
Method Article
High Throughput Yeast Strain Phenotyping with Droplet-Based RNA Sequencing
In This Article
Summary
A bottleneck in the ‘design-build-test’ cycle of microbial engineering is the speed at which we can perform functional screens of strains. We describe a high-throughput method for strain screening applied to hundreds to thousands of yeast cells per experiment that utilizes droplet-based RNA sequencing.
Abstract
The powerful tools available to edit yeast genomes have made this microbe a valuable platform for engineering. While it is now possible to construct libraries of millions of genetically distinct strains, screening for a desired phenotype remains a significant obstacle. With existing screening techniques, there is a tradeoff between information output and throughput, with high-throughput screening typically being performed on one product of interest. Therefore, we present an approach to accelerate strain screening by adapting single cell RNA sequencing to isogenic picoliter colonies of genetically engineered yeast strains. To address the unique challenges of performing RNA sequencing on yeast cells, we culture isogenic yeast colonies within hydrogels and spheroplast prior to performing RNA sequencing. The RNA sequencing data can be used to infer yeast phenotypes and sort out engineered pathways. The scalability of our method addresses a critical obstruction in microbial engineering.
Introduction
A primary goal of microbial engineering is to modify microbes to induce them to produce valuable compounds1,2. S. cerevisiae has been the primary organism for microbial engineering due to its ease of culture and the breadth of tools available for engineering its genome3,4,5. However, a hurdle remains in performing functional screens on the modified yeast: screening throughput lags behind genome engineering by orders of magnitude. Screening typically involves isolating strains in microwell plates and phenotyping them by measuring production of a specific compound6,7. The throughput of this process is limited by the large amounts of reagent needed for assaying individual strains in hundred microliter reactions. Droplet microfluidics provides an attractive solution to increase the throughput of yeast screening by orders of magnitude by downscaling reactions normally performed in well plates8. However, as with well plate screens, droplet screens typically detect single product compounds, which provides limited information into the global function of the engineered pathway9,10,11.
RNA sequencing (RNA-seq) may enable more comprehensive characterization of pathway operation by allowing expression levels of all relevant genes to be assessed simultaneously12,13. Moreover, droplet methods allow thousands of cells to be profiled per experiment, providing the throughput necessary to screen libraries of engineered variants14,15. However, RNA-seq methods are optimized for mammalian cells; yeast, by comparison, have less mRNA per cell and a cell wall that is difficult to remove16, precluding their sequencing by existing methods. If a high-throughput droplet method could be devised to enable yeast RNA-seq, it would provide a scalable, cost-effective, and information-rich phenotyping platform for yeast engineering.
We present a detailed protocol of our recently developed method for sequencing yeast cells using high-throughput droplet microfluidics17. To overcome the challenge of limited RNA, we encapsulate and culture single yeast cells in picoliter hydrogel spheres. Culture duplicates the cells, yielding hundreds of copies sharing the same engineered pathway; this reduces variation due to single cell gene expression while significantly increasing the amount of RNA available for sequencing. After culture-based amplification, we spheroplast the cells, removing the cell wall via bulk enzymatic digestion. Cell membranes remain intact, so that each isogenic colony and its associated mRNA remain encapsulated in their hydrogel spheres. This allows us to pair the individual colonies with mRNA capture reagents and lysis buffer, and the mRNA to be captured, barcoded, and sequenced following the Drop-Seq workflow14. Our method allows transcriptome-wide screening of thousands of isogenic yeast colonies per experiment.
Protocol
1. Microfluidic device fabrication
- SU-8 master fabrication
- Design the negative mask for the microfluidic channels for Device A and B (Supplemental File 1 and 2) using computer-assisted design software and have them printed on circuit board film with at least 10 µm resolution.
- Place a clean 75 mm silicon wafer on a spin coater and pour about 1 mL of SU-8 onto its center. Turn on the vacuum to secure the wafer to the chuck.
- For Device A, spin coat SU-8 2150 at 500 rpm for 30 s, followed by 30 s at 2,750 rpm. For Device B, spin-coat SU-8 2100 at 500 rpm for 30 s, followed by 30 s at 2,500 rpm. This will yield SU-8 layers of thickness 200 µm and 120 µm, respectively.
- Remove the wafer from the spin-coater and place on a hotplate at 95 °C for 60 min to soft bake.
- Remove the wafer from the hotplate and let it cool down to room temperature. Place the mask on top of the wafer, and expose under a collimator 190 mW, 365 nm UV LED for 2 min.
- Place the wafer on a hotplate set at 95 °C for 5 min for postexposure baking.
- Remove the wafer and let it cool down to room temperature. Place the wafer in a bath of propylene glycol monomethyl ether acetate (PGMEA) for 20 min.
- Rinse the wafer with PGMEA followed by isopropanol. If any opaque residue is visible during this process, repeat the rinsing with PGMEA and isopropanol. Air dry the wafer.
- Place the wafer on a hotplate at 95 °C for 3 min.
- Remove and place the wafer in a 90 mm diameter Petri dish.
- Polydimethylsiloxane (PDMS) casting on SU-8 master
- Mix together a 10:1 mass ratio of silicone base to curing agent. Degas the PDMS after mixing for about 30 min.
- Pour degassed PDMS on top of the SU-8 master until at least a 5 mm thick layer is formed on top of the wafer.
- Degas the PDMS on top of the wafer for about 30 min.
- Place the wafer in a 65 °C oven for at least 80 min to cure the PDMS.
- Cut out the cured PDMS slab from the wafer.
- Place the PDMS slab with the microfluidic features facing upwards, and punch inlet and outlet holes with a 0.75 mm biopsy punch.
- Clean a 50 mm x 75 mm glass slide with isopropanol and remove all dust from the microfluidic features side of the PDMS slab with tape.
- Expose the cleaned glass slide and the PDMS slab with the microfluidic features face up to 100 Pa (1 mbar O2) plasma for 1 min.
- Place the PDMS slab with the features face down onto the glass slide to allow for bonding. Place the slide in a 65 °C oven for at least 30 min to complete bonding.
- Treat all microfluidic channels by flushing with a fluorinated surface treatment fluid. Bake the device in a 65 °C oven for at least 10 min to evaporate the fluid.
2. Yeast encapsulation in hydrogels using Device A
- Take yeast growing in a suspension culture and count on a hemocytometer.
- Resuspend the cells in phosphate buffered saline (PBS) to a concentration of about 750 k/mL. This ensures that 30% of hydrogels will have one yeast cell in them. Only about half of yeast cells grow into colonies, leading to ~15% of hydrogels containing yeast colonies.
- Mix ultralow melting point agarose at 2% w/v in PBS and heat at 90 °C until melted. This takes ~10 min.
- Load the agarose mix into a syringe with an attached 0.22 µm filter into a syringe pump in front of the space heater set to 80 °C.
- Load a syringe filled with the yeast suspension and a syringe containing fluorinated oil with 2% w/v ionic fluorosurfactant18 into syringe pumps.
- Take the coflow drop splitter device made in section 1 and connect the tubing from the syringes into the device. Guide the tubing from the outlet into a 15 mL conical tube in an ice bucket for drop collection.
- Flow in the three solutions into the device with the following flow rates:
- Flow the yeast suspension with the flow rate of 3 mL/h.
- Flow the agarose mixture at the flow rate of 3 mL/h.
- Flow the fluorinated oil at the flow rate of 15 mL/h.
- Collect about 1 mL of emulsion. Wait an additional 5 min to allow the agarose to fully set.
3. Breaking and washing gel for culture
- Add an equal volume of 20% perfluorooctanol (PFO) in fluorinated oil to the emulsion. Invert the conical tube a few times to allow for the mixing.
- Spin down the broken emulsion at 2,000 x g for 2 min. The gels will pellet above the oil and PFO phases.
- Remove the oil phase and add 2 mL of TE-TW buffer (10 mM Tris pH = 8.0, 1 mM EDTA, 0.01% Tween-20) to resuspend the gels. Transfer the suspension into a new 15 mL conical tube.
- Pellet down the gels as in step 3.2 and wash one more time in TE-TW for a total of two washes.
- Remove supernatant and resuspend gels in 2 mL of media. Transfer to a 5 mL culture tube.
- Incubate at 30 °C overnight under shaking.
NOTE: After the overnight incubation, yeast hydrogels can be kept at 4 °C for several days.
4. Yeast colony lysis
- Transfer gels to a 15 mL conical tube and pellet hydrogels at 2,000 x g for 2 min.
- Wash hydrogels in PBS 2x.
- Wash in 1x spheroplasting buffer 1x.
- Perform a 2–50x dilution of spheroplasting enzyme in spheroplasting buffer and add 1 mL to the hydrogels.
- Incubate at 37 °C for 1 h. The treated yeast will look more transparent (Figure 3A).
- Take the bottom 0.8 mL of hydrogel suspension and transfer into a 1 mL uncapped syringe.
- Place the syringe in the 3D printed syringe holder (Supplemental file 3) and spin at 2,000 x g for 2 min. This will cause the hydrogels to close pack in the syringe head.
5. mRNA capture from lysed yeast colonies using Device B
- Take 240,000 Drop-Seq beads and transfer into a 15 mL conical tube.
- Pellet Drop-Seq beads by spinning down at 1,000 x g for 1 min.
- Remove the supernatant and resuspend beads in 2 mL of 0.9x yeast lysis buffer with 500 mM sodium chloride for a bead suspension concentration of 120,000 beads/mL.
- Transfer the bead suspension to a 3 mL syringe with a stir bar inserted.
- Prepare a syringe containing several milliliters of 2% w/v perfluoropolyether-polyethylene glycol (PFPE-PEG) surfactant in fluorinated oil.
- Evacuate all the aqueous head of the syringe containing close-packed hydrogels and cap the syringe.
- Insert the hydrogel, bead suspension, and oil syringes into syringe pumps and connect via tubing into the encapsulation device made in section 1.
- Connect from the outlet tubing into a 50 mL conical tube on ice.
- Flow in the three solutions into the device with the following flow rates:
- Flow the hydrogels at 0.4 mL/h.
- Flow the bead suspension at 0.4 mL/h.
- Flow the fluorinated oil at 1.6 mL/h.
- Collect ~1 mL of emulsion or run the device until there are no more hydrogels left.
6. cDNA generation, sequencing library preparation, and sequencing
- Add 30 mL of 6x SSC buffer and 1 mL of PFO to the collected emulsion as stated in the Drop-Seq protocol14.
- Continue to follow the Drop-Seq protocol to generate cDNA from mRNA captured on beads, sequencing library preparation, and sequencing data analysis.
Results
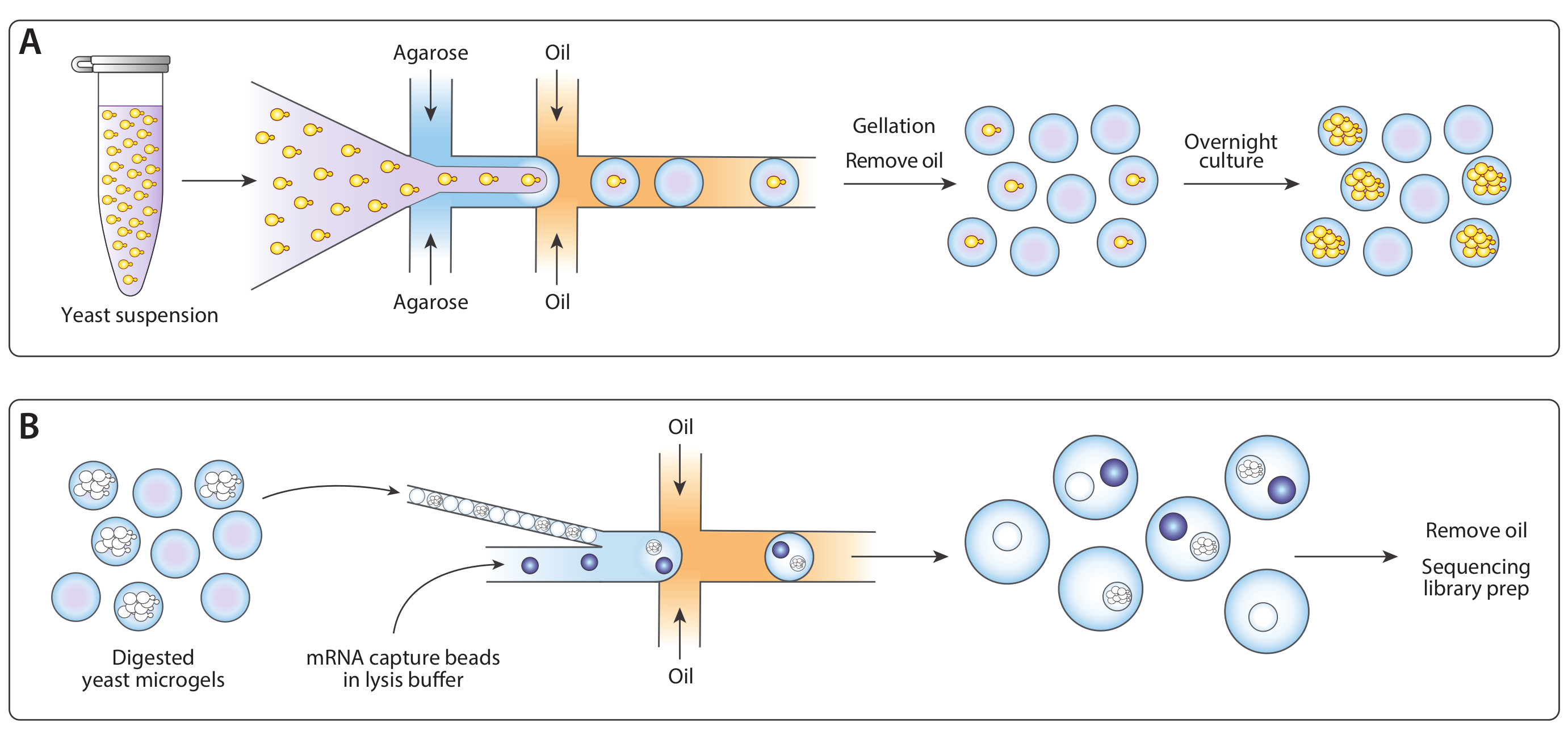
We adapted the previously published Drop-Seq workflow14 for isogenic colony sequencing (ICO-seq) to perform gene expression profiling of isogenic yeast colonies. We isolated single yeast cells and encapsulated them into agarose microgels (Figure 1A). Following overnight incubation of microgels, these encapsulated yeast cells grew into isogenic colonies. Before loading gels into a second microfluidic device for mRNA capture, we digested the yeast cell wall to make the mRNA more accessible (Figure 1B, left). We close-packed these microgels and merged the mRNA capture beads and lysis buffer. Some droplets contained exactly one bead paired with a lysed yeast colony. All beads in the emulsion were collected and the cDNA synthesized and sequenced following the Drop-Seq protocol.
We generated isogenic yeast colonies through single yeast cell encapsulation within agarose microgels using a coencapsulation microfluidic device with an eight drop splitter attached (Figure 2A). We diluted the input yeast suspension to a concentration of ~750,000/mL so that ~30% of microgels have exactly one yeast in them. Prior to inserting the ultralow melting temperature agarose into the device, we dissolved it at an elevated temperature and maintained the syringe at this temperature to prevent premature gelation. At the drop-generation junction (Figure 2B), yeast cells were initially encapsulated into 160 µm droplets. Following the drop-generation junction an eight fold splitter divided these droplets into eight 80 µm droplets (Figure 2C). A syringe filter was attached to the molten agarose to prevent clogs from forming within the channels, which can be as narrow as 37 µm during the drop-splitting. We collected the emulsion on ice, which immediately began the agarose gelation process. We calculated the polydispersity of a typical emulsion to be ~6% (Supplemental Figure 1), though polydispersity values up to 10% are acceptable. Once the agarose gels set, we broke the emulsion and removed the oil phase. The gels were washed in aqueous buffer before immersion in growth media. Overnight incubation of the microgels resulted in isogenic colonies growing within some of the microgels (Figure 2D). The percentage of hydrogels containing colonies of at least 20 cells depended on the culture conditions, including incubation time and media composition. In our demonstration using C. albicans, we determined that about 15% of hydrogels contained a colony after 20 h of suspension culture.
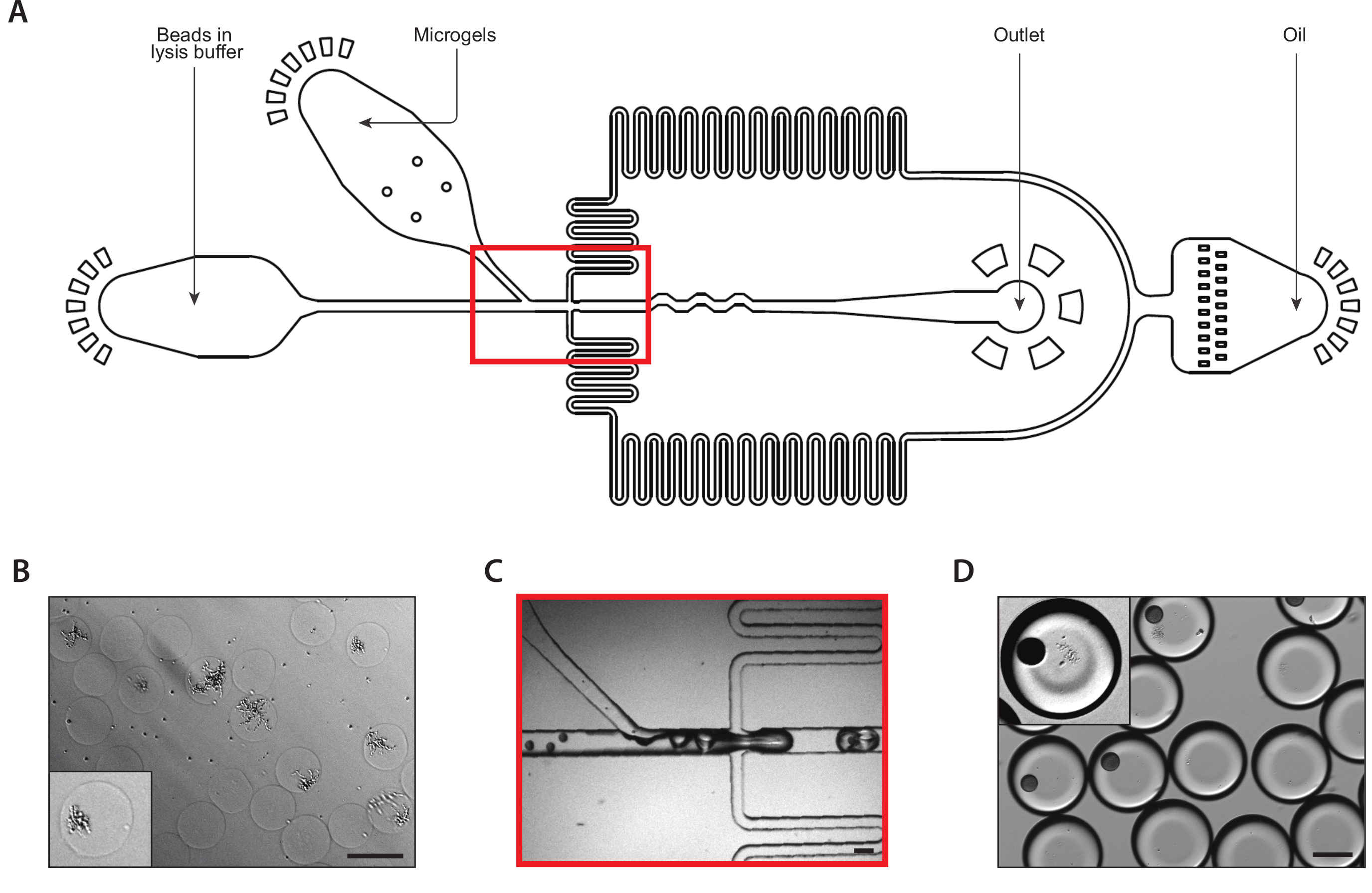
A second coencapsulation device extracted the mRNA from isogenic colonies (Figure 3A). Prior to loading the yeast microgels into the microfluidic device, we washed and immersed the gels in a solution to digest the yeast cell walls. Proper digestion of the yeast cells was verified by microscopy, with treated yeast having a more reflective morphology (Figure 3B). We close-packed the microgels in a syringe and tuned the gel input flow rate such that one gel was in each drop. A stream of mRNA capture beads in lysis buffer mixed with the close-packed gel stream prior to the drop-making junction (Figure 3C). We collected a resulting emulsion of 160 µm droplets, and colonies began to lyse and release their cellular contents. We loaded beads at a limiting dilution to minimize the number of drops containing multiple beads, but close-packing of the gels during drop-making resulted in about 10% of collected drops containing one bead with a lysed colony (Figure 3D).
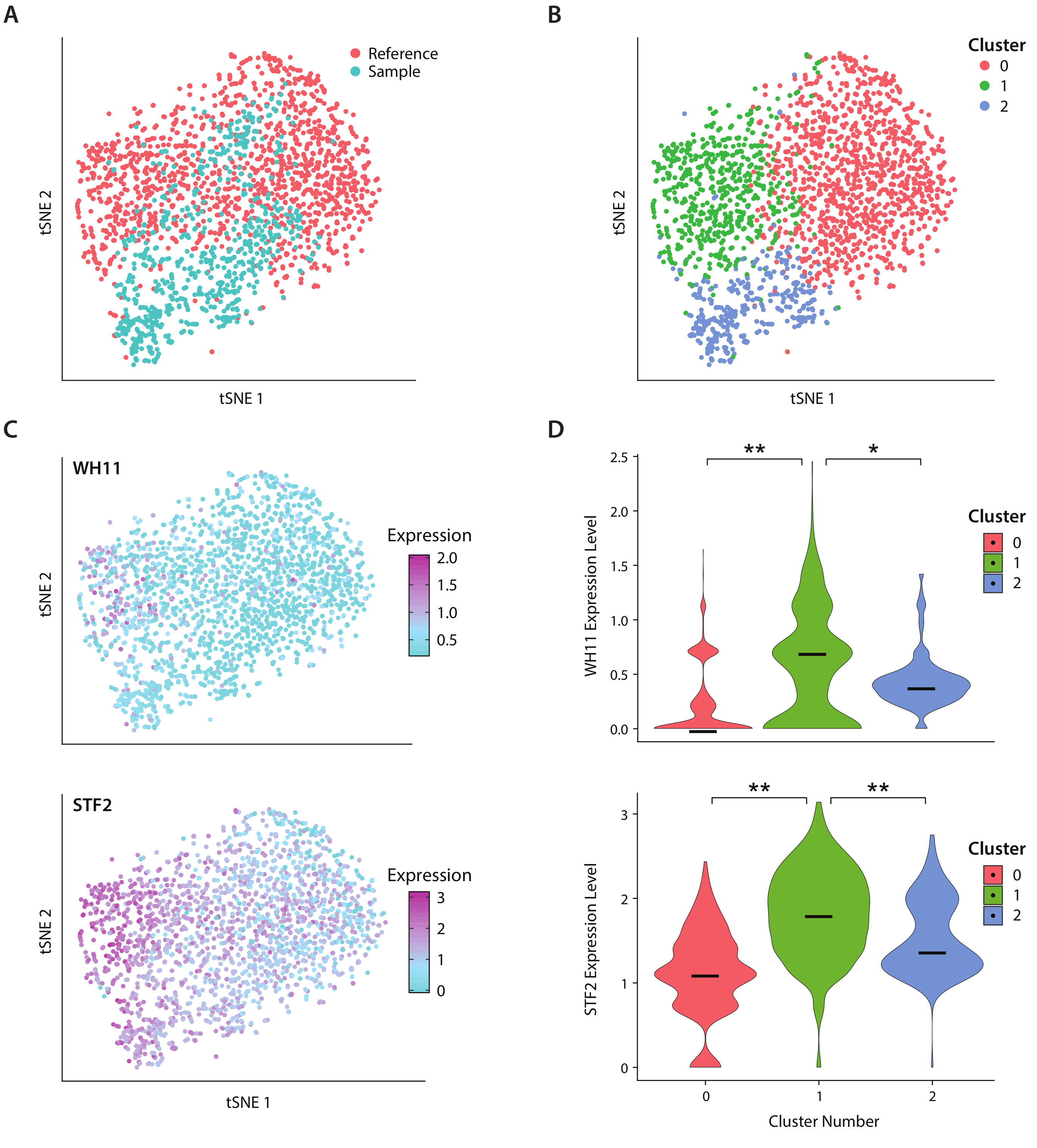
We analyzed gene expression of C. albicans, a species of yeast present in the human gut microbiome, using the ICO-seq workflow. C. albicans is noted for its ability to switch between two different cell states, termed white and opaque19. We use an engineered C albicans strain, strain RZY122, which replaces one copy of the WH11 gene, only active in white cells with YFP20. We obtained a set of gene expression profiles using the workflow and used them for analysis of colonies expressing at least 300 unique genes. As a reference dataset, we used C. Albicans expression data obtained from a previously published study17 and filtered out colonies expressing fewer than 600 unique genes. After performing principal component (PC) analysis and a t-stochastic neighbor embedding (tSNE) dimensionality reduction21, we found general concordance between our sample dataset and the reference (Figure 4A). PC analysis revealed that YFP and WH11 significantly contributed to the first two PC’s. Furthermore, tSNE analysis revealed three clusters (Figure 4B). While cluster 2 was predominantly comprised of cells from the sample dataset, clusters 0 and 1 were comprised of cells from both samples. By overlaying WH11 expression on the tSNE (Figure 4C, upper panel), we determined that cluster 1 likely contained white colonies. We also found that STF2 expression increased in cluster 1 (Figure 4C, lower panel), consistent with previously obtained data17. In clusters 0 and 2, WH11 and STF2 were significantly downregulated compared with cluster 1 (Figure 4D). Genes involved in fermentation, such as ADH1, were upregulated in cluster 0, consistent with previous studies of opaque cells22. We found that colonies in cluster 2 had decreased ribosomal RNA compared with colonies in clusters 0 and 1. Though the sample and reference datasets were obtained using the same stock of cells, this result suggests that even subtle differences in experimental handling can affect gene expression.

Figure 1: Overview of ICO-seq workflow. (A) Yeast growing in a suspension culture were diluted in buffer and coencapsulated with molten agarose in a flow-focusing droplet generator device to enable the Poisson loading of agarose microgels with single yeast cells. The gels set when the agarose cooled, the oil/water suspension was broken, and the oil was removed, yielding a suspension of gel beads in water. Following overnight culture, yeast cells grew into isogenic colonies within the microgels. (B) Colonies were subjected to a cell wall degradation buffer, after which they were close-packed and coencapsulated with mRNA capture beads in a second microfluidic device. Close packing of the microgels ensured each drop had one gel, while Poisson loading of the beads reduced the chance of multiple beads within one drop. Collected drops were processed for cDNA synthesis and generation of a sequencing library. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Generation of isogenic yeast colonies within agarose microgels using Device A. (A) Schematic of microfluidic device, showing locations of the three inputs and output ports. The drop-making junction is highlighted in red. (B) Close-up of the drop-making junction during normal device operation. (C) Micrograph of collected droplets, with a close-up of a droplet containing an encapsulated cell (inset). (D) Micrograph of isogenic yeast colonies in agarose microgels following a 24-hour incubation, with a close-up of two colonies (inset). All scale bars = 100 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Lysis and mRNA capture from isogenic colonies using Device B. (A) Schematic of microfluidic device, showing locations of the three inputs and output ports. The drop-making junction is highlighted in red. (B) Micrograph of yeast colonies following cell wall digestion, with a close-up of one colony (inset). (C) Close-up of the drop-making junction during normal device operation. (D) Micrograph of collected emulsions following microgel and bead pairing, with a close-up showing a drop with a bead and a lysed colony (inset). All scale bars = 100 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Analysis of white-opaque switching response in C. albicans. (A) tSNE plot of a sample dataset combined with a reference dataset from Liu17. (B) Clustering of transcriptomes reveals three clusters visualized on a tSNE plot. (C) Key genes involved in the white-opaque switching response contributed to variation as determined through principal component analysis. (D) Violin plots of normalized expression levels of YFP and WH11 by clusters marked on tSNE plot. **indicates p <<< 0.05 and * indicates p << 0.05. Please click here to view a larger version of this figure.
{kind=link}
Supplemental Figures 1 and 2. Please click here to download these filgures.
Supplemental Files 1-3. Please click here to download these files.
Discussion
Our method for isogenic yeast colony RNA sequencing (ICO-seq) adapts a published single cell RNA sequencing platform, Drop-Seq, for high-throughput screening of engineered yeast strains. Yeast cells contain less than 10% of the copies of mRNA of a typical mammalian cell and have a cell wall that needs to be degraded prior to mRNA capture16. These two factors preclude the direct application of yeast to Drop-Seq or other droplet-based scRNA-seq platforms. To address these issues, we encapsulate single cells within hydrogels and grow them into colonies to provide enough input material for RNA sequencing and we digest the yeast cell wall to generate spheroplasts prior to lysis and mRNA capture. These changes add additional complexity in the ICO-seq workflow when compared with the original Drop-Seq workflow and are critical steps that users must ensure proceed smoothly.
Proper operation of Device A is necessary for encapsulating single yeast cells within agarose hydrogels. Proper counting of the input yeast suspension must be followed to minimize the number of hydrogels with more than one yeast cell, while ensuring that enough hydrogels contain a single cell to ensure a reasonable cell capture efficiency during mRNA capture. During microfluidic device operation, the agarose gel mixture must be well dissolved and passed through a syringe filter to minimize the chance of device clogging. The agarose gel mixture is viscous and the region in which a single channel splits into eight is especially prone to clogging. By centering a high-speed camera to visualize device operation in that region of the device, users can monitor the uniformity of droplets emerging from each of the eight channels and quickly react if the uniformity changes due to clogs in any of the channels. Inspection of a small quantity of collected emulsion under the microscope provides a secondary method for confirming a high-quality emulsion.
Following growth of yeast colonies within hydrogels, several precautions are necessary for ensuring quality mRNA extraction at the single colony level. It is important to optimize the time yeast are in hydrogel culture, because if the yeast are left in culture for too long, many will escape the confines of the hydrogels, leading to a higher background signal during RNA sequencing and lower sensitivity when discriminating between cell types. Proper generation of spheroplasts using Zymolyase ensures that mRNA will be released following cell exposure to lysis buffer. Visual inspection of yeast colonies following Zymolyase should yield shinier yeast cells. Improper cell wall digestion will lead to lower RNA capture efficiency. Lastly, the hydrogels should be close-packed as they are injected into Device B. Monitoring the hydrogel input with a high-speed camera will allow for the termination of emulsion collection once the hydrogels are no longer close-packed upon input into the device, otherwise capture efficiency will be impacted.
A potential concern with our method is that microgel culture of yeast may significantly alter gene expression. Previous work investigating yeast gene expression in microgels and on agar demonstrate differences in gene expression averages but overall a positive correlation17, though further investigation of this claim on a variety of yeast strains is prudent. The method also has limited cell capture efficiency due to stochastic loading of mRNA capture beads following Poisson statistics14. Currently about 10% of drops contain a bead and a colony, and the rate of double encapsulations is expected to be under 1%. Double encapsulations lead to confounding elements during RNA-seq data analysis and their filtering remains challenging23; a capture rate of 25% would lead to a corresponding increase of double encapsulations to 5% (Supplemental Figure 2). Although we demonstrate ICO-seq using the Drop-Seq platform, there are other droplet RNA-seq platforms that introduce mRNA capture beads deterministically rather than statistically, such as the commercially available 10x Genomics Chromium platform15,24. The integration of those platforms with ICO-seq could boost capture efficiencies beyond what Poisson statistics allow. Lastly, a fundamental limitation of droplet RNA-seq is the inability to recover cells of interest after sequencing. This limitation should be taken into account when considering the types of yeast libraries to analyze using this method.
Cell-to-cell heterogeneity has been demonstrated at the clonal level for microbes such as E. coli25 and S. cerevisiae26 revealing new cell states that a bulk-level analysis would otherwise mask. Bulk RNA-seq analyses performed on C. albicans tend to either look at population-wide transcriptome changes, or white and opaque cells as two separate populations27,28. The application of ICO-seq could lead to the discovery of additional substates and provide an analytical framework for discovering new cell states within other yeast species. However, the growth of cells within hydrogels is not limited to yeast: other cell types, such as mammalian, bacterial, and other fungal cells may also be cultivated within hydrogels29,30. The sequencing of isogenic colonies versus single cells leads to the averaging out of biological noise due to cell-to-cell variation, improving discrimination between cell types. This may help when analyzing cells where genetic diversity centers on specific synthesis pathways. The expanded possibilities of cell type input to ICO-seq and its potential integration with commercially available droplet RNA-seq platforms positions ICO-seq as a promising platform for dissecting cellular heterogeneity at the genetic level.
Disclosures
The authors declare no conflicts of interest.
Acknowledgements
This project was supported by National Science Foundation Career Award DBI-1253293, National Institutes of Health New Innovator Award DP2AR068129 and grant R01HG008978, the National Science Foundation Technology Center grant DBI-1548297, and the UCSF Center for Cellular Construction. ARA and ZJG are Chan-Zuckerberg Biohub Investigators.
Materials
| Name | Company | Catalog Number | Comments |
| 0.22 um syringe filter | Milipore Sigma | SLGP033RS | |
| 0.5M EDTA, pH 8.0 | Thermo-Fisher | 15575020 | Used to make TE-TW buffer |
| 0.75 mm biopsy punch | World Precision Instruments | 504529 | |
| 1 mL syringes | BD | 309628 | |
| 1H,1H,2H-Perfluoro-1-Octanol (PFO) | Sigma-Aldrich | 370533 | |
| 1M Tris-HCI, pH 8.0 | Thermo-Fisher | 15568025 | Used to make TE-TW buffer |
| 27 gauge needles | BD | 305109 | |
| 3 mL syringes | BD | 309657 | |
| 3" silicon wafers, P type, virgin test grade | University Wafers | 447 | |
| 3D-printed centrifuge syringe holder | (custom) | (custom) | See Supplemental Files for 3D print file |
| Agarose, low gelling temperature | Sigma-Aldrich | a9414 | |
| Aquapel (hydrophobic glass treatment) | Pittsburgh Glass Works | 47100 | |
| Drop-Seq Beads | ChemGenes | MACOSKO-2011-10 | |
| Glass microscope slides (75 mm x 50 mm) | Corning | 294775X50 | |
| Ionic Krytox Surfactant | Synthesis instructions in ref 14. Can substitute with PEG-PFPE surfactant. | ||
| Isopropanol | Sigma-Aldrich | 109827 | |
| NaCl | Sigma-Aldrich | S9888 | |
| Novec 7500 | 3M | 98-0212-2928-5 | Commonly knowns as HFE 7500 |
| PBS | Fisher Scientific | BP243820 | |
| PE-2 polyethylene tubing | Scientific Commodities | B31695-PE/2 | |
| PEG-PFPE surfactant | Ran Biotechnologies | 008-FluoroSurfactant | |
| PGMEA developer | Sigma-Aldrich | 484431 | |
| Photomasks | CadArt Servcies | (custom) | See Supplemental Files for mask designs |
| Spin coater | Specialty Coating Systems | G3P-8 | |
| SSC Buffer | Sigma-Aldrich | S6639 | |
| SU-8 2100 | MicroChem | Y111075 | |
| SU-8 2150 | MicroChem | Y111077 | |
| Sylgard 184 silicone elastomer kit | Krayden | 4019862 | |
| Tween-20 | Sigma-Aldrich | P1379 | Used to make TE-TW buffer |
| YR Digestion buffer | Zymo Research | R1001-1 | Spheroplasting buffer |
| YR Lysis Buffer | Zymo Research | R1001-2 | |
| Zymolyase | Zymo Research | E1005 | Spheroplasting enzyme mixture |
References
- Peralta-Yahya, P. P., Zhang, F., Del Cardayre, S. B., Keasling, J. D. Microbial engineering for the production of advanced biofuels. Nature. 488 (7411), 320-328 (2012).
- Curran, K. A., Alper, H. S. Expanding the chemical palate of cells by combining systems biology and metabolic engineering. Metabolic Engineering. 14 (4), 289-297 (2012).
- Botstein, D., Chervitz, S. A., Cherry, J. M. Yeast as a model organism. Science. 277 (5330), 1259-1260 (1997).
- Mager, W. H., Winderickx, J. Yeast as a model for medical and medicinal research. Trends in Pharmacological Sciences. 26 (5), 265-273 (2005).
- Krivoruchko, A., Siewers, V., Nielsen, J. Opportunities for yeast metabolic engineering: Lessons from synthetic biology. Biotechnology Journal. 6 (3), 262-276 (2011).
- Vanella, R., et al. Yeast-based assays for screening 11β-HSD1 inhibitors. Microbial Cell Factories. 15 (1), (2016).
- Zhuang, X., Chappell, J. Building terpene production platforms in yeast. Biotechnology and Bioengineering. 112 (9), 1854-1864 (2015).
- Agresti, J. J., et al. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proceedings of the National Academy of Sciences. 107 (9), 4004-4009 (2010).
- Wang, G., et al. RNAi expression tuning, microfluidic screening, and genome recombineering for improved protein production in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences. 116 (19), 9324-9332 (2019).
- Beneyton, T., et al. Droplet-based microfluidic high-throughput screening of heterologous enzymes secreted by the yeast Yarrowia lipolytica. Microbial Cell Factories. 16 (1), 18(2017).
- Sjostrom, S. L., et al. High-throughput screening for industrial enzyme production hosts by droplet microfluidics. Lab on a Chip. 14 (4), 806-813 (2014).
- Nadal-Ribelles, M., et al. Sensitive high-throughput single-cell RNA-seq reveals within-clonal transcript correlations in yeast populations. Nature Microbiology. 4 (4), 683-692 (2019).
- Gasch, A. P., et al. Single-cell RNA sequencing reveals intrinsic and extrinsic regulatory heterogeneity in yeast responding to stress. PLoS Biology. 15 (12), 2004050(2017).
- Macosko, E. Z., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 161 (5), 1202-1214 (2015).
- Klein, A. M., et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 161 (5), 1187-1201 (2015).
- von der Haar, T. A quantitative estimation of the global translational activity in logarithmically growing yeast cells. BMC Systems Biology. 2, 87(2008).
- Liu, L., Dalal, C. K., Heineike, B. M., Abate, A. R. High throughput gene expression profiling of yeast colonies with microgel-culture Drop-seq. Lab on a Chip. 19 (10), 1838-1849 (2019).
- Fallah-Araghi, A., Baret, J. C., Ryckelynck, M., Griffiths, A. D. A completely in vitro ultrahigh-throughput droplet-based microfluidic screening system for protein engineering and directed evolution. Lab on a Chip. 12 (5), 882-891 (2012).
- Berman, J., Sudbery, P. E. Candida albicans: A molecular revolution built on lessons from budding yeast. Nature Reviews Genetics. 3 (12), 918-930 (2002).
- Srikantha, T., Soll, D. R. A white-specific gene in the white-opaque switching system of Candida albicans. Gene. 131 (1), 53-60 (1993).
- van der Maaten, L., Hinton, G. Visualizing data using t-SNE. Journal of Machine Learning Research. 9, 2579-2605 (2008).
- Sun, Y., et al. Deletion of a yci1 domain protein of Candida albicans allows homothallic mating in MTL heterozygous cells. mBio. 7 (2), 00465-00516 (2016).
- McGinnis, C. S., Murrow, L. M., Gartner, Z. J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Systems. 8 (4), 329-337 (2019).
- Baran-Gale, J., Chandra, T., Kirschner, K. Experimental design for single-cell RNA sequencing. Briefings in Functional Genomics. 17 (4), 233-239 (2018).
- Silander, O. K., et al. A Genome-Wide Analysis of Promoter-Mediated Phenotypic Noise in Escherichia coli. PLoS Genetics. 8 (1), 1002443(2012).
- Newman, J. R. S., et al. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 441 (7095), 840-846 (2006).
- Tuch, B. B., et al. The Transcriptomes of Two Heritable Cell Types Illuminate the Circuit Governing Their Differentiation. PLoS Genetics. 6 (8), 1001070(2010).
- Romo, J. A., et al. Global Transcriptomic Analysis of the Candida albicans Response to Treatment with a Novel Inhibitor of Filamentation. mSphere. 4 (5), 00620(2019).
- Huang, H., et al. Generation and manipulation of hydrogel microcapsules by droplet-based microfluidics for mammalian cell culture. Lab on a Chip. 17 (11), 1913-1932 (2017).
- Lin, X., Nishio, K., Konno, T., Ishihara, K. The effect of the encapsulation of bacteria in redox phospholipid polymer hydrogels on electron transfer efficiency in living cell-based devices. Biomaterials. 33 (33), 8221-8227 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved