
Method Article
Confocal Laser Scanning Microscopy of Calcium Dynamics in Acute Mouse Pancreatic Tissue Slices
In This Article
Summary
We present the preparation of acute pancreatic tissue slices and their use in confocal laser scanning microscopy to study calcium dynamics simultaneously in a large number of live cells, over long time periods, and with high spatiotemporal resolution.
Abstract
The acute mouse pancreatic tissue slice is a unique in situ preparation with preserved intercellular communication and tissue architecture that entails significantly fewer preparation-induced changes than isolated islets, acini, ducts, or dispersed cells described in typical in vitro studies. By combining the acute pancreatic tissue slice with live-cell calcium imaging in confocal laser scanning microscopy (CLSM), calcium signals can be studied in a large number of endocrine and exocrine cells simultaneously, with a single-cell or even subcellular resolution. The sensitivity permits the detection of changes and enables the study of intercellular waves and functional connectivity as well as the study of the dependence of physiological responses of cells on their localization within the islet and paracrine relationship with other cells. Finally, from the perspective of animal welfare, recording signals from a large number of cells at a time lowers the number of animals required in experiments, contributing to the 3R-replacement, reduction, and refinement-principle.
Introduction
The mammalian pancreas is a large exocrine and endocrine gland. The exocrine part makes up 96-99% of the total pancreas volume and consists of acini and ducts. The endocrine part is made up of a large number of islets of Langerhans accounting for the remaining 1-4% of the total pancreas volume1. The exocrine part secretes major digestive enzymes that break down energy-rich polymers in food, as well as a bicarbonate-rich fluid, which combines with other gastrointestinal secretions to provide an environment suitable for the action of enzymes. The endocrine part secretes hormones that regulate postprandial distribution, storage, and interprandial release of energy-rich nutrients. Although the exocrine tissue is relatively underdeveloped and the endocrine relatively well-developed at birth, the former quickly overgrows the latter upon weaning2,3,4. Early studies of pancreatic function marked the birth of modern physiology, and major methodological advancements in the field have been followed by major scientific breaktroughs5. Working with the pancreas is technically challenging due to the intricate structure of the gland, but is also a big motivation because of diseases such as pancreatic cancer, pancreatitis, and diabetes that present major threats to public health, and for which novel therapeutic approaches are needed.
Isolated islets6, acini7,8, and ductal fragments had been developed and used for decades as gold standard methods owing to their advantages compared with cell lines and primary dispersed endocrine, acinar, and ductal cells9,10. Despite the markedly improved function of isolated cell collectives, these methods still involve considerable mechanical and enzymatic stress, isolate cells from the surrounding tissue and thus lack paracrine interactions and mechanical support, and most importantly, are accompanied by significant changes to normal physiology11,12,13. The acute mouse pancreatic tissue slice was developed in 2001 out of a perceived need to develop an experimental platform similar to brain, pituitary, and adrenal slices with preserved intercellular contacts, paracrine interactions, mesenchyme, and tissue architecture, as well as without some of the most important shortcomings of the gold standard method in islet research of that time-the isolated islets12,14. Among these shortcomings are damage to the outermost layers, lack of accessibility of core islet areas, and the need for cultivation with possibly important effects on cell identity and physiology12,15. Moreover, the tissue slice method enables studies on animal models with grossly deranged islet architecture where it is impossible to isolate islets, or when islet yield is extremely low by traditional isolation16,17,18,19,20,21.
Additionally, the slice is more suitable for studying morphological changes during the development of diabetes and pancreatitis, for instance, as it enables a better overview of the whole tissue and is also compatible with studying regional differences. Importantly, despite the early focus on the endocrine part, the tissue slice method inherently enables the study of the exocrine components9,22,23. During the first decade after its introduction, the method was employed for electrophysiological studies of beta14,24,25,26,27,28,29 and alpha30,31 cells as well as for examining the morphological and functional maturation of the pancreas2,3. A decade later in 2013, the method was successfully adapted for live-cell calcium imaging of islet cells using CLSM to characterize their responses to glucose32, their functional connectivity patterns33, and the relationship between membrane potential and intracellular calcium by combining a fluorescent calcium dye with a membrane potential dye34. Later in the same year, the method was also used to assess calcium dynamics in acinar cells22,35. Over the following years, pancreatic tissue slices have been used in a number of different studies and successfully adapted to pig and human tissue9,36,37,38,39,40,41. However, taken together, calcium imaging-in mouse pancreatic tissue slices in general and in islets in particular-is still mostly performed by this group. One of the main reasons for this may lie in the combination of a technically challenging tissue slice preparation, the need for a confocal microscope, and rather complex data analysis. The main aim of the present paper is to make this powerful method more accessible to other potential users.
There are already some excellent methodological articles dealing in detail with tissue slice preparation and the use of slices for structural and secretion studies, but not for confocal calcium imaging9,42,43. Therefore, this paper focuses on some additional tips and tricks during the preparation of slices, on steps critical for successful dye loading, image acquisition, as well as on the main steps of basic calcium data analysis. Therefore, this contribution should be viewed as being complementary to rather than as an alternative for the abovementioned method. Similarly, calcium imaging in mouse pancreatic tissue slices shall be viewed as an experimental approach to be used to answer specific questions and is thus complementary to rather than an absolute alternative for other calcium imaging approaches in pancreatic physiology such as isolated ducts or acini, isolated islets, organoids, islets transplanted into the anterior chamber of the eye, and recordings in vivo11,44,45,46,47,48. The promise of calcium imaging in mouse pancreatic tissue slices is probably best illustrated by recent successful recordings of calcium dynamics in islet mesenchymal cells such as pericytes49 and macrophages50, as well as in ductal cells23.
Protocol
NOTE: All experiments were performed in strict accordance with institutional guidelines for the care and use of animals in research. The protocol was approved by the Administration of the Republic of Slovenia for Food Safety, Veterinary Sector and Plant Protection (permit number: 34401-35-2018/2).
1. Preparation of pancreatic tissue slices
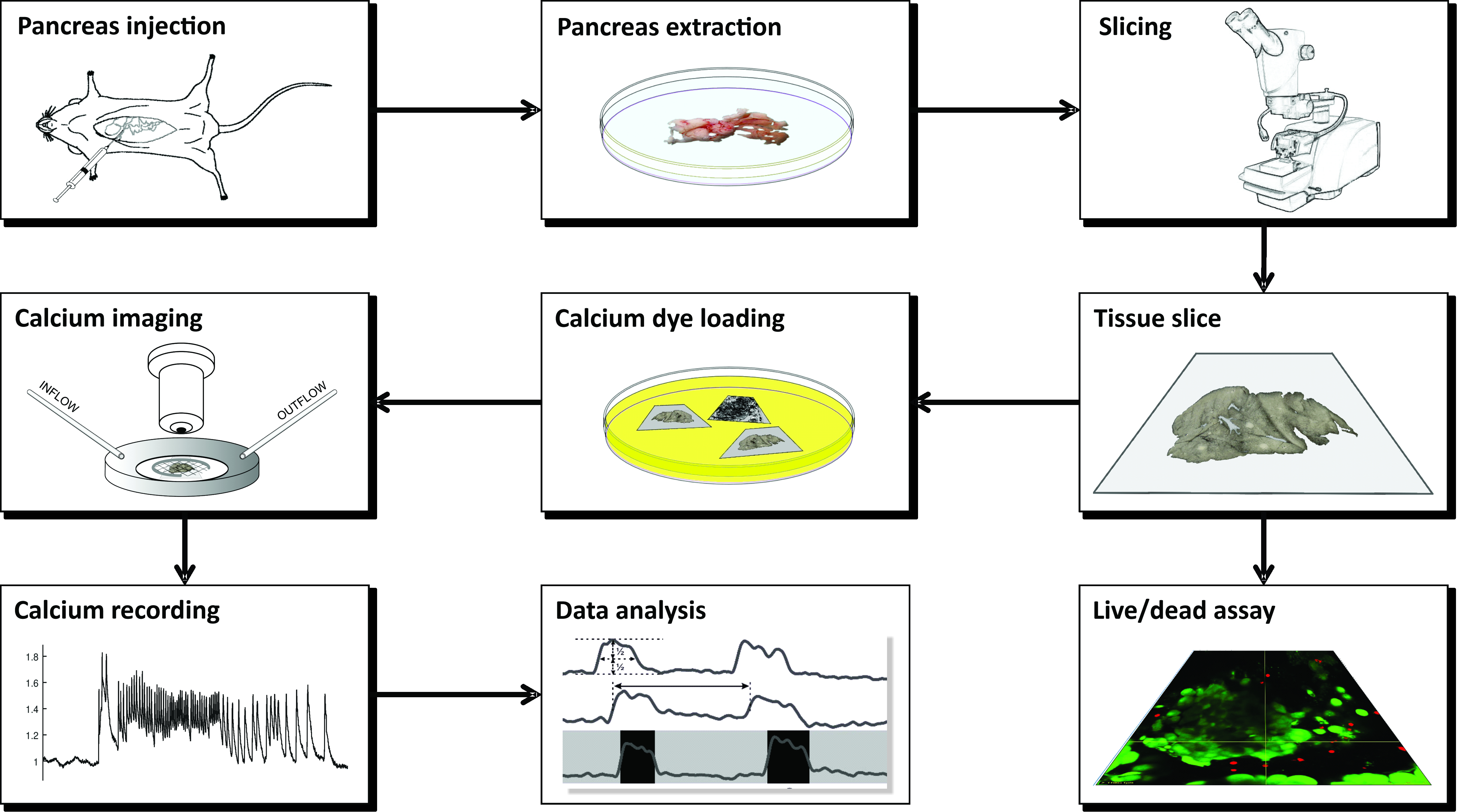
NOTE: The preparation of acute mouse pancreas tissue slices for calcium imaging using CLSM requires a number of instruments, different solutions, and proceeds in a series of critical steps that are schematically presented in Figure 1 and described in detail below.

Figure 1: Workflow diagram. Schematic representation of all steps in the process of pancreatic tissue slice preparation, beginning with the injection of agarose into the common bile duct, followed by extraction of the pancreas and slicing. The prepared slices can be used for assessing the viability of the tissue with a Live/Dead kit or stained with a calcium sensor. Once stained, they are ready for imaging. Recordings obtained from the imaging process are then used for data analysis. Please click here to view a larger version of this figure.
{kind=link}
- Preparation of solutions
NOTE: All solutions should be prepared in advance and can be stored in the refrigerator at 4-8 °C for up to one month. For the preparation and storage of tissue slices, approximately 0.5 L of extracellular solution (ECS) with 6 mM glucose and 0.3 L of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer is needed. For 1 day of calcium imaging with the perifusion system set at 1-2 mL/min flow rate, approximately 0.5 L of ECS is needed.- Extracellular solution with 6 mM glucose
- Prepare 1 L of ECS containing 125 mM NaCl, 26 mM NaHCO3, 6 mM glucose, 6 mM lactic acid, 3 mM myo-inositol, 2.5 mM KCl, 2 mM Na pyruvate, 2 mM CaCl2, 1.25 mM NaH2PO4, 1 mM MgCl2, and 0.5 mM ascorbic acid. Mix thoroughly until all ingredients completely dissolve. Take 50 μL of ECS into an 0.5 mL microcentrifuge tube, place it onto the osmometer according to the manufacturer's instructions, and check the osmolarity.
NOTE: The osmolarity should be 300-320 mOsm. For stimulation of beta cells, use solutions with higher glucose concentrations. To ensure physiological pH value of 7.4 during slicing and experiments, constantly bubble the ECS with carbogen (i.e., a gas mixture of 95% O2 and 5% CO2) at barometric pressure. A simple bubbling system can be set up by attaching one end of a 5 mm silicon tubing to the source of carbogen (i.e., a pressurized gas cylinder) and the other end of the tubing placed directly into the bottle containing ECS. - Alternatively, prepare a 10x stock containing 1250 mM NaCl, 260 mM NaHCO3, 30 mM myo-inositol, 25 mM KCl, 20 mM Na pyruvate, 12.5 mM NaH2PO4, and 5 mM ascorbic acid. When the ECS containing 6 mM glucose is needed, mix 100 mL of the stock with 2 mL of 1 M CaCl2, 1 mL of 1 M MgCl2, 0.455 mL of 13.2 M lactic acid, and 1.08 g of glucose, and fill with double-distilled water up to 1 L. If needed, use different amounts of glucose to obtain other glucose concentrations.
- Prepare 1 L of ECS containing 125 mM NaCl, 26 mM NaHCO3, 6 mM glucose, 6 mM lactic acid, 3 mM myo-inositol, 2.5 mM KCl, 2 mM Na pyruvate, 2 mM CaCl2, 1.25 mM NaH2PO4, 1 mM MgCl2, and 0.5 mM ascorbic acid. Mix thoroughly until all ingredients completely dissolve. Take 50 μL of ECS into an 0.5 mL microcentrifuge tube, place it onto the osmometer according to the manufacturer's instructions, and check the osmolarity.
- HEPES buffer with 6 mM glucose
- Prepare 0.5 L of HEPES-buffered solution (HBS) containing 150 mM NaCl, 10 mM HEPES, 6 mM glucose, 5 mM KCl, 2 mM CaCl2, and 1 mM MgCl2; titrate to pH = 7.4 with 1 M NaOH.
NOTE: If carbogen is not available, this buffer can be used for all steps instead of ECS.
- Prepare 0.5 L of HEPES-buffered solution (HBS) containing 150 mM NaCl, 10 mM HEPES, 6 mM glucose, 5 mM KCl, 2 mM CaCl2, and 1 mM MgCl2; titrate to pH = 7.4 with 1 M NaOH.
- Agarose (1.9% w/w)
- Prewarm a water bath to 40 °C.
- Add 0.475 g of low-melting-point agarose and 25 mL of ECS containing 6 mM glucose to an Erlenmeyer flask, and place the flask in a microwave oven at maximum power for a few seconds until it starts to boil. Take the flask out of the oven and swirl it a few times, until the agarose dissolves completely. Transfer the flask with the liquid agarose to the prewarmed water bath at 40 °C to cool the agarose to the desired temperature and to keep it liquid until injection. Secure the flask with a stabilizing lead ring.
NOTE: The agarose can be prepared in advance and kept in the refrigerator. Before use, warm the agarose in the microwave oven until it liquefies, and transfer the Erlenmeyer flask into a water bath prewarmed to 40 °C. The agarose can be re-used up to 5x. If re-used beyond 5x, it will become dense and harder to inject.
- Extracellular solution with 6 mM glucose
- Injection of pancreas with agarose
NOTE: Sections 1.2 and 1.3 explain the preparation of tissue slices that can be used for different experimental purposes such as calcium imaging, electrophysiology, immunohistochemistry, secretion studies, and structural/microanatomical studies.- Fill a 5 mL syringe with the liquid agarose from the Erlenmeyer flask in the water bath from step 1.1.3.2, remove any bubbles, and mount a 30 G needle. Protect the needle with a cap, and keep the filled syringe back in the water bath with the needle facing downward and the entire volume of agarose below the water surface. Secure the syringe with a stabilizing lead ring in such way that the ring presses the syringe against the wall of the water bath.
NOTE: Be careful not to push any agarose into the needle as it will harden quickly and block the needle. If the room temperature is low, and if the injection is performed by a less experienced person, increase the temperature of the water bath up to 42 °C to gain some additional time for injection. - Fill an ice bucket with ice, and place the bottle containing ECS in it. Bubble the ECS constantly at 1.5 mL/min with carbogen at barometric pressure and room temperature to ensure oxygenation and a pH of 7.4.
- Sacrifice a mouse by administering a high concentration of CO2 followed by cervical dislocation. Make all efforts to minimize animal suffering.
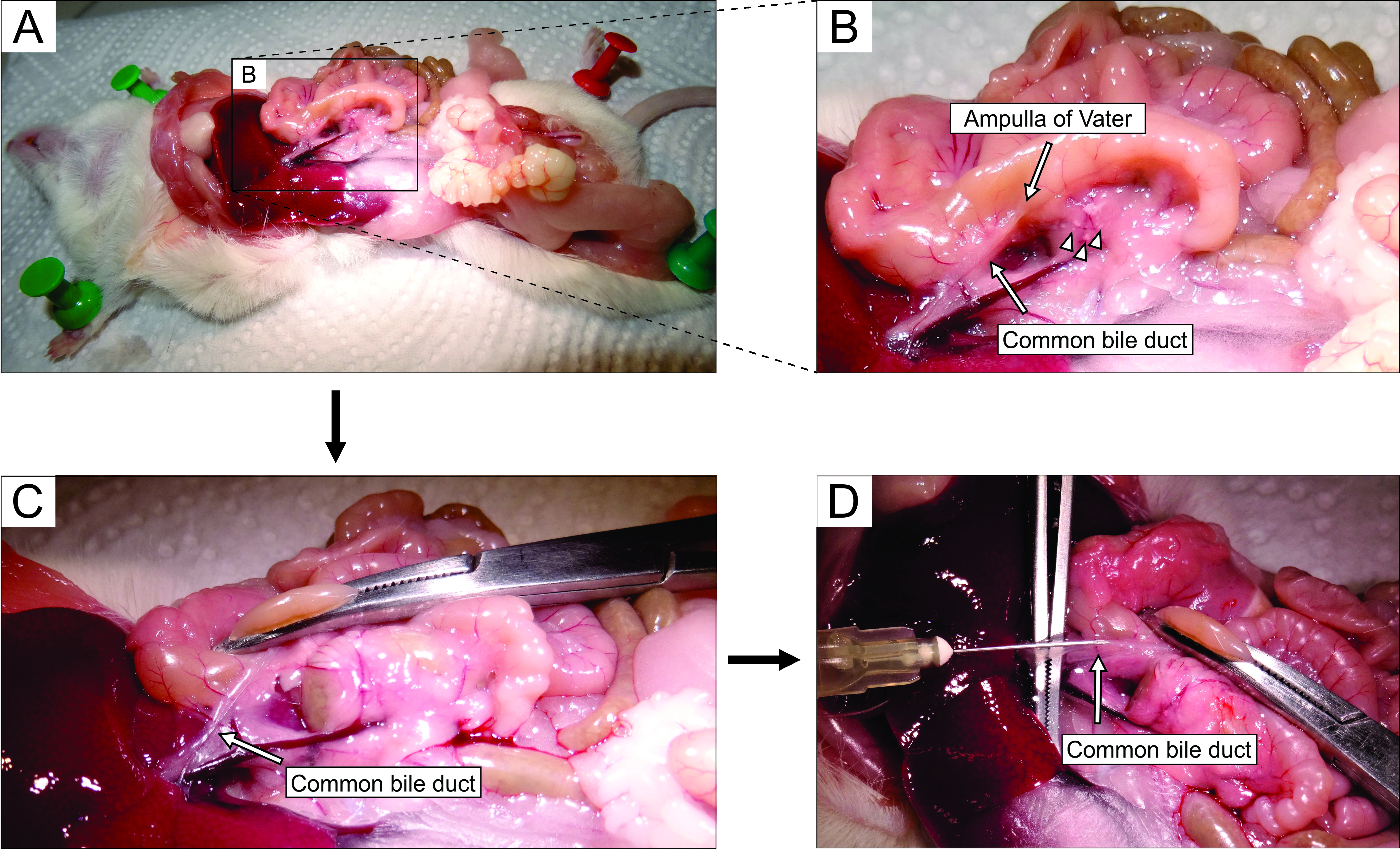
- Working under a stereomicroscope, access the abdomen via laparotomy (Figure 2A). Gently flip the gut to the left side of the mouse (from the anatomical perspective of the mouse) to expose the common bile duct. Use forceps to slightly lift the duodenal part, and find the major duodenal papilla-papilla of Vater. Clamp the common bile duct at the duodenal papilla using a hemostat (Figure 2B,C) to prevent leakage of agarose from the duct into the duodenum.
NOTE: To prevent agarose leakage into the duodenum and further up and down in the gastrointestinal tract, place the hemostat in such a way that it also clamps the duodenum both proximally and distally from the papilla. It is best to use a curved hemostat for this purpose. - With small sharp forceps, reach under the common bile duct, and break the membrane that attaches the duct to the pancreatic tissue. For better visual control and easier injection, clear as much fat and connective tissue from the duct as possible.
- Place the duct perpendicularly onto large forceps (Figure 2D), and inject the prepared liquid agarose into the proximal part of the common bile duct (Figure 2D). Be sure to squeeze the syringe hard as the agarose is viscous. Keep filling the pancreas until it becomes whitish and slightly distended or for at least 20-30 s.
NOTE: This is the most critical step in slice preparation. If there are any kinks in the ductal tree of the pancreas, gently elevate or pull the pancreas away from the syringe to level them out. Do not decide when to stop injection based on the volume injected from the syringe as the backflow at the point of injection and forward leakage into the duodenum are typically much higher than the volume injected into the ductal tree of the pancreas. Importantly, successful injections can be performed with practically unnoticeable changes in syringe volume. - Remove the syringe, and slowly pour 20 mL of the bubbled ice-cold ECS at 0-4 °C from the bottle onto the pancreas to cool the tissue and harden the agarose.
- Gently extract the pancreas using forceps and fine tough-cut scissors. Place the extracted pancreas into a 100 mm Petri dish containing ~40 mL of ice-cold ECS, and gently move it around to wash it. Transfer the pancreas into a fresh 100 mm Petri dish containing ~40 mL of ice-cold ECS.
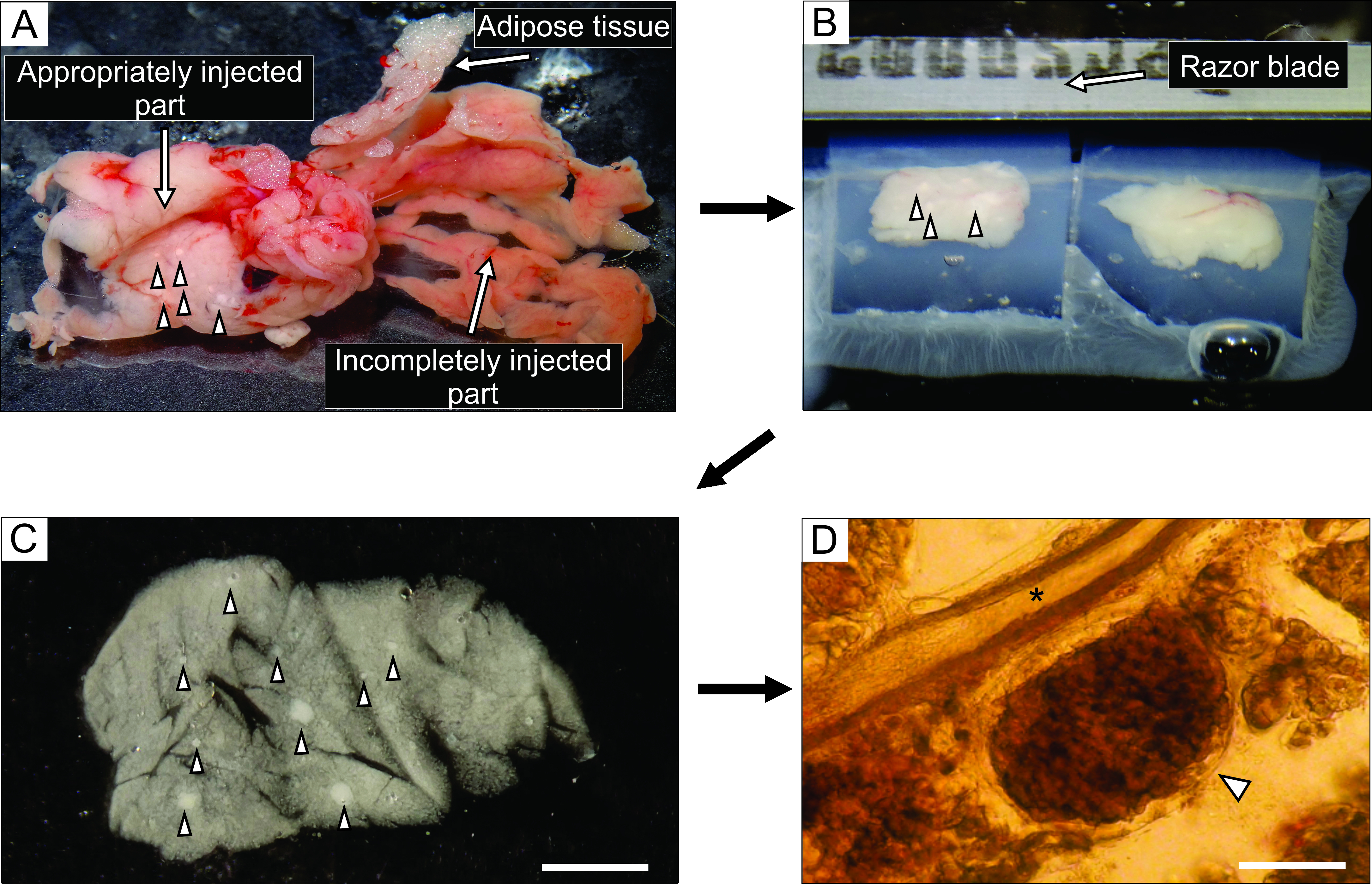
- From the well-injected part of the pancreas, which appears whitish (Figure 3A), cut up to 6 blocks of tissue, 0.1-0.2 cm3 in size, using forceps and tough-cut scissors. Clear them of any connective and fatty tissue.
- Fill a 35 mm non-sticky bottom-Petri dish with approximately 5 mL of liquid agarose at 40 °C, transfer the tissue blocks into it, and immediately put the Petri dish on ice to cool it down and harden the agarose.
NOTE: The way the blocks of pancreas are trapped in agarose determines the way they are cut during slicing. Experienced experimentalists can try and fine-tune the position of the blocks during the few moments before the agarose hardens when placed on ice. - After the agarose with the tissue blocks hardens, turn the Petri dish upside down onto a flat smooth surface such as the lid of a 100 mm Petri dish, and remove the agarose by gently cutting with one half of a razor blade into the margin between the lateral wall of the Petri dish and the agarose. With a razor blade, cut individual agarose cubes, each containing one tissue block, taking care that each tissue block is surrounded by agarose. Glue the agarose blocks onto the sample plate of the vibratome with cyanoacrylate glue (Figure 3B).
- Fill a 5 mL syringe with the liquid agarose from the Erlenmeyer flask in the water bath from step 1.1.3.2, remove any bubbles, and mount a 30 G needle. Protect the needle with a cap, and keep the filled syringe back in the water bath with the needle facing downward and the entire volume of agarose below the water surface. Secure the syringe with a stabilizing lead ring in such way that the ring presses the syringe against the wall of the water bath.

Figure 2: Injection of agarose into the common bile duct. (A) Open the abdominal cavity, and expose the organs in the peritoneal cavity. (B) The magnified part of the area enclosed by the rectangle in panel A. The white spot on the duodenum (indicated by the arrow) indicates the ampulla of Vater. Islets of Langerhans are denoted by arrowheads. (C) Clamp the ampulla of Vater by a curved hemostat, and raise it slightly to expose and gently stretch the common bile duct (arrow). (D) Cannulation of the common bile duct and the injection of 1.9% agarose solution using a 5 mL syringe and a 30 G needle. Please click here to view a larger version of this figure.
{kind=link}
- Slicing
- Fill the cutting chamber of the vibratome with ~0.15 L of ice-cold ECS, and bubble constantly with carbogen. Surround the cutting chamber with ice, and add 2 ice cubes (~10 mL each) made of ECS with 6 mM glucose into the cutting chamber. Mount the razor blade for cutting onto the vibratome, and screw-fix the sample plate with agarose blocks into its place.
- Set the slicer to cut agarose blocks at 0.05 to 1 mm/s and 70 Hz into 140 µm-thick slices with a surface area of 20-100 mm2. For slicer settings, follow the manufacturer's instructions.
- Immediately after each cutting step, pause the slicer, gently collect the slices with a fine paint brush, and transfer them into a 100 mm Petri dish filled with 40 mL of HEPES buffer with 6 mM glucose at room temperature (Figure 3C).
NOTE: The slices can be kept in HEPES buffer at room temperature for at least 12 h, and the buffer should be exchanged every 2 h.

Figure 3: Pancreas tissue preparation and slicing. (A) The extracted mouse pancreas after agarose injection. White tissue on the left indicates a well-injected part (duodenal part), while the more reddish part on the right shows the insufficiently injected part of the pancreas (splenic part). (B) Vibratome slicing of two blocks of pancreas tissue embedded in agarose. (C) Acute pancreatic tissue slice with islets of Langerhans indicated by arrowheads. Scale bar = 3000 µm. (D) Acute pancreatic tissue slice under the light microscope with the islet of Langerhans indicated by an arrowhead, asterisk indicates a pancreatic duct. Scale bar = 100 µm. Please click here to view a larger version of this figure.
{kind=link}
2. Live/dead assay using LIVE/DEAD Viability/Cytotoxicity Kit for mammalian cells
NOTE: For some experiments, it is useful to check the viability of cells in the slices (Figure 4) by the live/dead assay as follows.
- Follow the manufacturer's instructions to thaw vials with reagents of the LIVE/DEAD Viability/Cytotoxicity Kit, and prepare working solutions of calcein AM just before use. Use the solutions within one day.
- In a 15 mL centrifuge tube, mix 5 µL of 4 mM calcein AM (Component A), 20 µL of 2 mM ethidium homodimer-1 (EthD-1, Component B), and 10 mL of Dulbecco's Phosphate-buffered Saline (D-PBS) to prepare a working solution containing approximately 2 µM calcein AM and 4 µM EthD-1. Vortex thoroughly.
- Using a fine paint brush, gently transfer the tissue slices into a 3 mL Petri dish with fresh HEPES buffer to dilute serum esterase activity. Remove the HEPES buffer, and cover the slices with 100-200 µL (or more if necessary) of the working solution from step 2.2.
- Incubate the slices for 30-45 min at room temperature in a closed Petri dish. Image the tissue slices using excitation/emission filters as recommended by the manufacturer.
3. Calcium dye loading
NOTE: Fluorescent dyes should be shielded from light exposure during the whole process of preparation and loading of the dye, as well as during handling of the stained tissue slices. Tin foil can be used to cover tubes or Petri dishes containing the calcium dye.
- Dye preparation
- Dissolve the contents of one vial (50 µg) of the cell permeable Ca2+ indicator dye (excitation/emission 495/523 nm; see the Table of Materials), 7.5 µL of dimethylsulfoxide (DMSO), and 2.5 µL of the polaxamer (20% solution in DMSO; Table of Materials) in 6.667 mL of HBS containing 6 mM glucose in a 15 mL screw cap tube.
NOTE: This final solution contains 6 µM of the Ca2+ indicator dye, 0.11% DMSO, and 0.037% polaxamer. - Aspirate and expel the solution in the screw cap tube repeatedly with a pipette for 20 s; submerge the tube in an ultrasonic bath chamber for 30 s, and vortex for 30 s to improve solubilization. Aliquot 3.333 mL of the final Ca2+ indicator dye solution prepared in step 3.1.1 into 5 mL Petri dishes.
- Dissolve the contents of one vial (50 µg) of the cell permeable Ca2+ indicator dye (excitation/emission 495/523 nm; see the Table of Materials), 7.5 µL of dimethylsulfoxide (DMSO), and 2.5 µL of the polaxamer (20% solution in DMSO; Table of Materials) in 6.667 mL of HBS containing 6 mM glucose in a 15 mL screw cap tube.
- Dye loading
- Transfer the prepared tissue slices from the 60 mL Petri dish with HBS into 5 mL Petri dishes filled with the dye solution by gently lifting each tissue slice using a thin, soft paint brush and placing it in the dye solution. Incubate up to 10 tissue slices per Petri dish.
- Place the slice-loaded Petri dish on an orbital shaker at room temperature set to orbital motion at 40 turns per minute for 50 min. Incubate the slices in the dye solution exposed to ambient air at room temperature, but shielded from light by covering the Petri dish with tin foil.
- Storing of slices
- Transfer the stained tissue slices from the 5 mL Petri dish into a 60 mL Petri dish filled with dye-free HBS by gently lifting them using a fine, soft paint brush. Store up to 20 slices per Petri dish.
NOTE: Use the tissue slices for imaging at this point. The tissue slices will retain the Ca2+ indicator dye for several hours. The survival of slices and retention of the dye can be improved by placing the Petri dish in an insulated container, surrounded by ice. This is especially important if the dye-loaded slices are to be transported. Additionally, exchange the HBS every 2 h.
- Transfer the stained tissue slices from the 5 mL Petri dish into a 60 mL Petri dish filled with dye-free HBS by gently lifting them using a fine, soft paint brush. Store up to 20 slices per Petri dish.
4. Calcium imaging
- Setup of the confocal microscope
- Choose an appropriate objective magnification depending on the interest of the study. Select 20x and 25x (numerical aperture [NA] 0.77-1.00) for visualizing a whole islet, several acini simultaneously, or larger ducts. Select higher magnifications to study intracellular dynamics.
- Choose the acquisition mode for time-lapse imaging (e.g., time-lapse, xyt, or similar mode). Set the pinhole to 100-200 µm.
- Set the light path for green fluorophores: excitation at 488 nm, and collection of emission at 500-700 nm. Preferably select detectors with a high quantum efficiency (e.g., gallium arsenide phosphide) over photomultiplier detectors.
- Setup of the recording chamber and the perifusion system
- Mount the recording chamber on the temperature-controlled stage of the microscope and the perifusion system (either gravity-fed or peristaltic-pump-based setup, volume 1 mL). Position the inlet and the outlet on the far edges of the recording chamber to avoid meandering of the perfusate within the chamber, and set the inflow and the outflow to equal values (1-2 mL/min). Avoid drifting of the liquid meniscus height and droplets in the perifusate.
- Set the temperature control of the perifusion system to 37 °C.Initiate the perifusion with the non-stimulatory solution, and prepare the stimulating solutions. Change the solutions via motorized valves or by manually switching the solutions that feed the perifusion system.
- Record calcium dynamics
- Transfer a single tissue slice into the recording chamber. Immobilize the tissue slice with a U-shaped platinum weight with taut nylon mesh (e.g., from nylon stockings). Avoid positioning nylon threads over the structure of interest.
- Locate an islet/acinus/duct using the brightfield option. Run live imaging to position the studied structures into the field of view, and set up the imaging parameters. Optimize the signal-to-noise ratio by adjusting the laser power, detector amplification, and line averaging/binning to allow visualization of the cells while keeping the laser power minimal.
- Adjust the focal plane of the recording to ~15 µm below the cut surface (Figure 5) to avoid recording from potentially damaged cells at the cut surface.
- Acquire images. Set the sampling frequency to 1-2 Hz to detect individual oscillations initially, and use a resonant scanner capable of fast line averaging (8-20) at a higher acquisition rate (>10 Hz) to record intracellular Ca2+ ([Ca2+]IC) activity. Allow an interval (e.g., 30% of the total sampling time) between consecutive point illuminations to prevent phototoxicity. Record a high-resolution image (e.g., 1024 x 1024 pixels, line averaging > 50) before the time series acquisition (see section 5).
NOTE: The sampling frequency of 1-2 Hz is below the Nyquist criterion for acquisition frequency for most cells, and the shape of the signal will be undersampled by default. - Refer to an online chart, if available in the imaging software, to obtain instant feedback on the preparation response, over-illumination, photobleaching, and mechanical drift. In case of a high rate of bleaching during acquisition, stop the recording and decrease laser power while increasing the detector gain to maintain the signal-to-noise ratio. In the case of mechanical drift, check for tension between tubing/cables and the microscope stage as well as liquid leakage or changes of the volume in the recording chamber. Optionally, attempt to correct the drift during acquisition manually; however, note that this will inherently yield limited results.
NOTE: Endocrine cells are highly heterogeneous at near-threshold concentrations. A sufficient length of stimulation is needed to detect the range of activation/deactivation delays in individual cells. This is especially important for accurate detection of off-responses following highly stimulatory protocols. - Use calcium imaging to functionally discriminate between endo- and exocrine cells (Figure 6). To record transient activity during activation and deactivation, apply stimuli without stopping the recording.
- Save the data after the end of experimentation (consider using an auto-save function). Allow a cooling period before switching off the laser power so as not to damage the lasers during the shutdown procedure.
5. Analysis of data
- Visually inspect the recording qualitatively by replaying the time-lapse video. Check for cell drifts from the field of view or the optical plane. If a drift within the optical plane has occurred, employ the drift correction plugin in ImageJ.
- Select regions of interest (ROIs) using microscope software or third-party software. Use the high-resolution image, maximal projection, or frame average as the reference to select ROIs. Replay the time-lapse imaging to visualize responding cells that are not visible in reference images. Position the ROIs such that the selected area of a ROI does not overlap with neighboring cells to avoid signal crosstalk between ROIs.
- Export the time series data as ROI average value per frame. Export ROI coordinates.
- Correct the time series data for bleaching (Figure 7A) by employing a combination of an exponential and linear fit, as described by
 (1)
(1)
where x(t) denotes the fluorescence signal at a time point t; xcorr(t) the corrected signal at the corresponding time points; and a, b, and c the parameters of the fit calculated as the least sum of squares between the corr(t) and x(t). - Analyze the activation and deactivation phase of the response (Figure 7B). Calculate the first derivative of the time series data, and determine the zenith and the nadir of the derivative corresponding to the activation and deactivation, respectively. Alternatively, manually select the start of the phasic increase. Save and export the activation/deactivation times and corresponding cell coordinates.
- Analyze the plateau phase (Figure 7C). Detect individual oscillations by thresholding the raw data or by thresholding the first derivative of the time-series data. Define the beginning and end of an individual oscillation as the time corresponding to the oscillation's half amplitude.
- Calculate the duration and the frequency of individual oscillations for each cell. Calculate the inverse value of the interspike interval (suitable for regular activity patterns). Alternatively, divide the number of oscillations by the time interval of the record (suitable for irregular activity patterns).
- Calculate the active time. Express the active time as the sum of durations, and divide this value by the time interval. Alternatively, multiply the frequency and the duration that correspond with an oscillation.
NOTE: Dividing the sum of durations by the time interval provides robust results, but has low statistical discrimination as a single data point per cell is obtained. Multiplying the frequency and duration of an oscillation provides an oscillation-to-oscillation temporal resolution.
Results
The injection of the agarose solution into the pancreatic duct is the most critical step in pancreas tissue slice preparation. A successful injection can be recognized by a whitening of the pancreas tissue, as seen on the left side of Figure 3A, while an incompletely injected part of the pancreas is presented on the right side of Figure 3A. The islets of Langerhans can be recognized by the naked eye or under a stereomicroscope, and this aids in cutting the appropriate parts of the pancreas for subsequent embedding in agarose blocks (Figure 3B). In a freshly cut mouse pancreatic tissue slice, islets of Langerhans can be easily distinguished from the surrounding exocrine tissue and mesenchyme as white spots under the stereomicroscope (Figure 3C) or as brownish structures under the light microscope (Figure 3D). The pancreatic tissue slices can be used for distinct types of experiments for at least 12 h after slicing. In addition to the gross morphological assessment under the stereomicroscope, the light microscope, and the functional responses of cells during calcium imaging, the viability of the pancreatic tissue slices can be assessed (Figure 4).

Figure 4: Viability of cells within the tissue slice. Viability of cells was determined with the Live/Dead assay. Live cells are stained by Calcein AM (shown in green), while dead cells are stained with ethidium homodimer-1 (shown in red). Yellow lines denote the position of the X-Y cross section of the Z-stack displayed at the bottom and the right. The full depth of the Z-stack is 88 µm. Please click here to view a larger version of this figure.
{kind=link}
For calcium imaging experiments, the fluorescent calcium indicator needs to penetrate through a few layers of cells. Figure 5A presents successful loading of the cell-permeable Ca2+ indicator dye into the pancreatic tissue slice in which individual islet and acinar cells can be recognized. In contrast, slices in Figure 5B-D are not optimal due to unsuccessful penetration of the dye (Figure 5B), lack of islet cells (Figure 5C), and a lot of necrotic tissue on the surface (Figure 5D). Such slices can be discarded, checked for the presence of additional islets that are cut or stained better (see Table 1 for troubleshooting), or used for recording the responses of exocrine cells.

Figure 5: Examples of usable and unusable preparations. (A) An example of a successful preparation of the pancreas tissue slice with well-stained cells in the islets of Langerhans, as well as ductal cells and surrounding acinar tissue. (B) An example of a poorly stained tissue slice. (C) Example of an islet of Langerhans with structural discontinuations. (D) An example of an islet of Langerhans containing many dead cells and a lot of debris. The "glow-over, glow-under" lookup table on the right displays 0 intensity in green and saturation in blue. Scale bar = 100 µm. Please click here to view a larger version of this figure.
{kind=link}
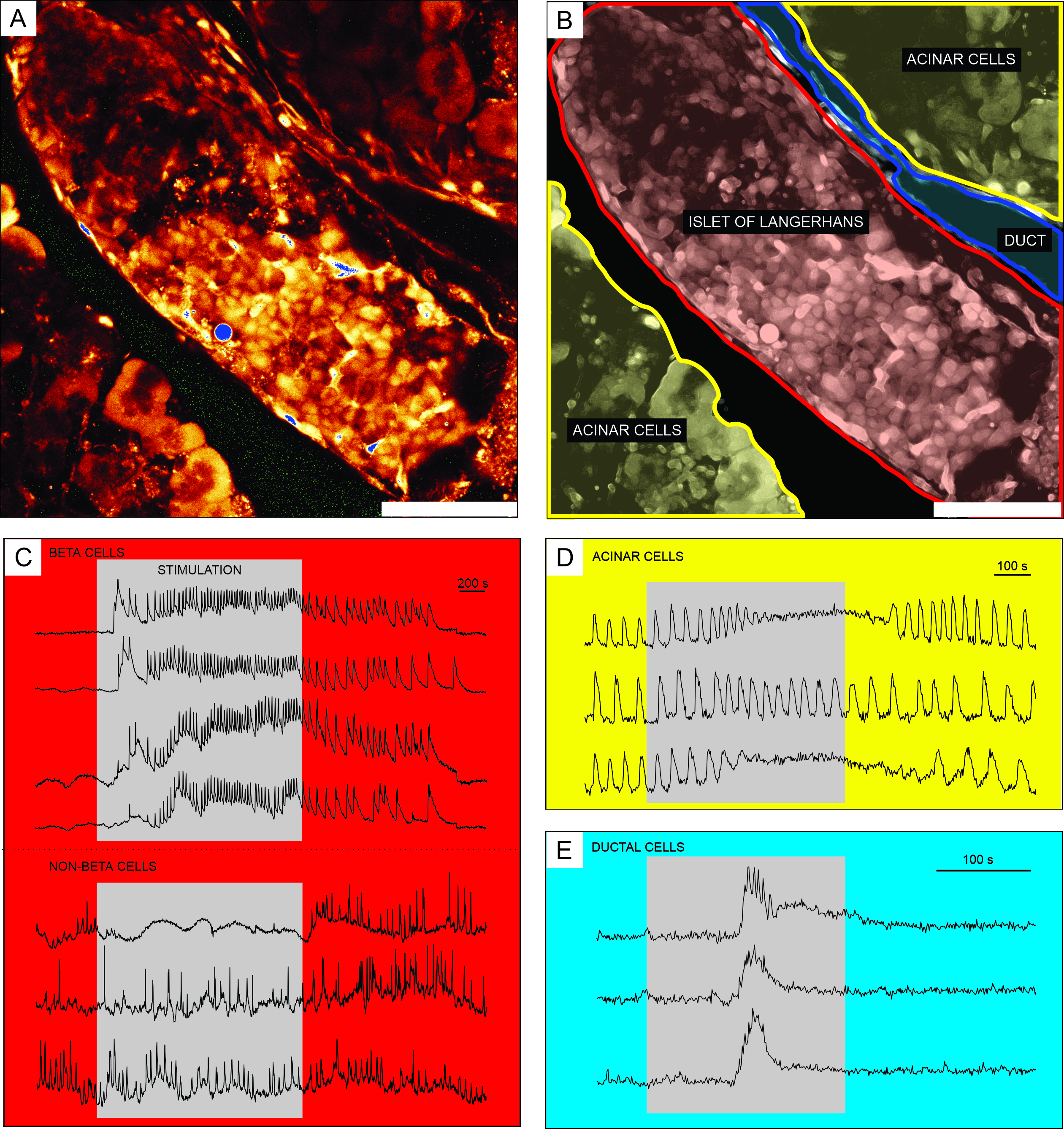
Representative results from calcium imaging using the cell-permeable Ca2+ indicator dye are shown in Figure 6. In Figure 6A, a high-resolution image of a pancreatic tissue slice is presented, containing an islet of Langerhans, acinar tissue, and a pancreatic duct. For better distinction, the endocrine, exocrine, and ductal part of the pancreatic tissue slice presented in Figure 6A are colored in Figure 6B. Using appropriate stimuli can functionally discriminate between different islet cells, or islet and non-islet cells51. Beta cells will typically respond to a square-pulse stimulation by glucose with a transient increase in [Ca2+]IC followed by fast calcium oscillations on a sustained plateau (Figure 6C, upper panel).
As all beta cells are coupled into a single, large, functional syncytium, these oscillations are also very well synchronized among different cells by means of spreading [Ca2+]IC waves32,34,52,53,54 (Figure 7C). Slower [Ca2+]IC oscillations with a period of 5-15 minutes may underlie the fast oscillations or even be the predominant type of reponse55,56. The same simple protocol may reveal other types of responses, especially at the periphery of islets (Figure 6C, lower panel). As these cells are not synchronized with beta cells and respond with faster and more irregular oscillations that are already present in low glucose conditions or with a decrease in activity, such responses are highly suggestive of non-beta cells21,32,57,58. However, their definitive functional characterization requires more complex protocols with additional stimulation steps or alternative approaches, which are discussed below. Typical responses of acinar and ductal cells are presented in Figure 6D and Figure 6E, respectively. Refer to the literature for more details on acinar and ductal cells22,23,35.

Figure 6: Representative results of calcium dynamics in distinct types of pancreatic cells. (A) A high-resolution image of an islet of Langerhans with surrounding tissue. Scale bar = 100 µm. (B) Delineation of distinct parts of pancreatic tissue with acinar tissue shown in yellow, an islet of Langerhans shown in red, and a segment of the ductal tree in blue. Scale bar = 100 µm. (C) Typical traces of calcium dynamics in beta and putative non-beta cells during stimulation with 12 mM glucose; 3 mM glucose was used for non-stimulatory conditions. Protocols that can be used for more specific discrimination of non-beta cells are described in the discussion section. (D) A typical trace of calcium dynamics of acinar cells stimulated by 25 nM acetylcholine. (E) A typical trace of calcium dynamics of ductal cells stimulated by 1 mM chenodeoxycholic acid. Please click here to view a larger version of this figure.
{kind=link}
After successful calcium imaging, the data are first exported and corrected for bleaching by a combination of an exponential and linear fit, as described in the protocol section. A time series before and after bleaching correction is presented in Figure 7A. Thereafter, several parameters in the activation and deactivation phase of the response as well as the plateau phase can be analyzed. A delay in the onset of [Ca2+]IC increase after stimulation can be measured as represented by delayA in Figure 7B and the heterogeneity in delays among individual cells (delayA1). The same parameters (delayD and delayD1) can be used to describe the deactivation phase. Following the initial transient [Ca2+]IC increase, the plateau phase in most pancreatic beta cells in an islet is characterized by relatively regular high frequency [Ca2+]IC oscillations. The plateau phase can be described by analyzing the classical functional parameters. The schematic presentation of [Ca2+]IC oscillations duration, frequency, and percentage of active time are presented in Figure 7C. In calcium imaging with acquisition rates higher than 10 Hz, calcium waves repeatedly spreading across the islet can also be recognized clearly (Figure 7C).

Figure 7: Analysis of time-series data. (A) Correction of time-series data for photobleaching. (B) Analysis of delays to activation after stimulation and to deactivation after cessation of stimulation with 12 mM glucose. Duration of stimulation is denoted by the light gray, shaded bar in the image. (C) Analysis of several parameters of the plateau phase: I) Duration of the oscillation determined at half-height, II) frequency of oscillations determined by inter-oscillation intervals. III) active time as a product of frequency and duration of oscillations. I-IV) Delays between oscillations in any given wave of oscillations that spread across the islet of Langerhans determined by the delays (Δt) in time at which a single cell reaches half-height of the oscillation. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The pancreatic tissue slice method is a fast experimental method to study the morphology and physiology of the endocrine and exocrine parts of the pancreas in a more conserved, in situ preparation. Many of the advantages have already been pointed out in the Introduction. It is worth pointing out that in general (i.e., not only for calcium imaging), the slice approach to study pancreatic physiology saves time because it does not involve a recovery period after isolation. The latter is not absolutely necessary with all types of experiments and uses of isolated islets from different species, but is typically employed to increase purity, restore viability and functionality, and sometimes to collect islets from several donors59,60,61,62,63,64. However, in the context of calcium imaging, beta cell responses have been found to depend on culture duration and conditions, and this is an important source of variation that should be taken into account when using isolated islets15,65. The same issue should be considered for tissue slices if their long-term culture becomes a widely used option in the future22,36. The tissue slice method also has a high yield and thus potentially reduces animal suffering and increases statistical power. Moreover, as many slices can be prepared from a single animal and because the slices survive for long periods, including the same animal or even the same islet in both the experimental and the control groups becomes feasible.
As the original architecture and cell-to-cell communications are preserved, and because it is compatible with a number of structural analyses, electrophysiological, imaging methods, and hormone secretion assays, this method is especially useful to study pancreatic functions that depend on undisturbed interactions between individual cells, e.g., sensitivity to secretagogues, paracrine and immune interactions between different cell types, patterns of electrical activity, properties of calcium dynamics, and the secretion of different hormones. For calcium imaging specifically, the main advantages of using slices are the exposure of the islet core and the possibility to acquire signals from many different cell types with high resolution. Depending on the requirements of the experiment and the age of animals, the thickness can be varied, the slices can be transfected, or obtained from animals with genetically encoded reporters. As explained in more detail below, the latter two approaches also enable specific functional identification and characterization of responses from non-beta cells 31,66. Moreover, islets from well-defined parts of the organ can be studied for differences in responsiveness or susceptibility to disease. Although they do not require a recovery incubation period, they can easily be incubated with different pharmacological agents, fatty acids, high glucose, and cytokines.
Most importantly, as high resolution is achievable in combination with single-cell or even subcellular resolution, confocal calcium imaging in slices is one of the most suitable methods for analyzing calcium waves, functional connectivity, and the different functional roles of cells in distinct parts of an islet54,67. Despite a number of advantages, the tissue slice approach has important limitations. First, it is still at least partly disruptive to islet and exocrine architecture, especially at the cut surface, and precautions, such as low temperature, frequent exchange of solutions, and gentle and quick manipulation, are needed during preparation to prevent additional mechanical and endogenous enzymatic damage. Second, the patterns of nutrient and secretagogue delivery are still inferior to the in vivo route, the preparation is detached from systemic innervation, and inter-organ feedback, such as between the islet and its target tissues, is impossible, in contrast to in vivo approaches. Third, maximum slice thickness is limited by oxygenation, nutrient delivery, and pH regulation at ~200 µm9. Further, both preparation of slices and imaging need a lot of training, and in-depth analyses of calcium data from long time series and from many cells require specialized knowledge that is often not included in the toolkit of a classical physiologist and requires help from physicists or data scientists. The advantage that homo- and heterotypic interactions are preserved can also complicate the analysis of samples due to the presence of signals from other cells in regions of interest. Depending on protocols, activation of other cells can lead to indirect additional stimulation or inhibition of an observed cell.
This can only be resolved conclusively by deconvolution approaches, by more complex stimulation protocols including substances that block some of the indirect effects, by using specific knock-out animals, and by careful comparison of results with results from other studies employing more reductionist methodologies. Additionally, if secretion measurements are necessary, it should be kept in mind that some slices may lack islets, and the total mass of endocrine tissue in a single slice is typically low. The preparation of acute pancreatic tissue slices for imaging involves several critical steps discussed in the following sections and summarized in Table 1, where the reader can also find short, but important tips for troubleshooting. First, when preparing the agarose solution, the agarose powder must dissolve completely, otherwise the undissolved particles may obstruct the injection. Keep the homogeneous agarose solution at 37-45 °C to prevent hardening of agarose due to too low a temperature on the one hand and to prevent tissue damage due to too high temperatures on the other hand. After use, the remaining agarose may be stored at 4 °C and reheated, although repeated reheating can result in increased density due to water evaporation, eventually making the injection difficult or impossible.
The next critical step in the preparation is clamping the major duodenal papilla correctly. A white spot on the duodenum indicates the junction of the common bile duct and the duodenum. A clamp placed too proximally will result in obstruction of some lateral pancreatic branches of the common duct, disabling the injection of these parts, whereas a clamp placed too distally will result in agarose leaking through the lower resistance path directly into the duodenum. Before cannulation of the common bile duct, the surrounding adipose tissue can be carefully removed for better visualization of the duct and greater control during injection. Insufficient precision during removal of the surrounding tissue may result in perforation of the duct. The selection of the needle diameter used for agarose injection is also important. In mice, a 30 G needle is preferably used; smaller (32 or 33 G) needles require more effort due to high viscosity of the agarose solution and are more prone to obstruction. However, if used in combination with a lower-density agarose solution, they can be very helpful in smaller mouse strains and younger animals. During the initial postnatal days, agarose may alternatively be injected subcapsularly rather than intraductally2. Using needles with greater diameter in mice will most probably result in damaging the common bile duct. This can also happen with the correct needle diameter, and a forceps can help in keeping the needle in place during injection. Larger diameter needles may be the only solution in case of larger ducts, as found in rats. If the needle is too narrow to ensure a tight seal preventing back-leakage, a ligature may be placed around it upon successful entry into the duct.
Agarose injection takes some effort due to the solution's viscosity, and once the injection process has started, it should not be interrupted as the low-melting-point agarose solution may solidify in the needle or the largest parts of the ductal tree before the injection is completed. This will result in poor tissue penetration and worse support during cutting. The duct should always be cannulated at the point where the left hepatic duct and the cystic duct join to form the common bile duct.. If the common bile duct gets perforated, repeatedly try cannulating closer to the duodenum. When the pancreas is sufficiently stabilized with agarose solution and extracted from the peritoneal cavity, small pieces of well-injected tissue are cut. Before embedding them into the agarose, it is crucial to remove all the adipose and connective tissues as their residues make slicing more challenging. The same applies to blood vessels and duct residues, except when they are the focus of the experiment. In this case, make sure to position them in such a way that the desired cross-section will be obtained. When embedding the tissue in agarose, ensure that the temperature is appropriate (37 °C), and that the tissue is completely surrounded by agarose, as forces during vibratome slicing can rip out the pancreas tissue from the agarose blocks.
Quickly drying the tissue blocks before placing them in agarose by placing them briefly on a paper tissue can help prevent poor contact between tissue and agarose during this step. During solidification of agarose blocks, place the Petri dish horizontally, and prevent contact between the pancreas tissue and the bottom of the Petri dish. If the pancreas is not fully injected, the cutting process will be challenging. Therefore, try to reduce the cutting speed to obtain tissue slices. To minimize cell damage during vibratome slicing, replace the ECS (and the ice cubes made of ECS) in the slicing chamber regularly. The latter will reduce the activity of pancreatic enzymes released from acinar tissue during slicing. The thickness of the slices is also of crucial importance. For calcium dynamics and electrophysiological experiments, 140 µm slices are usually cut; however, according to the aim of the study, slice thickness can range from 90 µm to 200 µm. Keep in mind that in thicker slices, the diffusion of oxygen and nutrients will be limited, but they will include more tissue. Additionally, the proportion of uncut islets may be expected to increase with increasing slice thickness. Slices can be stored in a regularly exchanged ECS at room temperature for several hours or even cultivated in an appropriate cell medium for several days; however, this may eventually affect the normal islet cell physiology3,22.
When preparing the dye solution, ensure careful mixing of all components, and avoid exposure to ambient light. The pancreatic slice is composed of many cell layers, and the uptake of calcium dye is limited to the first few most superficial cell layers, as described previously for isolated islets58,68, and pituitary slices69. However, in contrast to isolated islets where the surrounding capsule and outer cell layers hinder the penetration of the dye into deeper layers, tissue slices permit access to the entire cross-sectional surface of the islet, enabling simultaneous measurement of calcium dynamics in hundreds of cells from all layers of an islet. Fluorescent Ca2+ indicators are the most widely used for measuring calcium dynamics, and together with CLSM, they enable recordings with high temporal resolution, reaching several hundred Hertz. When selecting the most appropriate fluorescent Ca2+ indicator, consider different factors, including the indicator form, which influences the cell loading method, measurement mode (qualitative or quantitative), and dissociation constant (Kd) that needs to be in the Ca2+ concentration range of interest and depends on pH, temperature, presence of Mg2+ and other ions, as well as protein binding. As cellular Ca2+ signals are usually transient, the Ca2+ binding rate constant should also be considered. For measuring [Ca2+]IC dynamics in pancreatic cells, this group mainly uses the cell-permeable Ca2+ indicator dye described in this protocol (Table of Materials) as it is a long wavelength indicator with the emission wavelengths in the spectrum where cellular autofluorescence is usually less problematic, and the energy of excitation light is low, which reduces the potential for cellular photodamage. Because this dye is fluorescent at low Ca2+ concentrations, this facilitates the determination of baseline [Ca2+]IC and increases cellular visibility before stimulation. After binding Ca2+, the fluorescence intensity of the dye increases 14-fold, enabling detection of even slight changes in [Ca2+]IC.
For successful live-cell calcium imaging, several crucial hardware parameters need to be considered, as described in the protocol section. For live-cell imaging wherein signal amplitudes are low and chances of phototoxicity are high, objectives with a higher NA are preferably used to collect more light from the specimen. If calcium dynamics must be recorded with a high temporal resolution, use the resonant scanner instead of linear galvanometers. Besides choosing the right objective, the use of highly sensitive detectors-such as hybrid detectors that require less laser power-avoids phototoxicity and photobleaching. This is of special importance for long-lasting calcium imaging. Other important steps in calcium imaging are parameter settings of image quality for time series acquisitions. The most important are the temporal and the spatial resolution. As the calcium dynamics per se determines the lowest acceptable temporal resolution, the sampling rate needs to be at least two-fold higher than the expected signal frequency to detect the signal or even 10 times higher to detect the shape of the signal reliably. In acute pancreatic tissue slices, calcium dynamics can be measured in hundreds of cells simultaneously and therefore, the spatial resolution is also important. This can be enhanced by increasing the number of pixels or by increasing the line averaging during live acquisition. However, because of the inverse relationship between the spatial and the temporal resolution, a trade-off between both settings is needed.
If calcium imaging has to be performed in a specific cell population within the pancreas, a stimulus able to functionally differentiate the cells within the slice is necessary. High glucose reliably and quickly activates beta cells to an oscillatory pattern that is superimposed on an elevated calcium level and is highly synchronized among all cells within an islet32,58,70. The beta cells are the most numerous cell type within an islet and are located mostly in the islet core in mice. The same stimulation protocol decreases and sometimes does not appreciably change the bursting in alpha cells30,32,58,70,71,72. To discriminate alpha cells functionally, low (3 mM) glucose, glutamate or adrenalin can be used to increase their frequency or basal [Ca2+]IC21,72,73,74,75. They represent 10-20% of islet cells and will be detected on the islet periphery1. Delta cells are also found on the periphery. They make up only ~5% of the total number of endocrine cells in an islet and are typically active in 6 mM glucose and respond to glucose stimulation with an increased irregular bursting activity from the baseline or a slightly elevated calcium level1,32,71,76. Ghrelin can be used for specific stimulation of delta cells21,77,78,79 in calcium imaging experiments. However, protocols for specific functional identification of PP and epsilon cells remain to be defined. Further, 25 nM acetylcholine reliably activates acinar cells into bursting activity35,80,81. Additionally, a number of other secretagogues, such as cerulein, cholecystokinin, and carbamylcholine, can be used to evoke calcium responses in acinar cells22,40,82,83.
Finally, 1 mM chenodeoxycholic acid reliably evokes calcium responses in ductal cells in tissue slices; angiotensin II, ATP, and some other secretagogues can also be used11,23,84,85. Whenever a functional identification based on characteristic responses to specific secretagogues and inhibitors is not sufficient, genetically labelled animals31, transfected cells73, or immunocytochemistry can be employed for the identification of different cell types9,22,71,86. During the last couple of years, the tissue slice method has been successfully adapted to human tissue, opening many new important research avenues in both exocrine41 and endocrine physiology9,36,37,39. Interestingly, a detailed assessment of calcium dynamics in human islets has been notoriously difficult and remains to be investigated in greater detail87. Combined with advanced confocal microscopy, the pancreatic tissue slice method has enabled many new insights into the calcium dynamics in mice and will hopefully do the same for human tissue.
Disclosures
The authors declare that the research was conducted in the absence of any commercial or financial interest.
Acknowledgements
The work presented in this study was financially supported by the Slovenian Research Agency (research core funding nos. P3-0396 and I0-0029, as well as research projects nos. J3-9289, N3-0048, and N3-0133) and by the Austrian Science Fund / Fonds zur Förderung der Wissenschaftlichen Forschung (bilateral grants I3562--B27 and I4319--B30). We thank Maruša Rošer, Maša Čater, and Rudi Mlakar for excellent technical assistance.
Materials
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Analytical balance KERN ALJ 120-4 | KERN & SOHN GmbH | ALJ 160-4A | |
| Confocal microscope Leica TCS SP5 II Upright setup | Leica | 5100001578 | |
| Confocal microscope Leica TCS SP5 AOBS Tandem II setup | Leica | ||
| Cork pad 15 cm x 15 cm | |||
| Corning 15 mL centrifuge tubes | Merck KGaA, Darmstadt, Germany | CLS430790 | |
| Corning Round Ice Bucket with Lid, 4 L | Fischer Scientific, Leicestershire, UK | 432124 | |
| Double edge razor blade | Personna, USA | ||
| Dumont #5 - Fine Forceps | FST, Germany | 11254-20 | |
| Eppendorf Safe-Lock Tubes 0.5 mL | Eppendorf | 0030 121.023 | |
| Erlenmeyer flask 200 mL | IsoLab, Germany | 027.01.100 | |
| Fine Scissors - ToughCut | FST, Germany | 14058-11 | |
| Flat orbital shaker IKA KS 260 basic | IKA | Ident. No.: 0002980200 | |
| Glass lab bottle 1000 mL | IsoLab, Germany | 091.01.901 | |
| Hartman Hemostat, curved | FST, Germany | 13003-10 | |
| HCX APO L 20x/1.00 W HCX APO L (water immersion objective, 20x, NA 1.0) | Leica | 15507701 | |
| Measuring cylinder 25 mL | IsoLab, Germany | 015.01.025 | |
| Micromanipulator Control box SM-7, Keypad SM-7 | Luigs & Neumann | 200-100 900 7311, 200-100 900 9050 | |
| Microwave owen | Gorenje, Slovenia | MO20MW | |
| Osmometer Gonotec 010 | Gonotec, Berlin, Germany | OSMOMAT 010 Nr. 01-02-20 | |
| Paint brush | Faber-Castell, No.2 | Any thin soft round paint brush No.2, preferably black | |
| Paper towels | |||
| Perifusion pumps | Ismatec | ISM 827 | Reglo Analog MS - 4/8 |
| Petri dish 100/20 mm | Sarstedt | 83.3902 | |
| Petri dish 35/10 mm | Greiner bio-one | 627102 | |
| Petri dish 35 x 10 mm Nunclon Delta | Thermo Fischer Scientific, Waltham, MA USA | 153066 | NON-STICKY for agarose blocks |
| pH meter inoLab pH Level 1 | WTW, Weilheim, Germany | E163694 | |
| Pipette 1000 mL | Eppendorf | 3121 000.120 | |
| Pipette 50 mL | Eppendorf | 3121 000.066 | |
| Push pins 23 mm | Deli, Ningbo, China | E0021 | |
| Screw cap tube, 15 mL | Sarstedt | 62.554.502 | |
| Semken Forceps | FST, Germany | 11008-13 | |
| Stabilizing ring for Erlenmeyer flask | IsoLab, Germany | 027.11.048 | |
| Stereomicroscope Nikon SMZ 745 | Nikon, Melville, NY USA | ||
| Syringe Injekt Solo 5 mL | Braun, Melsungen, Germany | 4606051V | |
| Syringe needle 0.30 x 12 mm (30 G x 1/2") | Braun, Melsungen, Germany | 4656300 | |
| Temperature controller | Luigs & Neumann | 200-100 500 0150, 200-150-500-145 | Slice mini chamber, Temperature controller TC 07 |
| Tubings for perifusion system | Ismatec | SC0310 | Ismatec Pharmed 1.14 mm(ID) + silicone tubing 1.0 (ID) x 1.8 mm(OD) |
| Ultrasonic bath Studio GT-7810A | Globaltronics | ||
| Vibrotome Leica VT 1000 S | Leica, Nussloch, Germany | 14047235613 | |
| Volumetric flask 1000 mL | IsoLab, Germany | 013.01.910 | |
| Vortex mixer Neolab 7-2020 | Neolab | 7-2020 | |
| Water bath Thermo Haake open-bath circulator | Thermo Fisher Scientific | Z527912 | |
| Material/Reagent | |||
| Calcium chloride dihydrate - CaCl2.2H2O | Sigma Aldrich, Germany | C5080-500G | |
| D-(+)-glucose | Sigma Aldrich, Germany | G8270-1KG | |
| Dimethyl sulfoxide | Sigma Aldrich | D4540-100ML | |
| DL-lactic acid | Sigma Aldrich, Germany | L1250-500ML | |
| Dulbecco’s Phosphate Buffered Saline | Merck KGaA, Darmstadt, Germany | D8662-500ML | |
| Gas mixture containing 95% O2 and 5% CO2 at barometric pressure | |||
| Glue Wekem sekundenkleber WK-110 | Wekem GmbH, Bergkamen, Germany | WK 110-020 | |
| HEPES | Sigma Aldrich, Germany | H3375-250G | |
| L-(+)-ascorbic acid | Sigma Aldrich, Germany | A9,290-2 | |
| LIVE/DEAD Viability/Cytotoxicity Kit, for mammalian cells | Thermo Fischer Scientific, Waltham, MA USA | L3224 | |
| Magnesium chloride hexahydrate - MgCl2.2H2O | Sigma Aldrich, Germany | M2670-500G | |
| Myo-inositol | Sigma Aldrich, Germany | I5125-100G | |
| Oregon Green 488 BAPTA-1, AM | Invitrogen (Thermo FisherScientific) | O6807 | cell-permeable Ca2+ indicator (excitation/emission: 495/523 nm) |
| Pluronic F-127 (20% Solution in DMSO) | Invitrogen (Thermo Fisher Scientific) | P3000MP | polaxamer: nonionic triblock copolymer |
| Potassium chloride - KCl | Sigma Aldrich, Germany | 31248 | |
| SeaPlaque GTG agarose | Lonza, Rockland, USA | 50111 | |
| Sodium bicarbonate - NaHCO3 | Honeywell, Germany | 31437-500G | |
| Sodium chloride - NaCl | Honeywell, Germany | 31434-1KG | |
| Sodium hydroxide - NaOH | Sigma Aldrich, Germany | 30620 | |
| Sodium phosphate monobasic- NaH2PO4 | Sigma Aldrich, Germany | S0751-500G | |
| Sodium pyruvate | Sigma Aldrich, Germany | 15990-100G | |
| Software | |||
| FIJI | FIJI is an open source project | ||
| LASAF | Leica microsystems, Inc. | ||
| Matlab | Mathworks | ||
| Python | Python Software Foundation | Python is an open source project |
References
- Dolensek, J., Rupnik, M. S., Stozer, A. Structural similarities and differences between the human and the mouse pancreas. Islets. 7 (1), e1024405(2015).
- Meneghel-Rozzo, T., Rozzo, A., Poppi, L., Rupnik, M. In vivo and in vitro development of mouse pancreatic ß-cells in organotypic slices. Cell and Tissue Research. 316 (3), 295-303 (2004).
- Rozzo, A., Meneghel-Rozzo, T., Delakorda, S. L., Yang, S. B., Rupnik, M. Exocytosis of insulin: in vivo maturation of mouse endocrine pancreas. Annals of the New York Academy of the Sciences. 1152, 53-62 (2009).
- Dolenšek, J., Pohorec, V., Rupnik, M. S., Stožer, A. Pancreas physiology, challenges in pancreatic pathology. IntechOpen. Seicean, A. , (2017).
- Williams, J. A. The nobel pancreas: a historical perspective. Gastroenterology. 144 (6), 1166-1169 (2013).
- Lacy, P. E., Kostianovsky, M. Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes. 16 (1), 35-39 (1967).
- Williams, J. A., Korc, M., Dormer, R. L. Action of secretagogues on a new preparation of functionally intact, isolated pancreatic acini. American Journal of Physiology. 235 (5), 517-524 (1978).
- Peikin, S. R., Rottman, A. J., Batzri, S., Gardner, J. D. Kinetics of amylase release by dispersed acini prepared from guinea pig pancreas. American Journal of Physiology. 235 (6), E743-E749 (1978).
- Marciniak, A., et al. Using pancreas tissue slices for in situ studies of islet of Langerhans and acinar cell biology. Nature Protocols. 9 (12), 2809-2822 (2014).
- Skelin, M., Rupnik, M., Cencic, A. Pancreatic beta cell lines and their applications in diabetes mellitus research. Altex-Alternatives to Animal Experimentation. 27 (2), 105-113 (2010).
- Molnar, R., et al. Mouse pancreatic ductal organoid culture as a relevant model to study exocrine pancreatic ion secretion. Laboratory Investigation. 100 (1), 84-97 (2020).
- Rupnik, M. The physiology of rodent beta-cells in pancreas slices. Acta Physiologica (Oxford, England). 195 (1), 123-138 (2009).
- Blinman, T. A., et al. Activation of pancreatic acinar cells on isolation from tissue: cytokine upregulation via p38 MAP kinase. American Journal of Physiology. Cell Physiology. 279 (6), C1993-C2003 (2000).
- Speier, S., Rupnik, M. A novel approach to in situ characterization of pancreatic ß-cells. Pflügers Archive: European Journal of Physiology. 446 (5), 553-558 (2003).
- Gilon, P., Jonas, J., Henquin, J. Culture duration and conditions affect the oscillations of cytoplasmic calcium concentration induced by glucose in mouse pancreatic islets. Diabetologia. 37 (10), 1007-1014 (1994).
- Huang, C., Gu, G. Effective isolation of functional islets from neonatal mouse pancreas. Journal of Visualized Experiments: JoVE. (119), e55160(2017).
- Szot, G. L., Koudria, P., Bluestone, J. A. Murine pancreatic islet isolation. Journal of Visualized Experiments: JoVE. (7), e255(2007).
- Qi, M., et al. Human pancreatic islet isolation: Part I: digestion and collection of pancreatic tissue. Journal of Visualized Experiments: JoVE. (27), e1125(2009).
- Qi, M., et al. Human pancreatic islet isolation: Part II: purification and culture of human islets. Journal of Visualized Experiments: JoVE. (27), e1343(2009).
- Stull, N. D., Breite, A., McCarthy, R., Tersey, S. A., Mirmira, R. G. Mouse islet of Langerhans isolation using a combination of purified collagenase and neutral protease. Journal of Visualized Experiments: JoVE. (67), e4137(2012).
- Hamilton, A., Vergari, E., Miranda, C., Tarasov, A. I. Imaging calcium dynamics in subpopulations of mouse pancreatic islet cells. Journal of Visualized Experiments: JoVE. (153), (2019).
- Marciniak, A., Selck, C., Friedrich, B., Speier, S. Mouse pancreas tissue slice culture facilitates long-term studies of exocrine and endocrine cell physiology in situ. PLoS ONE. 8 (11), e78706(2013).
- Gal, E., et al. A Novel in situ approach to studying pancreatic ducts in mice. Frontiers in Physiology. 10, 938(2019).
- Speier, S., Yang, S. B., Sroka, K., Rose, T., Rupnik, M. KATP-channels in beta-cells in tissue slices are directly modulated by millimolar ATP. Molecular and Cellular Endocrinology. 230 (1-2), 51-58 (2005).
- Speier, S., Gjinovci, A., Charollais, A., Meda, P., Rupnik, M. Cx36-mediated coupling reduces β-cell heterogeneity, confines the stimulating glucose concentration range, and affects insulin release kinetics. Diabetes. 56 (4), 1078-1086 (2007).
- Rose, T., Efendic, S., Rupnik, M. Ca2+-secretion coupling is impaired in diabetic Goto Kakizaki rats. The Journal of General Physiology. 129 (6), 493-508 (2007).
- Paulmann, N., et al. Intracellular serotonin modulates insulin secretion from pancreatic β-cells by protein serotonylation. PLoS Biology. 7 (10), e1000229(2009).
- Mandic, S. A., et al. Munc18-1 and Munc18-2 proteins modulate β-cell Ca2+ sensitivity and kinetics of insulin exocytosis differently. Journal of Biological Chemistry. 286 (32), 28026-28040 (2011).
- Dolensek, J., Skelin, M., Rupnik, M. S. Calcium dependencies of regulated exocytosis in different endocrine cells. Physiological Research. 60, S29-S38 (2011).
- Huang, Y. C., Rupnik, M., Gaisano, H. Y. Unperturbed islet α-cell function examined in mouse pancreas tissue slices. Journal of Physiology. 589 (2), 395-408 (2011).
- Huang, Y. C., et al. In situ electrophysiological examination of pancreatic α cells in the streptozotocin-induced diabetes model, revealing the cellular basis of glucagon hypersecretion. Diabetes. 62 (2), 519-530 (2013).
- Stožer, A., Dolenšek, J., Rupnik, M. S. Glucose-stimulated calcium dynamics in islets of Langerhans in acute mouse pancreas tissue slices. PLoS ONE. 8 (1), e54638(2013).
- Stožer, A., et al. Functional connectivity in islets of Langerhans from mouse pancreas tissue slices. PLoS Computational Biology. 9 (2), e1002923(2013).
- Dolenšek, J., Stožer, A., Skelin Klemen, M., Miller, E. W., Slak Rupnik, M. The relationship between membrane potential and calcium dynamics in glucose-stimulated beta cell syncytium in acute mouse pancreas tissue slices. PLoS ONE. 8 (12), e82374(2013).
- Perc, M., Rupnik, M., Gosak, M., Marhl, M. Prevalence of stochasticity in experimentally observed responses of pancreatic acinar cells to acetylcholine. Chaos. 19 (3), 037113(2009).
- Qadir, M. M. F., et al. Long-term culture of human pancreatic slices as a model to study real-time islet regeneration. Nature Communications. 11 (1), 3265-3265 (2020).
- Panzer, J. K., et al. Pancreas tissue slices from organ donors enable in situ analysis of type 1 diabetes pathogenesis. JCI Insight. 5 (8), e134525(2020).
- Cohrs, C. M., et al. Vessel network architecture of adult human islets promotes distinct cell-cell interactions in situ and is altered after transplantation. Endocrinology. 158 (5), 1373-1385 (2017).
- Cohrs, C. M., et al. Dysfunction of persisting beta cells is a key feature of early type 2 diabetes pathogenesis. Cell Reports. 31 (1), 107469(2020).
- Dolai, S., et al. Pancreatitis-induced depletion of syntaxin 2 promotes autophagy and increases basolateral exocytosis. Gastroenterology. 154 (6), 1805-1821 (2018).
- Liang, T., et al. Ex vivo human pancreatic slice preparations offer a valuable model for studying pancreatic exocrine biology. Journal of Biological Chemistry. 292 (14), 5957-5969 (2017).
- Panzer, J. K., Cohrs, C. M., Speier, S. Using pancreas tissue slices for the study of islet physiology. Methods in Molecular Biology. 2128, 301-312 (2020).
- Klemen, M., Dolenšek, J., Stožer, A., Rupnik, M. Exocytosis Methods. Thorn, P. 7, Humana Press. 127-146 (2014).
- Speier, S. Experimental approaches for high-resolution in vivo imaging of islet of Langerhans biology. Current Diabetes Reports. 11 (5), 420-425 (2011).
- Leibiger, I. B., Berggren, P. O. Intraocular in vivo imaging of pancreatic islet cell physiology/pathology. Molecular Metabolism. 6 (9), 1002-1009 (2017).
- Reissaus, C. A., et al. A Versatile, portable intravital microscopy platform for studying beta-cell biology in vivo. Scientific Reports. 9 (1), 8449(2019).
- Jacob, S., et al. In vivo Ca(2+) dynamics in single pancreatic beta cells. FASEB Journal. 34 (1), 945-959 (2020).
- Fernandez, J., Valdeolmillos, M. Synchronous glucose-dependent [Ca2+]i oscillations in mouse pancreatic islets of Langerhans recorded in vivo. FEBS Letters. 477 (1-2), 33-36 (2000).
- Almaca, J., Weitz, J., Rodriguez-Diaz, R., Pereira, E., Caicedo, A. The pericyte of the pancreatic islet regulates capillary diameter and local blood flow. Cell Metabolism. 27 (3), 630-644 (2018).
- Weitz, J. R., et al. Mouse pancreatic islet macrophages use locally released ATP to monitor beta cell activity. Diabetologia. 61 (1), 182-192 (2018).
- Tian, G., Sandler, S., Gylfe, E., Tengholm, A. Glucose- and hormone-induced cAMP oscillations in α- and β-cells within intact pancreatic islets. Diabetes. 60 (5), 1535-1543 (2011).
- Benninger, R. K., Zhang, M., Head, W. S., Satin, L. S., Piston, D. W. Gap junction coupling and calcium waves in the pancreatic islet. Biophysical Journal. 95 (11), 5048-5061 (2008).
- Santos, R. M., et al. Widespread synchronous Ca oscillations due to bursting electrical activity in single pancreatic islets. Pflügers Archive: European Journal of Physiology. 418 (4), 417-422 (1991).
- terk, M., et al. Assessing the origin and velocity of Ca2+ waves in three-dimensional tissue: Insights from a mathematical model and confocal imaging in mouse pancreas tissue slices. Communications in Nonlinear Science and Numerical Simulation. 93, 105495(2021).
- Gosak, M., et al. Critical and supercritical spatiotemporal calcium dynamics in beta cells. Frontiers in Physiology. 8, 1106(2017).
- Satin, L. S., Butler, P. C., Ha, J., Sherman, A. S. Pulsatile insulin secretion, impaired glucose tolerance and type 2 diabetes. Molecular Aspects in Medicine. 42, 61-77 (2015).
- Tengholm, A., Gylfe, E. Oscillatory control of insulin secretion. Molecular and Cellular Endocrinology. 297 (1-2), 58-72 (2009).
- Quesada, I., et al. Glucose induces opposite intracellular Ca2+ concentration oscillatory patterns in identified α- and β-cells within intact human islets of Langerhans. Diabetes. 55 (9), 2463-2469 (2006).
- Ferguson, J., Allsopp, R. H., Taylor, R. M., Johnston, I. D. Isolation and long term preservation of pancreatic islets from mouse, rat and guinea pig. Diabetologia. 12 (2), 115-121 (1976).
- Andersson, A. Isolated mouse pancreatic islets in culture: effects of serum and different culture media on the insulin production of the islets. Diabetologia. 14 (6), 397-404 (1978).
- Ramirez-Dominguez, M. Isolation of mouse pancreatic islets of Langerhans. Advances in Experimental Medicine and Biology. 938, 25-34 (2016).
- Carter, J., Dula, S., Corbin, K., Wu, R., Nunemaker, C. A practical guide to rodent islet isolation and assessment. Biological Procedures Online. 11 (1), 3-31 (2009).
- Daoud, J., Rosenberg, L., Tabrizian, M. Pancreatic islet culture and preservation strategies: advances, challenges, and future outlook. Cell Transplantation. 19 (12), 1523-1535 (2010).
- Zawalich, W. S., Yamazaki, H., Zawalich, K. C. Biphasic insulin secretion from freshly isolated or cultured, perifused rodent islets: comparative studies with rats and mice. Metabolism. 57 (1), 30-39 (2008).
- Roe, M. W., et al. Absence of effect of culture duration on glucose-activated alterations in intracellular calcium concentration in mouse pancreatic islets. Diabetologia. 38, 876-877 (1995).
- Shuai, H., Xu, Y., Yu, Q., Gylfe, E., Tengholm, A. Fluorescent protein vectors for pancreatic islet cell identification in live-cell imaging. Pflügers Archive. European Journal of Physiology. 468 (10), 1765-1777 (2016).
- Dolenšek, J., et al. Glucose-dependent activation, activity, and deactivation of beta cell networks in acute mouse pancreas tissue slices. bioRxiv. , (2020).
- QZhang, Q., et al. Cell coupling in mouse pancreatic beta-cells measured in intact islets of Langerhans. Philosophical Transactions. Series A, Mathematical, Physical, and Engineering Sciences. 366 (1880), 3503-3523 (2008).
- Sánchez-Cárdenas, C., Hernández-Cruz, A. GnRH-induced Ca2+-signalling patterns in mouse gonadotrophs recorded from acute pituitary slices in vitro. Neuroendocrinology. 91 (3), 239-255 (2010).
- Asada, N., Shibuya, I., Iwanaga, T., Niwa, K., Kanno, T. Identification of alpha- and beta-cells in intact isolated islets of Langerhans by their characteristic cytoplasmic Ca2+ concentration dynamics and immunocytochemical staining. Diabetes. 47 (5), 751-757 (1998).
- Nadal, A., Quesada, I., Soria, B. Homologous and heterologous asynchronicity between identified α-, β- and δ-cells within intact islets of Langerhans in the mouse. Journal of Physiology. 517 (1), 85-93 (1999).
- Shuai, H., Xu, Y., Yu, Q., Gylfe, E., Tengholm, A. Fluorescent protein vectors for pancreatic islet cell identification in live-cell imaging. Pflugers Archive: European Journal of Physiology. 468 (10), 1765-1777 (2016).
- Cabrera, O., et al. Glutamate is a positive autocrine signal for glucagon release. Cell Metabolism. 7 (6), 545-554 (2008).
- Li, J., et al. Submembrane ATP and Ca2+ kinetics in α-cells: unexpected signaling for glucagon secretion. FASEB Journal. 29 (8), 3379-3388 (2015).
- Hamilton, A., et al. Adrenaline stimulates glucagon secretion by Tpc2-dependent Ca(2+) mobilization from acidic stores in pancreatic α-cells. Diabetes. 67 (6), 1128-1139 (2018).
- Arrojo, E. D. R., et al. Structural basis for delta cell paracrine regulation in pancreatic islets. Nature Communications. 10 (1), (2019).
- DiGruccio, M. R., et al. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Molecular Metabolism. 5 (7), 449-458 (2016).
- Adriaenssens, A. E., et al. Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia. 59 (10), 2156-2165 (2016).
- Rorsman, P., Huising, M. O. The somatostatin-secreting pancreatic δ-cell in health and disease. Nature reviews. Endocrinology. 14 (7), 404-414 (2018).
- Petersen, C. C., Toescu, E. C., Petersen, O. H. Different patterns of receptor-activated cytoplasmic Ca2+ oscillations in single pancreatic acinar cells: dependence on receptor type, agonist concentration and intracellular Ca2+ buffering. The EMBO journal. 10 (3), 527-533 (1991).
- Thorn, P., Lawrie, A. M., Smith, P. M., Gallacher, D. V., Petersen, O. H. Local and global cytosolic Ca2+ oscillations in exocrine cells evoked by agonists and inositol trisphosphate. Cell. 74 (4), 661-668 (1993).
- Behrendorff, N., Floetenmeyer, M., Schwiening, C., Thorn, P. Protons released during pancreatic acinar cell secretion acidify the lumen and contribute to pancreatitis in mice. Gastroenterology. 139 (5), e1711-e1715 (2010).
- Criddle, D. N., et al. Cholecystokinin-58 and cholecystokinin-8 exhibit similar actions on calcium signaling, zymogen secretion, and cell fate in murine pancreatic acinar cells. American Journal of Physiology. Gastrointestinal and Liver Physiology. 297 (6), G1085-G1092 (2009).
- Venglovecz, V., et al. Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut. 57 (8), 1468-3288 (2008).
- Maleth, J., Hegyi, P. Calcium signaling in pancreatic ductal epithelial cells: an old friend and a nasty enemy. Cell Calcium. 55 (6), 337-345 (2014).
- Nadal, A., Quesada, I., Soria, B. Homologous and heterologous asynchronicity between identified alpha-, beta- and delta-cells within intact islets of Langerhans in the mouse. Journal of Physiology. 517 (Pt 1), 85-93 (1999).
- Skelin Klemen, M., Dolenšek, J., Slak Rupnik, M., Stožer, A. The triggering pathway to insulin secretion: Functional similarities and differences between the human and the mouse β cells and their translational relevance. Islets. 9 (6), 109-139 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved