
Method Article
Single Particle Cryo-Electron Microscopy: From Sample to Structure
In This Article
Summary
Structure determination of macromolecular complexes using cryoEM has become routine for certain classes of proteins and complexes. Here, this pipeline is summarized (sample preparation, screening, data acquisition and processing) and readers are directed towards further detailed resources and variables that may be altered in the case of more challenging specimens.
Abstract
Cryo-electron microscopy (cryoEM) is a powerful technique for structure determination of macromolecular complexes, via single particle analysis (SPA). The overall process involves i) vitrifying the specimen in a thin film supported on a cryoEM grid; ii) screening the specimen to assess particle distribution and ice quality; iii) if the grid is suitable, collecting a single particle dataset for analysis; and iv) image processing to yield an EM density map. In this protocol, an overview for each of these steps is provided, with a focus on the variables which a user can modify during the workflow and the troubleshooting of common issues. With remote microscope operation becoming standard in many facilities, variations on imaging protocols to assist users in efficient operation and imaging when physical access to the microscope is limited will be described.
Introduction
Single particle CryoEM
To investigate life at a molecular level we must understand structure. Many techniques to probe protein structure are available, such as NMR, X-ray crystallography, mass spectrometry and electron microscopy (EM). To date, the majority of structures deposited to the Protein Databank (PDB) have been solved using X-ray crystallography. However, from ~2012 onwards, cryo-electron microscopy (cryoEM) became a mainstream technique for protein structure determination and its use increased dramatically. The total number of EM maps deposited to the Electron Microscopy Databank (EMDB) (as of Dec 2020) was 13,421 compared with 1,566 in 2012 (Figure 1, www.ebi.co.uk). In 2012 the number of atomic coordinates modelled in cryoEM density maps, deposited to the PDB was just 67 but as of Dec 2020, 2,309 structures have been deposited so far, a 35-fold increase. This underlying growth in the quality and quantity of cryoEM density maps produced, sometimes referred to as the ‘resolution revolution'1, was caused by a coalescence of advances in multiple areas: the development of new cameras for imaging known as direct electron detectors; new software; and more stable microscopes2,3,4.

Figure 1: Cumulative submissions to the EMDB from 2012 to December 2020. Please click here to view a larger version of this figure.
{kind=link}
Single particle analysis (SPA) is a powerful tool to generate biological insight in a wide variety of sample types by elucidating high resolution structures of isolated complexes5, 6 including viruses7,8, membrane proteins9, 10, helical assemblies11 and other dynamic and heterogeneous macromolecular complexes12, 13, the sizes of which vary by orders of magnitude (from 39 kDa14, 15 to tens of megadaltons). Here, a protocol for a standard pipeline for cryoEM SPA from sample to structure is described.
Prior to embarking upon this pipeline, a purified sample should be subjected to biochemical analysis to assess its chances of downstream success. Preparation of a suitable sample is arguably the biggest barrier to SPA, particularly for transient and heterogeneous (both compositional and conformational) complexes. The macromolecular complex preparation should contain as few contaminants as possible, at sufficient concentration to yield many particles in each cryoEM micrograph, and in a buffer composition well suited to cryoEM analysis. Certain buffer constituents, including sucrose, glycerol and high (~> 350 mM concentrations of salts, depending on the sample size, properties and other buffer constituents) can interfere with the process of vitrification or reduce the signal-to-noise ratio in images, hindering structure determination16.
Typically, as a minimum, size exclusion chromatography (SEC) and SDS- PAGE gel analysis should be used to assess sample purity17, 18, but circular dichroism, functional assays, SEC coupled with multi-angle light scattering, and thermal stability assays are all useful tools for qualitative analysis of macromolecular complex preparations prior to cryoEM analysis. However, the results from these biochemical analyses may yield little insight into the structural heterogeneity of the sample and its behaviour on a cryoEM grid. For this reason, negative stain EM is routinely used as a quick, cheap and powerful tool for assessing compositional and conformational heterogeneity, and therefore a good way of ascertaining which elution fraction from a purification is most promising, or screening different buffer compositions19, 20. Once a promising sample has been identified, we can proceed to the SPA cryoEM pipeline. Negative stain does not always align with the subsequent results seen in cryoEM; sometimes a sample looks poor by negative stain but improves when seen in vitreous ice in cryoEM. In contrast, sometimes samples look excellent during negative stain steps but require significant further optimisation when progressing to cryoEM. However, in the majority of cases negative stain provides a useful quality control step.
Vitrification
The harsh environment within the vacuum system of the electron microscope causes both dehydration and radiation damage to unfixed biological specimens21. Therefore, to image the sample in a native-like state, the biological specimen must be preserved prior to imaging. For purified preparations of macromolecular complexes, vitrification is the method of choice to enable its visualization by cryoEM whilst preserving the atomic details of the complex. The discovery of vitrification as a method of sample preparation was a fundamental advancement in electron microscopy of biological specimens, for which Dubochet was recognized in the 2017 Nobel Prize in Chemistry. Sample vitrification involves creating a thin layer of solution containing the specimen of interest, typically tens of nm thick, suspended on a cryoEM grid support. The thin film is then frozen extremely rapidly in a cryogen such as liquid ethane at ~-175 °C. The freezing rate is ~106 °C/s, fast enough that amorphous, or vitreous ice forms, suspending the specimen in a thin, solid film22.
The initial variable to consider is the cryoEM grid support chosen23. An EM grid typically consists of an amorphous carbon film with perforations (either regular or irregular), over a support structure. The support structure is typically a circular metal grid 3.05 mm in diameter, usually made from copper, but other metals such as gold, or molybdenum (which has preferred thermal expansion properties24) can be used. Sometimes, an additional thin, continuous support is applied across the grid, such as graphene, graphene oxide or a thin (~1-2 nm) amorphous carbon layer. While standard cryoEM grids (most commonly 400-200 mesh copper with a perforated (1.2 µm round holes separated by 1.3 µm (r1.2/1.3), or 2 µm separated by 2 µm of carbon (r2/2)) carbon support- although many different patterns are available) have been used in the vast majority of structures reported to date, novel grid technologies with improved conductivity and reduced specimen movement have been reported25. Selected grids are subjected to a glow-discharge/plasma cleaning treatment to render them hydrophilic and amenable to sample application26.
Following glow-discharge, the next stage is thin film formation. This thin film is most commonly formed using filter paper to remove excess liquid from the grid. While this can be carried out manually, a number of plunge freezing devices are commercially available, including the Vitrobot Mk IV (Thermo Fisher Scientific), EM GP II (Leica) and CP3 (Gatan). With these devices, ~3-5 µL of sample in solution is applied to the EM grid, followed by blotting away excess solution using filter paper. The grid, with a thin film suspended across it, is then plunged into liquid ethane cooled by liquid nitrogen (LN2) to ~-175 °C. Once frozen, the grid is maintained at a temperature below the devitrification point (-137 °C) prior to, and during imaging.
Specimen screening and data collection
Following vitrification of a cryoEM grid, the next stage is to screen the grid to assess its quality and to determine whether the grid is suitable to proceed to high resolution data collection. An ideal cryoEM grid has vitreous ice (opposed to crystalline ice) with the ice thickness just sufficient to accommodate the longest dimension of the specimen, ensuring the surrounding ice contributes as little noise to the resulting image as possible. The particles within the ice should have a size and (if known) shape consistent with biochemistry, and ideally be monodisperse with a random distribution of particle orientations. Finally, the grid should have enough areas of sufficient quality to satisfy the desired data collection length. Depending on the specimen, this may take many iterations of vitrification and screening until optimal grids are produced. Both fortunately and unfortunately, there are a huge range of variables that can be empirically tested to alter particle distribution on cryoEM grids (reviewed in16,27). In this manuscript, representative results for a membrane protein project10 are shown.
Once a suitable grid has been identified, data collection can proceed. Several models of cryo-transmission electron microscopes for biological specimens are optimized to collect high-resolution data in an automated fashion. Typically, data is collected on 300 kV or 200 kV systems. Automated data collection can be achieved using software including EPU (Thermo Fisher Scientific)28, Leginon29, JADAS30 and SerialEM31, 32. An automated data collection with modern detectors typically results in terabytes (TB) of raw data in a 24 h period (average datasets are ~ 4 TB in size).
Due to the COVID-19 restrictions in place on much of the world (time of writing December 2020), many microscopy facilities have moved to offering remote access. Once the grids have been loaded into the autoloader of a microscope, data acquisition can be conducted remotely.
Image processing and model building
Where a data collection session may be typically 0.5-4 days, subsequent image processing may take many weeks and months, depending on the availability of computing resources. It is standard for initial image processing steps, namely motion correction and contrast transfer function (CTF) estimation to take place 'on the fly' 33, 34. For downstream processing, there are a plethora of software suites available. Particles are 'picked' and extracted from micrographs35, 36. Once particles are extracted, a standard protocol would be to process the particles through several rounds of classification (in both two dimensions (2D) and three dimensions (3D) and/or focused on specific regions of interest) to reach a homogeneous subset of particles. This homogeneous subset of particles is then averaged together to produce a 3D reconstruction. At this point data is often corrected further to produce the highest quality map possible, for example through CTF refinement, distortion corrections37 and Bayesian polishing38 . The outcome of this image processing is a 3D cryoEM map of the biological specimen of interest. The resolution range reached in a 'standard' automated single particle experiment from a grid of sufficient quality, with data collected on a 300 kV microscope system is typically between 10 Å and 2 Å depending on the size and flexibility of the protein complex. With an ideal specimen, resolutions of ~1.2 Å have now been reached using SPA workflows5. While this protocol details steps towards obtaining an EM density map, once this is in hand it can be further interpreted through fitting and refining a protein model (if resolution is < 3.5 Å) or building de novo39. Data associated with structure determination experiments can be deposited into online public repositories, including EM density maps (Electron Microscopy Data Bank)40, resulting atomic coordinates (Protein Data Bank)41 and raw datasets (Electron Microscopy Public Image Archive)42.
In this protocol, the outer-membrane protein complex RagAB (~340 kDa) from Porphyromonas gingivalis is used as an example macromolecular complex10 (EMPIAR-10543). For those new to cryoEM, support for samples through this pipeline from sample to structure is available, subject to peer review, through funded access schemes such as iNEXT Discovery and Instruct.
Protocol
1. Grid Vitrification
NOTE: For all steps in steps 1 and 2, ensure that all tools are clean, dry and at room temperature before cooling them to LN2 temperature, using freshly decanted LN2 to reduce ice contamination. Where possible, work within a humidity-controlled environment with < 20% relative humidity. Ensure appropriate personal protective equipment and H&S documentation is in place before commencing work.
- Ensure the specimen of interest is ready for sample preparation.
- Choose appropriate cryoEM grids and ensure these have been rendered hydrophilic using glow discharge or plasma treatment. A wide variety of systems and variations to the protocol are available but all involve placing the grids into a glow discharge/plasma cleaning system and running a program which will pump the chamber to a desired vacuum level, before introducing a specific gas mixture/chemical vapor or air into the system. An electric current is passed through the system, ionizing the gas particles and inducing the surface of the grids to be rendered more hydrophilic.
- Start up the plunge freezing device for grid vitrification by turning on the system using the power switch on the back, and wait for the touch screen to load.
- Using the provided stylus or fingers, in Console, set the desired working temperature of the chamber (range available is 4-60 °C, recommended for most macromolecules 4-6 °C).
- Fill the humidifier with 50 mL of type II lab water using a syringe, via the rubber tubing at bottom of humidifier. Making sure to remove any trapped air in the syringe prior to filling. Be careful not to overfill the humidifier or water will exude into the chamber. Once the humidifier is filled, draw back the syringe plunger by 5-10 mL to create a vacuum seal.
- In Console, set the desired relative humidity for the chamber (range available is 0-100%, humidity of 95-100% is typically used). Leave humidity set to 'off' until immediately prior to grid making so that the chamber does not get too wet.
- Obtain the plunge freezing device tweezers and filter paper cut to the correct size to fit onto the pads, either purchased or using a stamp to cut an aperture of the appropriate size.
- Prepare the cryogen for plunge freezing.
- Place the metal cryo-grid box holder, cryogen cup and metal spider's legs into the coolant container.
- Cool down the container by filling up the outer chamber with LN2. Keep the outer chamber topped up to cover the top of the cryo-grid box holder. Add ~ 1 cm of additional LN2 to the cryogen cup to assist equilibration of the system to LN2 temperature.
NOTE: The anti-contamination ring can be used to limit humid air condensing around the cryogen cup and leading to cryo-coolant/ethane contamination. This is not generally required in a humidity-controlled environment. If using the anti-contamination ring, be careful not to overfill the container with LN2 or it may spill when the ring is pressed into the container later in the process. - Wait 3-5 min to observe the boiling of the spider legs, and then wait a further 3 min to ensure the cryogen cup is sufficiently cold to condense the vitrification medium.
- Liquefy the cryogen (liquid ethane) into the cryogen cup.
- Take the ethane cylinder pipe with thin tubing and a nozzle to dispense the gas. A P200 pipette tip with the aperture opened by cutting off the very tip using a razor blade is ideal here. A wider aperture is needed to prevent ethane solidifying at the tip and blocking the flow of gas.
- Ensuring the cryogen cup does not contain any remaining LN2, take the ethane gas nozzle and place it within the cryogen cup. Using the gas cylinder regulator, start a low flow and dispense cryogen gas into the cryogen cup to condense the gas. Keep the tip from which the gas flows directly pressed against the wall of the cryogen cup but move it gently back and forth in a tapping motion against the surface. Regulate the flow of the gas to allow a low, steady flow to begin to condense/liquify in a controlled fashion within the cryogen cup.
- Fill the cup to just below the silver spider rim and stop the gas flow, then remove gas line carefully to avoid contaminating the surrounding LN2 with ethane.
- Top up the coolant container with LN2, being very careful not to spill any into the liquid ethane.
- Leave the spiders legs in position for ~ 3-5 min to ensure the liquid ethane is equilibrated to a sufficiently cold temperature. The cryogen will begin to look cloudy/slightly opaque. This indicates that it is close to its freezing point. At this stage, use tweezers to remove the spider. As long as the LN2 is kept within the container surrounding the cryogen cup, the ethane will now stay liquefied and suitable for vitrification for 1-2 h. However, aim to complete the procedure as quickly as possible, especially in non-humidity-controlled rooms, to reduce ice contamination.
NOTE: If the spider appears to be 'stuck on', use a metal object such as a nut and hold against the spider's legs to warm them up slightly, and then remove the legs.
- Prepare the plunge freezing device and accessories for sample vitrification.
- Add grid storage boxes to the metal cryo-grid box holder and throughout the procedure ensure the LN2 is kept topped up to just above the level of the grid boxes (usually every ~ 5 min).
- On the plunge freezing device screen, in the Process Parameters box input the chosen parameters including: blot time (the time the plunge freezing device pads will come together), force (the distance of the blotting pads from the grid, which alters the gradient of ice formation) and total (number of times the blotting pads will come in to meet). Choose these parameters based on the individual plunge freezing device and behaviour of the macromolecule. Typical values are a blot force between 0 and 5, blot time from 1-6 s and a blot total of 1. Typical wait time (time between initiating the blot, and the blot beginning) and drain time (time after blotting before plunging) is 0-2 s.
NOTE: Depending on user preferences, additional options in the Options, Miscellaneous section can be selected, including Use Foot Pedal to move to the next step on each press, Skip Grid transfer (skips the final step where the tweezer arm is raised slightly), turn Humidity Off During Process (while the sample is being applied, stops active humidification of the chamber which can make it harder to see the grid) and Autoraise Ethanelift (combines the step of tweezers being raised into the chamber and raising the coolant container- skips Raise ethane container step). Here, all of these options are turned on. - Place the coolant container securely onto the moving platform arm under the chamber
- Insert fresh blotting paper onto each blotter arm making sure plastic ring clips are secured. Each filter paper will allow 16 blots (arms rotate blotting paper). Press the Reset blot paper button in Controls section.
- Run 1 full cycle of the plunge freezing device vitrification process to ensure that each moving part is behaving as expected.
- Press (or use foot pedal) to Place new grid, then Start process, then Process then Continue. At this stage, watch to ensure the blotting arms are contacting each other as expected.
- Turn 'on' the humidifier. Water vapor will be produced (as long as the set humidity is higher than currently in the chamber).
- The specimen of interest can now be vitrified. Use the foot pedal or Place New Grid and the plunging rod will descend out of the chamber allowing the tweezers to be attached into the mount.
- Using the plunge freezing device tweezers, pick up the desired glow discharged/plasma cleaned cryoEM grid, taking care to note which side is the correct side to be used for sample application according to the grid manufacturer. Pick up the grid by the rim, taking care to avoid excessive/unnecessary contact with the tweezers as this will damage the support. Secure the grid in the tweezers by moving the black clip down to the ridged part of the tweezers. The grid needs to be securely held, but the clip should not be too far down as it will contact the blotting pads, leading to irreproducible blotting and, later, the tweezers will need to be held below this point when releasing the clip.
- Place the plunge freezing device tweezers holding the cryoEM grid onto the pneumatic arm with the correct side facing your dominant hand. The design of the plunge freezing device tweezers and chamber are such that the sample can be applied through either the right or left-hand side of the chamber, according to the handedness of the user.
NOTE: Applying the sample on different sides with the same blotting parameters rarely results in comparable results, so left-handed researchers may need to tune their blotting parameters independently of their right-handed colleagues. - Press Start Process and the grid held in the tweezers will be taken into the chamber and the coolant container will be raised.
- Press Process and the tweezers will move the grid to the position where a pipette can be used to apply the specimen to the grid. Open the side port facing the correct side of the grid and apply sample by pipetting, making sure that the pipette tip does not touch the grid as it may lead to damage of the grid support/bending the grid, but dispense the liquid close enough so that the droplet dispenses onto the grid. Typically, 3-5 µL is applied.
- Press Continue and user predefined parameters will blot the grid and then plunge the tweezers with grid mounted into the coolant cup for sample vitrification. Tweezers will descend in conjunction with the arm holding the coolant container and coolant, keeping the grid submerged in the cryogen.
- Transfer the grid from the cryogen cup to the grid storage box submerged in LN2.
- Detach the tweezers from the tweezer arm, taking great care not to contact the vitrified grid with the sides of the cryogen cup. Adjust the grip so that the tweezers are held comfortably. As quickly and carefully as possible, move the grid from the cryogen to the LN2. With one hand, hold the tweezers shut using your fingers and with the other hand, slide the black clip upwards out of the way, holding the tweezers shut. Readjust the grip and manipulate the grid into the grid storage box.
- Repeat steps 1.10-1.19 until all grids are made (a typical session will involve making 4-12 grids). Store all grid storage boxes containing grids in LN2 dewar until the next stages.
2. Clipping grids for loading into an autoloader microscope
- Clip grids into autogrid assembly according to the protocol previously described 28.
3. Secure remote log in to microscopes
NOTE: With COVID-19 controls at the time of writing, but also with environmental concerns associated with international travel, more microscopy facilities have been offering services where the user operates remotely. The method of implementation for this will vary according to the local IT configuration of each facility, and the needs of its internal and external user community. Here the process for remotely accessing cryoEMs at eBIC and controlling the microscope through EPU software is described.
- Remotely log in to cryoEM's. Remote logon is mediated via NoMachine software to access the microscope support PC and is configured to only allow access to users who are registered on a visit via the users FedID logon credentials. Access remains active only for the duration of the session.
- Open NoMachine and start a new NX connection to nx-cloud.diamond.ac.uk with password authentication.
- Open the connection and log in with the username fedid@fed.cclrc.ac.uk and FedID password. Double-click the icon corresponding to relevant microscope from the available options to open a connection to the relevant support PC.
- Enter username clrc\FedID and password at the Windows logon screen.
- Open TeamViewer software from the desktop icon and connect to PartnerID: TEM with the supplied password. This establishes the connection from the support PC to the TEM PC. The Next Monitor button in the TeamViewer ribbon can be used to toggle between the microscope user interface and the EPU window.
- Microscope functions can then be controlled by users directly through the EPU interface.
4. Loading samples into an autoloader microscope and screening for ice and sample quality
NOTE: In this section a microscope with an autoloader and EPU software is used for sample screening, but this can be achieved using other software and a side entry system and cryoEMs from other manufacturers.
- Load clipped grids into the microscope autoloader as previously described28.
- In the autoloader tab of the microscope user interface, tab out the Options dialogue using the arrow and press the Inventory button. This will sequentially check each position in the cassette to determine if a cartridge is present. Occupied slots will be labelled in blue. If all occupied slots have been mapped, press the Inventory button again to stop after the current position, otherwise leave running until all occupied slots have been mapped. Label all occupied slots with the sample details in the boxes provided.
- Highlight the grid to be transferred to the microscope column and click Load. The slot label will turn from blue to yellow once the grid has been successfully loaded onto the stage. Proceed to screen the grids.
- Open EPU software. On the Preparation page, select Acquisition optics and settings then select the Atlas preset from the drop-down menu. Choose appropriate beam setting presets (e.g., 64x nominal mag, spot size 5, Microprobe, with an illuminated area in the parallel range for Falcon detector- for further information choosing beam setting presets see28). Press Set to push the parameters to the microscope.
- Press Open column valves and insert the FluScreen. Check that a beam is visible and sufficiently spread and centered to cover the detector. If necessary, navigate to a thinner region of the grid using the joystick or stage menu to control stage movements in X and Y.
- Lift the FluScreen and take an image using the Preview button in EPU. Based on the acquired image, the dose can be increased by moving to a lower number spot size, and vice versa.
- In EPU, go to the Atlas page and press New Session. Select MRC image format and enter a suitable folder name and location for saving the screening session, then click Apply.
- Select Screening from the menu on the left. Tick the checkboxes next to each grid to have an atlas montage acquired. Start the screening session in EPU. An atlas will be acquired for each checked grid, with a number of available grid squares listed upon completion. Each atlas can be viewed by highlighting it on the screening page, complete with a mark-up showing grid squares with a similar predicted ice thickness grouped by colour.
- On completion, review the collected atlases and identify the grids suitable for assessing sample quality at higher magnifications (i.e., those with an appropriate number of grid squares which are neither dry nor obscured by thick ice). Highlight the chosen grid on the EPU screening menu and click Load sample.
- Use the beam setting presets (See 28 for explanation of beam setting presets desired for each stage) and the preview function to examine the desired grid squares in greater detail.
- From the Atlas screening menu, select the grid presently loaded and move the stage to a grid square containing filled holes by right clicking over the desired location on the grid image and selecting move to grid square.
- Return to EPU, Preparation page and select the GridSquare preset.
- Open the EPU, Auto Functions page and run Auto-eucentric by stage tilt with the GridSquare preset to move the sample to eucentric height.
NOTE: Auto- eucentric by beam tilt is also available, which is faster but typically less accurate than auto-eucentric by stage tilt. - In EPU, Preparation, take a new GridSquare Preview image. Note the differing grey values across different holes indicating differing ice thicknesses. Move the stage over a hole using right click > move stage here. Select the Hole/Eucentric Height preset and Preview.
NOTE: Depending on the molecular weight and shape of the particle of interest, it may be possible to identify it at Hole/Eucentric Height magnification. - Select Data Acquisition preset and set a magnification that allows easy identification of the particles (corresponding to an object sampling of generally <2 Å/pixel). Set the defocus offset to ~-3 to -5 µm with an exposure electron dose of ~40-80 e-/ Å2.
- Iterate through steps in 4.4 to assess a range of ice thicknesses for particle distribution, orientation and contamination across the grid. The particle distribution may vary close to the edges versus the center of the hole and hence it is important to survey different locations with the hole.
- Screen all grids which show promise from atlases as having sufficient grid squares. Either keep these in the microscope and proceed to data acquisition using EPU, or unload the samples from the microscope and store under LN2 until data collection is scheduled.
5. Single particle cryoEM data collection (with a focus on remote operation)
NOTE: A detailed protocol for data acquisition with EPU is described in the manufacturers manual and elsewhere28. Here modifications of this protocol for remote operation (namely reducing use of the hand panels to conduct tasks and using software-based alternatives) are highlighted.
- Unless one has already been collected during the session, collect an Atlas for the grid.
- Define each of the beam setting presets according to the experimental needs of the project.
- Perform image shift calibrations28.
- Set up the EPU session.
- In EPU, select EPU page then Session Setup, select New session then New from preferences.
- Select New session a pop up will appear providing an option to use previous settings. Yes will automatically load the settings from the previous EPU (i.e., specimen carrier, defocus range, autofocus settings, grid type) into the current EPU session. Selecting New from preferences enables the user to pick a file with saved preferences (i.e., defocus range, autofocus settings, grid type) and this information will be preloaded into EPU.
- Fill in session name with something informative. The local facility may suggest a naming convention.
- In Type, select Manual.
- For Acquisition mode, select accurate hole centering or faster acquisition.
- In Image format, select desired format.
- Select an appropriate Storage Folder and EPU will create a directory with the session name.
- Select the appropriate Specimen Carrier according to which grid type and hole spacing being used (e.g., Quantifoil 1.2/1.3) and press Apply. This protocol describes the process for generating a template for a regular array of holes
- Select an initial grid square and set an acquisition template.
- Go Square selection, if all squares are green, click unselect all in top left.
- Open tiles (right click > open tile). Select a square (right click > add, right click > Move stage to grid square).
- Go to Hole selection and press Auto Eucentric. Wait until this is complete and a Grid Square image has been taken. If the autofunction fails this may be because the height is significantly off; if so it can be adjusted manually using the FluScreen at Grid Square magnification.
- Measure hole size. Move and adjust the yellow circles so they are over the holes with correct size and spacing.
- Press Find holes. Check that the holes have been found correctly. If not change the hole size and find holes again. Repeat this until it finds the hole correctly. If it consistently fails, consider moving to a lower number (brighter) spot size at grid square magnification.
- Use the Filter ice quality histogram on the right to adjust hole selection. This can be useful to exclude areas with thick ice and thin ice. This will be remembered for future grid squares selected during this session.
- Optimize hole selection with the tools in Select menu at the top. For example, click Remove holes close to grid bar.
- Go to Template definition and press Acquire.
- Click Find and centre hole. There will now be an image of a hole with a yellow circle around the hole.
NOTE: If it struggles to find the hole, insert the objective aperture. If it still cannot find the hole, try increasing the exposure time for the hole/eucentric height preset or increasing the defocus for this preset or bin the image. A large defocus change may alter the image shift alignment. - Change the Delay after Stage Shift and the Delay after Image Shift times to 1-5 s.
- Check Maximum Image shift value (if option is available) is as desired. If aberration free image shift collection is being used, this value is defined in the EPU configuration file, otherwise 5 µm is a standard value.
- Click Add acquisition area, then click anywhere on the image. Move the acquisition area to desired location (i.e., at the edge of a hole) so that areas of acquisition are not doubly exposed with the beam (the square in the green circle represents the detector area, the green circle is the beam diameter).
- On the top right, add the defocus range. Then add other acquisition areas. A typical defocus list for a membrane protein project is -0.8 to -3 µm defocus.
- Click Add autofocus area and click anywhere on the image. Move the autofocus area to the carbon surrounding a hole. Standard practice is to autofocus after centring when using AFIS, or every 5-15 µm, depending on the z-height variation across the square.
- Click Add drift measurement area, drift measurement performed once per grid square, with a set threshold of 0.05 nm/s is a standard setting. The drift measurement area can (and it is a good idea to) overlap directly with autofocus area. Make sure neither drift nor autofocus area overlap with an acquisition area.
NOTE: The template can be checked using the template execution function. This is a good idea to see if acquisition areas need moving (e.g., too much/not enough carbon in images), but is not necessary. - Go back to Square selection, and on the grid select the squares for acquisition. Use the number of acquisition areas and expected data acquisition rate (from facility based on detectors and experimental set up) to predict how many acquisition areas are required.
- When all desired squares are selected, press Prepare all Squares.
- Once each square is collected, navigate between the grid squares and fine tune the holes using the selection brush.
- Move to a stage location over specimen and use auto functions to set eucentric height. Perform microscope alignments as previously described28, but instead of performing Coma-free alignment and correcting for objective astigmatism manually, make use of alignment tools within the software. Briefly, set acquisition beam conditions, ensure the objective aperture (OA) is removed and the stage is positioned over a beam stable area of specimen at eucentric height. Perform coma-free alignment within the auto-functions before reinserting and centering the OA and correcting the objective lens astigmatism with EPU. Ensure that both alignments converge on suitable values (<150 nm of coma and close to zero astigmatism.
- Before starting the automated acquisition run, ensure the autoloader turbo pump is turned off, and the objective aperture is inserted.
- In Automated acquisition, press Start run to commence automated data acquisition.
6. Image processing to yield EM density map
NOTE: The majority of cryoEM facilities offer pre-processing of micrograph movies 'on the fly'. There are a wide variety of software packages and approaches available for this including RELION pipelines28,33, cryoSPARC43, Scipion34 and WarpEM44. A RELION based pipeline is described here and it is assumed that the user has moved the micrograph movies to an appropriate storage location with access to computing resources. An overview of the process and representative results for a membrane protein project are provided, a detailed description and step by step tutorial can be found on the RELION homepage: https://www3.mrc-lmb.cam.ac.uk/relion.
- Perform 'on the fly' analysis of micrograph motion correction and CTF estimation. Start RELION within the project directory. Schedule Import, Motion correction and CTF estimation jobs to loop such that they are concurrent with data collection and transfer. A micrograph analysis script28 provides real-time visual feedback on astigmatism and estimated defocus values (see representative results).
- Pick particles from pre-processed micrographs. There are a number of automated particle picking software packages to choose from. Reference free and template-based picking options are available within the Auto-picking tab of RELION37. Other programs may be used for various steps, for example using crYOLO for particle picking35.
- Extract particles from the CTF-corrected micrographs.
NOTE: To reduce the computational time required for early, 'clean-up', processing steps, down-scale/bin the particles upon extraction. Details on how run the extract job can be found in the RELION 3.1 tutorial. For this project, particles were initially binned by a factor of 2. - Perform 2D class averaging. Classifying across 100-200 classes works well for most datasets containing ≥100,000 particles. It is not recommended to use many more than 200 classes or fewer than 50 classes, even where datasets are small unless the sample has high symmetry (i.e., icosahedral virus) in which case fewer than 50 classes might still give a good result. Set the mask diameter large enough to accommodate the longest dimension of the particle, but tight enough to exclude any neighbouring particles (this may require some trial and error).
- Select good classes (i.e., those with structural details) using the subset selection job. Examples of good and bad 2D class averages can be found in the representative results section.
- Generate an initial model de novo from the data using the 3D initial model job in RELION.
NOTE: Less clean particle stacks may benefit from multi-reference ab initio SGD (stochastic gradient descent) refinement since this provides an additional opportunity to sift out junk/sub-optimal particles. Select a mask diameter that can accommodate the particle of interest, and leave the default values for fields in the 'SGD' tab since these routinely perform well. Ensure that the initial model looks reasonable in Chimera (or another appropriate visualisation program) (see representative results). - Perform 3D classification to address heterogeneity in the data using the output from step 6.6 as a reference model. Assess the resulting maps in Chimera. Process particle stacks corresponding to unique conformational states independently. Use the subset selection job to select a class/classes of interest and generate particles.star files for the associated particle stacks.
- Run 3D auto-refinement. Use the 3D class averages obtained in the previous step as references for refinement of their corresponding particle stacks. If the resolution of the refinement is approaching the Nyquist limit of the data, re-extract the particles without down-scaling. After re-extraction, repeat the 3D auto-refine job with the unbinned particle stack. In this case, the 3D reference models must be rescaled such that the pixel and box sizes are consistent with those of the re-extracted particle images. Use the relion_image_handler command line tool to carry out this operation.
- Utilize symmetry in refinement if appropriate. If a reconstructed map possesses symmetry, align the map on the appropriate symmetry axis using the relion_align_symmetry command line tool. Use the resulting aligned map as a reference in a new 3D auto-refinement job with the appropriate symmetry operator specified in the reference tab.
- Sharpen maps from 3D auto-refinement. This is done using the post-processing job in RELION, but first a suitable mask must be created from the refined map. The steps of mask creation and post-processing are detailed in the RELION tutorial (see also representative results).
NOTE: The resolution of many reconstructions can be further improved using the Bayesian polishing and CTF refinement functionalities in RELION. Use the CTF refinement job-type to estimate and correct for higher order aberrations (beam tilt, trefoil aberrations and 4th order aberrations) and, as separate jobs, anisotropic magnification and per-particle defocus. Following this, use the Bayesian polishing job (trained or with default values) to address beam-induced motion on a per-particle basis. As addressed in the RELION 3.1 tutorial, these jobs will likely benefit from an iterative approach (CTF-refinement → Bayesian polishing → 3D auto-refinement → post-processing →…loop) since both benefit from higher resolution models. - Correct the handedness of EM density maps if necessary. Examine the maps to determine whether the handedness is correct either by attempting to fit an existing atomic model, or assessing the handedness of the alpha helical regions. Where required, flip the map along the z-axis in UCSF Chimera45 using the 'vop zflip' command.
Results
When screening, grids can be discarded at the atlas stage, where features resolved at low magnification mark the grid as not suitable for data acquisition. For example, if a grid has been subject to significant mechanical damage with the majority of grid squares broken (Figure 2A), or where the grid appears to be 'dry', with no vitreous ice (Figure 2B). Such grids are typically identifiable as the edges of the grid squares appear sharp and distinct. Across the majority of grids made using the plunge freezing device, a gradient of ice is observed (Figure 2C,D). Particle distribution, depending on the specimen of interest, can vary dramatically with ice thickness and so screening a range of grid squares to assess particle distribution is recommended. Tools have been implemented within EPU software during the atlas screening step to help the user identify grid squares of similar or different ice thickness, which can be particularly useful to users who are new to examining cryoEM grids (Figure 2E, F).

Figure 2: Example low magnification 'atlas' montages from screening sessions. A) A grid which has suffered significant damage with the majority of grid squares broken - unsuitable for collection. B) A dry grid with no vitreous ice - unsuitable for collection. C) A grid demonstrating an ice gradient with ~ 50% of the grid useable. D) An ice gradient with ~ 33% of the grid useable. Both C and D, are suitable for data collection if the usable grid squares have an ice thickness appropriate for collection, and there are enough acquisition areas to satisfy the minimum duration of a collection (e.g., 24 h) E) An example atlas with range of ice thicknesses. F) The same atlas presented in E but with, grid squares categorized and coloured by EPU software according to ice thickness. Please click here to view a larger version of this figure.
{kind=link}
When screening particle distribution, ensure that imaging parameters, such as magnification and total electron dose, are similar to those expected to be used during data acquisition in order to provide an accurate picture of expected results. During screening, an ideal particle distribution is monodisperse with a range of particle orientations visible (depending on the specimen and existing knowledge of the particle's morphology, this may be challenging to ascertain) (Figure 3A). The ice should be as thin as possible while accommodating the particles largest dimension, if ice is too thin it can melt when illuminated with the electron beam. This causes excessive motion in the micrograph, and areas that display this characteristic should be avoided (Figure 3B). From collective experience, this effect is most commonly observed when there is detergent in the buffer. This can result in very thin ice at the centre of the hole and so particles can be physically excluded and forced towards the edge. This effect is observed in Figure 3C, but in this case it is not an extreme example and these images would still usefully contribute to a dataset. Finally, the ice needs to be vitreous; exclude any areas of the grid (or grids) where the majority or all of the images taken show crystalline ice (Figure 3D) from data acquisition. Often, non-vitreous ice is observed at the edge of grid squares. Readers are referred to detailed reviews of the variables that can be altered during grid vitrification16 and descriptions of particle behaviour in the thin film environment46, 47 for further information.

Figure 3: Representative micrographs showing differing particle distributions. A) An 'ideal' distribution of monodisperse particles adopting a range of orientations. B) Overly thin ice in the middle of the hole that it deforms upon exposure to the electron beam causing excessive motion in the micrograph. This effect is most often observed when detergent is present in the buffer C) Where ice is thinner in the centre of the hole, this physically excludes particles from the centre, causing crowding of particles towards the hole edge. In this case it is not extreme enough to prevent these images being useful, but it suggests it is worth screening slightly thicker areas. D) Ice is not vitreous, data should not be collected on areas which look like this example micrograph. Please click here to view a larger version of this figure.
{kind=link}
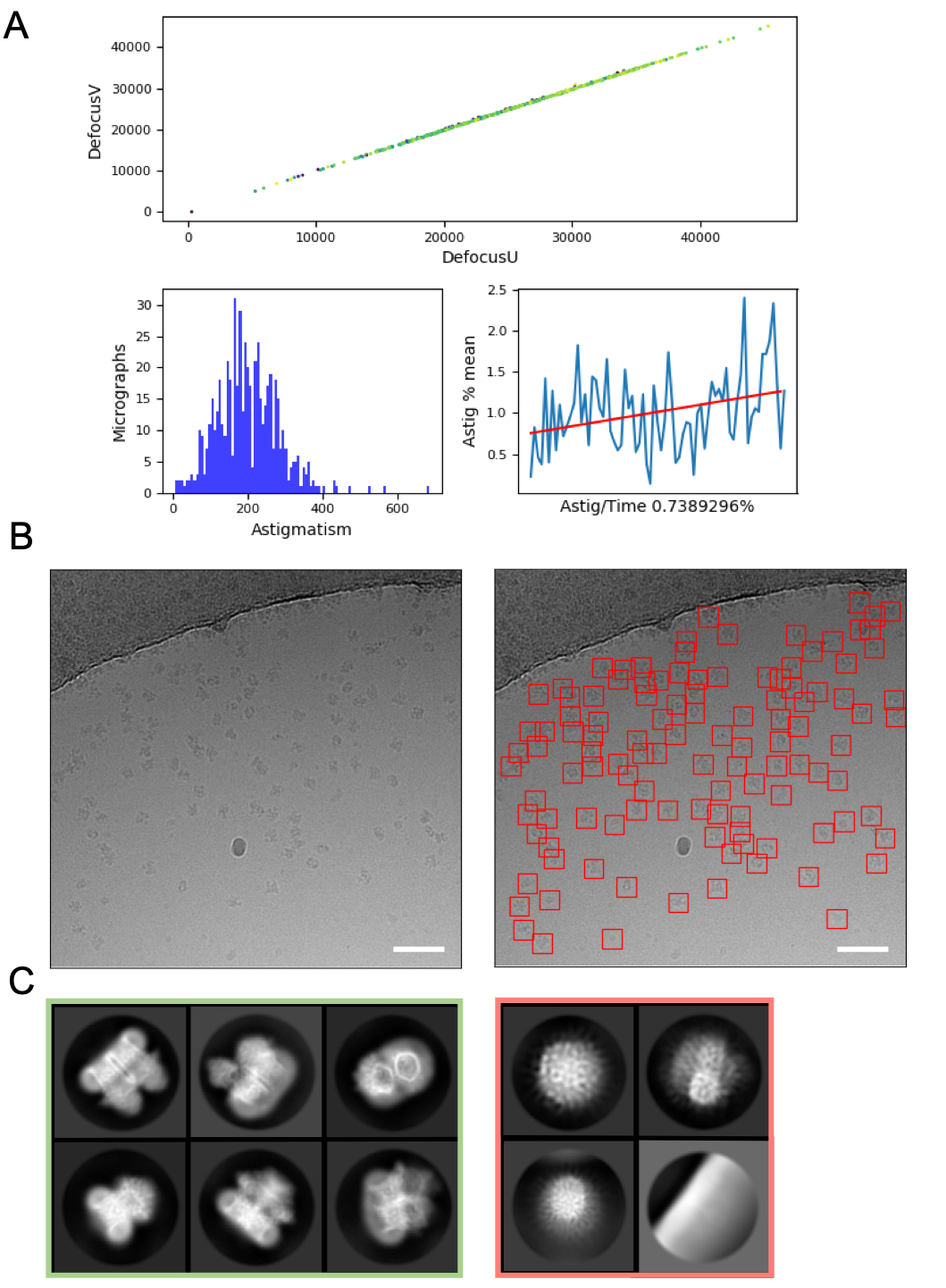
On-the-fly image processing can help to pick up errors and problems with data acquisition and so is always recommended where possible. For example, excessive motion within micrographs may indicate that the autoloader turbo pump is active, or data is being collected on a cracked grid square where ice is moving significantly in the electron beam, indicating the grid square should be skipped. On the fly CTF estimation can reveal circumstances where a positive focus point (rather than defocus) is applied (where CTF estimation programs and parameters to find these points are used), and determine the phase shift where a Volta phase plate48 is used. On the fly image processing pipelines often include a graphical summary of the data (Figure 4A) to make it easier for users to assess micrograph quality quickly and decide if data collection amendments are required.
Selection of particles from micrographs, whilst avoiding 'false positives' such as contamination or the grid support film can require optimisation. However, particle pickers such as crYOLO often work sufficiently well using default parameters for a 'first pass' of the data (Figure 4B), enabling progression to 2D class averaging where it can be easier to assess the quality of the data and the likelihood of downstream success. For most projects, 2D classification of ~> 10k particles should start to reveal classes which have secondary structure detail. To proceed to 3D, the 2D classification stage should typically reveal classes representing a range of particle orientations. If a preferred orientation is revealed, more iterations of sample preparation16 or further data acquisition with the sample tilted may be required49. All classes which show secondary structure detail should be chosen to take forward to 3D analysis, while 'junk' particles are discarded (Figure 4C).

Figure 4: Initial image processing steps. A) Output from an 'on the fly' image processing script. B) Example micrograph (left) with appropriately auto-picked particles identified using the crYOLO general model (right, with particles bounded by red squares) Scale bars (white) are 50 nm. C) Results from 2D classification showing classes which were discarded in the red square, and classes from which particles were selected for further processing in green. Please click here to view a larger version of this figure.
{kind=link}
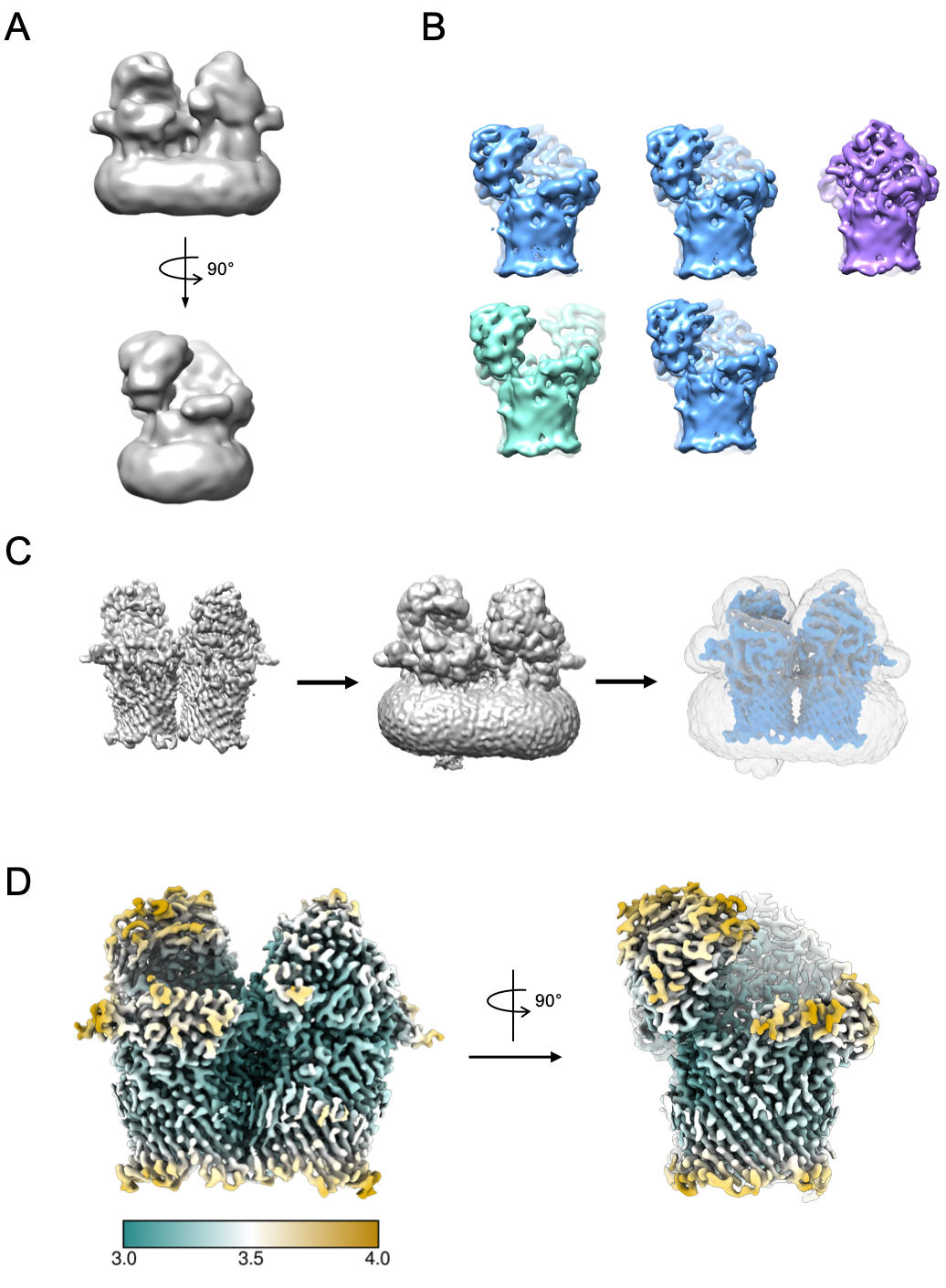
A small subset of particles can be used to generate an initial model (Figure 5A). This initial model can then be used as a starting model in 3D classification and refinement. In the case of RagAB, the dataset contained three distinct conformers which can be separated during 3D classification (Figure 5B). Particles contributing to each of these classes can then be treated independently and used to refine an EM density map which can then be subject to further interpretation and model building.

Figure 5: Generating 3D EM density map. A) Typical initial model generated using RELION. B) 3D classification over 5 classes showing separation of particles into three distinct conformational states: open-open (green), open-closed (blue), closed-closed (purple). C) Process of mask creation. The map from 3D refinement (left) should be visualized in chimera. The volume viewer can then be used to identify the lowest threshold at which the map is free from disjointed, noisy density (middle). This threshold value is input as initial binarization threshold in the RELION Mask creation job. An example mask output is shown in grey (right). D) High resolution EM density map of the open-closed state of RagAB (EMD-10245), filtered and colored by local resolution (Å). Please click here to view a larger version of this figure.
{kind=link}
Discussion
In this protocol we have described a basic pipeline applicable to specimens amenable to routine SPA. While this filter paper blotting method of thin film formation and vitrification is undoubtedly successful given its use in the vast majority of SPA projects to date, it comes with a number of disadvantages. These include sample wastage, the slow timescales (seconds) required to form the thin film and freeze the specimen, reported irreproducibility27 and reported negative effects of using filter paper to blot away excess liquid50. Recently, new technologies have been developed to improve reproducibility of thin film production51, 52. Other technologies have been developed which reduce the time between sample application and vitrification53,54,55. While filter paper-based methods for thin film formation remain most ubiquitous method of SPA cryoEM sample preparation at the time of writing, these new technologies may bring a range of benefits in terms of efficiency and reproducibility of grid vitrification, as well as creating new opportunities to bring in additional experimental dimensions, such as time resolution and rapid mixing prior to vitrification.
The process of grid screening for most users is presently a qualitative process which involves the acquisition of low magnification atlases followed by taking high-magnification images across the grid to assess particle distribution. While this is a sufficiently robust approach for some types of specimen, it can be difficult to assess by eye if the specimen is indeed what the researcher is hoping to image or has a preferred orientation, for example with small (<200 kDa) samples or where the low-resolution morphology makes it hard to identify by eye if a range of particle distributions are present. For some projects, it is impossible to determine if the specimen is as desired, for example where a ligand is bound or where the sample is being screened to assess if a small (e.g., 10 kDa) subunit is still present in association with a complex. For these projects, fully automated pipelines for data analysis combined with a 'short' 0.5 - 1-h collections, that can proceed through image processing steps to 2D classification or even 3D classification and refinement would help efficiently determine if a longer collection is warranted. These pipelines are still under development and are not widely implemented at present, but they have the potential to improve the efficiency of cryoEM grid screening, especially for challenging specimens.
Improvements in direct electron detectors, as well as modifications in microscopy combined with advances in image processing such as image shift data collection, have increased the throughput and quality of images produced during data acquisition. This increase in the rate of data being collected highlights the need for thorough screening of cryoEM grids ahead of many TB of data being acquired.
CryoEM SPA has become a truly mainstream structural biology technique, and in many cases the 'go to' approach for some classes of specimens, such as heterogeneous and labile macromolecular complexes. While the protocol here describes a basic overview of the SPA pipeline, each section covered here (grid vitrification and screening, cryoEM and image processing) is a topic in its own right and worthy of exploration during the development of an SPA project. As sample preparation and microscopy technologies progress, and new image processing algorithms and approaches come online, SPA will continue to develop as a pipeline, assisting researchers in gaining insight into complex biological systems.
Disclosures
No conflicts of interest are reported.
Acknowledgements
This work was supported by the iNEXT-Discovery (Grant 871037) funded by Horizon 2020 program of the European Commission. J B. R. White is funded by the Wellcome Trust (215064/Z/18/Z). The FEI Titan Krios microscopes were funded by the University of Leeds (UoL ABSL award) and Wellcome Trust (108466/Z/15/Z). We thank M Iadanza for use of his micrograph analysis script. We acknowledge Diamond Light Source for access and support of the cryo-EM facilities at the UK's national Electron Bio-imaging Centre (eBIC) funded by the Wellcome Trust, MRC and BBRSC.
Materials
| Name | Company | Catalog Number | Comments |
| Blunt tweezers | Agar Scientific | AGT5022 | |
| Cryo EM round storage box | Agar Scientific | AGG3736 | |
| CryoEM autogrid boxes | ThermoFisher Scientific | 1084591 | |
| CryoEM grids | Quantifoil | N1-C14nCu30-01 | |
| Ethane gas | Boc | 270595-F | |
| LN2 foam dewar | Agar Scientific | AG81760-500 | |
| LN2 storage dewar | Worthington industries | HC 34 | |
| Pipette | Gilson | 10082012 | |
| Pipette tips | Star labs | s1111-1706 | |
| Syringe | BD | BD 300869 | |
| Type II lab water | Suez | select fusion | |
| Vitrobot | ThermoFisher Scientific | 1086439 | |
| Vitrobot filter paper | Whatman | 1001-055 | |
| Vitrobot styrophome container assembly | ThermoFisher Scientific | 1086439 | |
| Vitrobot tweesers | ThermoFisher Scientific | 72882-D | |
| Software | |||
| EPU | ThermoFisher Scientific | 2.8.1.10REL | |
| TEM server | ThermoFisher Scientific | 6.15.3.22415REL | |
| Tia | ThermoFisher Scientific | 5.0.0.2896REL | |
| Titan krios microscope | ThermoFisher Scientific | Titan Krios G2 |
References
- Kuehlbrandt, W. The Resolution Revolution. Science. 343 (6178), 1443-1444 (2014).
- McMullan, G., Faruqi, A. R., Henderson, R. Direct Electron Detectors. Methods in Enzymology. , (2016).
- Elmlund, D., Le, S. N., Elmlund, H. High-resolution cryo-EM: the nuts and bolts. Current Opinion in Structural Biology. , (2017).
- Lyumkis, D. Challenges and opportunities in cryo-EM single-particle analysis. Journal of Biological Chemistry. , (2019).
- Nakane, T., et al. Single-particle cryo-EM at atomic resolution. Nature. , (2020).
- Yip, K. M., Fischer, N., Paknia, E., Chari, A., Stark, H. Atomic-resolution protein structure determination by cryo-EM. Nature. , (2020).
- Conley, M. J., et al. Calicivirus VP2 forms a portal-like assembly following receptor engagement. Nature. 565 (7739), 377-381 (2019).
- Hesketh, E. L., et al. The 3.3 Å structure of a plant geminivirus using cryo-EM. Nature communications. 9 (1), 2369 (2018).
- Malone, L. A., et al. Cryo-EM structure of the spinach cytochrome b6 f complex at 3.6 A resolution. Nature. 575 (7783), 535-539 (2019).
- Madej, M., et al. Structural and functional insights into oligopeptide acquisition by the RagAB transporter from Porphyromonas gingivalis. Nature Microbiology. , (2020).
- Gallardo, R., et al. Fibril structures of diabetes-related amylin variants reveal a basis for surface-templated assembly. Nature Structural and Molecular Biology. , (2020).
- Scarff, C., et al. Structure of the shutdown state of myosin-2. Nature. , (2020).
- Scarff, C. A., et al. Structure of the protective nematode protease complex H-gal-GP and its conservation across roundworm parasites. PLoS Pathogens. 16 (4), 1008465 (2020).
- Wu, M., Lander, G. C. How low can we go? Structure determination of small biological complexes using single-particle cryo-EM. Current Opinion in Structural Biology. , (2020).
- Khoshouei, M., Radjainia, M., Baumeister, W., Danev, R. Cryo-EM structure of haemoglobin at 3.2 Å determined with the Volta phase plate. Nature Communications. , (2017).
- Drulyte, I., et al. Approaches to altering particle distributions in cryo-electron microscopy sample preparation. Acta crystallographica. Section D, Structural biology. 74, 560-571 (2018).
- Thompson, R. F., Walker, M., Siebert, C. A., Muench, S. P., Ranson, N. A. An introduction to sample preparation and imaging by cryo-electron microscopy for structural biology. Methods. 100, 3-15 (2016).
- Cheng, Y., Grigorieff, N., Penczek, P. A., Walz, T. A primer to single-particle cryo-electron microscopy. Cell. 161 (3), 438-449 (2015).
- Scarff, C. A., Fuller, M. J. G., Thompson, R. F., Iadanza, M. G. Variations on negative stain electron microscopy methods: tools for tackling challenging systems. Journal of Visualized Experiments. (132), e57199 (2018).
- Ohi, M., Li, Y., Cheng, Y., Walz, T. Negative Staining and Image Classification - Powerful Tools in Modern Electron Microscopy. Biological procedures online. 6, 23-34 (2004).
- Baker, L. A., Rubinstein, J. L. Radiation Damage in Electron Cryomicroscopy. Methods in enzymology. 481, 371-388 (2010).
- Dubochet, J., et al. Cryo-electron microscopy of vitrified specimens. Quarterly Reviews of Biophysics. 21 (02), 129-228 (1988).
- Passmore, L. A., Russo, C. J. Specimen Preparation for High-Resolution Cryo-EM. Methods in enzymology. 579, 51-86 (2016).
- Carragher, B., et al. Current outcomes when optimizing ‘standard’ sample preparation for single-particle cryo-EM. Journal of Microscopy. , (2019).
- Naydenova, K., Jia, P., Russo, C. J. Cryo-EM with sub-1 Å specimen movement. Science. , (2020).
- Passmore, L. A., Russo, C. J. Specimen Preparation for High-Resolution Cryo-EM. Methods in Enzymology. , (2016).
- Thompson, R. F., Iadanza, M. G., Hesketh, E. L., Rawson, S., Ranson, N. A. Collection, pre-processing and on-the-fly analysis of data for high-resolution, single-particle cryo-electron microscopy. Nature protocols. 14 (1), 100-118 (2019).
- Suloway, C., et al. Automated molecular microscopy: the new Leginon system. Journal of Structural Biology. 151 (1), 41-60 (2005).
- Zhang, J., et al. JADAS: A customizable automated data acquisition system and its application to ice-embedded single particles. Journal of Structural Biology. , (2009).
- Mastronarde, D. N. SerialEM: A program for automated tilt series acquisition on Tecnai microscopes using prediction of specimen position. Microscopy and Microanalysis. , (2003).
- Schorb, M., Haberbosch, I., Hagen, W. J. H., Schwab, Y., Mastronarde, D. N. Software tools for automated transmission electron microscopy. Nature Methods. , (2019).
- Fernandez-Leiro, R., Scheres, S. H. W. A pipeline approach to single-particle processing in RELION. Acta crystallographica. Section D, Structural biology. 73, 496-502 (2017).
- Gómez-Blanco, J., et al. Using Scipion for stream image processing at Cryo-EM facilities. Journal of Structural Biology. , (2018).
- Wagner, T., et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Communications biology. 2 (1), 213-218 (2019).
- Bepler, T., et al. TOPAZ: A Positive-Unlabeled Convolutional Neural Network CryoEM Particle Picker that can Pick Any Size and Shape Particle. Microscopy and Microanalysis. , (2019).
- Zivanov, J., et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 7, 163 (2018).
- Zivanov, J., Nakane, T., Scheres, S. H. W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ. , (2019).
- Cianfrocco, M. A., Kellogg, E. H. What Could Go Wrong? A Practical Guide to Single-Particle Cryo-EM: From Biochemistry to Atomic Models. Journal of Chemical Information and Modeling. , (2020).
- Tagari, M., Newman, R., Chagoyen, M., Carazo, J. M., Henrick, K. New electron microscopy database and deposition system. Trends in Biochemical Sciences. , (2002).
- Berman, H. M., et al. The Protein Data Bank. Nucleic Acids Research. , (2000).
- Iudin, A., Korir, P. K., Salavert-Torres, J., Kleywegt, G. J., Patwardhan, A. EMPIAR: A public archive for raw electron microscopy image data. Nature Methods. , (2016).
- Punjani, A., Rubinstein, J. L., Fleet, D. J., Brubaker, M. A. CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods. , (2017).
- Tegunov, D., Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nature Methods. , (2019).
- Goddard, T. D., et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Science. 27 (1), 14-25 (2018).
- Klebl, D. P., et al. Need for Speed: Examining Protein Behavior during CryoEM Grid Preparation at Different Timescales. Structure. , (2020).
- Noble, A. J., et al. Routine single particle CryoEM sample and grid characterization by tomography. eLife. 7, 32 (2018).
- Danev, R., Buijsse, B., Khoshouei, M., Plitzko, J. M., Baumeister, W. Volta potential phase plate for in-focus phase contrast transmission electron microscopy. Proceedings of the National Academy of Sciences. , (2014).
- Zi Tan, Y., et al. Addressing preferred specimen orientation in single-particle cryo-EMthrough tilting. Nature Methods. , (2017).
- Armstrong, M., Han, B. -. G., Gomez, S., Turner, J., Fletcher, D. A., Glaeser, R. M. Microscale Fluid Behavior during Cryo-EM Sample Blotting. Biophysical Journal. 118 (3), 708-719 (2020).
- Arnold, S. A., et al. Blotting-free and lossless cryo-electron microscopy grid preparation from nanoliter-sized protein samples and single-cell extracts. Journal of Structural Biology. , (2017).
- Dandey, V. P., et al. Spotiton: New Features and Applications. Journal of Structural Biology. , (2018).
- Rubinstein, J. L., et al. Shake-it-off: a simple ultrasonic cryo-EM specimen-preparation device. Acta crystallographica. Section D, Structural biology. 75, 1063-1070 (2019).
- Tan, Y. Z., Rubinstein, J. L. Through-grid wicking enables high-speed cryoEM specimen preparation. bioRxiv. , (2020).
- Klebl, D. P., et al. Sample deposition onto cryo-EM grids: From sprays to jets and back. Acta Crystallographica Section D: Structural Biology. , (2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved