RNA in 200 µL of DEPC-treated H2O (65 °C incubation can be used, similar to the procedure in step 2.12).
- Measure the RNA concentration at A260 using a spectrophotometer; the sample may have to be diluted 1/10 to get it within the linear range of the spectrophotometer. Vortex this dilution for at least 10 s to ensure the viscous RNA is evenly dissolved.
NOTE: The efficiency of biotinylation can be assessed by dot blot13 if required. - Add equal amounts of RNA to a fresh tube and make up to 200 µL in DEPC-treated H2O. Use all of the sample with the lowest RNA concentration and use an appropriate volume of the other samples to have a similar amount of RNA for each.
NOTE: The spreadsheet 4tU experiment template.xlsx has a form to aid this calculation. - When the sample is at RT, add 25 µL of 10 x NaTM buffer (0.1 M Tris HCl pH 7.0, 2 M NaCl, 250 mM MgCl2), 25 µL of 1 M NaPi pH 6.8 (0.5 M NaH2PO4 0.5 M Na2HPO4), and 2.5 µL of 10% SDS. Mix thoroughly and spin gently (<30 s; approximately 100 x g).
NOTE: To avoid precipitation of the SDS and salts, the samples must be kept at RT throughout the following procedures up to step 4.13. - Make the bead buffer containing 1x NaTM buffer, 0.1 M NaPi, and 0.1% SDS, 2 mL per sample. Add the required amount of H2O first and the SDS last. This must be made fresh each time as a precipitate forms after 24 h.
NOTE: To avoid the formation of precipitates, the bead buffer must be kept at RT throughout the following procedures. Do not DEPC treat or autoclave. - Add 50 µL of streptavidin beads to a low retention 1.5 mL tube. Place the tube on the magnetic rack, wait for the beads to settle and then remove the fluid
- Wash the streptavidin beads.
- Add 200 µL of bead buffer and vortex until the bead pellet is fully resuspended. Usually 3−5 s is all that is required. For washes before the RNA sample is added it is sufficient to turn the tubes round so the beads travel across the tube to the other side. Then turn the tubes back to the original side so the beads travel across the tube once again.
- Spin the tube at low speed (approximately 100 x g) for a maximum of 5 s to spin down the fluid, but not the beads.
- Place in the magnetic rack to allow the beads to be captured by the magnet.
- Remove the fluid by aspiration for a small number of samples; pour off the liquid if many samples.
NOTE: With a large number of samples, removing all the fluid purely by aspiration can be problematic, as the beads in the first sample may be dried out before the last sample is finished, this increases background. Washing can be expedited by pouring off the fluid from all samples at once whilst on the magnet. They should be left a little longer on the magnet before pouring and the small amount of fluid that remains has to be aspirated away but, overall, it means less time on the magnet and without fluid. In this way, it is possible to do 24 or more extractions quickly.
- Block with 200 µL bead buffer, 10 µL 20 mg/mL glycogen, and 2.5 µL 5 mg/mL tRNA, 20 min rotating end over end at moderate speed at RT. The rotation is to keep the beads in suspension. Once the blocking is complete remove the fluid as steps 4.7.2−4.7.4 and wash again, as the steps in section 4.7.
- Resuspend the beads in the sample. Incubate at RT with rotation for 30 min.
- During the incubation, prepare a fresh 1.5 mL tube for each sample. Add 1/10 volume (approximately 10 µL) of 3 M sodium acetate pH 5.3 and 20 µg of glycogen, and spin at approximately 100 x g for 3 s. Store in a rack until needed.
- Remove the unbound RNA from the beads, as steps 4.7.2−4.7.4. The unbound RNA can be persevered in a fresh tube, but the salts and SDS make it very difficult to purify. Then wash the beads, as section 4.7 with vortexing, for a minimum of 3 to a maximum of 5 times.
- After the final wash take special care to aspirate all the liquid; return to each tube and aspirate the dregs of the buffer once more.
- To elute the RNA, add 50 µL of freshly prepared 0.7 M β-mercaptoethanol (βME) to the beads (1/20 dilution of the commercially supplied stock solution). Vortex and spin briefly, as steps 4.7.1 and 4.7.2. Place the slurry in the magnetic rack and pipette the RNA containing solution into the 1.5 mL centrifuge tube prepared in step 4.10.
- Elute once more as step 4.13 to recover residual RNA from the beads and add the eluted sample to the tube containing the first elution from these beads.
- Remove residual beads from the eluted RNA by placing the sample back in the magnetic rack and transferring the fluid to a fresh, low binding 0.5 mL centrifuge tube.
- Mix the sample and then precipitate the nsRNA by adding 2.5x volumes (280 µL) of ethanol and mix once more. Leave for 1 h to overnight at -20 °C. Spin in a pre-chilled centrifuge (4 °C) for 20 min at the maximum speed (at least 13,000 x g).
- Wash thoroughly with 200 µL of 70% ethanol at -20 °C. As residual βME will inhibit downstream applications, spin at every step to remove as much of the dregs as possible; at the end the sample should not smell of βME.
- Re-dissolve in 10−20 µL of DEPC-treated 1x TE with the equivalent of 0.005 µL RNase inhibitor.
NOTE: All subsequent stages should be performed on ice. - Measure the RNA concentration and purity.
- Measure the A260 and A225 in a low sample volume spectrophotometer.
NOTE: An absorbance maximum near λ = 225 nm is from an unavoidable contaminant from the beads. In the absence of RNA the signal from the contaminant declines to 35% at λ = 260 nm. Therefore, the actual amount of RNA is approximated by the formula: (A260-(A225*0.35))*40 ng/µL. - Alternatively, analyze the sample on a micro-fluidics electrophoresis system such as a bioanalyzer.
NOTE: This analysis is preferable to using a spectrophotometer as RNA integrity can be assessed, the contaminant does not interfere with the quantitation and less sample is required.
- Measure the A260 and A225 in a low sample volume spectrophotometer.
- Analyze the nsRNA.
NOTE: For example, specific RNAs can be quantified by standard reverse transcriptase qPCR techniques. RNA prepared this way is compatible with library preparation for RNA-seq. Removal of rRNA is not necessary for labelling times of less than 5 min. Recoding SLAMseq14 can also be performed on this RNA.
Results
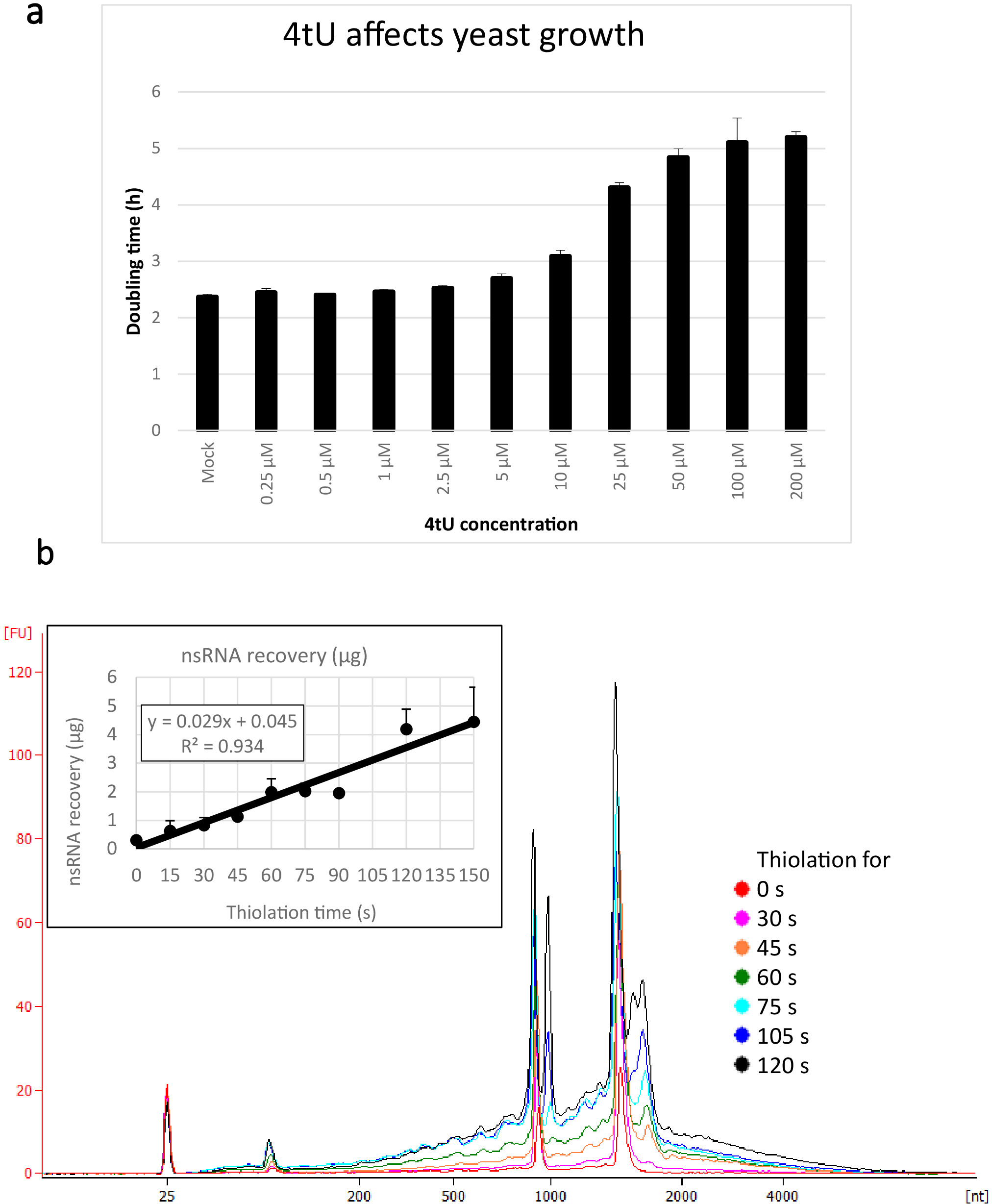
Typical yields for nsRNA recovered using this ers4tU protocol are displayed in Figure 1b, this has been produced by a bioanalyzer and the trace shows yield of RNA versus size (nucleotides [nt]). Note, in both the bioanalyzer trace and the inset graph, that RNA recovery from time point 0 is a very small portion of that recovered from longer time points - approximately 0.3 µg of RNA recovered from approximately 109 cells compared with over twice as much after just 30 s of labelling (0.8 µg of nsRNA) from the same number of cells. RNA recovery at 15 s is more variable as small differences in performing the sampling have a proportionately larger effect on RNA recovery. In the bioanalyzer trace, rRNA precursors can be seen as a peak near 1000 nt and a doublet of peaks at 1700−1800 nt. The abundance of these intermediates increases as thiolation continues.
Thio-labelling was used to quantify splicing of the ACT1 transcript (Figure 3). Thiolation was performed and samples taken at 15 s intervals from the start of thio-labelling and the processing of ACT1 RNA monitored (Figure 3a,b). As can be seen, pre-mRNA is generated (by transcription), and lariats (by the first step of splicing from pre-mRNA), even after just 15 s of labelling. After about 45 s to 1 min, the amounts of lariats and pre-mRNA reach equilibrium with as much of these RNA species being created by transcription as are processed away by splicing.
To produce the data shown in Figure 3c the strain was pulsed with 4tU for 25 s and then chased with uridine. The generation of pre-mRNA and lariats reaches a maximum at 1 minute. This compares well with Figure 3b; the maximum being achieved after 45 s to reach equilibrium plus the 25 s of the labelling. After the peak, the levels decline as the thio-labelled RNAs are chased through the splicing process.
Figure 3d shows depletion of a protein splicing factor and its effect on RNA metabolism, using the β-est AID 4U system6,7. Here, Prp16p is reduced from near physiological levels to 5% of this level after 25 min of depletion. Prp16p is an essential splicing factor for the second step of splicing15. Lariats are removed during the second step of splicing (Figure 3a), but here they increase above the level of pre-mRNA as Prp16 becomes limiting. At later depletion times, other factors become limiting due to secondary effects, so that levels of lariat decrease, and pre-mRNA levels rise. The level of spliced mRNA declines.

Figure 1: Growth in 4tU and RNA recovery. (a) 4-thiouracil affects growth. Increasing the concentration of 4tU in YMM drop-out growth medium without uracil increases the doubling time of S. cerevisiae (BY4741) carrying the p4FuiΔPEST plasmid. Growth of four replicate cultures was monitored at 30 °C in a Tecan Infinite Pro 200. All cultures were in log phase throughout, with OD600 between 0.1 and 0.6. Mock is a control culture with an equivalent amount of NaOH added, which does not by itself change the growth rate. This graph demonstrates that thio-labelling is compromise between rapid labelling and damage to the cell. Error bars are standard error of 4 replicates. (b) nsRNA yield increases linearly from about 15 s of labelling. The main figure shows the bioanalyzer traces of purified, nsRNA from 0 (not thiolated) to 2 min after addition of 4tU at 15 s intervals. Note that the 15 s sample is not shown, as it was indistinguishable from the unlabelled sample. The two large peaks correspond to ribosomal RNAs (rRNAs). The rRNA precursors and intermediates are visible as several peaks at greater molecular weight than mature rRNAs. The recovery of these precursors and intermediates increases with time. Results from one representative experiment are shown. The inset graph shows the recovery of nsRNA with increasing incubation with 4tU. The yield of nsRNA increases with increasing time of growth with 4tU. The recovery is remarkably linear (R2 = 0.934) throughout the timescale of this experiment and shows a slight increase over background even at 15 s labelling with 4tU even though not distinguishable from the unlabelled sample by eye from the bioanalyzer trace. Error bars show standard error for three biological replicates. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: β-est AID 4U β-est AID 4U graphical protocol. A graphical summary of the protocol of the β-est AID 4U protocol. β-estradiol (β-est) promotes the expression of the auxin inducible degron (AID) system which in turn depletes an AID* tagged target protein, refer to Barrass et al.7 for a detailed protocol. In this case, degron system expression is initiated 25 min before protein degradation commences and thiolation at each time point is for 1 min. Samples are taken before induction and every 2 minutes during depletion. An animated version appears in the Supplementary Figure 2. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Precursors and intermediates of ACT1 RNA splicing. Splicing of ACT1 pre-mRNA transcripts was monitored by quantitative reverse transcription PCR16. The levels of ACT1 precursor (pre-mRNA), intermediate lariat-exon2 (Lariat) and spliced product (mRNA) are shown normalized against the level of ACT1 Exon2 and steady state levels of these RNAs. (a) Location of qPCR products on the ACT1 transcript. Schematic of the locations of the qPCR products used to assay the levels of precursors, intermediates and products of the splicing reaction of the ACT1 transcripts16. Exons are represented by boxes, intron as a line and the qPCR products as lines with diamonds at either end, the color matches those used in the graphs. The pre-mRNA PCR is specific for pre-mRNA and not any intermediates of splicing as this product crosses the branch point which is disrupted after the first step of splicing. Lariat PCR will detect the product of the first step of splicing and the excised lariat produced after the second. The mRNA PCR is specific for the product of splicing, mRNA. Results from the exon PCR (present in all precursors, intermediates and products, except the excised lariat) is not shown in the graphs as this was used to normalize the data and is therefore always equal to 1. (b) Continuous thiolabelling. The amount of pre-mRNA increases with time as 4tU is incorporated by transcription and, after a short delay, splicing converts it to lariat-exon2 intermediate and spliced products. The levels of these pre-mRNA and lariat species are detectable above background after as little as 15 s of growth with 4tU and reach a maximum after approximately 45 s of continuous labelling with 4tU, at which point their production is balanced by conversion to spliced mRNA and/or degradation. Values are normalized to their steady state (left-most point of the graph), and exon 2 levels to show their appearance and processing in comparison to transcription of exon 2. As RNA splicing of ACT1 is largely co-transcriptional4,17 spliced mRNA rapidly becomes the most abundant species, its level is similar to that of exon 2. Standard error of three biological replicates, each assayed in triplicate. (c) Pulse/chase. Thiolation pulse of 25 seconds followed by chase with uridine. Compared to the steady state levels of these RNAs (left-most point), they are initially very abundant in the newly synthesized pool. The levels gradually decline as they are processed into mRNA (or degraded), approaching levels very similar to steady state levels by 5 min. Standard error of three biological replicates, each assayed in triplicate. (d) nsRNA and protein depletion. Splicing of ACT1 pre-mRNA transcripts monitored by quantitative reverse transcription PCR as in panel (a) upon depletion of the Prp16 protein using the auxin degron system as described in Figure 2. The Prp16 protein levels are also displayed in the graph plotted against the second Y-axis as percentage of levels prior to auxin depletion. Prp16 is a vital component of the spliceosome, particularly important for the second step of splicing shown in panel (a), after which lariats are degraded. When this step becomes limiting lariats accumulate initially. At later time points splicing fails completely, lariats are no longer produced and pre-mRNA levels rise. Error bars are standard error of three biological replicates, each assayed in triplicate. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Graphical summary of the protocol sections 1 to 3. The cells are thiolated with 4tU and allowed to grow to incorporate the modified nucleotide into the RNA. A thiolated S. pombe spike can be added to allow normalization across time points and experiments. The pulse of 4tU can be chased using un-thiolated uridine. Labelling can either be performed continuously from 4tU addition or from a change to growth conditions, the culture split and 4tU added to cultures at increasing times from the growth condition change, but each labelling only for a brief time. The cells are collected, and RNA prepared from the cells, preferably using a homogenizer and phenol-based methods. The RNA is biotinylated and then the biotinylated RNA purified from unincorporated biotin using a size exclusion column. The nsRNA is now ready for purification with streptavidin beads (section 4, Figure 5). Numbers in red correspond to the step numbers in the protocol. Please click here to view a larger version of this figure.
{kind=link}

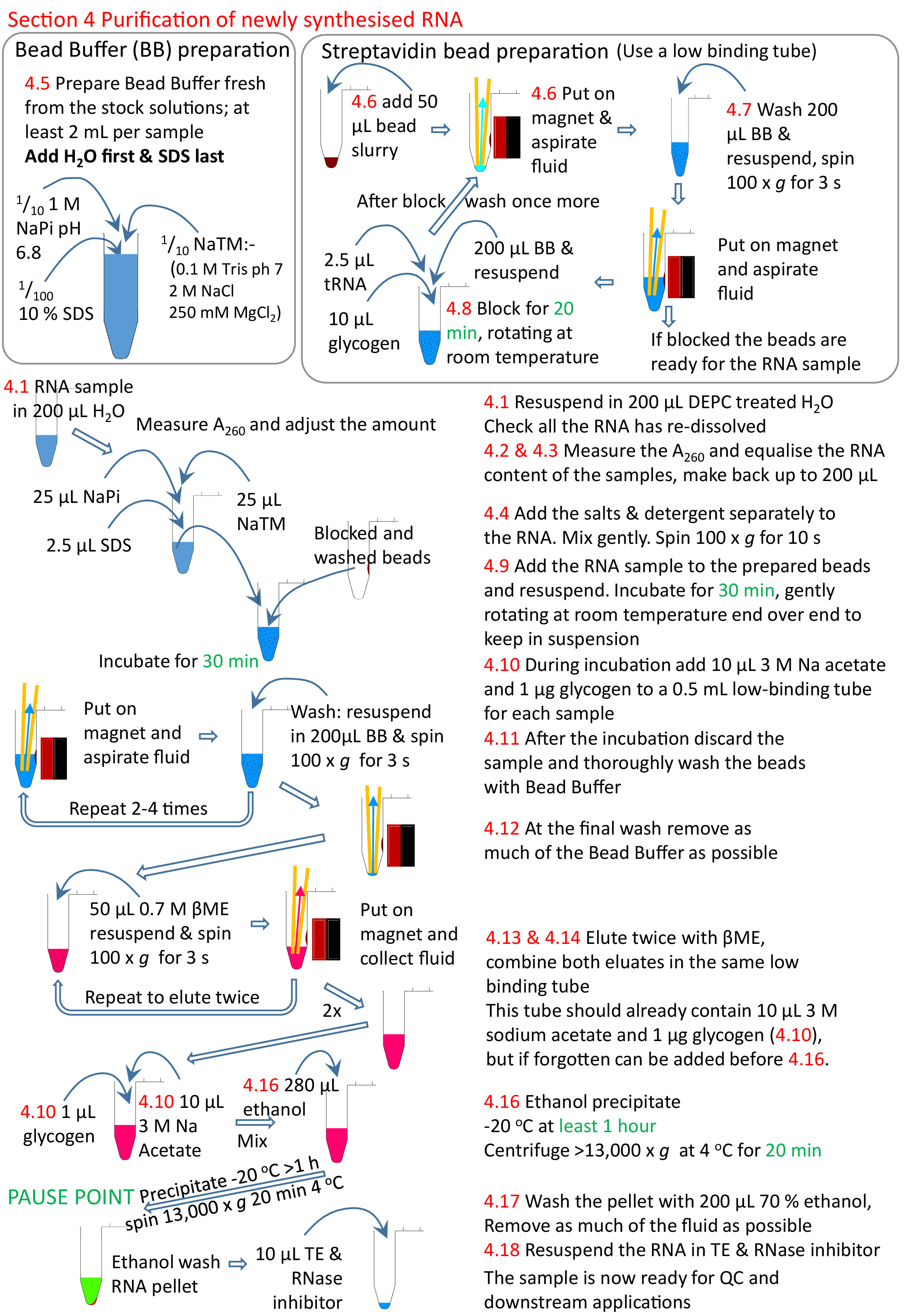
Figure 5: Graphical summary of the protocol section 4. Following on from sections 1 to 3 (Figure 4), the streptavidin beads are blocked and the biotinylated RNA sample added to the prepared beads. The biotinylated RNA binds to the streptavidin beads and the un-biotinylated RNA removed and washed. The biotinylated RNA is eluted from the beads using βME and precipitated ready for further research. Numbers in red correspond to the step numbers in the protocol. Please click here to view a larger version of this figure.
{kind=link}
Supplementary Figure 1: Improvement of nsRNA recovery from yeast cells with and without additional copies of the importer at 1 and 3 minutes of thio-labelling. Note that Fui1 is the yeast's own promoter expressed from a 2 µm plasmid. The genomic copy of this gene is present in both of these strains. Please click here to download this file.
Supplementary Figure 2: Animated version of the β-est AID 4U β-est AID 4U graphical protocol. Please click here to download this file.
Supplementary File 1: 4tU_experiment_template.xltx. Please click here to download this file.
| Plasmid Name | Importer/permease | Marker | Comment | |
| p4Fui | S. cerevisiae Fui1 | URA3 | Fui1 imports Uracil and Uridine, making it ideal for pulse/chase experiments. | |
| pAT2 | S. cerevisiae Fui1 | LEU2 | ||
| p4Fui-ΔPEST | S. cerevisiae Fui1 | URA3 | The PEST motif of Fui1 has been deactivated, so the permease is not degraded when there is sufficient intracellular uracil for the cell’s needs. Works well in labelling experiments and improves pulse/chase performance. | |
| p4Fur | S. cerevisiae Fur4 | URA3 | Uracil permease | |
| YEpEBI311 | H. Sapiens ENT1 | LEU2 | Miller et al.11. Also contains an HSV thymidine kinase gene. | |
| (equilibrative nucleoside transporter) | ||||
| All plasmids are 2 µm based. All p4 plasmids and pAT are based on the pRS16 series of plasmids. FUI1 and FUR4 are expressed from their own, endogenous promoters. | ||||
Table 1: Plasmids used with this protocol.
Discussion
This article presents a protocol for extremely rapid and specific 4tU labelling, for recovery of nascent, newly synthesized RNA from S. cerevisiae after as little as 15 s of labelling, with very low contamination by unlabeled RNA.
The user should always take care to maintain the integrity of the RNA by use of cold temperatures and DEPC-treated reagents. Streptavidin bead purification is generally reliable; however, the bead buffer is difficult to handle; it must be made freshly, with its components added in the right order, and not chilled or autoclaved. Common failings include the RNA being incompletely dissolved after the precipitation steps, and so being either not biotinylated or otherwise lost during the processing steps. There is extensive troubleshooting help in the supplementary material.
There are some limitations to be aware of in ers4tU. One already mentioned is that 4tU slows growth of the yeast (Figure 1a). Apart from endogenously thiolated RNAs9, only RNAs that have been transcribed during the labelling period can be purified by this method. Polymerases paused on genes throughout the thiolation time will not produce thiolated transcripts that can be purified, although transcripts that are partially labelled due to polymerases entering or leaving a paused state during thiolation can be recovered. Strains that transcribe poorly, either because of mutation or growth conditions, produce little nsRNA, although the techniques used here will nevertheless improve recovery of nsRNA compared to other methods. Longer times and increased culture volumes may be necessary in these strains and conditions. Note that uracil is a good source of nitrogen and so this method should be trialed before being used for studies involving nitrogen starvation.
The ers4tU protocol is particularly useful for analysis of short-lived RNAs, many of which are so rapidly degraded that they cannot be identified without crippling the degradation machinery. Examples include cryptic unstable transcripts (CUTs)4, and short transcripts produced by premature termination or promoter proximal pausing18 and antisense transcription "upstream" from a promoter (PROMPTs)19. The intermediates produced during processing of stable RNA species are also transient but can be enriched using ers4tU transcription4. The ers4tU protocol is therefore exceptional in permitting highly transient RNA species to be analyzed and captured under near physiological conditions, which is a huge advantage over other methods. This technique has been used to study transcription and downstream RNA processing kinetics in RNA polymerase mutants that elongate faster or slower than normal20.
Thiolation is also compatible with RNA-seq and SLAM-seq21, allowing all RNA produced within a very short time window to be characterized in exquisite detail.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by Wellcome funding to JB [104648]. Work in the Wellcome Centre for Cell Biology is supported by Wellcome core funding [092076]. The authors acknowledge members of the lab for their help: Bella Maudlin, Emanuela Sani, Susanna De Lucas-Arias and Shiney George. The authors would also like to thank Patrick Cramer for the plasmid YEpEBI31111.
Materials
| Name | Company | Catalog Number | Comments |
| β-mercaptoethanol (βME) | Sigma-Aldrich | M3148 | CAUTION toxic. Stock solutions are aproximatly 14 M, make at 1/20 dilution for use |
| Chloroform | Sigma-Aldrich | 25668 | CAUTION toxic |
| Diethyl pyrocarbonate (DEPC) | Sigma-Aldrich | D5758 | add 1/1000 volume to a solution, leave at room temperature for 24 h, then autoclave |
| DMF (N,N-dimethylformamide) | Sigma-Aldrich | 227056 | CAUTION toxic |
| EDTA | Sigma-Aldrich | 3609 | Make 0.5 M and pH to 8.0 with sodium hydroxide |
| Ethanol | Sigma-Aldrich | 29221 | |
| EZ-link HPDP Biotin | Thermo scientific | 21341 | Store protected from light. Disolve all the vial contents in 22.7 mL DMF (to make a 4 mM stock solution). Store away from water, in the dark & at -20 °C. Check the solution before using, as some batches of HPDP precipitate in storage; heat at 42 °C to resuspend. |
| Glucose | Fisher Scientific | G/0500/60 | |
| Glycogen [20 mg/mL] | Sigma-Aldrich | 10901393001 | Store at -20 °C |
| Immobilised TCEP Disulfide Reducing Gel | Thermo Scientific | 77712 | Optional |
| LiCl | Sigma-Aldrich | 793620 | 10 M solution. CAUTION: this gets very hot as is dissolves and can even boil at greater than 100 oC, add the LiCl crystals to the water slowly. |
| Magnesium chloride (MgCl2) | Sigma-Aldrich | 63033 | 1 M solution. CAUTION: this gets very hot as is dissolves and can even boil at greater than 100 oC, add the MgCl2 crystals to the water slowly. |
| Methanol | Fisher Scientific | M/4000/PC17 | CAUTION Toxic and flammable |
| NaH2PO4 | Sigma-Aldrich | S3139 | Make 1 M solutions of each and mix in equal amount to obtain a solution of the appropriate pH |
| Na2HPO4 | Sigma-Aldrich | S3264 | |
| NaCl | Sigma-Aldrich | S9888-M | 5 M solution |
| Phenol, low pH. | Sigma-Aldrich | P4682 | Store in the dark at 4 °C. CAUTION toxic |
| Phenol Chloroform 5:1 (125:24:1) low pH. | Sigma-Aldrich | P1944 | Store in the dark at 4 °C. CAUTION toxic |
| Pierce Spin Columns | Thermo Scientific | 69702 | Optional |
| SCSM single drop-out –ura | Formedium | DSCS101 | |
| Sodium Acetate | Sigma-Aldrich | 32318-M | Make a 3 M solution and pH to 5.3 with acetic acid |
| Sodium hydroxide | Sigma-Aldrich | 795429 | CAUTION corrosive |
| SDS (Sodium dodecyl sulfate) | Sigma-Aldrich | 436143 | CAUTION irritant, do not inhale |
| Streptavidin Magnetic beads | NEB | 1420S | Store at 4°C |
| SUPERase-In, RNase inhibitor | Life technologies | AM2696 | Store at -20°C |
| Thiolated Schizosaccharomyces pombe for spike | See section 1.7 of the protocol | ||
| 4-thiouracil (4tU) | ACROS ORGANICS | 359930010 | Store in the dark. Make 100 mM Stock in 1M NaOH, store solutions at -20°C. |
| Tris base | Sigma-Aldrich | 93362 | 1 M solutions at various pH |
| tRNA | Sigma-Aldrich | 10109541001 | 5mg/ml, store at -20°C |
| Uridine | Sigma-Aldrich | U3750 | Make 1 M solution in H2O. Split into 2 mL aliquots and store at -20 C. |
| Yeast nitrogen base without amino acids with amonium sulphate | Formedium | CYN0410 | |
| Zeba Columns 0.5ml | Thermo Scientific | 89882 | Store at 4 °C |
| Zirconia beads | Thistle Scientific | 110791052 | |
| Equipment and Consumables | |||
| Beadbeater | Biospec | 112011EUR | Other homogenisers can be used; the correct conditions for each homogeniser and strain must be established. |
| Bioanalyser (Agilent) or similar to assess RNA quality. If this is not important a spectrophotometer is useful to quantify the RNA. | |||
| Centrifuge: capable of spinning cultures at 4 °C and at least 3000 g. Pre-chill if possible. | |||
| Centrifuge: capable of spinning up to 2 mL tubes at variable speeds upto 13,000 g and down to 1000 g | |||
| Magnetic rack for separating the beads from the sample. The one used in the paper is 3D printed, available from Thingiverse (thing:3562952). Comercially available racks exist | |||
| PCR machine with a heated lid that will allow incubation in the dark. | |||
| Rotating wheel to rotate 1.5 mL tubes end over end | |||
| Shaking heating block (such as Eppendorf Thermomixer) is recomended | |||
| Tubes, centrifuge, Low retention, RNase free 0.5mL | Eppendorf | H179467N | |
| Tubes, centrifuge, Low retention, RNase free 1.5mL | Ambion | AM12350 | |
| Tubes, centrifuge, 50 mL | Sarstedt | 62.547.004 | Other centrifuge tubes are not gas proof allowing CO2 to disolve in the methanol, this comes out of solution vigorously on adding warm culture, leading to sample loss |
| Tubes, centrifuge, 15 mL | Sarstedt | 62.554.001 | |
| Tubes, 2 mL, screw cap | Greiner | 723361 | |
| Tubes 0.2 mL strip of 8 with integral lids | Brand | 781332 |
References
- Duffy, E. E., Schofield, J. A., Simon, M. D. Gaining insight into transcriptome-wide RNA population dynamics through the chemistry of 4-thiouridine. Wiley Interdisciplinary Reviews: RNA. 10 (1), e1513 (2018).
- Windhager, L., et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Research. 22, 2031-2042 (2012).
- Baptista, T., Devys, D. Saccharomyces cerevisiae Metabolic Labeling with 4-thiouracil and the Quantification of Newly Synthesized mRNA As a Proxy for RNA Polymerase II Activity. Journal of Visualized Experiments. (140), e57982 (2018).
- Barrass, J. D., et al. Transcriptome-wide RNA processing kinetics revealed using extremely short 4tU labeling. Genome Biology. 16, 282 (2015).
- Burger, K., et al. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biology. 10, 1623-1630 (2013).
- Mendoza-Ochoa, G. I., et al. A fast and tuneable auxin-inducible degron for depletion of target proteins in budding yeast. Yeast (Chichester England). 36 (1), 75-81 (2018).
- Barrass, J. D., Mendoza-Ochoa, G. I., Maudlin, I. E., Sani, E., Beggs, J. D. Tuning degradation to achieve specific and efficient protein depletion. Journal of Visualized Experiments. , (2019).
- Hobro, A. J., Smith, N. I. An evaluation of fixation methods: Spatial and compositional cellular changes observed by Raman imaging. Vibrational Spectroscopy. 91, 31-45 (2017).
- Gustilo, E. M., Vendeix, F. A. P., Agris, P. F. tRNA's Modifications Bring Order to Gene Expression. Current Opinion in Microbiology. 11, 134-140 (2008).
- Dolken, L., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
- Miller, C., et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Molecular Systems Biology. 7, 458 (2011).
- Schmitt, M. E., Brown, T. A., Trumpower, B. L. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Research. 18, 3091-3092 (1990).
- Rädle, B., et al. Metabolic Labeling of Newly Transcribed RNA for High Resolution Gene Expression Profiling of RNA Synthesis, Processing and Decay in Cell Culture. Journal of Visualized Experiments. (78), e50195 (2013).
- Herzog, V. A., et al. Thiol-linked alkylation of RNA to assess expression dynamics. Nature Methods. 14, 1198-1204 (2017).
- Ohrt, T., et al. Molecular dissection of step 2 catalysis of yeast pre-mRNA splicing investigated in a purified system. RNA. 19, 902-915 (2013).
- Alexander, R. D., et al. RiboSys, a high-resolution, quantitative approach to measure the in vivo kinetics of pre-mRNA splicing and 3′-end processing in Saccharomyces cerevisiae. RNA. 16, 2570-2580 (2010).
- Wallace, E. W. J., Beggs, J. D. Extremely fast and incredibly close: cotranscriptional splicing in budding yeast. RNA. 23, 601-610 (2017).
- Adelman, K., Lis, J. T. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nature Reviews Genetics. 13, 720-731 (2012).
- Preker, P., et al. RNA Exosome Depletion Reveals Transcription Upstream of Active Human Promoters. Science. 322, 1851-1854 (2008).
- Aslanzadeh, V., Huang, Y., Sanguinetti, G., Beggs, J. D. Transcription rate strongly affects splicing fidelity and cotranscriptionality in budding yeast. Genome Research. 28, 203-213 (2018).
- Schofield, J. A., Duffy, E. E., Kiefer, L., Sullivan, M. C., Simon, M. D. TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding. Nature Methods. 15, 221-225 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved