Method Article
Cryo-Electron Tomography Remote Data Collection and Subtomogram Averaging
* These authors contributed equally
In This Article
Summary
The present protocol describes high-resolution cryo-electron tomography remote data acquisition using Tomo5 and subsequent data processing and subtomogram averaging using emClarity. Apoferritin is used as an example to illustrate detailed step-by-step processes to achieve a cryo-ET structure at 2.86 Å resolution.
Abstract
Cryo-electron tomography (cryo-ET) has been gaining momentum in recent years, especially since the introduction of direct electron detectors, improved automated acquisition strategies, preparative techniques that expand the possibilities of what the electron microscope can image at high-resolution using cryo-ET and new subtomogram averaging software. Additionally, data acquisition has become increasingly streamlined, making it more accessible to many users. The SARS-CoV-2 pandemic has further accelerated remote cryo-electron microscopy (cryo-EM) data collection, especially for single-particle cryo-EM, in many facilities globally, providing uninterrupted user access to state-of-the-art instruments during the pandemic. With the recent advances in Tomo5 (software for 3D electron tomography), remote cryo-ET data collection has become robust and easy to handle from anywhere in the world. This article aims to provide a detailed walk-through, starting from the data collection setup in the tomography software for the process of a (remote) cryo-ET data collection session with detailed troubleshooting. The (remote) data collection protocol is further complemented with the workflow for structure determination at near-atomic resolution by subtomogram averaging with emClarity, using apoferritin as an example.
Introduction
Cryogenic electron microscopy (cryo-EM) is widely known to have experienced a renaissance period, accelerating it to become a core and centrally useful tool in structural biology. The development and utilization of direct electron detectors1,2,3, improved microscopes and electron sources3,4,5, improvements in automation/throughput6,7,8,9, and computational advances in single-particle analysis10,11,12,13,14 and tomography15,16,17 are all, in part, responsible for the recent success of the technique. These technological drivers have developed cryo-EM's capability to solve biological macromolecular structures under cryogenic and native conditions. The resolutions that are readily obtainable are sufficient for atomically accurate modeling and have brought the technique to the forefront of the structural biology arena. A reductionist approach to expressing and purifying a biological target of interest has long proven successful in macromolecular crystallography (MX) for basic biological research, drug discovery, and translational science. In the same approach, cryo-EM can now deliver results that parallel high-resolution MX studies. The current major success in the cryo-EM branch of structural biology is called single particle analysis (SPA), which acquires 2D projection images typically of a purified protein specimen18 to obtain thousands of views of a biological macromolecule19. These images (1) contain information from a range of views that fully represent the orientations of the target in 3D space and (2) capture the object conformational heterogeneity, which can later be separated and investigated.
An alternative approach to acquiring these 2D projection images of biological samples, even in situ and without purification, is cryo-electron tomography (cryo-ET). Cryo-ET takes a series of images of the same object at tilted angles by mechanically rotating the specimen. Thus, the 2D projections collected in SPA, representing the angular poses of the molecule of interest, are inherently collected as part of the cryo-ET imaging experiment20. Tomographic tilt series are then reconstructed into a tomogram that contains 3D representations of the imaged macromolecular complexes. The nature of tomographic data collection does, to a degree, decrease the reliance on averaging to achieve a full 3D representation of a molecule from a collection of 2D images. However, due to current stage designs, the specimen is typically tilted from −60° to +60°, leaving a missing wedge21 of information in the tomographic 3D reconstruction.
The 3D reconstructions in a single tomogram then have a missing wedge of information and low signal to noise. Individual macromolecules may be extracted as subtomograms and averaged together to tackle this. Where each macromolecule in a subtomogram is found at a different orientation, the missing wedge is oriented differently in each subtomogram of the target object, so averaging over many copies fills in information due to the missing wedge. Recent developments in image processing have also attempted to train artificial intelligence neural networks to fill in the missing wedge with meaningful data22. This averaging process also increases the signal to noise, akin to the goal of averaging in single particle analysis, so the reconstruction's quality and resolution improve. If the molecule of interest possesses symmetry, that too may be defined and employed during averaging, further improving reconstruction resolution. The extraction of 3D volumes of a macromolecule from a tomogram into a set of subtomograms and their subsequent processing is known as subtomogram averaging (STA)23. Where each subtomogram represents a unique copy of the molecule being studied, any structural heterogeneity may be interrogated using the STA workflow. As commonly utilized in the SPA workflow, classification techniques may be employed during STA to dissect the conformational states of the complex of interest. As well as STA enabling high-resolution reconstruction in cryo-ET, this approach makes the technique a powerful tool to interrogate the structural mechanisms of macromolecules in their native cellular environment or of targets often not amenable to SPA24,25,26.
Electron tomography has a long history of determining the 3D ultrastructure of cellular specimens at room temperature27. The acquisition of views by physical tilting of the specimen provides enough information for the 3D reconstruction of an object at cellular-length scales and is particularly important when cellular structures lack the regularity for averaging. Cells may also be frozen onto substrates for cryo-ET imaging at the cell edges where the specimen is thin enough to be electron transparent. Under these conditions, STA may be employed to determine macromolecular structures in a cellular environment, albeit when the specimen is thin enough to be electron transparent28. However, when combined with additional preparative techniques, including cryo-correlative light and electron microscopy (cryo-CLEM) and focused ion beam milling (cryo-FIB), cryo-ET can be used to image inside whole cells under cryogenic conditions29. This brings together the power of cryo-ET to study the cellular ultrastructure with the power of STA to determine the structures of macromolecular complexes in situ while identifying their cellular location30 and providing snapshots of complexes engaged in dynamic processes31. The ability of the technique to image cellular specimens and employ STA in several studies has highlighted the power of the technique to solve macromolecular structures in situ, even at resolutions comparable to SPA32. A further benefit is found in the knowledge of the original location of the macromolecule, represented by the final classified 3D reconstruction in the tomogram30. Therefore, the macromolecular structure can be correlated with the cellular ultrastructure. These observations across length scales will presumably lead to important findings where structural mechanisms may be correlated with cellular changes in the context of functional studies.
Cryo-ET and STA allow data collection in three major workflows: molecular, cellular, and lamella tomography. The structures of purified macromolecular complexes may be determined by cryo-ET by molecular tomography. Determining protein structures in their cellular environment where the cell is thin enough may be described as cellular tomography. More recently, with the development of cryogenic targeting and milling, these same techniques may be applied in lamella tomography workflows to determine the protein structures deep inside the cell in their native environment while revealing the cellular context in which those proteins are observed. Different data collection strategies can be used depending on the available software packages and, most importantly, depending on the requirement of the specimen. Molecular or non-adherent samples on a copper TEM grid of a purified protein typically require less handling and, thus, remain flat and undamaged in ideal cases. Electron tomograms can easily be set up in series across a holey-carbon grid to quickly acquire tens to hundreds of tomograms in a systematic manner. The simplest way for users to set up molecular tomography samples where proteins are abundantly present on the grid would be to use Tomo5 (software for 3D electron tomography used in the present study, see Table of Materials). Other tomography software such as Leginon9 and serialEM6 are also available; they offer more setup options for more personalized approaches for data collection but are more complex and consequently can be harder to navigate, particularly for users new to tomography and users accessing their session remotely. For a facility with a large and diverse user base, Tomo5 is easy to operate in a remote environment and to train users in. For adherent cells, grids typically require more handling steps, and the necessity to use fragile gold grids increases the need for improved care in handling and data collection strategies. To facilitate finding a cellular region of interest and avoid occlusion from the grid itself at high tilt angles, it is also beneficial to use larger mesh sizes, but at the cost that they are inherently more fragile. For lamella samples, the fragility of the sample is determined by the quality of the lamella, which can be variable. These factors increase the setup time and considerations, but the increased adaptability and robustness again make Tomo5 suitable for this type of data collection. However, specialized data collection scenarios exist for each workflow. BISECT and PACE-tomo (both run in SerialEM) introduces the possibility of scripted beam-image shifting during tomography acquisition to increase tomogram collection speed28, particularly in molecular tomography. Medium magnification montages (MMM) in SerialEM6,7,33 can better identify and precisely target molecular features in all workflows, although, at the time of writing, these features are beginning to be implemented in Tomo5.
Like SPA, cryo-ET and STA are becoming increasingly accessible through the improvements made to acquisition software and a wealth of available packages for subtomogram averaging16,17,32,34,35,36,37,38. In addition, during the pandemic, enabling remote access to cryo-EM instrumentation became essential to the continued operation of national facilities like the electron Bio-Imaging Centre (eBIC) at Diamond Light Source (DLS), UK. These developments have made cryo-ET more accessible and robust for researchers wishing to utilize the technique. Once data have been acquired, STA is an essential tool for analyzing recurrent objects to obtain maximum resolution reconstruction and allow the classification of macromolecular heterogeneity. The current protocol aims to provide a detailed walk-through of preparing a cryo-TEM microscope for cryo-ET data collection and how to perform subtomogram averaging using emClarity on a molecular tomography dataset of apoferritin as an example. The use of emClarity (software for high-resolution cryo-electron tomography and subtomogram averaging, see Table of Materials) requires running scripts from the command line, so a level of familiarity with Linux/UNIX systems is assumed.
The remote connection depends on the network environment in each institute/facility. At eBIC, the remote system uses programs that allow remote data collection on the specific network configuration used at Diamond. Remote connection to the microscope is facilitated by two platforms: NoMachine and TeamViewer (see Table of Materials). Using the program NoMachine, the user may log onto a remote Windows desktop. The remote Windows desktop provided by NoMachine resides on the same network as the microscope and, thus, acts as a virtual support PC to the microscope. From the virtual support PC, the user connects to the microscope via TeamViewer providing direct access and control to the microscope PC running TUI and Tomo.
The present protocol consists of two parts (step 1 and step 2). Step 1 focuses on remote cryo-ET data acquisition using Tomo5 (software for 3D electron tomography). The walk-through for a (remote) session captures images at increasingly higher magnifications to ultimately allow the user to direct the tomography software to target specimen areas for tomographic data collection. Figure 1 summarizes this process. Step 2 details cryo-ET STA data processing using emClarity (software for high-resolution cryo-electron tomography and subtomogram averaging). Figure 9 summarizes this process.
The protocol is intended for a remote audience. It assumes the person physically at the microscope and loading the samples has done the direct alignments and taken care of the camera tuning and gain reference acquisition. For this protocol, a three-condenser lens system with an autoloader is assumed. For further detailed guidelines on the tomography software, a detailed manual by the manufacturer is available in the Windows Start button where the software was loaded from.
Protocol
The software packages used in this study are partly freely available (see Table of Materials).
1. Remote cryo-ET data acquisition using Tomo5
- If the software is not loaded, begin by starting this software from the TEM server PC (see Table of Materials).
- Perform initial checks and configure the imaging condition by selecting Presets.
- Start the session setup by adjusting the image acquisition parameters in the "Preparation" tab under Presets (Figure 2A,B).
- Adjust the "Overview magnification" to suit the grid-square size and dose to get adequate counts.
NOTE: If the suitable magnification is unknown for the grid type, return to this step after loading a grid. - "Eucentric Height" and "Search Magnification" can be the same. Click on Search in the "Tomography" tab to set up positions and ensure the magnification is sufficient to show the hallmark of interest and to fit the "Exposure" and the "Tracking/Focus" area in the field of view (Figure 2C).
- Set "Exposure Magnification" to the desired target pixel size. If this is unknown, establish it by adjusting the magnification to the desired field of view to fit the region of interest.
- Adjust the "Overview magnification" to suit the grid-square size and dose to get adequate counts.
- Copy the parameters of image acquisition (Exposure) to all the other high-magnification presets (Tracking, Drift, Focus, Thon Ring, Zero Loss,). To do so, press Set to set "Exposure" to the microscope and Get to get the "Exposure Settings" to all other high magnifications after selecting them individually.
- Adjust the exposure times, especially for "Focus" and "Tracking" to give an appropriate number of counts, also considering the thickness at 60°, meaning both at 0° and 60°, for enough electrons to pass through the sample to provide a strong signal for cross-correlation.
NOTE: As an estimate, exposure times for "Tracking" and "Focus" equal to the "Exposure" preset are a good first measure. For a dose calculation example for the "Exposure" preset, see Figure 3. Set the calculated exposure time for the "Exposure" preset in the "Preparation" tab.
- Adjust the exposure times, especially for "Focus" and "Tracking" to give an appropriate number of counts, also considering the thickness at 60°, meaning both at 0° and 60°, for enough electrons to pass through the sample to provide a strong signal for cross-correlation.
- Adjust the "Zero Loss Preset" to use a brighter and larger spot size (a larger spot size corresponds to a smaller number), as this needs a higher dose.
NOTE: This preset is best used to align the slit of the energy filter if present on the microscope.
- Start the session setup by adjusting the image acquisition parameters in the "Preparation" tab under Presets (Figure 2A,B).
- Perform atlas collection following the steps below.
- To collect an atlas, click on New Session in the "Atlas" tab, set the session preferences, enter the storage path and the output format, and press Apply. Select Screening (Figure 4) and then tick all the atlases to be acquired. Select Close Col Valves to leave the microscope unsupervised; this will close the column valves after all the selected atlases are collected. To start the screening, press the Start button.
- Inspect the single or multiple atlases for targets by clicking on the Grid in the left panel under "Screening" (Figure 4), then left-click and drag the mouse to move around and middle scroll to zoom in and out. Choose grid for target setup, select it, and click on Load Sample from inside the software.
- For the image shift calibrations, continue using the atlas screening tab to navigate within the loaded grid. Find an identifiable feature that will be recognizable at all magnification presets, i.e., an ice crystal overlapping with an edge of a hole or another recognizable feature (Figure 5). Move to that square by a right mouse click, then "Move stage to Grid Square".
NOTE: The stage needs to be at eucentric height to implement image shift calibrations correctly.
- Perform image shift calibration.
- In the "Auto Functions" tab (Figure 6), set the preset to "Eucentric Height", navigate to "Auto-Eucentric by Stage Tilt", and press Start. Monitor the status window to ensure finding the eucentric height is successful and keep an eye on the positive and negative tilt images; they must correlate.
NOTE: From version 5.8 of the Tomo software, the criterion for eucentric height acceptance can be modified; the default is 0.25 µm, and setting it to 0.5-0.8 µm gives more leeway. Values depend on the microscope performance, but it is recommended for them to be as small as possible.- When at eucentric height and in focus, center the stage on a feature. Collect a preview using the "Atlas" preset. Set all "Presets" from the "Preparation" tab. Subsequently, increase the magnification to Overview > centre feature > Preview, then repeat "Search Magnification" and, finally," Exposure Magnification".
- If the feature remained centered during targeting, skip the image shift calibrations. Otherwise, to calibrate the image shift, go to the "Preparation" tab, select Calibrate Image Shift, and press Start (Figure 5). This will iteratively align lower magnifications to the feature centered at exposure magnification.
NOTE: If the initial preset "Exposure" is not centered at this stage, for recentering, the software will use stage shift for this step only.- Press Proceed for the "Exposure" preset. The next image shown will be the "Search preset". To calibrate the image shift between the presets, double left-click in the "Search" preview to where the center of the "Exposure" preview is, then press Re-acquire (Figure 5). Click on Proceed to move to the next pair of presets.
NOTE: If an image shift at atlas magnification was applied during this calibration, ensure to re-acquire the atlas after the image shift calibration has been completed. Select the desired atlas and press Reset Selected in the top panel in the "Atlas" tab. Confirm and start acquiring that atlas again.
- Press Proceed for the "Exposure" preset. The next image shown will be the "Search preset". To calibrate the image shift between the presets, double left-click in the "Search" preview to where the center of the "Exposure" preview is, then press Re-acquire (Figure 5). Click on Proceed to move to the next pair of presets.
- In the "Auto Functions" tab (Figure 6), set the preset to "Eucentric Height", navigate to "Auto-Eucentric by Stage Tilt", and press Start. Monitor the status window to ensure finding the eucentric height is successful and keep an eye on the positive and negative tilt images; they must correlate.
- Perform the tomography setup.
- Create the tomography data collection setup in the "Tomography" tab.
NOTE: Unless specifically stated, the setup is done wholly in the "Tomography" tab. - Start a New Session. In "Session Setup" for biological samples, choose Slab-like as the sample type and select Batch and Low Dose, select the output format and storage folder, optionally add an email recipient, and then press Apply.
NOTE: A "Batch Positions" menu item now becomes available (Figure 7A). The currently loaded atlas automatically gets imported. "Overview" and "Search images" can be acquired and revisited until a new image is acquired. Additionally, from version 5.8, search maps of 3 x 3, 4 x 4, and 5 x 5 tiles can be acquired with "Acquire Search Map" to ease target finding and setup. They correspond to a version of medium magnification montages. - To set up targets, go to the "Atlas arrow", find a region of interest, and move there by selecting the options that pop up with a right mouse click. Take an overview image to confirm a good position for eucentric height adjustment, then press on Auto Eucentric; this will run the eucentric by stage tilt routine. Then, re-acquire a new overview image to update the eucentric height.
NOTE: The "Auto Focus" button should not be necessary if the position is at eucentric height. If the eucentric height image appears far out of focus, the eucentric height is probably not correct and has to be redone (for eucentric height troubleshooting, see the discussion section). - Check the target using the "Overview" preset before setting up the first position; select the "Overview" preset and tilt the stage to ±60° to check which distance from the grid bars the positions can be set up (Figure 8). Perform this by entering the desired tilt angle value into the "Set Tilt" window (Figure 8A).
- Inspect the square "Overview" or the acquired "Search Map", move to a region of interest, and press Acquire Search. Inspect the "Search" image. If the region of interest is not centered, right-click at the desired position and then repeat Move Stage Here and Acquire Image. Set Tilt (°) to 0.00 (or any other starting angle that the stage can handle) to define the starting angle of the tomogram.
- For the first tomogram, enter the Name for the desired tomogram naming convention and the Defocus to set the desired defocus value for each tomogram.
- Optionally, from Tomo version 5.8 onwards, the defocus values can be systematically changed all in one go; for that, click on Select Positions, select the positions to be changed or tick the checkmark to select all positions, and then click on Update Defocus and adjust the parameters (Figure 7A).
- Adjust the "Focus" and "Tracking" area (Figure 2C). Left-click to drag the tracking and focus areas (yellow and blue circles). Ensure the tracking/focus area is (mostly) on carbon or another suitable tracking feature, i.e., a similar feature to the region of interest; avoid cracks, ice contamination, overly thick regions, and empty holes.
- When choosing blank carbon without many tracking features, ensure that the area starts burning halfway through the tomogram by choosing a high enough dose for focus, and tracking to slowly burn the sample at higher tilts. This can be beneficial to maintain the tracking accuracy. Ensure the tracking/focus area does not expose a later acquisition area.
NOTE: Be cautious of what is close and what will come into the beam. Avoid areas with features that will intervene with tracking and/or exposure, including grid bars.
- When choosing blank carbon without many tracking features, ensure that the area starts burning halfway through the tomogram by choosing a high enough dose for focus, and tracking to slowly burn the sample at higher tilts. This can be beneficial to maintain the tracking accuracy. Ensure the tracking/focus area does not expose a later acquisition area.
- Press Add Position once all the parameters are set and the desired region of interest and "Focus" and "Tracking" are defined. Repeat for new targets.
NOTE: To check the eucentric height is correctly calibrated for the batch positions that have been set up, there are several available strategies. Choosing one based on the type of tomography session performed (molecular, cellular, lamella) is recommended. - For molecular tomography, assuming the grids are fairly flat and eucentric height has been done in the center of each square containing target positions, skip the "Refine All" (or Refine Selected if positions have been selected, Figure 7A) procedure. If Tomo struggles with the auto-eucentric by stage tilt routine, possibly click on Skip Eucentric.
- For cellular or lamella samples, each batch position may be at a different z-height. Either use "Refine All" to be cautious or do not tick the "Skip Eucentric" option.
NOTE: "Refine all" will iterate through all the tomograms that have been set up or selected via "Select Positions"; refine the eucentric height and perform tracking and focus before data acquisition. This procedure can expose any overlapping exposure from the next tomogram focus/tracking region (Figure 7B), which should be considered before proceeding. - Check and revisit the squares that have failed refinement. With the right mouse button, click on the failed position to see options. If eucentric height fails, find "Auto-eucentric by stage-tilt" (step 1.4.1.), acquire a new "Search" image to update the z-height, and "Add Position". Delete the failed/previously initialized position. Click on the Skip Eucentric option, which has been done with "Refine all".
NOTE: If "Refine All" is not run, then "Skip Eucentric" must not be checked. Tomo5 will run eucentric refinement before each tomogram is acquired; in this way, positions that fail eucentric refinement will be skipped.
- For cellular or lamella samples, each batch position may be at a different z-height. Either use "Refine All" to be cautious or do not tick the "Skip Eucentric" option.
- Create the tomography data collection setup in the "Tomography" tab.
- Perform auto functions following the steps below.
- Check the settings to perform the alignments via the "Auto Functions" tab by navigating in the atlas to an area of carbon. Bring that area to eucentric height and follow the order of alignments as stated below.
- Run Auto-eucentric by stage tilt with "Eucentric Height" preset.
- Run the Autofocus routine with the "Focus" preset.
- Run the Autostigmate routine with the "Thon Ring" preset.
- Run the Autocoma routine with the "Thon Ring" preset.
- Insert the desired objective aperture (Figure 6B), likely the 100 µm aperture, as a good compromise for increased sample contrast and only a small cut-off in signal at very high resolution.
- Repeat the Autostigmate routine after aperture insertion.
NOTE: At magnifications with pixel sizes around 3 Å and lower resolutions, the Autocoma routine might fail; in this case, check and align Tomo Rotation Center under the Direct Alignments in the TEM user interface.
- Check the Auto Zero-Loss centering routine works on your sample. With the "Zero Loss" preset at a reasonably high dose (step 1.2.3), the routine in Auto Functions will more likely succeed.
- Check the settings to perform the alignments via the "Auto Functions" tab by navigating in the atlas to an area of carbon. Bring that area to eucentric height and follow the order of alignments as stated below.
- Perform data collection.
- To start the automated acquisition in the "Tomography" tab, select the Data Acquisition slab-out and set the desired parameters (Figure 7C). Set up data acquisition parameters: Tilt Step (°), Max. Positive Angle (°), Max. Negative Angle (°), tracking scheme (recommended: select Track/Focus Before Acquisition). Select Close Column Valves for eucentric height scheme.
NOTE: Rarely used parameters are "Adjust Exposure Time", "Adjust ZLP", "Use Phase Plate", and "Pause After Tilt".- Use the Adjust Exposure Time for tomograms that are not intended for STA to increase the exposure time at a specified ratio at higher tilts.
- The Adjust ZLP option will run a ZLP refinement after each tomogram, no matter the periodicity set. It is slow, occasionally fails without obvious reasons, and will skip that position acquisition. However, it can be useful for very narrow slit width, i.e., 3-5 eV on a Selectris(X) filter.
- Use Phase Plate is used rarely since the introduction of DEDs with energy filters.
- Pause After Tilt does pause the acquisition but does not allow any changes in the software.
- Double-check the data collection parameters (in the "Preparation" tab), agree with the dose calculation and desired tilt scheme, specify the number of Fractions (Nr.) in the "Exposure" preset, and then begin acquisition.
NOTE: The number of fractions depends on the sample, the acquisition parameters, and the planned post-processing steps, i.e., STA vs. morphological analysis, and must be kept to values where motion correction and CTF estimation get enough signal. A range of 4-10 fractions is a good estimate, where 4 is more suitable for thicker samples and 10 for thinner molecular samples.- Then, click on Start to commence data collection and monitor the first tomogram acquisition to ensure the tomograms are acquired as intended.
NOTE: Recent tomography software versions have an additional Movie Player slab-out in the "Tomography" tab where the acquired tomograms can be watched. Be patient during the loading process.
- Then, click on Start to commence data collection and monitor the first tomogram acquisition to ensure the tomograms are acquired as intended.
- To start the automated acquisition in the "Tomography" tab, select the Data Acquisition slab-out and set the desired parameters (Figure 7C). Set up data acquisition parameters: Tilt Step (°), Max. Positive Angle (°), Max. Negative Angle (°), tracking scheme (recommended: select Track/Focus Before Acquisition). Select Close Column Valves for eucentric height scheme.
2. Cryo-ET STA of apoferritin using emClarity
NOTE: Here, emClarity software17 is used to illustrate cryo-ET structure determination by STA. Figure 9 summarizes this process. Six tilt series of apoferritin (EMPIAR-10787) were taken as an example. Octahedral symmetry was applied, and the final map had a resolution of 2.86 Å, obtained from only 4,800 particles and close to the Nyquist frequency (2.68 Å).
- Ensure that the emClarity software package39 is downloaded and installed (see Table of Materials). Install IMOD for the software processing40.
NOTE: Basic knowledge of Linux commands is required. A more detailed tutorial can be found in reference41, which takes a ribosome dataset (EMPIAR-10304) as an example and describes the step-by-step procedures. For different types of protein samples, a previously published report is also available42. - Prepare the input files and directories.
NOTE: The raw frames were motion-corrected by MotionCor2 (patch 5 x 5)43 (see Table of Materials). The motion-corrected images were stacked to generate the tilt-series using newstack (IMOD) and manually aligned with patch tracking using Etomo (see Table of Materials), followed by emClarity. To get better alignments, it is recommended to carry out offsets and tilt compensation in Etomo. The six tilt series are renamed as TS1 to TS6. Below are the instructions and commands for the first tilt series (TS1) as an example.- Establish a project folder.
NOTE: The latest version of the software is 1.5.3.11. All the related software logs can be found in the local project folder (.../logFile/emClarity.logfile). - Under the project folder, make another new folder, "fixedStacks", and prepare the input files: TS1.fixed, TS1.xf, and TS1.tlt.
NOTE: TS1.fixed: the original tilt-series, also known as TS1.st in Etomo. TS1.xf: this file contains the transformation coordinates after Etomo tiltalign. TS1.tlt: this file contains tilt-angles.
- Establish a project folder.
- Estimate the defocus following the steps below.
- Calculate the defocus. Update the parameter file with the microscope and imaging parameters following Supplementary Table 1. Copy a parameter file to the project folder, change its name to param_ctf.m, and run: emClarity ctf estimate param_ctf.m TS1.
NOTE: The parameter file template can be found in the Supplementary File 1 or the local software installation directory (…/emClarity_1.5.3.11/docs/exampleParametersAndRunScript/).
- Calculate the defocus. Update the parameter file with the microscope and imaging parameters following Supplementary Table 1. Copy a parameter file to the project folder, change its name to param_ctf.m, and run: emClarity ctf estimate param_ctf.m TS1.
- Check the CTF estimation results for each stack.
- Check the aligned stacks (for example, aliStacks/TS1_ali1.fixed) using 3dmod and ensure that the fiducial beads are erased correctly.
- Ensure that the logfile reports the handedness is correct, which can be found in logfile/emClarity.logfile (Figure 9).
- Check the defocus value (for example, fixedStacks/ctf/TS_ali1_psRadial_1.pdf), and ensure it matches the theoretical CTF estimate.
- Define the sub-regions.
- Generate a binned tomogram. In the project folder, run: sh recScript2.sh -1.
NOTE: A smaller (in size) tomogram for each stack will be stored in a new folder "bin10". To speed up the process, the coordinates of one or several sub-regions can be defined in a bin10 tomogram. The script file will be in the local software installation directory (…/emClarity_1.5.3.11/docs/). - Determine the boundaries by choosing six points, xmin, xmax, ymin, ymax, zmin, and zmax, to create one sub-region. Under the folder "bin10", run: 3dmod TS1_bin10.rec.
NOTE: If four sub-regions are to be created in one tilt-series, choose 6 × 4 = 24 points. Each stack must have one model file (*.mod) under the folder "bin10". - Convert the model files to the emClarity format. This will create a new folder recon. Store all the coordinates of each sub-region under this folder. In the project folder, run: sh recScript2.sh TS1.
- Generate a binned tomogram. In the project folder, run: sh recScript2.sh -1.
- Pick particles.
- Find an apoferritin template for picking particles. Download a template from the Electron Microscopy Data Bank (EMD-10101)44. Ensure that the pixel size of the template matches that of the raw data. In the project folder, run: emClarity rescale ApoF.mrc ApoF_Template_rescale.mrc 3.60 1.34 cpu.
- Generate CTF-corrected tomograms for particle picking. In the project folder, run: emClarity ctf 3d param_ts.m templateSearch.
- Run particle picking for each sub-region. For the apoferritin dataset, carry out a template search at bin6. Modify the Tmp_angleSearch parameters (θout, Δout, θin, Δin) to determine the angle range and intervals for in-plane or out-of-plane search in degrees. In the project folder, run: emClarity templateSearch param_ts.m TS1 1 ApoF_Template_rescale.mrc O 1.
NOTE: A folder "convmap_wedge_Type2_bin6" will be generated in this step. The csv file under this folder (for example, TS1_1_bin6.csv) includes all the information about the particles picked. - Remove the incorrect particles using 3dmod. Under the folder "convmap_wedge_Type2_bin6", run: 3dmod ../cache/TS1_1_bin6.rec TS1_1_bin6.mod.
NOTE: It is common to find wrong particles next to carbon edges or ice contaminations in those areas. Perform a right mouse click on the incorrect points and press backspace on the keyboard to delete them. In this step, ensure that most of the false positive points are removed (Figure 10). - Change the folder name "convmap_wedge_Type2_bin6" to "convmap", as emClarity will go and find sub-region information under convmap in the following steps.
- Initialize the project. In the project folder, run: emClarity init param0.m, to create a database for emClarity, ApoF.mat.
- Perform tomograms reconstruction before STA and alignment. To generate CTF-corrected sub-region tomograms at bin4, run: emClarity ctf 3d param0.m.
NOTE: CTF-corrected tomograms (for example, cache/TS1_1_bin4.rec) will be generated in a new folder "cache". - Perform STA and alignment.
- Carry out the averaging using the CTF-corrected sub-tomograms starting at bin4. In the project folder, run: emClarity avg param0.m 0 RawAlignment.
- Continue to do alignments and run: emClarity alignRaw param0.m 0.
NOTE: The averaging step generates a reference, which will be used by emClarity in this step to align the particles. The Raw_angleSearch parameters (θout, Δout, θin, Δin) can be changed to set the angle range and step sizes for each cycle. As most incorrect particles are removed from the database (Step 2.6.4), these angular settings for alignment can start with a relatively small degree. - Update the Raw_angleSearch parameters and run a few more cycles (Steps 2.9.1 and 2.9.2). To speed up the process, perform in-plane and out-of-plane alignments separately.
NOTE: For each binning, it is recommended to run multiple cycles and gradually reduce the angular settings. For the ApoF dataset, five more cycles at bin 4 were carried out, and further details is available in Supplementary Table 2. For cycle001, in the project folder, run: emClarity avg param1.m 1 RawAlignment, followed by emClarity alignRaw param1.m 1. - Clean the overlapped particles. In the project folder, run: emClarity removeDuplicates param5.m 5.
- Perform Tilt-series refinement with tomoCPR.
NOTE: This step is optional.- Run tomoCPR to refine the stack geometry. In the project folder, run: emClarity tomoCPR param5.m 5.
- Generate newly aligned stacks and update the geometry files. In the project folder, run: emClarity ctf update param6.m.
NOTE: Ensure that the new geometry files (for example, fixedStacks/ctf/TS1_ali2_ctf.tlt) and the newly aligned stacks (for example, aliStacks/TS1_ali2.fixed) are correctly generated. - Create new sub-tomograms at bin2. In the project folder, run: emClarity ctf 3d param6.m.
NOTE: This will be followed by a few cycles for averaging and alignment at bin2 (steps 2.9.1, 2.9.2, and 2.9.3), duplicate cleaning (step 2.9.4), and optional tomoCPR (step 2.10). Due to high symmetry, another five cycles at bin2 were carried out for apoferritin. Update the Raw_angleSearch parameters for each cycle.
- Perform final reconstruction.
- Continue to run a few cycles at bin1 and tomoCPR (Step 2.9 and Step 2.10). Another 10 more cycles at bin1 were carried out for the apoferritin dataset. More details regarding the commands and parameters can be found in Table 2.
- Perform final reconstruction by combining the two half datasets. In the project folder, run: emClarity avg param21.m 21 RawAlignment, followed by emClarity avg param21.m 21 FinalAlignment.
NOTE: The final map (for example, with a B-factor of 10, cycle021_ApoF_class0_final_bFact-10.mrc) will be generated, Figure 11.
Results
For cellular and lamella samples, the data collection strategy largely depends on the sample and the goal of the imaging study (Figure 1). The targeting approach depends on whether the molecular target is in situ or prepared from the purified macromolecular complex for high-resolution reconstruction specimens containing molecular targets. For purified complexes vitrified onto holey (carbon) grids, targeting can simply be based on imaging in the holes of the (carbon) support film. For in situ work, the targeting approach requires knowledge of the location of the molecular entity based on correlative data or known low-magnification cellular landmarks. Cellular landmarks would ideally be identifiable when taking overview images and, if sufficient to roughly localize regions of interest, can provide a quick way to confirm target identity with a search image. However, if the observable events are rare, then medium magnification search images may be needed to qualify the target is correct. Search maps are medium magnification montages of search images and can, thus, make target finding much easier, where they can be acquired at a magnification at which the feature of interest is visible. Search maps can then be screened to find and set up target batch positions. For specimens containing cellular features for ultrastructural reconstruction, the targeting approach is similar, though equally dependent on the visibility of the cellular event at various magnifications and its prevalence in the sample.
The data acquisition strategy must also be considered; in all cases, the study's goal largely determines how the data is collected. For reconstruction of the cellular ultrastructure, a low magnification (20-5 Å/px) and large field of view may be appropriate, but high magnifications are required to reconstruct molecular or high-resolution detail (5-1 Å/px). A dataset collected at 1.5 Å/px under ideal conditions will only physically be able to produce a reconstruction at the Nyquist frequency of 3.0 Å/px; however, in reality, many factors, including but not limited to specimen thickness, size, and heterogeneity, all affect the obtained reconstruction quality. The exact imaging parameters also balance the magnification based on the study's goal with the field of view to contain enough information. This article presents a subtomogram averaging case reaching 2.86 Å, but additional studies17,42,43,44,45 are presented in Table 1 to illustrate the collection parameters associated with studies targeting different outcomes17,42,45,46.
Once a targeting workflow and data acquisition regime for cryo-ET is established, data collection of many different sample types is possible. Representative tomograms of a variety of specimens are presented here: molecular samples such as apoferritin (Movie 1), thin cellular processes (Movie 2), and FIB milled lamella of the thick cellular specimen (Movie 3).

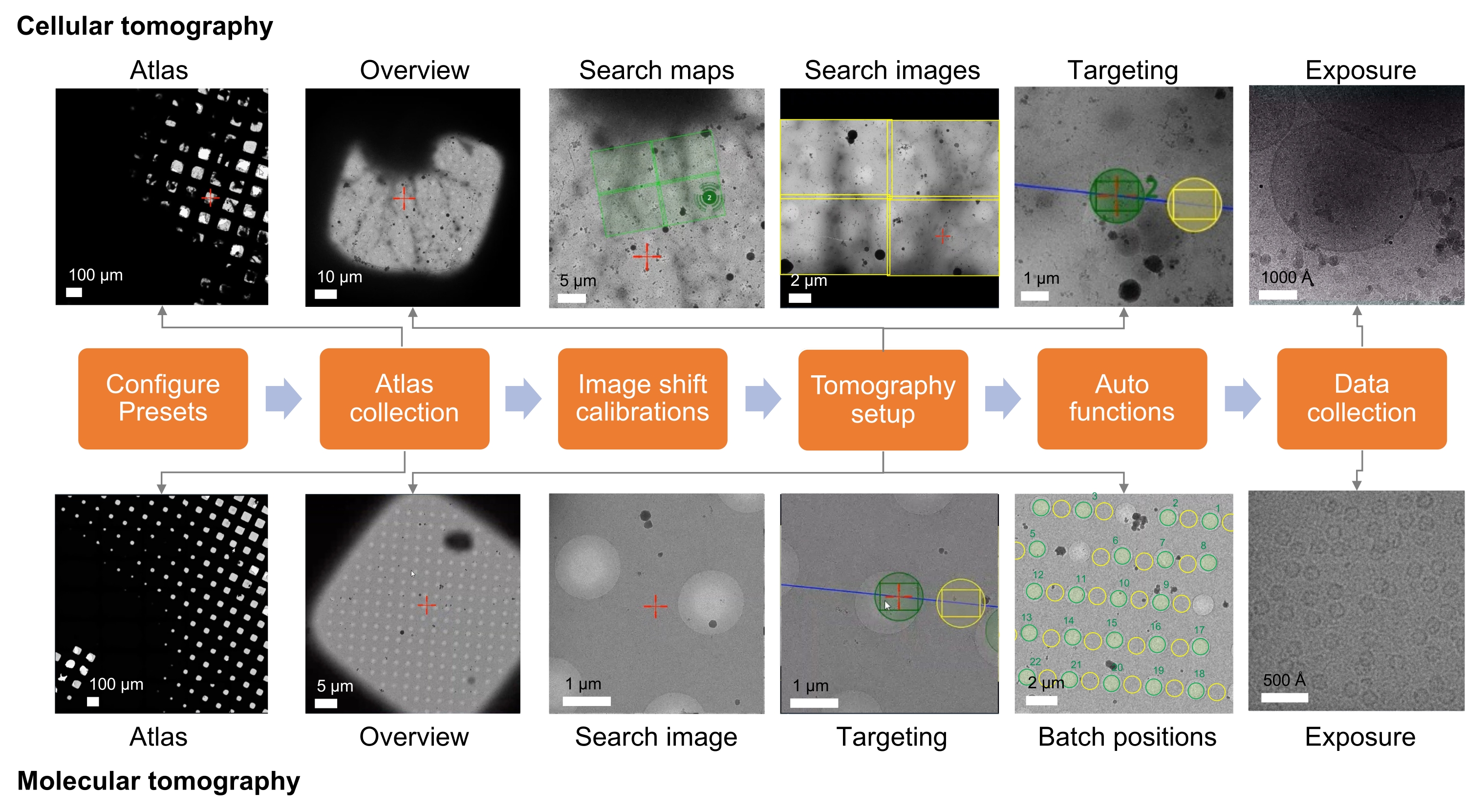
Figure 1: Overview of the tomography workflow setup. The cryo-ET imaging workflow described in the protocol is shown as a flow chart. The images expected to be acquired are shown for the cellular and molecular workflow. The presented naming follows the Tomo5 convention, although most tomography acquisition software shares common principles for collecting these images. Please click here to view a larger version of this figure.
{kind=link}

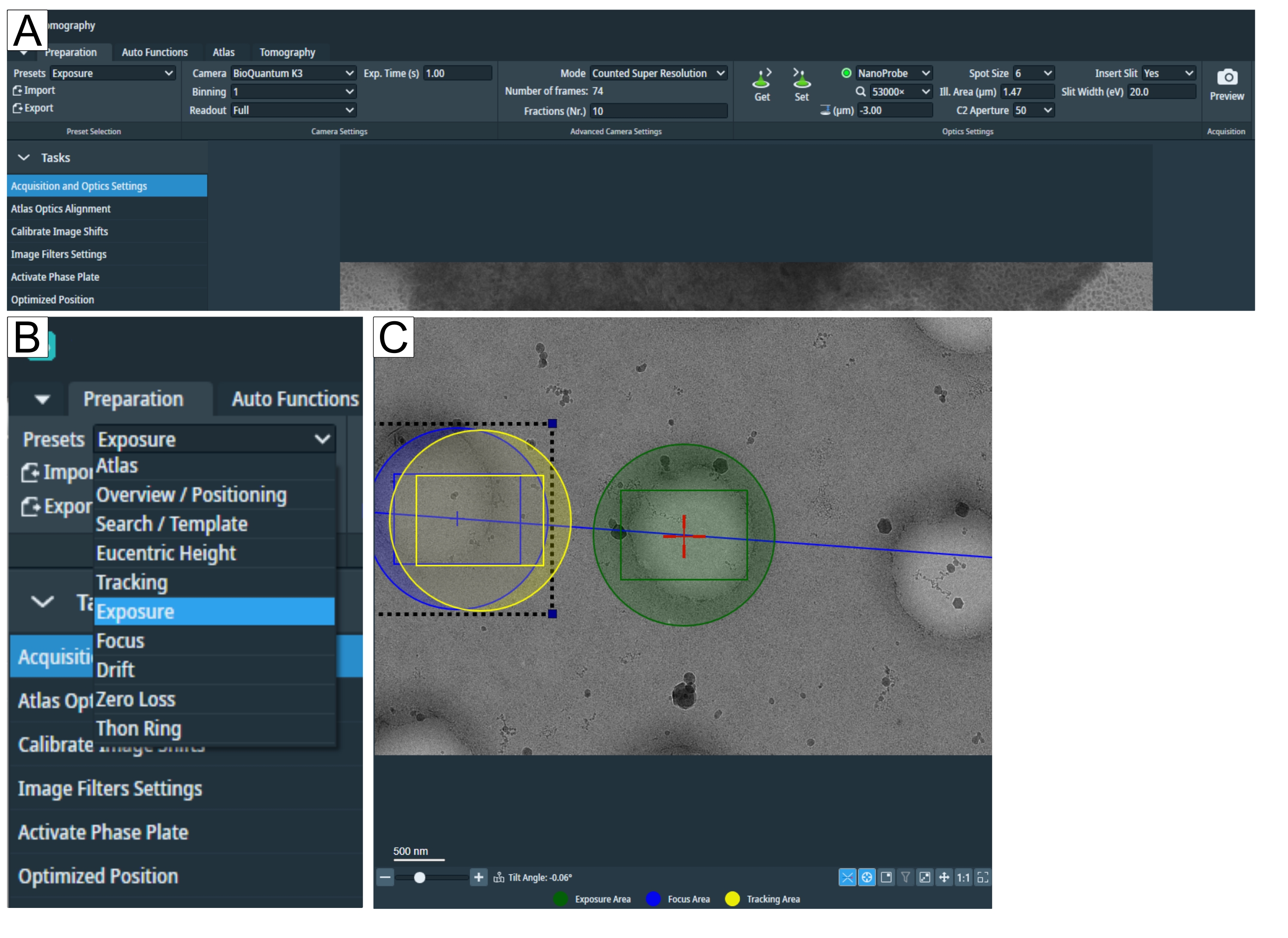
Figure 2: Preparation tab and search preset conditions. (A) Overview image of the whole tab. Imaging condition presets are set in this tab, and the "Calibrate Image Shift" and "Image Filter Settings" can be found in the "Tasks" drop-down. (B) Zoom-in of the "Presets" drop-down where each preset can be selected for individual imaging conditions setup. (C) Image depicting an adequate magnification to fit both the exposure and the "Focus and Tracking" area into the field of view. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Dose calculations. Example dose calculations for possible tomogram acquisition schemes where the dose rate has been measured over vacuum. The two calculations determine the exposure time (s) for each tilt, whether targeting an optimal dose per tilt or an optimal total dose for the full tilt series. In subtomogram averaging, it is common practice to target an optimal dose per tilted image in the range of 3.0-3.5 e-/Å2. In both cases, the "Fractions (Nr.)" are set to 6 to achieve ~0.5 e-/Å2 of dose per movie frame of the tilt for sufficient signal to perform motion correction. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Atlas tab. Overview image of the "Atlas" tab. The image has been cropped to avoid empty grid position spaces. The "Tasks" menu contains session setup preferences and grid selection spaces for individual selection of all grids inventoried and an option to acquire a single atlas after the cassette has been removed from the autoloader. A selected grid can be reset and then re-acquired. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Calibrated image shifts. Image depicts an exposure and search image. The red cross in the search image is the shifted marker to correct the offset between exposure and search. One should redo image shift calibrations at the session start or after changing the imaging condition presets. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Auto functions and apertures. (A) The "Auto Functions" tab depicts the "Presets" drop-down and the "Task" selection. Underlined in blue is the "Thon Ring" preset required for "Autostigmate" (also underlined in blue) and "Autocoma". One should select respective presets for each task and then press the Start button. (B) Apertures are found in the TEM User Interface. Select desired "Objective aperture" after the auto functions have been performed and run "Autostigmate" with the aperture in. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Tomography tab overview. Images show the Tomo 5.8 user interface. (A) Batch positions. The latest functions depicted in the image: the "Acquire Search Map" option; tomogram positions are shown in atlas view with the zoom-in option; highlighted at the top right is "Select Positions"; below all four positions have been selected to "Update Defocus" parameters. (B) Three positions are selected on the search map, labeled 1, 2, and 3. Position 1 will not pose a problem when "Refine All" is run. However, if in a high target density setting such as when lamellae positions are selected, as depicted for position 2 and position 3, then "Refine All" will run the "Tracking" and "Focus" routine on the "Exposure" region of position 2, exposing the target before the tomogram is acquired. The grid type is R1.2/1.3. Scale bar = 1.2 µm. (C) Data acquisition. Selected positions setup in "Batch Positions" can now be individually acquired. Please click here to view a larger version of this figure.
{kind=link}

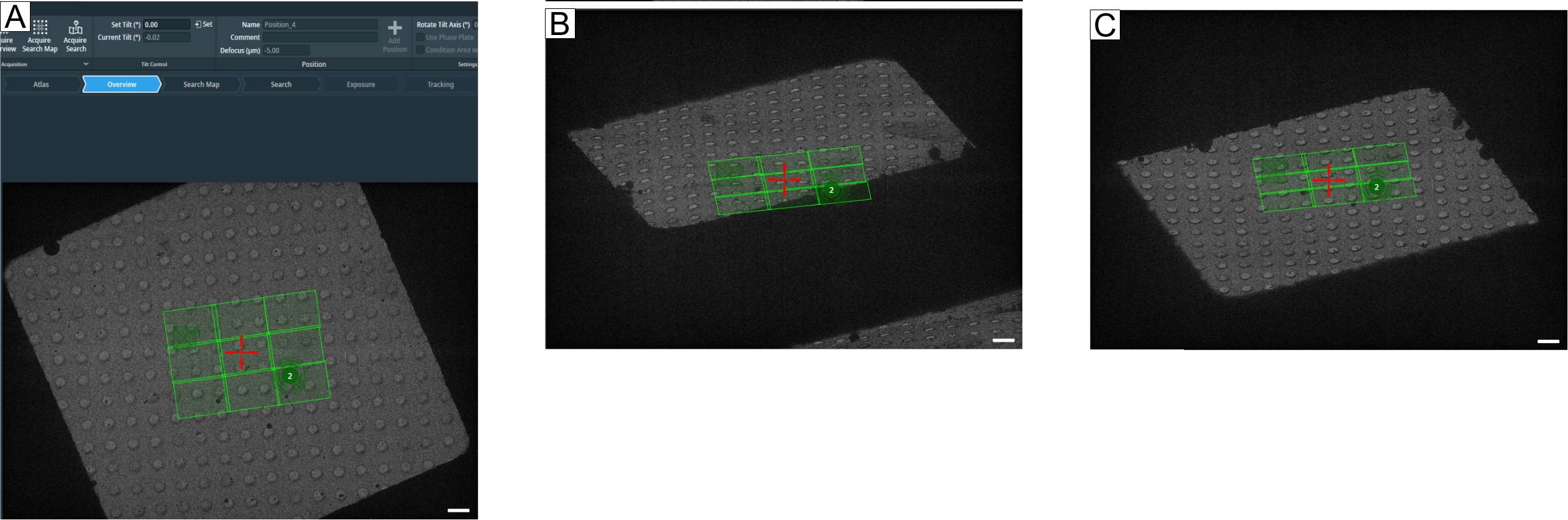
Figure 8: Defining the maximum tilt angle. Figure depicts a step-by-step approach to determining the maximum tilt range for tomogram acquisitions. (A) 0° overview and a search map in the center of the grid square. To test the tilt range, the angle can be set in the tomography software with "Set Tilt (°)" by typing the desired value and then pressing Set. (B) The stage has been tilted to −60°, showing that a corner of the search map would not be fully acquired at −60°. (C) The stage has been tilted to 60°. By counting the holes that have disappeared at ±60°, one can get an idea about the tilt range of a grid square. Scale bars = 2.5 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 9: emClarity flowchart. The flowchart described the various steps for cryo sub-tomogram averaging. Scale bars = 50 nm. Please click here to view a larger version of this figure.
{kind=link}

Figure 10: Template matching using emClarity. (A) A typical micrograph of apoferritin on a graphene-coated grid. Defocus: −3.430 µm. (B) A slice of the tomogram overlaid with model points after template searching. (C) A top and (D) a side projection views of model points, indicating a single flat layer of monodispersed apoferritin particles. Scale bars = 50 nm. Please click here to view a larger version of this figure.
{kind=link}

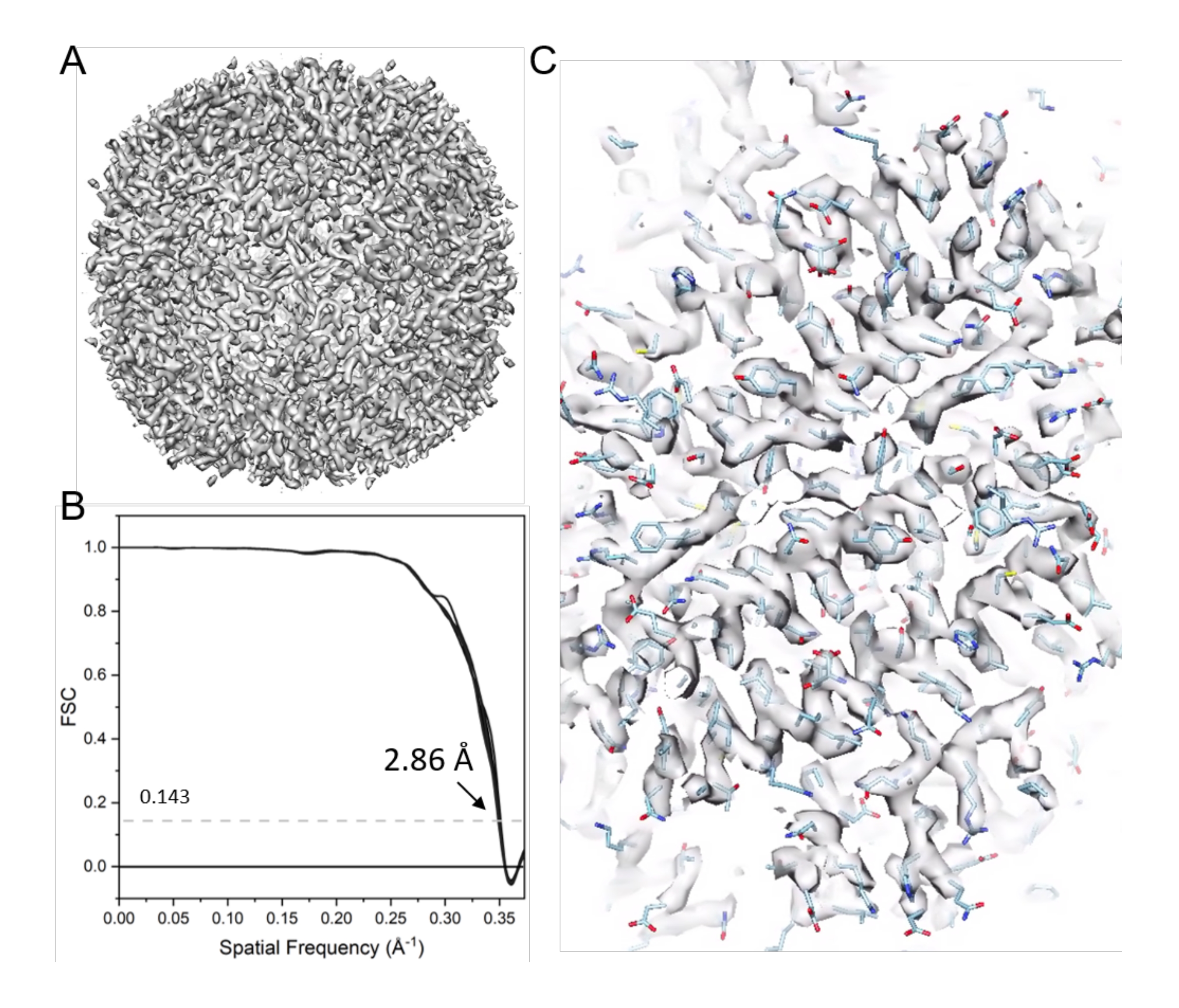
Figure 11: Cryo-ET STA of apoferritin. (A) The final map after 21 cycles of sub-tomogram alignment. (B) The final map's Fourier shell correlation (FSC) plot with a reported resolution of 2.86 Å, containing 38 cone FSCs. (C) Representative density maps (fitted with PDB model 6s6147). Please click here to view a larger version of this figure.
{kind=link}
| Sample | Cryo-ET type | Å/px | Tilt range (+/-) | Tilt step (°) | Defocus range (µm) | Total dose (e-/Å2) | Resolution | Raw data | Reference |
| Apoferritin | Purified/Molecular (STA) | 1.34 | 60 | 3 | 1.5 – 3.5 | 102 | 2.86 | EMPIAR-10787 | 17 & this paper |
| HIV-1 Gag | Purified/Molecular (STA) | 1.35 | 60 | 3 | 1.5 – 3.96 | 120 | 3.1 | EMPIAR-10164 | 17 |
| Ribosome | Purified/Molecular (STA) | 2.1 | 60 | 3 | 2.2 – 4.3 | 120 | 7 | EMPIAR-10304 | 42 |
| SARS-CoV-2 spike | Lamella/Molecular (STA) | 2.13 | 54 | 3 | 2 – 7 | 120 | 16 | EMPIAR-10753 | 45 |
| Neuron axon structure | Cellular (ultrastructural) | 5.46 | 60 | 2 | 3.5 – 5 | 90 | N.D. | EMPIAR-10922 | 47 |
Table 1: Collection parameters for several cryo-ET studies. Studies targeting the reconstruction of molecular detail from purified or in situ proteins compared to a study aiming to resolve and segment the ultrastructural cellular features.
Movie 1: A tomogram of apoferritin samples on a normal EM grid and then imaged with a cryo-TEM equipped with an ultra-high-resolution camera with a compatible filter. Tilt series were acquired with a dose-symmetric scheme, with a tilt span of 54° and a total dose of 134 e-/A2 in the electron tomography software. Scale bar = 50 nm. Please click here to download this Movie.
Movie 2: A tomogram of a primary neuron grown on an EM grid and then directly imaged with a cryo-TEM equipped with an ultra-high-resolution camera with a compatible filter. The tilt series were acquired with a dose-symmetric scheme, with a tilt span of 60° and a total dose of 120 e-/A2 in the electron tomography software. Scale bar = 100 nm. Please click here to download this Movie.
Movie 3: A tomogram of a Cyanobacteria on an EM grid, subjected to FIB milling and then imaged with a cryo-TEM equipped with a high-speed camera and a compatible filter. The tilt series were acquired with a dose-symmetric scheme, with a tilt span of 50° and a total dose of 120 e-/A2 in the electron tomography software. Scale bar = 87.2 nm. Please click here to download this Movie.
Supplementary File 1: The parameter file template for estimating the defocus. Please click here to download this File.
Supplementary Table 1: Data collection and microscope setup details. Please click here to download this Table.
Supplementary Table 2: List of commands in the order of execution. Please click here to download this Table.
Discussion
Tomo5
The workflow description of the tomography software highlights one potential and most streamlined way for a (remote) batch tomography session setup. While the software is easy for beginners, some initial cryo-EM experience and basic tomography understanding can help with the setup. Critical steps are highlighted in the protocol and should help troubleshoot even if a different setup approach has been used. The advancement of the software will ease (remote) data collection and make cryo-ET more accessible to a wide user base. A few tips and tricks that can help troubleshoot commonly encountered problems are described below.
One important point to discuss is the choice of grids because, when tilting the specimen to ±60°, grid bars at high tilts can obscure the view (Figure 8). On a TEM grid, the mesh size refers to the number of grid squares per unit length of the grid. Larger mesh numbers have more grid squares per unit length, a higher density of grid squares, and smaller grid squares, i.e., a 400-mesh grid has smaller squares than a 200-mesh grid. A good choice of grids for tomography is 200-mesh or 300-mesh grids. As shown in Figure 8, the available area to collect is reduced as the grid is tilted. At ±60° tilt, a 300-mesh grid will have a small field of view on which a full tomogram can be acquired. The advantages of 200-mesh grids are that the larger grid squares make molecular tomography setup faster, and with the increased grid-square area, one square will likely be enough for an overnight collection. The disadvantage is that 200-mesh grids are more fragile, so handling and clipping require more finesse.
Moreover, if using holey support film (see Table of Materials) on EM grids, the hole spacing must be considered for the setup of the focus and tracking region in relation to the exposure region. Ideally, the beam diameter at the desired magnification should be small enough to cover the carbon area adjacent to the exposure area along the tilt axis for optimal and fast setup. This way, potential regions of interest in each hole can be acquired.
As the software's eucentric height routine is currently not as robust, such as the serialEM routine, the following tips can work around that problem. If the eucentric height determination fails using the eucentric height preset, one can use the overview preset instead and re-run "Auto-eucentric by stage tilt"; this can solve issues if the eucentric height is far away from 0. If this succeeds, one can re-run "Auto-eucentric by stage tilt" with "Eucentric Height" presets to improve precision. If it fails, one can run "Auto-Eucentric by beam tilt" with the eucentric height preset and then re-run "Auto-Eucentric by stage tilt" or manually set the z-height consolidated by "Auto-Eucentric by beam tilt" in the TEM User Interface under "Stage" settings. In case grids with a repeating pattern of holes are used, they may prevent the identification of a single cross-correlation peak. One can try altering the eucentric height preset to a lower defocus offset such as −25 µm and/or a shorter exposure time to reduce cross-correlation from the hole patterns. On the other hand, using lacey grids/lamella may not provide sufficient signal for a strong cross-correlation peak. One can try altering the eucentric height preset to a greater defocus offset such as −75 µm and/or an extended exposure time to enhance the cross-correlation peak. Another option is to adjust image filter settings; they can be found in the "Preparation" tab. Options to adjust the filter settings can be set for low (Overview/Gridsquare), medium (Eucentric Height), and high magnification (Tracking/Focus) to find the optimal cross-correlation peak for each preset. The required input is one image, i.e., at 0° and one at 5°, followed by clicking Compare to compare both images. Recommended starting value for the longest wavelength is one-quarter of the scale bar in the image and for the shortest wavelength is one-fortieth of the scale bar. If the peak is not robustly identified, one can optimize the settings until a convincing peak can be found. There is no need to re-acquire images every time; simply pressing "Compare" is enough. If TOMO still fails to automatically find the eucentric height, the manual eucentric height calibration can be used. One should center over a reasonably large ice crystal in overview magnification in the "Preparation" tab, then go to the the "Stage control" of the TEM User Interface, set alpha to −30°, and adjust the stage z-value to re-center the crystal using the fluorescent screen image. Selecting "High Resolution" and "High Contrast" settings in the TEM User Interface will make this simple (buttons at the bottom of the fluorescent screen window). Optionally, if there is access to a camera with live mode, then this can be used to determine the eucentric height; it will be easier than on the fluorescent screen.
The biggest limitations in Tomo5 versions prior to 5.8 are the missing medium magnification montages, missing dose symmetric scheme, and problems related to eucentric height finding. These exist in serialEM, a freeware with rapid development and community support, a robust eucentric height routine, and the option to script, i.e., a custom-built dose symmetric scheme. From version 5.8 onwards in Tomo5, the most commonly encountered problem for finding the eucentric height, i.e., an unsuccessful looping around the target z-value, has been solved by implementing the option to set a eucentric height acceptance criterion. However, with different grid and sample types, it is highly recommended to adjust the image filter settings to reflect the unique imaging conditions of individual sessions and to give the best possible cross-correlation peak to find the eucentric height and for the focus and tracking region to work reliably during tomogram acquisition.
Overall, many facilities have rapidly adapted to remote operation during the pandemic. The Tomo5 software provides an easy access and user-friendly route to tomography which is well suited for remote operation. The advances made in the software will no doubt continue to make remote data collections and tomography collection in general more mainstream in the community.
emClarity
As emClarity uses a template-based particle picking method, it needs a template for the object of interest. The particle picking (step 2.6) is very sensitive and key to the final structure. Before averaging and alignment (step 2.9), one must ensure to check carefully and manually remove the false positives. When a template is not available, emClarity might not be easy to use, but it is possible to use other software, for example, Dynamo37 and PEET48, to create an initial model.
For heterogeneous samples, emClarity is equipped with a classification method that enables users to focus on specific features with different scales. It is helpful to run a few cycles of alignments before classification and run it at a higher binning (such as bin 4 or bin 3).
The up-to-date version of the software (V1.5.3.11) has significant updates compared with the first release (V1.0)17. These include, but are not limited to, a handedness check during CTF estimation (step 2.3); symmetry for alignments (CX, I, I2, O); calculation of per-particle 3D sampling functions (3DSF); a switch to MATLAB 2019a for compatibility and stability; and reconstruction using the raw projection images (cisTEM). The software will continue to improve for various samples, and the newest announcements can be found online (see Table of Materials).
Disclosures
The authors have no conflicts of interest.
Acknowledgements
We acknowledge Diamond Light Source for access to and support of the cryo-EM facilities at the UK's national Electron Bio-Imaging Centre (eBIC), funded by the Wellcome Trust, MRC, and BBRSC. We would also like to thank Andrew Howe for the acquisition of the Apoferritin tomogram (Movie 1), Ishika Kumar for the preparation and acquisition of the neuron tomogram (Movie 2), and Craig MacGregor-Chatwin for the Cyanobacteria lamella-tomogram (Movie 3).
Materials
| Name | Company | Catalog Number | Comments |
| Software | |||
| Tomography | Thermo Fisher Scientific | 5.9.0 | Internal terminology: Tomo5 in document |
| TEM server | Thermo Fisher Scientific | 7.10.1 | |
| TIA | Thermo Fisher Scientific | 5.10.1 | |
| DigitalMicrograph | Gatan | 3.44 | |
| emClarity | Open-Source software | 1.5.3.11 | Software for high-resolution cryo-electron tomography and subtomogram averaging |
| IMOD | Open-Source software | 4.11 | Modeling, display and image processing programs used for 3D reconstruction and modeling of microscopy images with a special emphasis on electron microscopy data |
| MotionCor2 | Free for academic use | 1.1.0 | A multi-GPU program that corrects beam-induced sample motion recorded on dose fractionated movie stacks |
| ETomo | Open-Source software | 4.11 | ETomo is an interface for running a subset of IMOD and PEET commands. |
| NoMachine | NoMachine, freeware | 7.9.2 | Remote desktop software |
| TeamViewer | TeamViewer AG | - | Remote access and remote control computer software |
| Materials | |||
| Quantifoil (holey support film) EM grids | Quantifoil | - | A flat film of carbon with pre-defined hole size, shape and arrangement |
| Instrumentation | |||
| Titan Krios microscope | Thermo Fisher Scientific | Titan Krios G2 | |
| K3 camera and GIB energy filter | Gatan | - | |
| Falcon 4 camera and Selectris X energy filter | Thermo Fisher Scientific | - | |
| Website | |||
| Website 1: https://github.com/bHimes/emClarity/ | - | - | Link to download the emClarity software package |
| Website 2: https://bio3d.colorado.edu/imod/ | - | - | Link to download IMOD |
| Website 3: https://github.com/ffyr2w/emClarity-tutorial | - | - | Link to the emClarity online tutorial |
| Website 4: https://emcore.ucsf.edu/ucsf-software | - | - | Link to download MotionCor2 |
| Website 5: https://github-wiki-see.page/m/bHimes/emClarity/wiki | - | - | Link to the newest announcements including updates and bug fixs for emClarity |
References
- Bai, X. -. C., Fernandez, I. S., McMullan, G., Scheres, S. H. W. Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. eLife. 2, 00461 (2013).
- Li, X., et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nature Methods. 10 (6), 584-590 (2013).
- Nakane, T., et al. Single-particle cryo-EM at atomic resolution. Nature. 587 (7832), 152-156 (2020).
- Kato, T., et al. CryoTEM with a cold field emission gun that moves structural biology into a new stage. Microscopy and Microanalysis. 25, 998-999 (2019).
- Yip, K. M., Fischer, N., Paknia, E., Chari, A., Stark, H. Atomic-resolution protein structure determination by cryo-EM. Nature. 587 (7832), 157-161 (2020).
- Mastronarde, D. N. SerialEM: A program for automated tilt series acquisition on Tecnai microscopes using prediction of specimen position. Microscopy and Microanalysis. 9, 1182-1183 (2003).
- Schorb, M., Haberbosch, I., Hagen, W. J. H., Schwab, Y., Mastronarde, D. N. Software tools for automated transmission electron microscopy. Nature Methods. 16 (6), 471-477 (2019).
- Deng, Y., et al. Smart EPU: SPA getting intelligent. Microscopy and Microanalysis. 27 (1), 454-455 (2021).
- Carragher, B., et al. Leginon: An automated system for acquisition of images from vitreous ice specimens. Journal of Structural Biology. 132 (1), 33-45 (2000).
- Bell, J. M., Chen, M., Baldwin, P. R., Ludtke, S. J. High resolution single particle refinement in EMAN2.1. Methods. 100, 25-34 (2016).
- Punjani, A., Rubinstein, J. L., Fleet, D. J., Brubaker, M. A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods. 14 (3), 290-296 (2017).
- Grant, T., Rohou, A., Grigorieff, N. cisTEM, user-friendly software for single-particle image processing. eLife. 7, 35383 (2018).
- Zivanov, J., et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 7, 42166 (2018).
- Kimanius, D., Dong, L., Sharov, G., Nakane, T., Scheres, S. H. W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochemical Journal. 478 (24), 4169-4185 (2021).
- Galaz-Montoya, J. G., Flanagan, J., Schmid, M. F., Ludtke, S. J. Single particle tomography in EMAN2. Journal of Structural Biology. 190 (3), 279-290 (2015).
- Bharat, T. A. M., Scheres, S. H. W. Resolving macromolecular structures from electron cryo-tomography data using subtomogram averaging in RELION. Nature protocols. 11 (11), 2054-2065 (2016).
- Himes, B. A., Zhang, P. emClarity: Software for high-resolution cryo-electron tomography and subtomogram averaging. Nature Methods. 15 (11), 955-961 (2018).
- Weissenberger, G., Henderikx, R. J. M., Peters, P. J. Understanding the invisible hands of sample preparation for cryo-EM. Nature Methods. 18 (5), 463-471 (2021).
- White, J. B. R., et al. Single particle cryo-electron microscopy: From sample to structure. Journal of Visualized Experiments. (171), e62415 (2021).
- Orlova, E. V., Saibil, H. R. Structural analysis of macromolecular assemblies by electron microscopy. Chemical Reviews. 111 (12), 7710-7748 (2011).
- Turk, M., Baumeister, W. The promise and the challenges of cryo-electron tomography. FEBS Letters. 594 (20), 3243-3261 (2020).
- Liu, Y. -. T., et al. Isotropic reconstruction of electron tomograms with deep learning. bioRxiv. , (2021).
- Briggs, J. A. G. Structural biology in situ-The potential of subtomogram averaging. Current Opinion in Structural Biology. 23 (2), 261-267 (2013).
- Grünewald, K., et al. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science. 302 (5649), 1396-1398 (2003).
- Allegretti, M., et al. In-cell architecture of the nuclear pore and snapshots of its turnover. Nature. 586 (7831), 796-800 (2020).
- Wang, Z., et al. The molecular basis for sarcomere organization in vertebrate skeletal muscle. Cell. 184 (8), 2135-2150 (2021).
- Winey, M., Meehl, J. B., O'Toole, E. T., Giddings, T. H. Conventional transmission electron microscopy. Molecular Biology of the Cell. 25 (3), 319-323 (2014).
- Davies, K. M., et al. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proceedings of the National Academy of Sciences. 108 (34), 14121 (2011).
- Wagner, J., Schaffer, M., Fernández-Busnadiego, R. Cryo-electron tomography-The cell biology that came in from the cold. FEBS Letters. 591 (17), 2520-2533 (2017).
- Mahamid, J., et al. Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science. 351 (6276), 969-972 (2016).
- Erdmann, P. S., et al. In situ cryo-electron tomography reveals gradient organization of ribosome biogenesis in intact nucleoli. Nature Communications. 12 (1), 5364 (2021).
- Tegunov, D., Xue, L., Dienemann, C., Cramer, P., Mahamid, J. Multi-particle cryo-EM refinement with M visualizes ribosome-antibiotic complex at 3.5 Å in cells. Nature Methods. 18 (2), 186-193 (2021).
- Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. Journal of Structural Biology. 152 (1), 36-51 (2005).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology. 116 (1), 71-76 (1996).
- Heymann, J. B., Belnap, D. M. Bsoft: Image processing and molecular modeling for electron microscopy. Journal of Structural Biology. 157 (1), 3-18 (2007).
- Tang, G., et al. EMAN2: An extensible image processing suite for electron microscopy. Journal of Structural Biology. 157 (1), 38-46 (2007).
- Castaño-Díez, D., Kudryashev, M., Arheit, M., Stahlberg, H. Dynamo: A flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments. Journal of Structural Biology. 178 (2), 139-151 (2012).
- Hrabe, T., et al. PyTom: A python-based toolbox for localization of macromolecules in cryo-electron tomograms and subtomogram analysis. Journal of Structural Biology. 178 (2), 177-188 (2012).
- Himes, B. A. emClarity. GitHub. , (2021).
- . The IMOD Home Page Available from: https://bio3d.colorado.edu/imod/ (2020)
- . GitHub: emClarity-tutorial Available from: https://github.com/ffyr2w/emClarity-tutorial (2021)
- Ni, T., et al. High-resolution in situ structure determination by cryo-electron tomography and subtomogram averaging using emClarity. Nature Protocols. 17 (2), 421-444 (2022).
- . MotionCor2 Available from: https://emcore.ucsf.edu/ucsf-software (2016)
- . Apoferritin from mouse at 1.84 angstrom resolution Available from: https://www.emdataresource.org/EMD-10101 (2022)
- Nedozralova, H., et al. In situ cryo-electron tomography reveals local cellular machineries for axon branch development. Journal of Cell Biology. 221 (4), 202106086 (2022).
- Mendonça, L., et al. Correlative multi-scale cryo-imaging unveils SARS-CoV-2 assembly and egress. Nature Communications. 12 (1), 4629 (2021).
- . Apoferritin from mouse at 1.84 angstrom resolution Available from: https://www.rcsb.org/structure/6s61 (2019)
- Nicastro, D., et al. The molecular architecture of axonemes revealed by cryoelectron tomography. Science. 313 (5789), 944-948 (2006).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved