A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
Bacterial Expression and Purification of Human Matrix Metalloproteinase-3 using Affinity Chromatography
* These authors contributed equally
In This Article
Summary
His-tag purification, dialysis, and activation are employed to increase yields of soluble, active matrix metalloproteinase-3 catalytic domain protein expression in bacteria. Protein fractions are analyzed via SDS-PAGE gels.
Abstract
Matrix metalloproteinases (MMPs) belong to the family of metzincin proteases with central roles in extracellular matrix (ECM) degradation and remodeling, as well as interactions with several growth factors and cytokines. Overexpression of specific MMPs is responsible in several diseases such as cancer, neurodegenerative diseases, and cardiovascular disease. MMPs have been the center of attention recently as targets to develop therapeutics that can treat diseases correlated to MMP overexpression.
To study the MMP mechanism in solution, more facile and robust recombinant protein expression and purification methods are needed for the production of active, soluble MMPs. However, the catalytic domain of most MMPs cannot be expressed in Escherichia coli (E. coli) in soluble form due to lack of posttranslational machinery, whereas mammalian expression systems are usually costly and have lower yields. MMP inclusion bodies must undergo the tedious and laborious process of extensive purification and refolding, significantly reducing the yield of MMPs in native conformation. This paper presents a protocol using Rosetta2(DE3)pLysS (hereafter referred to as R2DP) cells to produce matrix metalloproteinase-3 catalytic domain (MMP-3cd), which contains an N-terminal His-tag followed by pro-domain (Hisx6-pro-MMP-3cd) for use in affinity purification. R2DP cells enhance the expression of eukaryotic proteins through a chloramphenicol-resistant plasmid containing codons normally rare in bacterial expression systems. Compared to the traditional cell line of choice for recombinant protein expression, BL21(DE3), purification using this new strain improved the yield of purified Hisx6-pro-MMP-3cd. Upon activation and desalting, the pro domain is cleaved along with the N-terminal His-tag, providing active MMP-3cd for immediate use in countless in vitro applications. This method does not require expensive equipment or complex fusion proteins and describes rapid production of recombinant human MMPs in bacteria.
Introduction
Most complex eukaryotic proteins undergo elaborate posttranslational modifications after expression, requiring highly assisted protein folding and co-factors to be functional1. Producing large amounts of soluble human protein in a bacterial host remains a significant challenge due to high costs and the lack of robust expression and purification methods, even for smaller-scale laboratory experiments2,3. MMPs, human endopeptidases with large molecular weight, are usually expressed as insoluble inclusion bodies when expressed in E. coli. Extraction of soluble human MMPs often leads to a laborious, time-consuming solubilization and refolding process4.
MMPs have critical roles in both physiological and pathogenic processes. Human MMPs are a family of 23 zinc endopeptidases, categorized by structure and substrate specificity, and differentially expressed in spite of a highly conserved catalytic domain5,6. MMPs are secreted as inactive zymogens, regulated via posttranslational activation and their endogenous inhibitors, tissue inhibitors of metalloproteinases (TIMPs)7,8,9,10. Though initially recognized for their role in ECM turnover, MMPs have also been implicated in development, morphogenesis, tissue repair, and remodeling8. Dysregulation of MMPs has been notably linked to cancer along with neurodegenerative, cardiovascular, and fibrotic diseases, among other illnesses5,7.
The development of robust large-scale MMP production methods is critical to ensure the success of future studies of MMP mechanisms through biochemical and cell-based assays. Various MMPs have been previously expressed in bacteria11, including Hisx6-tagged MMPs, without altering MMP activity12,13,14,15. However, these methods include tedious, long steps that might be difficult to replicate.
Mammalian cells can also be used to express many different human proteins while ensuring the proper posttranslational modifications16. Although the mammalian expression system is an ideal choice to produce recombinant human proteins with proper post-translational modifications, the main disadvantages of this method are initial low yields, costly growth media and reagents, long timelines to reach stable expression lines, and risk of contamination with other species such as fungi or bacteria2,11. Moreover, MMP production in mammalian cell lines yields impurities from associated cellular proteins such as TIMPs or fibronectins11. Unlike the slow cell growth observed in mammalian cells, the bacterial expression system offers large-scale protein production in a short period along with simpler media and growth requirements. However, due to the lack of other associated cellular proteins (i.e., TIMPs) in bacterial expression systems, active MMPs at higher concentrations are subject to degradation through autoproteolysis, resulting in poor MMP yield17.
This paper describes a detailed method for bacterial expression, purification, and activation of recombinant Hisx6-pro-MMP-3cd using E. coli as an expression host due to its affordability, simplicity, and success in producing higher yields of MMPs2,3,18. Since E. coli lacks the protein folding machinery and posttranslational processing required for recombinant MMPs and other complex proteins, many E. coli strains have been engineered to overcome these limitations, making E. coli a more suitable host for expression of recombinant human MMP-3cd,19,20. For instance, the R2DP strain used in this study enhances eukaryotic expression by supplying a chloramphenicol-resistant plasmid containing codons rarely used in E. coli.
As described in this protocol, after overexpression of relatively pure inclusion bodies from the pET-3a vector (Figure 1) in R2DP cells, Hisx6-pro-MMP-3 catalytic domain (MMP-3cd) proteins are extracted and denatured4. Hisx6-pro-MMP-3cd3,19 was purified using affinity tag chromatography. Upon refolding and dialysis, the pro-MMP-3cd (zymogen) was activated by 4-aminophenylmercuric acetate (APMA), and SDS-PAGE analysis is used to evaluate yields and the need for further purification5,21. This protocol describes expression, purification, and activation of soluble MMP-3cd as an example. However, it may be also used as a guide for expression of other MMPs and human proteases with similar expression, and activation mechanisms (Figure 2). For other proteins other than MMP-3cd, the reader is advised to determine optimal buffer compositions and methods for their target protein before attempting this protocol.

Figure 1: Plasmid map of the pET-3a-Hisx6-pro-MMP-3cd plasmid. The pET-3a vector includes an ampicillin resistance gene. An N-terminal Hisx6-tag sequence is cloned into the pET-3a-based vector, including pro-MMP-3cd, to yield the pET-3a-Hisx6-pro-MMP-3cd construct under control of T7 promoter between BamHI and NdeI restriction sites. Please click here to view a larger version of this figure.
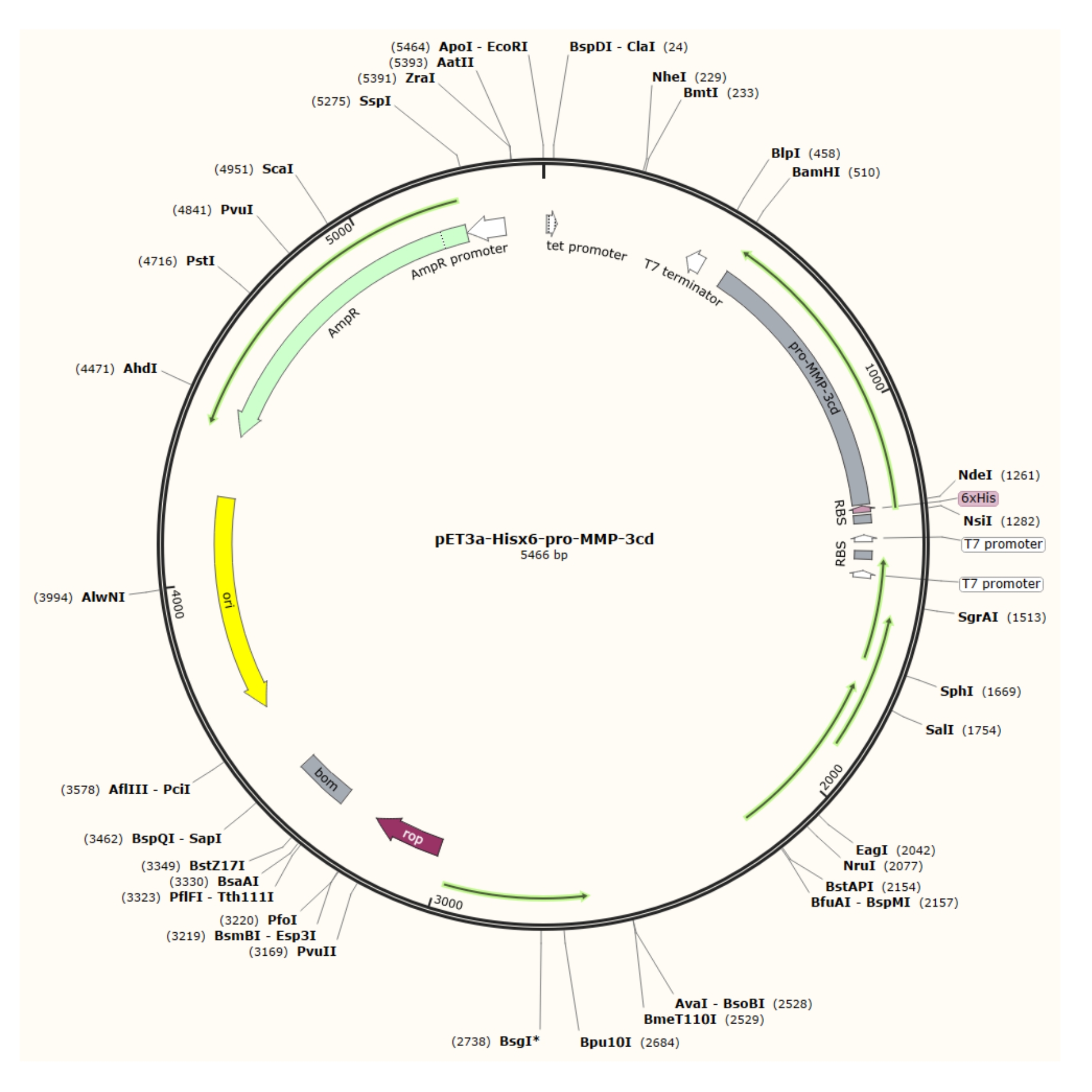{kind=link}

Figure 2: Bacterial expression of pro-MMP-3cd, purification, refolding, and activation. 1.1: pET-3a-Hisx6-pro-MMP-3cd plasmid was transformed into BL21(DE3) or R2DP Cells. 1.2: Pro-MMP-3cd protein expression was induced using IPTG. 1.3: Chemical lysis and sonication are used to extract Hisx6-pro-MMP-3cd proteins that are mainly insoluble and found in the inclusion bodies. Urea was used to denature and solubilize protein from inclusion bodies. 2.1. Denatured Hisx6-pro-MMP-3cd protein was purified via affinity chromatography purification. 3. The eluted Hisx6-pro-MMP-3cd was slowly refolded during dialysis through gradual removal of urea from the buffer. 4. Finally, refolded MMP-3cd protein was activated using APMA by removing the N-terminal pro-peptide domain. APMA is later removed from the solution through desalting. The numbers correspond to protocol sections describing these steps. Abbreviations: MMP-3cd = Matrix metalloproteinase-3 catalytic domain; APMA = 4-aminophenylmercuric acetate. Please click here to view a larger version of this figure.
{kind=link}
Protocol
1. MMP expression
- Cloning and transformation of pET-3a-Hisx6-pro-MMP-3cd into R2DP cells
- Digest the pET-3a plasmid (see the Table of Materials) with NdeI and BamHI restriction enzymes in Digest Buffer (see the Table of Materials). In a total reaction volume of 40 µL, add 4 µL of Digest Buffer, 33 µL of 100 ng/µL plasmid, and 1.5 µL of each restriction enzyme and allow the reaction to proceed for ~2 h until completion at 37 °C.
- Perform a PCR reaction on the MMP-3cd sequence to insert an N-terminal His-tag. Use 25 µL of PCR Mix (see the Table of Materials), 2.5 µL of 10 µM primers (Supplemental Figure S1), and 1.25 µL of the 100 ng/µL insert sequence. Add sterile water to a final reaction volume of 50 µL.
- Run the PCR product and digested vector on a 1% agarose gel. Purify the gel bands using a Gel Recovery Kit (see the Table of Materials) per the manufacturer's protocol.
- Clone the amplified PCR product into the digested vector between the NdeI and BamHI restriction sites using DNA Assembly Mix (see the Table of Materials). Use online tools to determine the required volume of the insert and cut vector for a total reaction volume of 15 µL.
- Thaw a 50 µL aliquot of high-transformation efficiency cells (see the Table of Materials) on ice until thawed. Prewarm SOC Growth Medium (see the Table of Materials) to 37 °C and LB-ampicillin (LB Amp) plates (see the Table of Materials).
- Add 1-2 µL of the pET-3a-Hisx6-pro-MMP-3cd assembly reaction to the 50 µL aliquot. Incubate on ice for 30 min.
- Heat-shock the cells by incubating at 42 °C for 30 s. Incubate on ice for 2 min.
- Add 950 µL of SOC growth medium to each transformant mixture. Shake for 1 h at 250 rpm and 37 °C.
- Plate 100 µL of the transformants on LB Amp plates and incubate overnight at 37 °C.
- Inoculate each isolated colony in 10 mL of LB Amp medium. Shake overnight at 250 rpm and 37 °C.
- Extract plasmid DNA per manufacturer's protocol for the miniprep kit (see the Table of Materials). Confirm the sequence of the construct using T7 forward and reverse primers (Supplemental Figure S1).
NOTE: The pET-3a-Hisx6-pro-MMP-3cd construct DNA can be stored at -20 °C. When ready, proceed with transformation into R2DP cells. - Thaw one 20 µL aliquot of R2DP cells (see the Table of Materials) on ice for 2-5 min. Prewarm SOC growth medium to room temperature and LB Amp CamR plates to 37 °C (see the Table of Materials).
- Add 1 µL of 100 ng/µL sequence-confirmed pET-3a-Hisx6-pro-MMP-3cd to the 50 µL aliquot. Stir gently to mix and return the tube to ice.
- Incubate the tube on ice for 5 min.
- Heat-shock the cells by incubating at 42 °C for exactly 30 s. Do not shake.
- Place the cells on ice for 2 min.
- Add 80 µL of room-temperature SOC medium to the transformant mixture. Shake for 1 h at 250 rpm and 37 °C.
- Plate the transformants on LB Amp CamR plates and incubate overnight at 37 °C.
- Growth and induction
- Inoculate a single, isolated colony of R2DP pET-3a-Hisx6-pro-MMP-3cd transformant from an LB Amp CamR plate in 10 mL of LB Amp CamR media at 37 °C. Shake at 250 rpm overnight (~16 h). Save aliquots from each culture and prepare 40% (v/v) glycerol (see the Table of Materials) stocks if desired.
- Per overnight culture, inoculate a 1 L flask containing 500 mL of LB Amp CamR medium to an optical density at 600 nm (OD600) of 0.05-0.1.
NOTE: This should return the cells to logarithmic growth. - Measure the OD600 at several time points, typically for 3-4 h, until it falls between 0.4 and 0.6.
- Before induction, aliquot a fraction of culture into a 1.5 mL microfuge tube (see the Table of Materials) and label it Un-induced Fraction. Store it at -80 °C for gel analysis. If not running an SDS-PAGE gel, skip this step and proceed to step 1.2.5.
- Induce the cultures to a final concentration of 1 mM using 1 M isopropyl-ß-D-thiogalactopyranoside (IPTG) stock (see the Table of Materials). Continue to incubate in the 37 °C shaker for an additional 3-4 h.
NOTE: During expression, the reader should determine the optimal OD600 at the time of induction and the IPTG concentration. If the yield drops substantially after purification, the imidazole concentration in purification buffers may require adjustment, or the cell pellet may need to be sonicated further. - Before centrifuging the cultures, aliquot a fraction of culture into a second 1.5 mL microfuge tube and label it Induced Fraction. Store it at -80 °C for gel analysis. If not running an SDS-PAGE gel, skip this step and proceed to step 1.2.7.
- Centrifuge the cell culture in 250 mL conical bottles (see the Table of Materials) at maximum speed and 4 °C for 10 min.
- Repeat step 1.2.7 until the cultures are completely pelleted.
NOTE: PAUSE: Cell pellets can be frozen at -80 °C and thawed later for further processing. Otherwise, skip this step and proceed to step 1.3.1.
- Inclusion body extraction and solubilization
NOTE: Prepare fresh 10 M urea, preferably no earlier than one day in advance, stirring thoroughly until dissolved completely. Do not heat or autoclave urea; store it at room temperature.- Resuspend the pellet (from step 1.2.8) in lysis buffer (see the Table of Materials). Per gram of pellet, add 3 mL of lysis buffer and resuspend by vortexing or pipetting. Shake overnight at 4 °C.
- Add 1.25 mL of 10% (w/v) sodium deoxycholate (see the Table of Materials) per 1 L of culture. Shake at room temperature for 30 min at 150 rpm.
- Add 10 µL of DNase I (see the Table of Materials) per 1 L of culture. Shake at room temperature for 30 min at 150 rpm.
- Centrifuge for 10 min at 13,000 × g and 4 °C.
- Set aside a fraction of Lysed MMP for gel analysis. Store it at -80 °C. If not performing gel analysis, proceed to step 1.3.6.
NOTE: After centrifugation, the pellet may be stringy and not compactly packed, making it risky to discard the supernatant. If this is the case, then skip step 1.3.6 and proceed to step 1.3.7. - Discard the supernatant from the centrifuged samples.
NOTE: The protocol can be paused at this point and the cell pellets frozen at -80 °C and thawed later. Otherwise, skip this step and proceed to step 1.3.7. - Resuspend the pellet in 100 mL/L culture of Inclusion Body Buffer (see the Table of Materials) by pipetting up and down.
- During sonication, keep the samples on ice to prevent overheating. Sonicate each sample for 6 cycles of 15 s, output 5, and 50% pulse. Allow 15 s rest periods for cooling between cycles.
NOTE: If necessary, transfer the samples into 50 mL conical tubes for further centrifugation (see the Table of Materials). Centrifuge for 10 min at 13,000 × g and 4 °C. - Set aside a fraction of Sonicated MMP for gel analysis. Store it at -80 °C. If not performing gel analysis, proceed to step 1.3.11.
- Check the pellet. If stringy, repeat steps 1.3.8-1.3.10. If the pellet is compact, discard the supernatant and proceed to step 1.3.12.
NOTE: Sonication in Inclusion Body Buffer can be repeated to recover more protein from the lysed cell debris. However, too much sonication can cause shearing, which harms MMP yield. The protocol can be paused at this stage, and the cell pellets can be frozen at -80 °C and thawed later. - Resuspend each pellet from a 1 L culture in 5 mL of Solubilization Buffer (see the Table of Materials) by pipetting. Incubate for at least 30 min on ice to allow the proteins to solubilize.
- Set aside a fraction of Solubilized MMP for gel analysis. Store it at -80 °C. If not performing gel analysis, proceed to step 1.3.14.
- Centrifuge the cells for 10 min at 13,000 × g and 4 °C. DO NOT DISCARD THE SUPERNATANT.
- If a pellet forms/remains after centrifugation, pour the supernatant into a separate 50 mL conical tube. Resuspend the pellet in another 5 mL of Solubilization Buffer (per 1 L of culture) by pipetting up and down.
- Centrifuge for 10 min at 13,000 × g and 4 °C. DO NOT DISCARD THE SUPERNATANT.
- Repeat steps 1.3.13 and 1.3.14 until a little to no pellet forms after centrifugation or only gray precipitate remains. Pool the supernatants. Discard or store the pellet at -80 °C for additional sonication.
2. MMP purification and refolding
- His-tag (HT) affinity purification
- Per the manufacturer's protocol, fill a gravity-flow column (see the Table of Materials) with well-mixed Ni-NTA resin (see the Table of Materials). Allow the resin to settle and separate from the storage buffer such that a distinct line forms between the two layers.
NOTE: Never allow the resin to dry, as air will penetrate the resin and harm protein yield. In between uses, perform the resin regeneration procedure described in section 2.2. - Allow the storage buffer to drain. Fill the column with two resin-bed volumes of HT Equilibration Buffer.
- Drain the HT Equilibration Buffer and discard. As the column is draining, centrifuge the protein extract at 13,000 × g for 1 min and filter-sterilize using a 0.22 µm filter (see the Table of Materials).
- Swap the waste container for a 50 mL conical tube labeled HT Flowthrough. Add the prepared protein extract to the column.
- Reapply the flowthrough to maximize binding.
- Set aside a fraction of Flowthrough Fraction for gel analysis. Store it at -80 °C. If not performing gel analysis, proceed to step 2.1.7.
- Immediately wash the resin with 15 mL of HT Wash Buffer (see the Table of Materials). Collect the flowthrough in 15 mL conical tubes (see the Table of Materials) labeled HT Wash.
NOTE: Absorbance values at 280 nm (A280) were obtained via spectrophotometry and used along with molecular weight and extinction coefficient, ε, to estimate protein concentrations. For denatured Hisx6-pro-MMP-3cd, the molecular weight is 29.86 kDa, and ε is 34.38 M-1 cm-1. - Blanking against HT Wash Buffer, measure and record the A280. Repeat steps 2.1.7 and 2.1.8 with additional wash fractions. Once the A280 approaches baseline and impurities have been minimized, proceed to step 2.1.9.
- Set aside a fraction of Wash Fraction for gel analysis. Repeat for multiple wash fractions. Store the fractions at -80 °C. If not performing gel analysis, proceed to step 2.1.10.
- Immediately elute His-tagged proteins by adding 5 mL of HT Elution Buffer (see the Table of Materials). Collect the flowthrough as 0.5-1 mL fractions in microfuge tubes labeled HT Elution.
- Set aside a fraction of Elution Fraction for gel analysis. Repeat for multiple elution fractions. Store the fractions at -80 °C. If not performing gel analysis, proceed to step 2.1.12.
- If the A280 is >0.3 mg/mL, dilute the fraction with HT Equilibration Buffer (see the Table of Materials).
NOTE: The eluted fraction must be diluted to an A280 of 0.3 mg/mL or less to prevent precipitation during dialysis. The protocol can be paused here and the pooled fractions frozen at -80 °C and thawed later. Otherwise, skip this step and proceed to step 2.2.1.
- Per the manufacturer's protocol, fill a gravity-flow column (see the Table of Materials) with well-mixed Ni-NTA resin (see the Table of Materials). Allow the resin to settle and separate from the storage buffer such that a distinct line forms between the two layers.
- Resin regeneration
- Wash the resin with ten resin-bed volumes of HT Regeneration Buffer (see the Table of Materials) and ten resin-bed volumes of sterile water.
- Store the resin as a 50% slurry in 20% (v/v) ethanol in water.
3. Protein refolding
NOTE: For smaller volumes, dialysis cassettes can be used at a lower risk of sample loss. Dialysis tubing is required if larger volumes are used (see the Table of Materials).
- Dialysis
NOTE: The optimum protein concentration for dialysis is ~0.3 mg/mL. If significant precipitation occurs during dialysis, reduce the urea concentration gradient between each dialysis using a stepwise dialysis method and add more intermediate steps (e.g., from 6 M to 5 M, and then 5 M to 4 M, rather than skipping the 5 M stage). As freeze-thaw cycles damage the cellular and protein structure, it is vital to minimize pauses in the protocol.- Per the manufacturer's protocol, use the appropriate amount of dialysis tubing according to the volume of Elution Fraction samples.
- Submerge the eluted MMP fractions in dialysis tubing in 1 L of Dialysis Buffer 1 (see the Table of Materials). Stir the tubing and its contents on a magnetic stirrer for no less than 8 h at 4 °C.
- Transfer to 1 L of Dialysis Buffer 2 (see the Table of Materials). Stir the tubing and its contents on a magnetic stirrer for no less than 8 h at 4 °C.
- Transfer to 1 L of Dialysis Buffer 3 (see the Table of Materials). Stir the tubing and its contents on a magnetic stirrer for no less than 8 h at 4 °C.
- Transfer the sample into new 50 mL conical tubes and label them as Dialyzed MMP.
- Examine the tube for any precipitate. If precipitate has formed, centrifuge the sample for 1 min at 13,000 × g and 4 °C.
- Transfer the supernatant into new 15 mL conical tubes and label them as Refolded MMP.
- Set aside a fraction for gel analysis and label it as Refolded MMP. Store it at -80 °C. If not performing gel analysis, proceed to step 3.1.9.
NOTE: If yields are low, the precipitate can be dissolved in HT Equilibration Buffer and steps in section 3.1 repeated with dialysis tubing. If gel analysis is not to be performed or if the protocol must be paused here, freeze the samples at -80 °C and thaw them later. If yields are in the desired range, proceed to step 3.2.1.
- Reconcentration
NOTE: The extinction coefficients for refolded and denatured Hisx6-pro-MMP-3cd are expected to be the same; hence, A280 calculations are not affected.- Reconcentrate the sample up to 0.5 mg/mL. Use a 400 mL stirred cell (see the Table of Materials) to concentrate the sample to 15 mL. To prevent foaming, use a 50 mL reconcentration tube to concentrate further if needed.
NOTE: If a precipitate forms, it can be pelleted and dissolved in HT Equilibration Buffer. Then, repeat sections 3.1 and step 3.2.1. Otherwise, continue to step 3.2.2. - Set aside a fraction for gel analysis and label it as Concentrated MMP.
NOTE: The protocol can be paused here and the samples frozen at -80 °C and thawed later.
- Reconcentrate the sample up to 0.5 mg/mL. Use a 400 mL stirred cell (see the Table of Materials) to concentrate the sample to 15 mL. To prevent foaming, use a 50 mL reconcentration tube to concentrate further if needed.
4. Activation
- 4-Aminophenylmercuric acetate (APMA) activation
NOTE: APMA is highly toxic. Make a fresh stock solution of 20 mM APMA before activation, and always work under a fume hood when using APMA. Discard the APMA waste into its container.- Per 1 mL aliquot of MMP (1 mg/mL), add 50 µL of 20 mM APMA (see the Table of Materials) to reach a final APMA concentration of 1 mM. Incubate overnight at 37 °C.
- If a precipitate forms, centrifuge it at maximum speed for 10 min at 4 °C. Store the supernatant in a 1.5 mL microfuge tube labeled Activated MMP. Discard the precipitate into a container marked for APMA waste.
- Set aside a fraction for gel analysis and label it Activated MMP.
NOTE: The protocol can be paused here and the samples frozen at -80 °C and thawed later. If not performing gel analysis, proceed to step 4.2.1. After activation, the molecular weight and extinction coefficient of MMP-3cd are 19.40 kDa and 28.42 M-1 cm-1, respectively.
- Desalting
- Remove APMA from the activated MMP-3cd sample with a 2 mL desalting column (see the Table of Materials), following the manufacturer's protocol.
- Set aside a fraction for gel analysis and label it Desalted MMP. Store it at -80 °C. If not performing gel analysis, proceed to section 4.3 with the remaining samples.
NOTE: The protocol can be paused here and the samples frozen at -80 °C and thawed later.
- Running the SDS-PAGE gels
- Run all protein fractions on SDS-PAGE gels: Un-induced Fraction, Induced Fraction, Lysed MMP, Sonicated MMP, Solubilized MMP, Flowthrough Fraction, Wash Fraction, Elution Fraction, Refolded MMP, Concentrated MMP, Activated MMP, and Desalted MMP.
- Long-term storage of MMP-3
- Add 0.05% (v/v) nonionic surfactant (see the Table of Materials) to the desalted MMP-3cd samples and store them at -80 °C.
Results
When running samples on SDS-PAGE, because the protein is expressed in the form of insoluble inclusion bodies, the lysed and sonicated fractions should contain little to no Hisx6-pro-MMP-3cd extract, as the protein has not yet been resolubilized in urea. Figure 3 compares the His-tag purification elution fractions of Hisx6-pro-MMP-3cd from BL21(DE3) cells and R2DP cells. Elution fractions were pooled separately for both BL21(DE3) and R2DP cells before dialysis. Fractions from each step were r...
Discussion
The large-scale production of soluble, human, recombinant MMPs remains a challenging task. Mammalian cells can express functional MMPs at high costs and long wait times, whereas E. coli rapidly produce high quantities of MMP inclusion bodies that must be purified and refolded11,16. R2DP cells significantly increase the yield of MMP inclusion bodies, enabling a more cost-effective and productive MMP refolding process. However, E. coli lack the po...
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgements
The authors would like to acknowledge Dr. Evette Radisky and Alexandra Hockla at the Mayo Clinic in Jacksonville, Florida, for providing the pET-3a-pro-MMP-3cd plasmid as the template for cloning the Hisx6pro-MMP-3cd gene, and their comments, along with Dr. Paul Hartley from the Nevada Genomics Center at the University of Nevada, Reno, for DNA sequencing. The authors would also like to thank Cassandra Hergenrader for helping with part of protein expression. M.R.-S. would like to thank the NIH-P20 GM103650-COBRE Integrative Neuroscience grant and the UNR R&D mICRO SEED Grant Award.
Materials
| Name | Company | Catalog Number | Comments |
| 0.22 µm sterile filter | Sigma Aldrich | SLGP033RS | Used to remove some contaminants from the protein extract before purification, and prevent the Ni-NTA column from clogging |
| 1 L Erlenmeyer flasks | Thermo Fisher Scientific | S76106F | n/a |
| 1 L glass bottles | Thermo Fisher Scientific | 06-414-1D | n/a |
| 1.5 mL microfuge tubes | Thermo Fisher Scientific | 02-682-002 | n/a |
| 15 mL conical tubes | Thermo Fisher Scientific | 339650 | n/a |
| 18 G, 1-in. beveled needle | Amazon | B07S7VBHM2 | Used in combination with the dialysis casette |
| 2 mL desalting column | Thermo Fisher Scientific | 89890 | Removes APMA following activation |
| 2-(N-Morpholino)ethanesulfonic acid (MES) | Thermo Fisher Scientific | AAA1610422 | n/a |
| 250 mL conical bottle cushions | Thermo Fisher Scientific | 05-538-53A | Stabilize conical bottles during large-volume centrifugation |
| 250 mL conical bottles | Thermo Fisher Scientific | 05-538-53 | n/a |
| 400 mL stirred cell | Sigma Aldrich | UFSC40001 | Re-concentrates a much larger volume than the centrifugal filter unit. Rosetta2(DE3)pLysS cells produce high volumes of protein that may exceed the 15 mL limit of the centrifugal filter unit |
| 4-aminophenylmercuric acetate (APMA) | Sigma Aldrich | A9563-5G | Activates MMP-3 by cleaving the propeptide |
| 5 mL syringe | Thermo Fisher Scientific | NC0829167 | Used in combination with the dialysis casette |
| 50 mL conical tubes | Thermo Fisher Scientific | 339650 | Used for storage in many purification steps |
| 50 mL re-concentration tube | Sigma Aldrich | UFC901024D | Used for re-concentrating protein samples after dialysis or removing contaminants |
| Agar | Thermo Fisher Scientific | BP1423-500 | Buffer ingredient that solidifies autoclaved LB media upon cooling |
| Ampicillin | Thermo Fisher Scientific | BP1760-25 | Antibiotic used with pET3a vector; used at 100 µg/mL in LB media |
| BamHI | NEB | R3136S | Restriction enzyme to be used with the pET3a vector |
| Calcium chloride (CaCl2) | Thermo Fisher Scientific | 600-30-23 | The calcium ion stabilizes MMP structure |
| Cell spreaders | Thermo Fisher Scientific | 50-189-7544 | Can be used to spread cells across a petri dish after transformation |
| Chloramphenicol | Thermo Fisher Scientific | 22-055-125GM | Antibiotic used with pET3a vector; used at 34 µg/mL in LB media |
| Dialysis Buffer 1 | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 mM CaCl2, 1 µM ZnCl2, 4 M Urea. |
| Dialysis Buffer 2 | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 mM CaCl2, 1 µM ZnCl2, 2 M Urea. |
| Dialysis Buffer 3 | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 mM CaCl2 , 1 µM ZnCl2. |
| Dialysis clips | Thermo Fisher Scientific | 68011 | Used in combination with snakeskin dialysis tubing |
| Dialysis tubing | Thermo Fisher Scientific | 88243 | Alternative dialysis method that holds much larger sample volumes, but with higher risk of sample loss |
| Digest buffer | NEB | B7204S | Buffer used in digesting the pET3a vector |
| Disposable cuvettes | Thermo Fisher Scientific | 21-200-257 | Used to measure the bacterial culture OD during growth and expression |
| Dithiothreitol (DTT) | Thermo Fisher Scientific | D107125G | Assists with protein denaturation by reducing any disulfide bonds |
| DNA assembly mix | NEB | E2621S | Used to ligate the Hisx6-pro-MMP-3cd PCR product and digested pET3a vector |
| DNase I | NEB | M0303S | Endonuclease for degrading unfavorable DNA contaminants that could later affect protein purification |
| Ethanol | Thermo Fisher Scientific | A995-4 | n/a |
| Ethylenediaminetetraacetic acid (EDTA) | Thermo Fisher Scientific | J15694-AE | Used in denaturation. Prevents oxidation and subsequent formation of disulfide bonds |
| Gel recovery kit | Promega | A9281 | Isolates and purifies DNA from agarose gels |
| Glycerol | Thermo Fisher Scientific | G33-500 | Used for making glycerol stocks, which are frozen at -80 °C |
| Gravity flow column | BioRad | 7321010 | Used for Ni-NTA purification of recombinantly His-tagged proteins |
| High-transformation efficiency cells | NEB | C2987 | High-transformation efficiency cells with greater chance of success for cloning the N-terminal His-tag into the pET3a-pro-MMP-3cd construct |
| HT Elution Buffer | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 6 M urea, 250 mM imidazole. Adjust pH to 7.4 |
| HT Equilibration Buffer | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 6 M urea. Adjust pH to 7.4 |
| HT Regeneration Buffer | n/a | n/a | 20 mM MES, 0.1 M NaCl. Adjust pH to 5.0 |
| HT Wash Buffer | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 6 M urea, 25 mM imidazole. Adjust pH to 7.4 |
| Hydrochloric acid (HCl) | Thermo Fisher Scientific | A144C-212 | Used to pH buffers |
| Imidazole | Thermo Fisher Scientific | AAA1022122 | Mimics the histidine side group. Used to separate non-specifically binding proteins from the his-tagged target protein |
| Inclusion Body Buffer | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 1 mM EDTA, 100 mM NaCl, 5 mM DTT, 2% v/v Triton X 100, 0.5 M Urea. Adjust pH to 8.0 |
| Isopropyl-ß-D-thiogalactopyranoside (IPTG) | Thermo Fisher Scientific | FERR0392 | A reagent that induces target gene expression in pET3a. Make 0.5 mL 1 M aliquots, filter sterilize and store in -20 °C |
| LB Amp CamR media | n/a | n/a | To be poured into a sterible 1 L bottle or 1 L flask. For 1 L, add 25 g LB Broth. Sterilize by autoclaving. Once cooled to below 50 °C, add ampicillin to 100 µg/mL and chloramphenicol to 34 µg/mL |
| LB Amp CamR plates | n/a | n/a | To be poured into sterile petri dishes. Pour until the petri dish lid is completely covered. 1 L of media yields 40-60 plates. For 1 L: 25 g LB Broth, 16 g Agar. Sterilize by autoclaving. Once cooled to below 50 °C, add ampicillin to 100 µg/mL and chloramphenicol to 34 µg/mL |
| LB Broth | Thermo Fisher Scientific | BP1426-2 | Pre-mixed with tryptone, yeast extract, and sodium chloride |
| Lysis Buffer | n/a | n/a | 50 mM Tris-HCl (pH 8.0), 1 mM EDTA, 100 mM NaCl, 0.133 g/mL lysozyme, 0.49% v/v Triton X-100. Adjust pH to 8.0 |
| Lysozyme | MP Biomedicals | 195303 | Used in protein extraction. Enzyme that lyses bacterial cell walls |
| Miniprep kit | Promega | A1330 | If successful, extracts the pET3a-pro-MMP-3cd construct from transformants |
| NdeI | NEB | R0111S | Restriction enzyme to be used with the pET3a vector |
| Ni-NTA resin | Thermo Fisher Scientific | PI88221 | Used to bind recombinant his-tagged proteins. This strong interaction can be displaced with higher concentrations of imidazole |
| Nonionic surfactant | Thermo Fisher Scientific | PI28316 | Storage detergent for preventing MMP aggregation. Minimizes interactions between hydrophobic residues on the MMP surface and water molecules, without disrupting catalytic activity. |
| PCR mix | NEB | M0492S | A PCR reagent for inserting an N-terminal his-tag into the pET3a-pro-MMP-3cd vector |
| pET plasmid | Addgene | n/a | The pET3a vector offers ampicillin resistance, inducible expression of a target gene, and sequencing with T7 primers |
| Petri dishes | VWR | 25384-342 | Used for plating transformants on LB agar media |
| R2DP cells | Novagen | 714033 | BL21 derivatives with enhanced expression of eukaryotic proteins. Contain tRNAs of codons found to be rare in e. coli |
| SOC growth media | NEB | B9020S | Non-selective growth media for rapid growth during transformation |
| Sodium chloride (NaCl) | Thermo Fisher Scientific | BP358-1 | Used in buffers and helps with protein stability |
| Sodium deoxycholate | Thermo Fisher Scientific | PI89905 | Detergent used in protein extraction. Lyses cell walls |
| Solubilization Buffer | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 10 mM DTT, 6 M Urea. Adjust pH to 8.0 |
| Tris base | Thermo Fisher Scientific | BP152-1 | Common buffer used in the physiological pH range. Temperature-sensitive |
| Triton X-100 | Thermo Fisher Scientific | M1122980101 | Detergent used for cell lysis |
| Urea | Thermo Fisher Scientific | AAJ75826A7 | First chaotropic agent for disrupting protein secondary structure |
| Zinc chloride (ZnCl2) | Thermo Fisher Scientific | AAA162810E | Stabilizes MMP structure. The zinc ion is found in the catalytic site of MMP-3 |
References
- Portolano, N., et al. Recombinant protein expression for structural biology in HEK 293F suspension cells: A novel and accessible approach. Journal of Visualized Experiments: JoVE. (92), e51897 (2014).
- Subedi, G. P., Johnson, R. W., Moniz, H. A., Moremen, K. W., Barb, A. High yield expression of recombinant human proteins with the transient transfection of HEK293 cells in suspension. Journal of Visualized Experiments: JoVE. (106), e53568 (2015).
- Nilvebrant, J., Alm, T., Hober, S. Orthogonal protein purification facilitated by a small bispecific affinity tag. Journal of Visualized Experiments: JoVE. (59), e3370 (2012).
- Yang, Z., et al. Highly efficient production of soluble proteins from insoluble inclusion bodies by a two-step-denaturing and refolding method. PLoS One. 6 (7), 22981 (2011).
- Hu, X., Beeton, C. Detection of functional matrix metalloproteinases by zymography. Journal of Visualized Experiments: JoVE. (45), e2445 (2010).
- Radisky, E. S., Raeeszadeh-Sarmazdeh, M., Radisky, D. C. Therapeutic potential of matrix metalloproteinase inhibition in breast cancer. Journal of Cellular Biochemistry. 118 (11), 3531-3548 (2017).
- Raeeszadeh-Sarmazdeh, M., Do, L. D., Hritz, B. G. Metalloproteinases and their inhibitors: Potential for the development of new therapeutics. Cells. 9 (5), 1313 (2020).
- Nagase, H., Visse, R., Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovascular Research. 69 (3), 562-573 (2006).
- Raeeszadeh-Sarmazdeh, M., et al. Directed evolution of the metalloproteinase inhibitor TIMP-1 reveals that its N- and C-terminal domains cooperate in matrix metalloproteinase recognition. Journal of Biological Chemistry. 294 (24), 9476-9488 (2019).
- Batra, J., et al. Matrix metalloproteinase-10 (MMP-10) interaction with tissue inhibitors of metalloproteinases TIMP-1 and TIMP-2. Journal of Biological Chemistry. 287 (19), 15935-15946 (2012).
- Singh, K. K., Jain, R., Ramanan, H., Saini, D. K., Galea, C. A. Expression and purification of matrix metalloproteinases in Escherichia coli. Matrix Metalloproteases. , 3-16 (2017).
- Manka, S. W., et al. Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proceedings of the National Academy of Sciences of the United States of America. 109 (31), 12461-12466 (2012).
- Gomis-Ruth, F. X., et al. Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature. 389 (6646), 77-81 (1997).
- Shirian, J., et al. Converting a broad matrix metalloproteinase family inhibitor into a specific inhibitor of MMP-9 and MMP-14. FEBS Letters. 592 (7), 1122-1134 (2018).
- Li, C., et al. Purification of recombinant histidine-tagged catalytic domain of MMP-13 in one step using affinity column and renaturation of it with histidine tag. Journal of Liquid Chromatography & Related Technologies. 37 (15), 2118-2130 (2014).
- Aydin, H., Azimi, F. C., Cook, J. D., Lee, J. E. A convenient and general expression platform for the production of secreted proteins from human cells. Journal of Visualized Experiments: JoVE. (65), e4041 (2012).
- McNiff, M. L., Haynes, E. P., Dixit, N., Gao, F. P., Laurence, J. S. Thioredoxin fusion construct enables high-yield production of soluble, active matrix metalloproteinase-8 (MMP-8) in Escherichia coli. Protein Expression and Purification. 122, 64-71 (2016).
- Maity, R., et al. GST-His purification: A two-step affinity purification protocol yielding full-length purified proteins. Journal of Visualized Experiments: JoVE. (80), e50320 (2013).
- Stefan, A., Ceccarelli, A., Conte, E., Montón Silva, A., Hochkoeppler, A. The multifaceted benefits of protein co-expression in Escherichia coli. Journal of Visualized Experiments: JoVE. (96), e52431 (2015).
- Yadavalli, R., Sam-Yellowe, T. HeLa based cell free expression systems for expression of Plasmodium rhoptry proteins. Journal of Visualized Experiments: JoVE. (100), e52772 (2015).
- Zeytuni, N., Zarivach, R. Purification of the M. magneticum strain AMB-1 magnetosome associated protein MamAΔ41. Journal of Visualized Experiments: JoVE. (37), e1844 (2010).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved