
Method Article
Bioluminescent Optogenetics 2.0: Harnessing Bioluminescence to Activate Photosensory Proteins In Vitro and In Vivo
In This Article
Summary
Bioluminescence-light emitted by a luciferase enzyme oxidizing a small-molecule substrate, a luciferin-can be harnessed to activate photosensory proteins, thereby adding another dimension to light stimulation and enabling the manipulation of a multitude of light-mediated functions in cells across temporal and spatial scales.
Abstract
Bioluminescence - light emitted by a luciferase enzyme oxidizing a small molecule substrate, a luciferin - has been used in vitro and in vivo to activate light-gated ion channels and pumps in neurons. While this bioluminescent optogenetics (BL-OG) approach confers a chemogenetic component to optogenetic tools, it is not limited to use in neuroscience. Rather, bioluminescence can be harnessed to activate any photosensory protein, thus enabling the manipulation of a multitude of light-mediated functions in cells. A variety of luciferase-luciferin pairs can be matched with photosensory proteins requiring different wavelengths of light and light intensities.
Depending on the specific application, efficient light delivery can be achieved by using luciferase-photoreceptor fusion proteins or by simple co-transfection. Photosensory proteins based on light-dependent dimerization or conformational changes can be driven by bioluminescence to effect cellular processes from protein localization, regulation of intracellular signaling pathways to transcription. The protocol below details the experimental execution of bioluminescence activation in cells and organisms and describes the results using bioluminescence-driven recombinases and transcription factors. The protocol provides investigators with the basic procedures for carrying out bioluminescent optogenetics in vitro and in vivo. The described approaches can be further extended and individualized to a multitude of different experimental paradigms.
Introduction
Photosensory proteins can be activated by light from either a physical light source or from a luciferase enzyme in the presence of its substrate, luciferin, to generate bioluminescence. For applications that require milli- or even femtosecond timescales and/or single-cell spatial resolution, physical light sources (lasers and light-emitting diodes (LEDs)) are the only ones tunable to these scales. Examples are the spatial restriction of light used for stimulating opposite poles in developing Drosophila larvae with millisecond temporal control1 or the precise stimulation of single subcellular structures such as mitochondrial tubules2. However, many other applications for optical switches have different priorities, including extended spatial control and repeated application non-invasively and without light damage but with defined temporal control in minute timescales and tunable intensities. Here, using luciferases as an alternative light source to activate light-sensing domains has several advantages. In contrast to optical fiber light activation, bioluminescence reaches every light-sensing domain expressed in the target cell population as the light source is genetically encoded. Using bioluminescence alleviates concerns over tissue and cell damage by fiber optics and extended physical light exposure. The light is turned on with the application of the luciferase substrate. The onset is immediate in vitro and in vivo depending on the route of administration and lasts for ~15-30 min; extended presence or phasic stimulation of light can be achieved with different luciferins and with additional or repeated applications of substrate3. Lastly, bioluminescence emission can be tuned by varying the concentration of luciferin.
The use of bioluminescence to activate ion-moving photoreceptors, i.e., optogenetic elements, such as channelrhodopsins or pumps, has been extensively demonstrated4,5,6,7,8. This BioLuminescent OptoGenetics (BL-OG) approach has been employed in in vivo experiments in mice and rats5,6,7,9,10,11,12. BL-OG activation of opsins was found to require an amount of bioluminescence of at least ~33 µW/mm2, with the efficiency of activation increasing with higher light emission6,9. Ion-moving sensory photoreceptors are a subgroup of the large contingent of sensory photoreceptors found in nature that are non-ion moving13,14. The extension of bioluminescence to activating non-ion moving photoreceptors, such as photosensing domains from plants or bacteria, is encouraged by reports15,16 that non-ion moving photosensors are significantly more light-sensitive than channelrhodopsins, ensuring an even better drive of light sensors with bioluminescence than already obtained with ion-moving optogenetic elements. Recently, several publications reported the use of bioluminescence as a light source for the activation of a variety of photoreceptors, including light-oxygen-voltage-sensing (LOV) domains, blue-light-using-flavin (BLUF) domains, and cryptochromes (CRYs)3,17,18,19,20,21,22 (Table 1). Applications for bioluminescence-driven activation of optical switches targeted intracellular processes ranging from reactive oxygen species-induced cell death, cAMP synthesis, protein recruitment and dissociation to genomic recombination and induction of transcription.
This protocol outlines the general design of bioluminescence-driven optogenetic tools and details the procedures for the experimental execution of bioluminescence activation in cells and organisms. It includes descriptions on how to set up a room, a tissue culture hood and incubator, and a microscope for work with bioluminescence, as well as the steps from preparing the luciferin to applying it. This protocol provides investigators with the basic procedures for carrying out BioLuminescent OptoGenetics (BL-OG) in vitro and in vivo. The described approaches can be further extended and individualized to different experimental paradigms. We anticipate this protocol to facilitate the adoption of the use of bioluminescence in optogenetic biological studies.
Protocol
All procedures in the current study were performed using Institutional Animal Care and Use Committee (IACUC) approved protocols for animal handling at Central Michigan University, MI.
1. Bioluminescence activation of photosensory proteins in vitro
- Constructs
- Select a luciferase sequence or luciferase-fluorescent protein fusion sequence that will result in the expression of a light emitter producing light of a wavelength matching the photoreceptor to be activated.
NOTE: For example, blue light-emitting luciferases, such as Gaussia luciferase variants or NanoLuc, can be paired with blue light-sensing photoreceptors such as CRY/Ca2+- and integrin-binding protein (CIB), LOV, or Vivid (VVD). - If not already available from other investigators or plasmid deposits, use standard molecular biology techniques to clone the DNA into a mammalian expression plasmid.
NOTE: The choice of promoters is dictated by the need to provide strong and constitutive expression of the light-emitting module, such as that provided by the CAG and CMV promoters. - For initial studies, use separate plasmids for co-transfection of the light emitter and the light sensor. Generate fusion proteins of the two moieties as needed and for subsequent studies.
- Obtain high-quality plasmid DNAs using mini-, midi-, or maxiprep kits according to manufacturer's protocols.
- Select a luciferase sequence or luciferase-fluorescent protein fusion sequence that will result in the expression of a light emitter producing light of a wavelength matching the photoreceptor to be activated.
- Cell culture and transfection
NOTE: HeLa cells and HEK293 cells are used as examples in this protocol.- Plate cells in formats and numbers according to the desired end use.
NOTE: Specific examples are given in Table 2. Cell density at the time of plating will determine how soon cells can be transfected.- For assessing bioluminescence-activated transcription by fluorescence microscopy, plate HEK293 cells on poly-D-lysine (PDL)-coated 12 mm coverslips placed in 24-well dishes.
- For assessing bioluminescence-activated transcription by measuring light emission from an orthogonal reporter luciferase in a luminometer, plate HeLa cells initially in 6- or 12-well dishes for transfection but re-plate them after transfection (see step 4).
- If repeated bioluminescence stimulation will be carried out in live-cell imaging chambers, select coverslips of the appropriate size and place them into multi-well plates of the appropriate size (24-well plates for 12 mm coverslips; 12-well plates for 15 mm and 18 mm coverslips). Seed the cells on top of the coverslips using the cell numbers specified in Table 2. If the cell type selected does not adhere well to the culture surface, plate the cells on PDL-coated dishes.
- Perform transfection by lipofection according to the manufacturer's recommendation or use any transfection method appropriate for the cell type selected.
NOTE: Table 3 details transfection experiments for two different photoreceptors, EL222 and CRY2/CIB, and their respective reporter plasmids, in addition to different light-emitting proteins. The ratios of the various plasmids work well for the selected examples but will have to be optimized for each light emitter/light sensor pair. - After transfection, place the cells in an incubator that is completely light-sealed (Figure 1).
- Depending on the desired end use, use the cells for bioluminescence stimulation the next day in their original wells/dishes, or re-plate them 3-4 h after lipofection. For reading transcription of a firefly luciferase reporter gene in a luminometer, re-plate the cells in white 96-well plates.
NOTE: Carry out all manipulations in a light-tight room in a laminar flow hood illuminated by red light (Figure 2).- Wash the transfected cells once with Dulbecco's modified Eagle medium (DMEM) or phosphate-buffered saline (PBS).
- Add the minimum volume of a trypsinizing reagent to the wells (24-well: 100 µL; 12-well: 150 µL; 6-well: 300 µL) and incubate the cells for 3 min at 37 °C.
- Add culture medium to achieve a cell concentration that will yield the appropriate cell density for the next plating step (for example, resuspend cells in a 24-well in a final volume of 1.2 mL for plating in 10 wells of a 96-well plate; resuspend cells in a 12-well in a final volume of 2.4 mL for plating in 20 wells of a 96-well plate). Pool the transfected cells from several wells depending on the number of wells needed in the end.
- Plate the transfected cells in their final format and return the plates to the light-protected incubator.
- Plate cells in formats and numbers according to the desired end use.
- Bioluminescence activation in vitro
- Prepare the luciferase substrate (luciferin).
- Prepare 50 mM stocks by dissolving 5 mg of lyophilized coelenterazine (CTZ) in 250 µL of its specific solvent. Ensure that all the CTZ along the walls of the vial is dissolved by pipetting or vortexing. Protect the vial from direct light.
- Prepare 50 µL aliquots in 0.5 mL black microcentrifuge tubes and store at -80 °C for future use.
NOTE: CTZ dissolved in solvent does not freeze at -80 °C. Aliquots can be removed from and returned to the freezer several times for making working solutions as long as exposure to light and room temperature is kept to a minimum.
- Single bioluminescence light stimulation
NOTE: All manipulations are carried out in a light-tight room in a laminar flow hood illuminated by red light (Figure 2).- Prepare a working solution of luciferin in cell culture medium (DMEM or NeuroBasal). Adjust the concentration of the luciferin such that the final concentration is 100 µM. Prepare all dilutions of CTZ in medium shortly before adding to the cells, as CTZ oxidizes over time.
NOTE: If the entire volume of medium will be replaced, the working solution will be 100 µM. If luciferin-containing medium is added to the cells, the concentration will be higher by the dilution factor (for example, adding 50 µL of medium containing 300 µM luciferin to 100 µL of medium in the well will result in a 1:3 dilution and thus in a 100 µM final concentration of luciferin). - Add luciferin-containing medium to the cells and incubate for the desired duration of light stimulation.
NOTE: This can be as short as 1 min or as long as 15 min and might be even shorter or longer. The length of time for leaving the luciferin-containing medium on the cells depends on the half-life and kinetics of the selected luciferase-luciferin combination. - Monitor light emission at 100 µM final luciferin concentration by eye after turning off the red light; wait for a few seconds until eyes have adjusted to complete darkness. Document the light emission by taking a photograph (even with a cell phone).
- Terminate the light stimulation by removing the luciferin-containing medium and replacing it with culture medium. Depending on the sensitivity of the experiments, wash the cells with culture medium once or twice after removing the luciferin-containing medium to eliminate all luciferin. If the cells do not adhere well to the culture surface, plate them on PDL-coated dishes to avoid losing the cells during washes.
- Return the cells to the light-protected incubator for 16-24 h.
- Prepare a working solution of luciferin in cell culture medium (DMEM or NeuroBasal). Adjust the concentration of the luciferin such that the final concentration is 100 µM. Prepare all dilutions of CTZ in medium shortly before adding to the cells, as CTZ oxidizes over time.
- Repeated bioluminescence light stimulation
NOTE: All manipulations are carried out in a room that can be made light-tight and be illuminated by red light.- Set up the live-cell imaging chamber. Create a light-tight compartment around the live-cell imaging microscope using a box and black plastic sheets or black drapes (Figure 3). Cover all the light sources present inside the light-tight compartment and the room (e.g., LED indicators on the microscope or instruments).
- Set up the perfusion system with the desired solution for intake and the chamber outport leading to a waste container.
NOTE: For example, the imaging solution can be Tyrode's Solution (sodium chloride (124 mM), potassium chloride (3 mM), HEPES (10 mM), calcium chloride dihydrate (2 mM), magnesium chloride hexahydrate (1 mM), D-glucose (20 mM)). - Prepare a working solution of luciferin in the imaging solution. Aliquot into as many microcentrifuge tubes as the number of repeat stimulations. Adjust the concentration of the luciferin such that the final concentration in the imaging chamber is 100 µM.
- Place a coverslip with transfected cells in the chamber.
- While keeping the pump running, remove the inlet tube of the pump from the intake beaker and quickly immerse it in the luciferin solution, keeping the transition time as short as possible to avoid any air void in the tubing.
- As soon as the luciferin solution has been taken up, place the inlet tube back into the intake beaker. Repeat this process as many times as needed and at intervals of several minutes to hours, depending on the physiological pattern to which the cells are supposed to be exposed.
- Return the cells to the light-protected incubator for 16-24 h for transcription, or for the length of time the effect of light stimulation is to be assessed.
- Prepare the luciferase substrate (luciferin).
2. Bioluminescence activation of photosensory proteins in vivo
- Constructs
- Select a luciferase sequence or luciferase-fluorescent protein fusion sequence that will result in the expression of a light emitter producing light of a wavelength matching the photoreceptor to be activated.
- Use standard molecular biology techniques to clone the DNA into a pAAV plasmid, if not already available from other investigators or plasmid deposits.
- Choose strong promoters for the expression of the light-emitting modules, such as CAG or CMV.
- Use standard approaches for preparing high-titer viral stocks6 or have viral vectors commercially prepared.
- For initial studies, use separate viral vectors for co-transduction of the light emitter and the light sensor to allow for adjustment of the ratios of the different components if needed.
- AAV transduction
- Inject the target organ of the experimental animal with viral vectors of the light emitter, light sensor, and reporter analogous to the concentration ratios used for the in vitro transfections (Table 3).
- Return the animals to their home cages for at least 2 weeks to allow maximal expression of all the components.
NOTE: If the target organ is inside the body and protected from ambient light, the animals can be housed under normal light conditions.
- Bioluminescence activation in vivo
- Prepare the luciferase substrate (luciferin).
- Take out a vial of water-soluble CTZ from the -80 °C freezer and let it warm to room temperature. Keep it protected from light.
- Per 500 µg vial, add 250 µL of sterile water, using either a syringe or by opening the vial and adding water with a pipette, then putting the rubber stopper back on the glass vial.
- Incubate the reconstituted glass vial in a 55 °C water bath for a few minutes to completely dissolve the powder.
- Transfer the solution into a black microcentrifuge tube. Rinse the walls of the glass vial to retrieve all CTZ.
- Remove the amount of solution needed for the day. Store the remaining solution at 4 °C for use the next day. Do not freeze!
- Carry out the same steps (2.3.1.1.-2.3.1.5) for a vial of vehicle.
- Bioluminescence light stimulation
- Remove the volume of luciferin/vehicle needed for the size of the animal and application route chosen (Table 4).
- Inject the animals with luciferin or vehicle. Repeat the bioluminescence light stimulation as per the experimental design. For example, if activation of a recombinase is desired during a specific behavioral paradigm, inject the animals just before the behavioral testing. If phasic transcription of a molecule is the goal, inject the animals repeatedly over days.
- Collect data from the bioluminescence-stimulated animals as designed.
- Prepare the luciferase substrate (luciferin).
Results
There are numerous intracellular events that can be manipulated with actuators responding to light, and that are amenable to bimodal activation with physical and biological light sources. Below are examples employing a photosensing calcium (Ca2+) integrator, light-induced protein translocation, a light-sensing transcription factor, and a photosensitive recombinase. The examples illustrate the feasibility of using bioluminescence to activate various kinds of photoreceptors. The experiments presented were not specifically optimized with respect to light-emitting diode (LED) application, the luciferase chosen, or with respect to concentrations and timing of luciferin application.
Fast light- and activity-regulated expression (FLARE) is an optogenetic system that allows the transcription of a reporter gene with the co-incidence of increased intracellular Ca2+ and light23 (Figure 4A). The presence of Ca2+ is required to bring the protease in proximity to the protease cleavage site accessible only with light stimulation, resulting in the release of the transcription factor. HEK293 cells were co-transfected with the original FLARE components, a dual Firefly (FLuc)-dTomato reporter construct, and a membrane-anchored Gaussia luciferase variant sbGLuc6. In the presence of increased intracellular Ca2+ through the exposure of cells to 2 µM ionomycin and 5 mM calcium chloride (CaCl2), the application of blue LED led to robust expression of the fluorescence reporter compared to cells left in the dark, as well as to the expression of FLuc determined by measuring luminescence upon adding the FLuc substrate, D-luciferin. Similar levels of FLuc expression were achieved with bioluminescence emitted by sbGLuc upon the application of the sbGLuc substrate (CTZ) together with ionomycin and CaCl2. Note that the luciferases used for light activation (sbGLuc) and for reporting the effect of light activation (transcription of FLuc) only produce light with their respective luciferins (CTZ vs. D-luciferin) and do not cross-react.
Different components were combined to generate a light-induced transcription system based on the heterodimerization of cryptochromes23,24 (Figure 4B). CRY2 was fused to a protease while the membrane-bound CIB was fused to the protease cleavage site and transcription factor. Light-induced protein translocation released the transcription factor, leading to the expression of FLuc and dTomato, as shown in Figure 4A. While the presence of the transcription factor component alone resulted in considerable background signal possibly due to spontaneous proteolysis, both physical light (LED) and bioluminescence (CTZ) robustly increased the expression of FLuc as measured in an in vivo imaging system (IVIS).
In another set of experiments, NanoLuc (luciferin: furimazine or hCTZ) was employed for the optogenetic regulation of transcription through the dimerization of CRY/CIB and the photosensitive transcription factor, EL22225,26,27. Figure 5A,B show the schematics of the different components in the dark and light states and the luciferase co-transfected or fused to the light sensor. Various comparisons are shown in Figure 5C. Bioluminescence, induced by adding hCTZ to HEK293 cells expressing the constructs and removing it after 15 min, was more efficient in driving reporter transcription than 20 min of LED light exposure for both CRY/CIB and EL222. For CRY/CIB, an hour of LED exposure was sufficient to reach a level of transcription comparable to 15 min of bioluminescence. In contrast, for EL222, even 60 min of LED were barely half as effective as a brief exposure to bioluminescence. There were no significant differences in the transcription efficacy between the two systems when co-transfected, although the fusion proteins of CRY/CIB were more efficient than those of EL222. For both systems, the fusion proteins led to significantly higher transcription levels than the co-transfected components. CRY/CIB showed consistently higher background levels with vehicle application compared to EL222, which had negligible background transcription. Increasing concentrations of hCTZ alone had no effect on the transcription of the reporter gene.
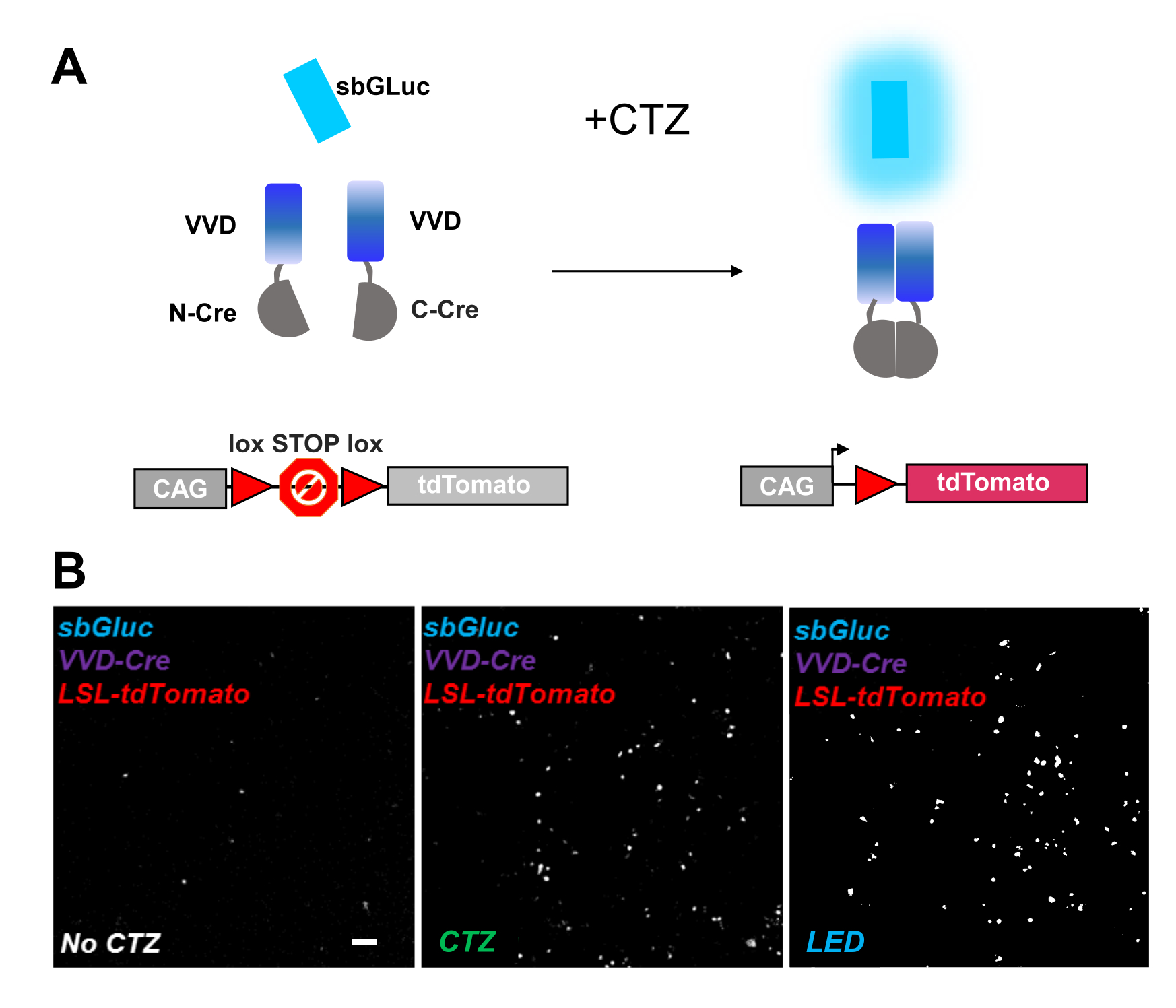
Photoactivatable recombinases provide a versatile tool for optogenomic manipulations. We tested bioluminescence activation of a photosensitive split Cre recombinase based on the Vivid LOV protein, iCreV28. Figure 6A shows a schematic of the different components, sbGLuc, iCreV, and a lox-stop-lox fluorescence reporter (tdTomato) before and after the application of CTZ. The results from CTZ application relative to controls (no CTZ or LED) are shown in Figure 6B. There is some background expression even in the dark (no CTZ); however, in the presence of CTZ, expression is robustly increased over the background and similar to that induced with LED application.

Figure 1: Light sealed incubator. Cardboard box flap covering the light from the illuminated control panel (top arrow). Light-impermeable cover over the glass door of the incubator (bottom arrow) to protect the cells from light exposure. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Laminar flow hood illuminated by red light. Setup showing a standard laminar flow tissue culture hood being illuminated by red light. Arrow indicates a standard desktop lamp with a red bulb. All manipulations under red light are carried out in an otherwise dark light-tight room. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Light-tight compartments around live-cell imaging microscopes. Two examples of live-cell imaging microscope setups showing the use of either a solid box with plastic drapes only on the front side (left panels: top and bottom) or black drapes all around the imaging setup (right panels: top and bottom). The front sides in both examples remain open and rolled up when not in use (top panels: left and right). The front black drapes are rolled down to prevent any light in the room (e.g., computer screens) from entering the imaging area when performing live-cell bioluminescence stimulation and/or imaging (bottom panels: left and right). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Bioluminescence for integrating intracellular signaling events. (A) Schematics of the FLARE components co-transfected with sbGLuc. In the presence of Ca2+ and the resulting proximity of the protease to the protease cleavage site, either bioluminescence or LED will lead to the unfolding of LOV, exposure of the cleavage site, and release of the transcription factor. Cells were exposed to LED (duty cycle 33%, 2 s on/4 s off for 40 min; 3.5 mW light power, 4.72 mW/cm2 irradiance) or to bioluminescence (100 µM CTZ final concentration for 15 min) or left in the dark. Microscopic images of HEK293 cells expressing the above components after treatment to increase Ca2+ levels and exposure to LED (left). FLuc luminescence measured in a luminometer comparing exposure to LED, bioluminescence (CTZ), or left in the dark (right). (B) Schematics of a non-Ca2+-dependent transcription system co-transfected with sbGLuc. HEK293 cells in 4-well plates were transfected with four different arrangements of components as depicted in the schematic. Plates were exposed to either LED (duty cycle 33%, 2 s on/4 s off for 40 min; 3.5 mW light power, 4.72 mW/cm2 irradiance) or bioluminescence (100 µM CTZ final concentration) by adding CTZ and leaving it on for 15 min; control plates were left in the dark. Transcription of the FLuc reporter was measured in an IVIS. IVIS images of representative dishes are shown on the left; radiance measurements from several replicates baselined to the dark controls are shown on the right. Scale bar = 100 µm. Abbreviations: FLARE = Fast light- and activity-regulated expression; LOV = light-oxygen-voltage-sensing; LED = light-emitting diode; CTZ = coelenterazine; FLuc = firefly luciferase; dTom = dTomato; CRY2 = cryptochrome 2; CRY2PHR = CRY2 photolyase homology region; CIB1 = Ca2+- and integrin-binding protein 1; CIBN = N-terminus of CIB1; IVIS = in vivo imaging system. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Bioluminescence for driving transcription. (A) Schematics of two photoactivatable transcription systems in their dark and light states. (B) NanoLuc was either co-transfected or fused to the light-sensing moieties as depicted (N-NanoLuc-CRY-GalDD-C; N-NanoLuc-VP16-EL222-C). (C) Comparisons using both systems regarding light sources, construct design, and signal to noise. Cells were exposed to LED (duty cycle 33%, 2 s on/4 s off for 40 min; 3.5 mW light power, 4.72 mW/cm2 irradiance) or to bioluminescence for 15 min (100 µM hCTZ final concentration; except where different concentrations are noted). Dark, plates were left untouched in the incubator between the initial transformation of plasmids and FLuc measurement; VEH, plates were handled the same as those receiving hCTZ, but received vehicle instead. Differences in transcription levels: hCTZ, co-transfected CRY vs. EL222 - not significant; hCTZ, luciferase - photoprotein fusion CRY vs. EL222 - p < 0.005; hCTZ, CRY co-transfection vs. fusion - p < 0.005; hCTZ, EL222 co-transfection vs. fusion - p < 0.01; vehicle, CRY vs. EL222 - p < 0.05. Abbreviations: UAS = upstream activating sequence; LED = light-emitting diode; CTZ = coelenterazine; FLuc = firefly luciferase; CRY = cryptochrome; CIB = Ca2+- and integrin-binding protein; VEH = vehicle. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Bioluminescence for optogenomic manipulation. (A) Schematics of bioluminescence-driven optogenomic manipulation using sbGLuc, the split iCreV components, and an LSL reporter cassette, before and after application of light. (B) HEK293 cells were lipofected with plasmids, then kept in the dark. Twenty-four hours later, the cells were treated for 30 min with just medium (no CTZ) or with CTZ (100 µM final concentration) or with LED (duty cycle 25%, 5 s on/15 s off for 5 min; 14.81 mW light power, 20 mW/cm2 irradiance) as a positive control. Microscopic images of tdTomato fluorescence using conditions as indicated. Scale bar = 100 µm. Abbreviations: LSL = lox-stop-lox; CTZ = coelenterazine; LED = light-emitting diode; VVD = Vivid. Please click here to view a larger version of this figure.
{kind=link}
Table 1: Bioluminescence activation of photoreceptors. Please click here to download this Table.
Table 2: Guidelines for plating and transfecting cells in different formats. Please click here to download this Table.
Table 3: Ratios of various plasmids for transfection. Please click here to download this Table.
Table 4: Injection routes, volumes, and concentrations of luciferin for in vivo applications (25 g mouse). Please click here to download this Table.
Discussion
There is a range of luciferases and luciferins with light emission wavelengths matching the activation spectra of photosensory proteins from blue to red light14,29. Apart from aligning emission and excitation wavelengths, there is no reliable way to determine a priori which pairing will work best. Thus, the need to experimentally determine how luciferin-luciferase pairs work in cells and organisms in driving photosensory systems.
The protocols outlined in this presentation describe how to prepare the luciferin and how to apply it in vitro and in vivo, together with guidelines on setting up rooms, tissue culture hoods, incubators, and microscopes for experiments utilizing bioluminescence. In the representative experiments, different luciferases (NanoLuc, Gaussia luciferase) with several photosensory proteins (CRY/CIB, EL222, VVD, LOV) were used, demonstrating the effects of bioluminescence versus physical light, co-transfection versus fusion proteins, signal-to-noise comparisons, and different readout assays. More applications of bioluminescence activating photosensory proteins are described in publications from several groups, targeting the induction of cell death, cAMP synthesis, and protein movement in addition to transcription (Table 1).
Simply co-transfecting light-emitting and light-sensing components is a good start. Variables are the molar ratios of emitter and sensor; unknowns are background levels of sensor activity in the dark, sensor activity in relation to light intensity and duration, and the efficiency of sensor activation comparing physical and biological light. While fusion constructs have the advantage of keeping the molar ratio of emitter and sensor at 1:1 and bringing the light emitter close to the light-sensing domain, other considerations come into play, such as where to tether (N- or C-terminus) and how to link (linker length and composition) without impacting the performance of the photosensory actuator.
For experiments both in vitro and in vivo, there are multiple options for tuning bioluminescent light emission, either by varying the concentration of the luciferin, and/or by varying the time the luciferin is made available to the respective sensor. The minimum amount and time are determined by the presence or absence of the effect expected with light activation. In contrast, the respective maxima are mainly determined by the tolerance of cells to high concentrations of luciferin over prolonged times. The concentration of CTZ chosen in the above examples, 100 µM, is close to the upper limit for various cell types, from HEK293 cells to neurons. The goal is to use as low a concentration as possible for the shortest time to achieve activation of the targeted photosensing domain. This will be achieved more readily using luciferases with high light emission and photoreceptors with high light sensitivity.
Bioluminescence for driving photoreceptors has been used in rodents (mice, rats) with photosensing proteins expressed in the liver, muscle, spinal cord, and brain as well as via photoreceptor-expressing cells transplanted subcutaneously or intraperitoneally. In principle, there are no limits preventing the approach from being applied to different species, from non-human primates to fish or flies. Depending on the permeability of the organism for the luciferin, the application may be as easy as applying the luciferin to the surrounding water (e.g., in fish larvae30). Before using BL-OG in any new organism, pilot experiments must be conducted to ensure that the luciferin reaches its targets via the chosen application route.
Critical aspects of the experimental design are the various controls that are important for the interpretation of results. Cells expressing a reporter driven by a luciferase acting on a photosensory protein should be compared to cells lacking the luciferase or lacking the photosensory protein. Further, comparisons should be made between cells exposed to luciferin, vehicle, or kept in the dark. It is also important to realize the limitations of different assays for assessing the effects of bioluminescence-driven photoreceptor activation. For example, the efficacy of bioluminescence-activated transcription can be tested in different ways, depending on whether the reporter gene is an orthogonal luciferase (luminometer, IVIS), or a fluorescent protein (fluorescence-activated cell sorting, microscopy image analysis). While the basic effects should be reproducible across testing platforms, the quantitative aspects of the effects might vary considerably.
The bioluminescence activation of photoreceptors has been demonstrated thus far for a limited number of luciferases and photosensory proteins, respectively, both in vitro and in vivo. It can be extended to the large class of photoreceptors for activating many more biological processes. Such expansion of the approach is further promoted by the continuous development of novel luciferases and luciferase-fluorescence protein pairs with much higher light emission than naturally occurring luciferases and with kinetic features tunable to different applications. These advances are paralleled by the generation of novel luciferins, further adding to increased brightness and color palettes29. This tool platform offers applications to manipulate and investigate intracellular dynamics and cell interactions inside living cells, tissues, and organisms.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgements
We thank our colleagues for constructs, specifically A. Ting for the Ca-FLARE protease, transcription factor and reporter (Addgene # 92214, 92213, 92202), H. Kwon for TM-CIBN-BLITz1-TetR-VP16 and NES-CRY2PHR-TevC (Addgene # 89878, 89877), C. Tucker for CRY-GalΔDD (B1013) and CIB-VP64 (B1016) (Addgene # 92035, 92037), M. Walsh for pGL2-GAL4-UAS-Luc (Addgene #33020), K. Gardner for VP-EL222 and C120-Fluc, and A. Cetin and H. Zeng for making iCreV available before publication. This work was supported by grants from NSF (NeuroNex 1707352), NIH (U01NS099709), the W.M. Keck Foundation, and the Swedish Research Council to A.B. (2016-06760).
Materials
| Name | Company | Catalog Number | Comments |
| ABI 25W Deep Red 660 nm LED Light Bulb | Amazon | to be used with any lamp stand | |
| Black Microcentrifuge Tubes, 0.5 mL, Argos Technologies | Fisher Scientific | 03-391-166 | |
| Black Microcentrifuge Tubes, 1.5 mL, Argos Technologies | Fisher Scientific | 03-391-161 | |
| Black Nylon, Polyurethane-Coated Fabric (1.5 m x 2.7 m) x 0.12 mm (thick) | THOR LABS | BK-5 | |
| C120-Fluc | K. Gardner | ||
| CaCl2 | Sigma | C8106; CAS: 10035-04-8 | |
| Ca-FLARE protease, transcription factor and reporter | Addgene # 92214, 92213, 92202 | A. Ting | |
| CIB-VP64 (B1016) | Addgene # 92037 | C. Tucker | |
| CRY-GalΔDD (B1013) | Addgene # 92035 | C. Tucker | |
| CTZ | Prolume Inc. (NanoLight) | 303 | formulation for in vitro applications with Gaussia luciferases |
| CTZ (Water soluble native coelenterazine) | Prolume Inc. (NanoLight) | 3031 | formulation for in vivo applications with Gaussia luciferases |
| D-(+)-Glucose | Sigma | G8270; CAS: 50-99-7 | |
| D-Luciferin, Potassium Salt | Gold Biotechnology | LUCK | |
| DMEM | Thermo Fisher | 11960044 | |
| D-PBS, no calcium, no magnesium | Thermo Fisher | 14190144 | |
| hCTZ | Prolume Inc. (NanoLight) | 301 | formulation for in vitro applications with Oplophorus luciferases |
| HEK293 | ATCC | CRL-1573 | |
| HeLa | ATCC | CCL-2 | |
| HEPES | Sigma | H3375; CAS: 7365-45-9 | |
| iCreV | A. Cetin and H. Zeng | ||
| In Vivo Imaging System (IVIS) | Perkin-Elmer | Lumina LT | |
| KCl | Sigma | P5405; CAS: 7447-40-7 | |
| LED Array Driver | Amuza | LAD-1 | |
| LED Array for Multiwell Plates | Amuza | LEDA-x | |
| Lipofectamine 2000 Reagent | Invitrogen | 11668-019 | Transfection reagent |
| Luminometer | Molecular Devices | SpectraMax L | |
| MgCl2 Hexahydrate | Sigma | M2670; CAS: 7791-18-6 | |
| NaCl | Sigma | S7653; CAS: 7647-14-5 | |
| NanoFuel Solvent | Prolume Inc. (NanoLight) | 399 | for dissolving CTZ preparations for in vitro use |
| NaOH | Sigma | 221465; CAS: 1310-73-2 | |
| NES-CRY2PHR-TevC | Addgene # 89877 | H. Kwon | |
| Opti-MEM | Thermo Fisher | 11058021 | transfection medium |
| PDL coated coverslips (12 mm, 15 mm, 18 mm) | Neuvitro Corporation | GG-12-PDL, GG-15-PDL , GG-18-PDL | |
| pGL2-GAL4-UAS-Luc | Addgene #33020 | M. Walsh | |
| Prizmatix USB Pulser TTL Generator for Optogenetics | Goldstone Scientific | ||
| TM-CIBN-BLITz1-TetR-VP16 | Addgene # 89878 | H. Kwon | |
| TrypLE Express | Gibco | 12604-013 | |
| Vehicle (Water-soluble carrier without CTZ) | Prolume Inc. (NanoLight) | 3031C | control for in vivo applications with CTZ |
| VP-EL222 | K. Gardner |
References
- Johnson, H. E., et al. The spatiotemporal limits of developmental Erk signaling. Developmental Cell. 40 (2), 185-192 (2017).
- Wang, Y., et al. Photostimulation by femtosecond laser triggers restorable fragmentation in single mitochondrion. Journal of Biophotonics. 10 (2), 286-293 (2017).
- Li, T., et al. A synthetic BRET-based optogenetic device for pulsatile transgene expression enabling glucose homeostasis in mice. Nature Communications. 12 (1), 615(2021).
- Berglund, K., Birkner, E., Augustine, G. J., Hochgeschwender, U. Light-emitting channelrhodopsins for combined optogenetic and chemical-genetic control of neurons. PLoS One. 8 (3), 59759(2013).
- Tung, J. K., Gutekunst, C. -A., Gross, R. E. Inhibitory luminopsins: genetically-encoded bioluminescent opsins for versatile, scalable, and hardware-independent optogenetic inhibition. Scientific Reports. 5, 14366(2015).
- Berglund, K., et al. Luminopsins integrate opto- and chemogenetics by using physical and biological light sources for opsin activation. Proceedings of the National Academy of Sciences of the United States of America. 113 (3), 358-367 (2016).
- Gomez-Ramirez, M., More, A. I., Friedman, N. G., Hochgeschwender, U., Moore, C. I. The BioLuminescent-OptoGenetic in vivo response to coelenterazine is proportional, sensitive and specific in neocortex. Journal of Neuroscience Research. 98 (3), 471-480 (2020).
- Moore, C. I., Berglund, K. BL-OG: BioLuminescent-OptoGenetics. Journal of Neuroscience Research. 98 (3), 469-470 (2020).
- Park, S. Y., et al. Novel luciferase-opsin combinations for improved luminopsins. Journal of Neuroscience Research. 98 (3), 410-421 (2020).
- Jaiswal, P. B., Tung, J. K., Gross, R. E., English, A. W. Motoneuron activity is required for enhancements in functional recovery after peripheral nerve injury in exercised female mice. Journal of Neuroscience Research. 98 (3), 448-457 (2020).
- Zenchak, J. R., et al. Bioluminescence-driven optogenetic activation of transplanted neural precursor cells improves motor deficits in a Parkinson's disease mouse model. Journal of Neuroscience Research. 98 (3), 458-468 (2020).
- Tung, J. K., Shiu, F. H., Ding, K., Gross, R. E. Chemically activated luminopsins allow optogenetic inhibition of distributed nodes in an epileptic network for non-invasive and multi-site suppression of seizure activity. Neurobiology of Disease. 109, Pt A 1-10 (2018).
- Hegemann, P. Algal sensory photoreceptors. Annual Review of Plant Biology. 59, 167-189 (2008).
- Losi, A., Gardner, K. H., Moglich, A. Blue-light receptors for optogenetics. Chemical Reviews. 118 (21), 10659-10709 (2018).
- Proshkina, G. M., Shramova, E. I., Shilova, O. N., Ryabova, A. V., Deyev, S. M. Phototoxicity of flavoprotein miniSOG induced by bioluminescence resonance energy transfer in genetically encoded system NanoLuc-miniSOG is comparable with its LED-excited phototoxicity. Journal of Photochemistry and Photobiology B: Biology. 188, 107-115 (2018).
- Kawano, F., Okazaki, R., Yazawa, M., Sato, M. A photoactivatable Cre-loxP recombination system for optogenetic genome engineering. Nature Chemical Biology. 12 (12), 1059-1064 (2016).
- Shramova, E. I., Proshkina, G. M., Chumakov, S. P., Khodarovich, Y. M., Deyev, S. M. Flavoprotein miniSOG cytotoxisity can be induced by bioluminescence resonance energy transfer. Acta Naturae. 8 (4), 118-123 (2016).
- Naim, N., et al. Luminescence-activated nucleotide cyclase regulates spatial and temporal cAMP synthesis. Journal of Biological Chemistry. 294 (4), 1095-1103 (2019).
- Kim, C. K., Cho, K. F., Kim, M. W., Ting, A. Y. Luciferase-LOV BRET enables versatile and specific transcriptional readout of cellular protein-protein interactions. Elife. 8, 43826(2019).
- Parag-Sharma, K., et al. Engineered BRET-based biologic light sources enable spatiotemporal control over diverse optogenetic systems. ACS Synthetic Biology. 9 (1), 1-9 (2020).
- Kim, E. H., et al. Self-luminescent photodynamic therapy using breast cancer targeted proteins. Science Advances. 6 (37), (2020).
- Kim, C. K., et al. A Molecular calcium integrator reveals a striatal cell type driving aversion. Cell. 183 (7), 2003-2019 (2020).
- Wang, W., et al. A light- and calcium-gated transcription factor for imaging and manipulating activated neurons. Nature Biotechnology. 35 (9), 864-871 (2017).
- Lee, D., Hyun, J. H., Jung, K., Hannan, P., Kwon, H. -B. A calcium- and light-gated switch to induce gene expression in activated neurons. Nature Biotechnology. 35 (9), 858-863 (2017).
- Pathak, G. P., et al. Bidirectional approaches for optogenetic regulation of gene expression in mammalian cells using Arabidopsis cryptochrome 2. Nucleic Acids Research. 45 (20), 167(2017).
- Nishio, H., Walsh, M. J. CCAAT displacement protein/cut homolog recruits G9a histone lysine methyltransferase to repress transcription. Proceedings of the National Academy of Sciences of the United States of America. 101 (31), 11257-11262 (2004).
- Motta-Mena, L. B., et al. An optogenetic gene expression system with rapid activation and deactivation kinetics. Nature Chemical Biology. 10 (3), 196-202 (2014).
- Yao, S., et al. RecV recombinase system for in vivo targeted optogenomic modifications of single cells or cell populations. Nature Methods. 17 (4), 422-429 (2020).
- Love, A. C., Prescher, J. A. Seeing (and using) the light: Recent developments in bioluminescence technology. Cell Chemical Biology. 27 (8), 904-920 (2020).
- Naumann, E. A., Kampff, A. R., Prober, D. A., Schier, A. F., Engert, F. Monitoring neural activity with bioluminescence during natural behavior. Nature Neuroscience. 13 (4), 513-520 (2010).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved