
Method Article
A Gut-on-a-Chip Model to Study the Gut Microbiome-Nervous System Axis
In This Article
Summary
neuroHuMiX is an advanced gut-on-a-chip model to study the interactions of bacterial, epithelial, and neuronal cells under proximal and representative co-culture conditions. This model allows unravelling of the molecular mechanisms underlying the communication between the gut microbiome and the nervous system.
Abstract
The human body is colonized by at least the same number of microbial cells as it is composed of human cells, and most of these microorganisms are located in the gut. Though the interplay between the gut microbiome and the host has been extensively studied, how the gut microbiome interacts with the enteric nervous system remains largely unknown. To date, a physiologically representative in vitro model to study gut microbiome-nervous system interactions does not exist.
To fill this gap, we further developed the human-microbial crosstalk (HuMiX) gut-on-chip model by introducing induced pluripotent stem cell-derived enteric neurons into the device. The resulting model, 'neuroHuMiX', allows for the co-culture of bacterial, epithelial, and neuronal cells across microfluidic channels, separated by semi-permeable membranes. Despite separation of the different cell types, the cells can communicate with each other through soluble factors, simultaneously providing an opportunity to study each cell type separately. This setup allows for first insights into how the gut microbiome affects the enteric neuronal cells. This is a critical first step in studying and understanding the human gut microbiome-nervous system axis.
Introduction
The human gut microbiome plays a crucial role in human health and disease. It has been extensively studied over the past decade and a half, and its potential role in modulating health and disease is now established1. A disruption in the microbiome leading to an unbalanced microbial community (dysbiosis) has been postulated to be involved in the pathogenesis of many chronic disorders, such as obesity, inflammatory bowel disease, and colorectal cancer, or even neurodegenerative diseases such as Parkinson's disease2,3.
Although the human gut microbiome has been associated with neurological conditions, it is still unclear how the gut microbiome communicates with and affects the enteric nervous system. As the human enteric nervous system is not easily accessible for immediate study, animal models have been used in experiments so far4. However, given the apparent differences between animal models and humans5, the development of in vitro models mimicking the human gut is of immediate interest. In this context, the burgeoning and advancing field of human induced pluripotent stem cells (iPSCs) has allowed us to obtain representative enteric neurons (ENs)6. iPSC-derived ENs allow for the study of the enteric nervous system in in vitro culture models, such as cell culture inserts, organoids, or organs-on-a-chip7,8.
The human-microbial crosstalk (HuMiX) model is a gut-on-a-chip model mimicking the human gut9. The initial HuMiX model (hereafter referred to as the initial device) accommodated epithelial cells (Caco-2) and bacterial cells10,11. However, to study the gut microbiome-nervous system link, iPSC-derived ENs6 have also been introduced in the system (Figure 1). The proximal co-culture of neuronal, epithelial, and bacterial cells allows the analysis of the different cell types individually and study of the interactions between the different cell types in an environment mimicking that of the human gut.
In recent years, advances have been made in the development of models to study organs in more physiologically representative ways by using organs-on-a-chip (e.g., gut-on-a-chip) models. These models are more representative of the human gut environment due to constant nutrient supply and waste removal, as well as real-time monitoring of, for example, oxygen levels or barrier integrity8,12. These models specifically allow study of the effects of gut bacteria on host cells. However, to be able to use organs-on-a-chip to study the interrelationships between the gut microbiome and the nervous system, neuronal cells need to be integrated into such systems. Therefore, the aim of further developing HuMiX and establishing the neuroHuMiX system (hereafter referred to as the device) was to develop a gut-on-a-chip model, which includes enteric neuronal cells in proximal co-culture with gut epithelial cells and bacteria.
Protocol
1. Cell culture and sorting
- Induced pluripotent stem cell-derived enteric neurons
NOTE: Culture iPSCs 6 weeks prior to the start of a run. The differentiation protocol for the ENs has been adapted from Fattahi et al.6.- Culture the iPSCs on a matrix gel-coated 6-well plate in 2 mL/well of iPSC culture medium supplemented with 1% penicillin-streptomycin (P/S).
- At 80%-90% confluency, passage the cells, seed in a matrix gel-coated plate using the same medium, incubate at 37 °C, 5% CO2, and 90% relative humidity (RH).
- For iPSC derivation, following two passages after thawing, at 80%-90% confluency, seed the cells in a matrix gel-coated 6-well plate at a density of 100,000 cells/well. Incubate in the above-described media + ROCK inhibitor Y-27632 (1:2,000) for 24 h at 37 °C.
- After 24 h, replace the supernatant medium with Day 0 medium, according to Table 1. Include one control well to be used as a negative control during cell sorting for the derivation process. Keep the control well in Day 0 media composition for the whole derivation period. Change the medium every other day from days 2 to 10, according to Table 1.
- On Day 11, sort the cells for CD49d-positive cells, which will be used in the following steps of this protocol.
- Pool the cells to be sorted based on CD49d-positive cells, as well as the control well. Centrifuge the cells for 3 min at 300 × g. Resuspend the cell pellet in 2% bovine serum albumin (BSA) + 1% P/S in 1x phosphate-buffered saline (PBS).
- Divide each cell lot (pooled and controls) in two: stain one fraction with anti-human CD49d antibody, and the other fraction with an isotype control antibody.
- Gate the main cell population and then gate single cells, based on which the cells presenting CD49d on their surface will be selected. Collect these cells for the next step of differentiation.
NOTE: Usually, 30%-40% of the sorted cells are CD49d-positive.
- Transfer two to four million of the sorted cells to one 6-well plate of an ultra-low attachment plate in N2B27 media (see Table 2) + FGF2 + CHIR for 4 days to allow spheroid formation.
- On day 15, replate the spheroids on a poly L-ornithine/laminin/fibronectin (P/L/F)-coated 6-well plate in 2 mL/well of N2B27 media with GDNF and ascorbic acid (AA). Replace the medium every 2-3 days.
NOTE: Coating ratio: poly L-ornithine, 15 µg/mL; laminin, 2 µg/mL; fibronectin, 2 µg/mL in 1x PBS. - After 3 weeks, use the cells for inoculation in the device.
- Caco-2
NOTE: Thaw Caco-2 cells at least 1 week prior to a run.- Seed a T75 flask with 1 × 106 Caco-2 cells in RPMI 1640 glutamine supplement + HEPES + 10% fetal bovine serum (FBS); incubate at 37 °C, 5% CO2, and 90% RH.
NOTE: For these experiments, ideally use Caco-2 at their first or second passage after thawing.
- Seed a T75 flask with 1 × 106 Caco-2 cells in RPMI 1640 glutamine supplement + HEPES + 10% fetal bovine serum (FBS); incubate at 37 °C, 5% CO2, and 90% RH.
2. HuMiX run preparation
- Polycarbonate lid preparation and coating
- Autoclave a pair of polycarbonate (PC) lids (Figure 2) together with four screws using the initial device instrument cycle (see Table 3 for details).
- In a biosafety cabinet, insert four screws in each corner of the bottom PC lid (see Figure 3) and transfer to a sterile square Petri dish for a two-step coating process.
- The first day, add 2 mL of 1.5% poly L-ornithine in PBS (1x) to the middle of the PC lid. Incubate overnight at 37 °C, 5% CO2, and 90% RH.
- The next day, remove the solution and replace it with a solution containing 0.2% laminin and 0.2% fibronectin in PBS (1x) to cover the surface of the neuronal cell chamber. Incubate the lid overnight at 37 °C, 5% CO2, and 90% RH.
- After incubation, keep the coating solution on the PC lid and close the Petri dish with a sealing film. Place them at 4 °C for storage until use.
- Before use, remove the coating solution, and air-dry in a biosafety cabinet for 30 min until the coating is completely dry.
- Gasket preparation and coating
NOTE: This section describes how to prepare gaskets by sticking semipermeable membranes to silicone gaskets. After an initial autoclaving step, the gaskets are coated with collagen or mucin to allow the adhesion of intestinal epithelial cells or bacteria. The mucin-coated membrane mimics the mucus barrier that is present in the human gut.- Attach a 1 µm pore size polycarbonate membrane to the collagen gasket and a 50 nm pore size polycarbonate membrane in between the bottom and top sandwich gaskets. Autoclave the gaskets with the attached membranes using the initial device instrument cycle (see Table 3).
- To coat the membranes, place each gasket in a sterile square dish.
NOTE: The coating is done in sterile conditions in a biosafety cabinet.- For the collagen gasket, add 3 mL of collagen (50 µg/mL) to the membrane and incubate for 3 h at 37 °C.
NOTE: Be careful not to touch the membrane with the pipette tip when adding the collagen to avoid damaging the membrane. - For the mucin coating, place the sandwich gasket with the top gasket side upward in a Petri dish to coat the top side of the membrane with 3 mL of mucin (0.025 mg/mL). Incubate the gaskets at 37 °C for 1 h.
- For the collagen gasket, add 3 mL of collagen (50 µg/mL) to the membrane and incubate for 3 h at 37 °C.
- After the incubation time, aspirate the collagen and mucin solutions from their respective gaskets and air-dry the membrane in a biosafety cabinet for 30 min. Close the dishes, seal with laboratory sealing film, and store the gaskets at 4 °C until use.
- Tubing assembly
NOTE: This section describes how to assemble the tubing for the perfusion of the device. Prior to assembly, autoclave all the pieces and/or buy those that are packed individually in a sterile package. Place all the components in a biosafety cabinet.- Use three tube lines for each device and for each tube line, one piece of pump tubing line, two pieces of long Marprene tubing (20 cm), two pieces of short Marprene tubing (8 cm), one 40 mm needle for the outflow bottle, one 120 mm needle for the 250 mL inflow bottles or 80 mm needle for smaller inflow bottles, three male as well as seven female Luer to barb connectors, three Luer adaptors, and two three-way stopcock valves.
- Assemble tubing lines from left to right using the autoclaved and sterilized components listed above (Figure 4).
NOTE: Spray each component with 70% ethanol between each connection step.
- Media preparation
NOTE: This section describes how to prepare the medium for the different cell types in the device, as well as how to transfer the medium to serum bottles in a sterile manner.- Supplement RPMI 1640 with 10% 0.22 µm filtered, heat-inactivated FBS. Supplement the N2B27 media freshly with GDNF and AA.
- Transfer the medium to serum bottles under sterile conditions. Transfer 200 mL of RPMI 1640 media to 250 mL bottles and 30 mL of N2B27 media to 50 mL bottles.
- Prepare bacterial medium (RPMI 1640 + 10% FBS + 5% De Man, Rogosa, and Shapre culture medium [MRS]) at a later stage, as this is only required during the final 24 h of the experiment.
- Close the bottles with a septum, aluminum crimp them, and autoclave.
- To transfer the media, remove the aluminum crimp using an appropriate decapper (diameter = 20 mm). Remove the septum as well. Carefully pour the prepared medium into the serum bottles without touching the bottle.
- To sterilize, flame the opening of the bottle using a portable Bunsen burner. Seal the bottles by spraying the septum with 70% ethanol, adding it to the bottle, and closing it with an aluminum crimp using a seal crimper.
- Place the closed bottles in the incubator at 37 °C, 5% CO2 for 24 h to warm up the medium to 37 °C, as well as to ensure there are no visible signs of contamination before using media bottles for the device.
3. HuMiX start
- NeuroHuMiX assembly
NOTE: This section explains how to assemble the device. In short, the clamping system is sterilized and retightened, after which the bottom PC lid is placed on the base of the clamp. Then, the coated gaskets and the top PC lids are stacked on each other, followed by the top part of the clamp. Finally, the clamp is tightened to compress the gaskets and render the system leak-free and gastight. Figure 4 depicts the different parts needed for the assembly.- Autoclave the clamps and the two PC lids (top and bottom) prior to assembly.
- Tighten the screws used to assemble the top and bottom parts of the clamp (Figure 4C,D). Open the screws prior to the autoclaving process and retighten them later.
- Transfer the coated and dried (step 2.2.3) bottom PC lid (Figure 4 A1) from the square Petri dish to the top of the clamp basis by holding the four screws in the corners of the PC lid.
NOTE: Avoid touching the PC lid to reduce the risk of contamination. - Place the epithelial chamber gasket facing up (i.e., with the membrane on top) on the PC lid using sterile tweezers. Use the screws to align the gasket and the PC lid to ensure the inlet and outlet ports in the corners of the lid are aligned with the openings in the gasket (Figure 3).
- Using the sterile tweezers, place the sandwich gasket on top of the collagen gasket, with the top side facing up.
- Place the top PC lid on top of the sandwich gasket. To reduce the risk of contamination, do this by only touching the edges of the lid (do not touch the top or bottom part of the lid). Ensure that the barbs on the clamp lid align with the inlet and outlet opening on the membrane assembly, and that the screws from the bottom PC lid fit into the opening of the top PC lid.
- To close the device, place the clamp lid (the top part of the clamp) on top of the PC lid. Ensure that the openings of the lid coincide with the inlet and outlet barbs of the top PC lid. Close the latches. Keep the closed device (Figure 5) under the hood to connect to the primed tubing lines (step 3.2.6).
- Priming of tubing lines
NOTE: This section describes how to connect the tubing to the in- and outflow bottles, as well as to the pump, subsequently prime the tubing with medium to remove any residual products used during cleaning and sterilization, and ensure that no bubbles are present in the tubing.- Priming of the tubing lines is performed in the biosafety cabinet. Insert aeration needles with filters in the septum of each inflow and outflow bottle. Using clean, sterile tweezers, insert the 120 mm needles in the 250 mL serum bottles. Insert the 80 mm needles in the 50 mL serum bottles.
- Insert the 40 mm needles, at the end of each tubing line, to an outflow serum bottle. Per device, there are two outflow bottles for the three tubing lines. The 40 mm needles of the epithelial and neuronal tubing lines are connected to the same outflow bottle for medium discarding. The bacterial tube line goes to the second outflow bottle.
- Insert the pump tubing lines into the pump cassettes. Ensure that the three-way stopcocks of the tubing lines are all open.
- At the beginning, set the peristaltic pump to direct media from the inflow to the outflow bottle at a speed of 5 rpm (see Table 4).
NOTE: During tubing line priming, the speed can be increased up to 10 rpm to accelerate the process. - Start the pump by pressing the start button and make sure the direction of the pumping action is clockwise. Once the media is dropping into the outflow bottles, make sure there are no leaks and no air bubbles are left in the tubing lines and connection points. Remove any remaining bubbles by tapping the tubing and connectors or by increasing the pump speed.
- When all the tube lines are dropping into the outflow bottles, set the flow rate to 2 rpm for connecting the initial device.
- Priming of the device
NOTE: This section describes how to prime the device with cell culture medium to ensure all the chambers are filled and precoated with medium, so that no bubbles are left in the system and the cells can easily adhere during the inoculation.- Priming of the device is performed in the biosafety cabinet under sterile conditions. To connect the device, start by connecting the neuronal line, the bottom chamber of the device.
- To connect a line, close the three-way stopcock valves (Figure 5C), starting first with the outflow side, where the short tubing is disconnected from the female Luer connector and connected to the outlet port from the device, by pushing the tubing over the barb. To ensure proper connection and reduce the chance of having leaks and/or contamination, ensure that the tubing is in contact with the lid by pushing it all the way down over the connector. Connect the inflow tubing the same way. Once one line is completely connected to the device (inflow and outflow), open the three-way stopcocks.
- Repeat the previous step with the epithelial line, and then with the bacterial line. Keep the pump at a flow rate of 2 rpm. Increase the pump speed to 2.5 rpm, but not higher, in order to avoid leakages due to pressure build-up. Allow the pump to prime the chambers in the device.
- Monitor the outflow serum bottles at all times. If air bubbles get stuck in the chambers, lines, or connector, a tubing line with an air bubble inside will not drop anymore in the outflow chamber. To get rid of air bubbles, first let the devices run for a few minutes. If this does not resolve the issue, close one of the other lines that are dropping for a short amount of time by closing the three-way stopcock of the outflow.
- Once all the chambers of the device are fully primed-all chambers are filled with cell culture medium and no bubbles remain in the device-reduce the pump speed to 0.5 rpm. The device is now ready for cell inoculation.
4. Cell preparation and inoculation
NOTE: This section describes how to prepare the different cell types needed to inoculate the device, as well as how to inoculate them in the device in a sterile manner and without introducing air bubbles. Furthermore, it describes how to perform medium refreshment for the neuronal cells, and how to prepare medium for the bacterial culture in the device.
- Epithelial cells
- Detach the Caco-2 cells from the flask using trypsin-ethylenediaminetetraacetic acid (EDTA), resuspend in RPMI 1640 + 10% FBS, and count in a Neubauer cell counter using the trypan blue exclusion assay. Centrifuge the Caco-2 cell suspension (3 min, 300 × g), and discard the supernatant to remove the remaining trypsin-EDTA. Resuspend the Caco-2 cells in RMPI 1640 + 10% FBS to obtain a suspension of 350,000 cells/mL. For each device, a volume of 1.5 mL is needed.
- Transfer 1.5 mL of the Caco-2 cell suspension into a sterile 2 mL syringe and remove any air or bubbles that remain in the syringe.
- Close the three-way stopcock valves of the tubing of the bacterial and neuronal chambers. Disconnect the tubing of the bacterial and neuronal chambers from the pump by removing the cassettes with the respective tubing from the rotor.
- Open the cap of the three-way stopcock valve of the inflow tubing leading to the epithelial chamber and turn the three-way stopcock valve to redirect the medium flow from the device to the 'open connector' (Figure 5C). Allow the medium to flow until a drop of medium appears at the open end of the three-way stopcock valve.
- Insert the syringe containing the epithelial cells into the 'open connector' using the drop-drop connection method to allow insertion of the syringe in the connector without the introduction of air bubbles. Turn the valve of the three-way stopcock to stop the flow of the medium from the inflow bottle (via the pump) and to allow flow from the connected syringe to the initial device. Disconnect the epithelial channel from the pump.
- Slowly press the syringe to inoculate the epithelial chamber with the cell suspension. Ensure that approximately one drop per 3 s drops into the outflow bottle.
- Add 1.5 mL of the cell suspension, then close the valve of the outflow three-way stopcock. Disconnect the syringe and close the open end of the three-way stopcock with the cap. Keep the chamber closed for at least 2 h. In the meantime, inoculate the neuronal cells.
- Neuronal cells
- Detach the neuronal cells from the well by resuspending the cells in the media from the respective wells using a pipette. Inoculate each device with fully confluent cells from one well (9.6 cm2) of a 6-well plate.
- Resuspend the cell pellet in the corresponding N2B27 medium volume (1.5 mL of media per device) + 1 µL of fibronectin/mL of cell suspension. Transfer 1.5 mL of the resuspended cell suspension into a 2 mL syringe and remove any air bubbles that remain in the syringe. Place the filled syringe in a sterile 50 mL conical tube and transfer to the biosafety cabinet containing the primed HuMiX device.
- Follow the same inoculation process as described previously for the epithelial chamber inoculation, except for the change of tube line. Here, disconnect the bacterial and epithelial tube lines from the pump.
- After neuronal cell inoculation, close all the three-way stopcocks of the tube lines and disconnect from the pump. Place the device in the incubator at 37 °C and 5% CO2. Keep all the channels closed for 2 h to allow the cells to attach.
- After 2 h, connect the bacterial and epithelial channels to the pump and open the inflow and outflow three-way stopcocks of both lines. Keep the neuronal chamber closed during the coming 14 days of the initial device run, except during medium change.
- During the 14 day run, change the medium of the neuronal chamber every 3-4 days. For medium refresh, prepare 3 mL of fresh N2B27 medium per device and transfer into a sterile 20 mL serum bottle. Close the serum bottle with medium using a sterile septum and an aluminum crimp seal.
- Insert an aeration and 80 mm needle into the septum of the new bottle. Replace the old media bottle with the new one in the incubator by disconnecting the male Luer from the needle of the old bottle to the needle of the new bottle.
- Connect the cassette with the pump tubing of the neuronal chamber to the pump and open the three-way stopcocks. Let the media flow at 0.5 rpm for 2 h before closing the three-way stopcocks of the neuronal tubing and disconnecting from the pump, until the next medium exchange.
5. Bacterial culture and inoculation
NOTE: In this study, on Day 12, a liquid culture of Limosilactobacillus reuteri strain F275 was reanimated from a glycerol stock. Depending on the needs or study designs, other bacterial species can be used.
- Prepare three tubes with 5 mL of MRS broth-one sterile control tube and two for L. reuteri inoculation. With an inoculation loop, scrape the top of the glycerol stock off and transfer it into one tube. Repeat with a second inoculation tube. Incubate the tubes at 37 °C, 170 rpm overnight.
NOTE: Do not let the glycerol stock thaw. - Prepare fresh medium for the initial devices by mixing Roswell Park Memorial Institute (RPMI) 1640 + 10% FBS with 5% MRS broth. Prepare 25 mL per device and transfer into 100 mL serum bottles under sterile conditions. Close the bottles with a sterile septum and aluminum crimp. Place the bottles in the incubator at 37 °C, 5% CO2 overnight.
- Prior to connecting the bottles to the bacterial tube line, add an aeration needle and 80 mm needle to each bottle of RPMI 1640/MRS media.
- Prepare new tubes, each with 3 mL of RPMI 1640 + 10% FBS + 5% MRS broth. Prepare at least two tubes, one control and one tube per device. Inoculate 15 µL of the L. reuteri overnight culture (optical density [OD] > 2) in two of the newly prepared tubes.
- Incubate the tubes for 1 h at 37 °C, 170 rpm, after which an OD of 0.05-0.10 is reached, corresponding to ~1 × 107 colony forming units (CFU)/mL. Transfer 1.5 mL into a 2 mL syringe (one per device). Use the rest of the bacterial suspension for CFU plating and live/dead staining.
- For bacterial inoculation of the device, follow the same procedure as mentioned in step 4.1, except that here the neuronal and epithelial tube lines are closed.
- After inoculation, close the bacterial tube line too, by closing the valves and disconnecting from the pump for 30 min. Connect the epithelial and bacterial tube lines and open again. Let the devices run for another 24 h at 37 °C and 0.5 rpm.
6. HuMiX opening and sampling
NOTE: The below section describes the sampling of different cell types. For example, neuronal cell pellets are used for RNA extraction and subsequent quantitative polymerase chain reaction (qPCR), bacterial pellets for DNA extraction and 16S rRNA gene sequencing, and supernatants for enzyme-linked immunosorbent assays (ELISAs) and other assays (e.g., lactate assay).
- On day 14, the opening day, close all the three-way stopcocks, disconnect the tubings from the pump, and take out the device of the incubator to the lab bench.
NOTE: When moving the device, make sure to keep the actual device horizontal. - Remove the tubing lines connected to the device before slowly opening the clamp and remove the clamp lid. With care, remove the top PC lid, collect the media in a 1.5 mL microcentrifuge tube, and place it on ice.
- Gently remove the sandwich gasket, while collecting the media from the epithelial chamber; be careful not to touch the cell layer. Place the sandwich gasket in a square Petri dish and add sterile 0.9% NaCl solution in H2O to the bacterial chamber until the chamber is fully covered (approximately 1 mL).
- Slowly remove the collagen gasket while collecting the media from the neuronal chamber and transferring it to a microcentrifuge tube. Place all the media tubes on ice. Place the collagen gasket into a square dish and gently add a few milliliters of 1x PBS to the Caco-2 layer until the cell layer is fully covered.
- Place the bottom PC lid in a square Petri dish and gently add approximately 2 mL of 1x PBS on top of the neuronal cells, so they do not dry out during the sampling process.
- Centrifuge the bacterial media tube at 5,000 × g for 5 min at 4 °C. Centrifuge the epithelial and neuronal media tubes at 300 × g for 5 min at 4 °C. After centrifugation, transfer the supernatant of each tube to a new microcentrifuge tube and immediately place it on dry ice.
- Epithelial cells
- Gently remove the PBS from the gasket and collect in a 15 mL conical tube. Add 2 mL of trypsin to the cells and incubate the gasket for 5 min at 37 °C, 5% CO2. Add 2 mL of RPMI 1640 to the gasket after incubation, resuspend the cells, and collect in another 15 mL conical tube.
- Centrifuge both tubes for 5 min at 300 × g. Discard the supernatant. Resuspend the pellet from the PBS wash in 300 µL of PBS and the cell pellet in 1 mL of PBS.
- Transfer 50 µL of each tube to a 0.5 mL microcentrifuge tube for the automated cell counter and trypan blue exclusion assay cell count. Transfer the resuspended cell pellet (1 mL) to a 1.5 mL tube and centrifuge at 300 × g for 5 min.
- Remove the supernatant and resuspend the cell pellet in 250 µL of lysis buffer + 1% beta-mercaptoethanol and place the tube on dry ice.
- Neuronal cells
- Take the square Petri dish, with the bottom, transparent PC lid with neuronal cells from the previous step (6.5) to an inverted phase contrast microscope.
NOTE: While moving the PC lid with PBS on top of it, be very gentle, as the neuronal network detaches very easily. - Perform a last quality check prior to inoculation by observing the morphology and cell density of the neuronal cells using brightfield microscopy. Verify that the cells have migrated from the spheroids and that a neuronal network has formed. The neuronal network should be approximately 90% confluent. For more details, refer to Fattahi et al.6.
- At the bench, resuspend the cells in PBS and collect in a 1.5 mL tube. Centrifuge the tube at 300 × g for 3 min. Discard the supernatant, resuspend the cell pellet in 250 µL of lysis buffer + 1% beta-mercaptoethanol, and place on dry ice.
NOTE: Immunofluorescence (IF) staining can be done on the neuronal cells on the PC lid. If IF staining is the preferred assay in order not to destroy the neuronal network, the cells should be fixed on the PC lid with 4% paraformaldehyde (PFA), which impedes reuse of the PC lid in future experiments.
- Take the square Petri dish, with the bottom, transparent PC lid with neuronal cells from the previous step (6.5) to an inverted phase contrast microscope.
- Bacterial cells
- Resuspend the bacterial cells attached to the membrane in 0.9% NaCl solution. If the cells are firmly attached, gently make use of a cell scraper to detach the bacterial cells from the membrane.
NOTE: Using a scraper can damage the cells, increasing the probability of seeing more dead cells. - Collect the cell suspension in a 15 mL conical tube. Add the pellet left over from the previously collected media to the 15 mL conical tube. Centrifuge at 5,000 × g for 5 min at 4 °C. Discard the supernatant and resuspend the cell pellet in 1 mL of 0.9% NaCl solution.
- Divide this volume into three parts: one for freezing a bacterial pellet for nucleic acid extraction (650 µL), one for CFU plating (50 µL) on MRS plates, and one for live/dead staining (300 µL). For preparation of the pellet for nucleic acid extraction, transfer the cell suspension into a microcentrifuge tube, centrifuge at 5,000 × g for 5 min at 4 °C and discard the supernatant. Place the cell pellet on dry ice.
- At the end of sampling, transfer all the tubes on dry ice to a -80 °C freezer for storing for later downstream analyses.
NOTE: Supernatant can further be used for gas chromatography-mass spectrometry (GC-MS) analysis. Furthermore, during the opening, the collagen-coated membrane can be divided in different parts for different analyses-one half to be used for IF occludin-staining, another part for RNA extraction for further gene expression analysis, and another part for cell counting.
- Resuspend the bacterial cells attached to the membrane in 0.9% NaCl solution. If the cells are firmly attached, gently make use of a cell scraper to detach the bacterial cells from the membrane.
Results
In neuroHuMiX, we co-cultured three different cell types together-bacterial, epithelial, and neuronal cells (Figure 1). To make sure the cells were all viable, we performed different assays on the different cell types. For example, we performed CFU counts on bacterial cells, cell count and cell viability assays on the epithelial cells, while the neuronal cells were assessed via microscopic analyses.

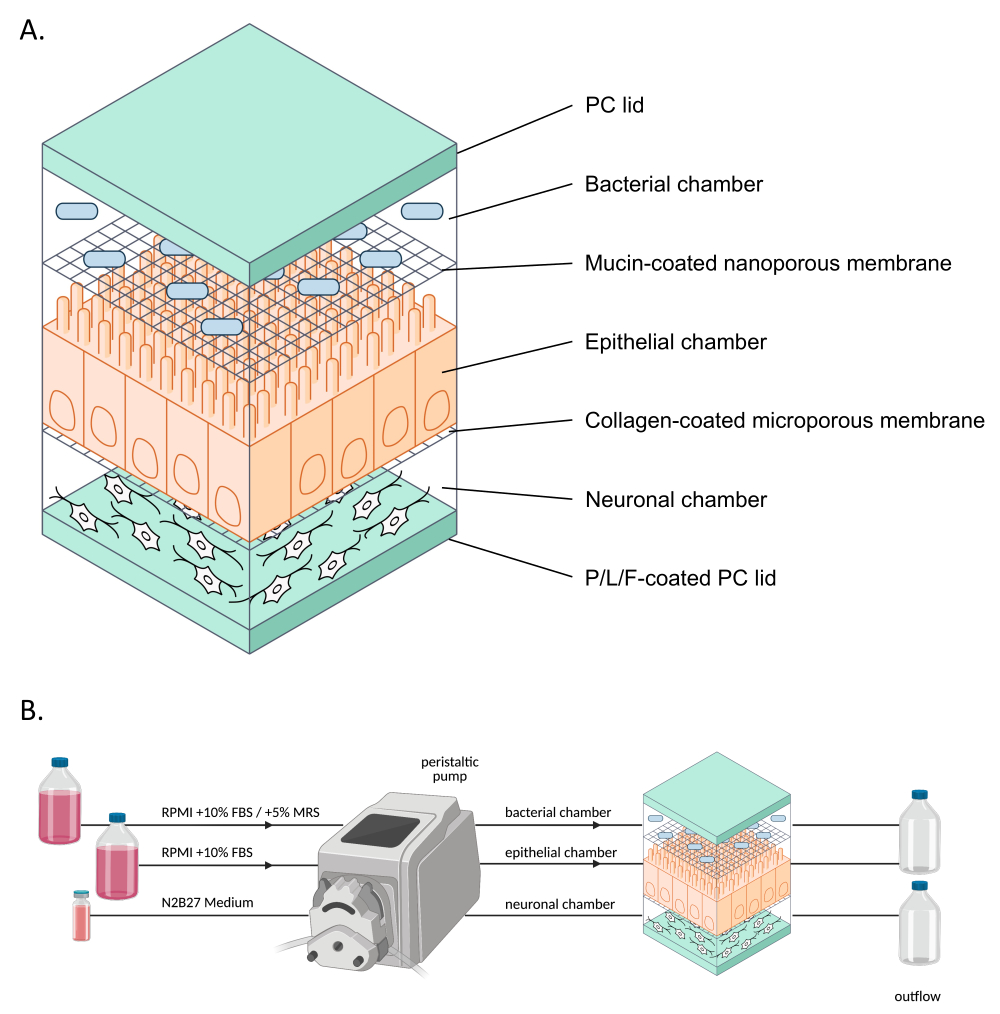
Figure 1: Schematic representation of neuroHuMiX and its experimental setup. (A) The three chambers are held between two PC lids to keep them closed. Each chamber is filled with a specific medium for the cells grown inside. The different chambers are separated by semi-permeable membranes allowing cell communication via soluble factors passing the membranes. (B) Representation of the neuroHuMiX setup. Each chamber is connected to different media bottles. For the bacterial chamber, for the first 12.5 days, the chamber is connected to RPMI + 10% FBS, before being changed for the last 36 h to RPMI + 10% FBS + 5% MRS. Abbreviations: PC = polycarbonate; P/L/F = poly L-ornithine/laminin/fibronectin; RPMI = Roswell Park Memorial Institute cell culture medium; MRS = De Man, Rogosa, and Shapre culture medium. Please click here to view a larger version of this figure.
{kind=link}
To determine whether the cells were appropriately attached, upon opening the devices, we assessed the formation of a cell layer on the collagen-coated membrane (Figure 6A). To make sure the cells in the device were viable, an automated cell counter count (Figure 6B) and a trypan blue exclusion assay cell count were performed (Figure 6C). The assays were performed on Caco-2 cells from three different HuMiX setups: (i) Caco-2 in culture with ENs, (ii) Caco-2 in culture with L. reuteri, and (iii) the device involving co-culture of all three cell types. Statistical testing using a one-way ANOVA did not yield any significant differences between the cell types, suggesting that the Caco-2 cells remained viable in all these initial device setups and conditions tested in this study. This underlines the fact that the bacterial density reached during the co-culture of L. reuteri and the two human cell types do not have cytotoxic effects on the human cells.

Figure 6: Assessment of Caco-2 cells on the collagen-coated membrane. (A) Layer of Caco-2 cells on the collagen-coated membrane after opening. The arrow indicates the collagen-coated membrane, which is surrounded by a dashed circle. The Caco-2 cells were growing on the spiral shape on the membrane. Cell viability of Caco-2 cells after 14 days in HuMiX. Cell counts were obtained using (B) the automated cell counter and (C) the trypan blue exclusion assay cell count. Caco-2 cell counts were determined from different culture setups in the initial device: co-culture with enteric neurons (ENs) (black), co-culture with L. reuteri (orange), and in the device (ENs and L. reuteri) (blue). A one-way ANOVA was performed, showing there is no significant difference between the different culture setups (one-way ANOVA, p = 0.1234 [ns]; error bars indicate standard error). Please click here to view a larger version of this figure.
{kind=link}
To be able to culture L. reuteri with mammalian cells, we first optimized and adapted the culture media for use in the device. We found that a 5% mix of MRS in RPMI 1640 (supplemented with 10% FBS) was optimally suited for the growth of L. reuteri, while not being cytotoxic for the mammalian cells used in these assays. Subsequently, a CFU count was performed to assess the growth of L. reuteri when cultured in the device for 24 h. The CFU count was assessed for two different initial device setups (Figure 7)-L. reuteri co-cultured with Caco-2 and L. reuteri in the device. In both setups, the CFU counts were significantly different from the HuMiX inoculum and the harvested cells (one-way ANOVA, p = 0.0002), indicating growth of the bacterial cells inside the initial device.

Figure 7: Limosilactobacillus reuteri CFU count of the inoculum (diluted 1:100,000) and after 24 h in HuMiX. Two different setups: Caco-2 cells in co-culture with L. reuteri and the device. A one-way ANOVA shows a significant difference (p = 0.0002 [***]) between the inoculum and the harvested cells, meaning the bacteria are growing inside HuMiX. Error bars indicate the standard error. Abbreviations: CB.HX = Caco-2 bacteria HuMiX; nHX = neuroHuMiX. Please click here to view a larger version of this figure.
{kind=link}
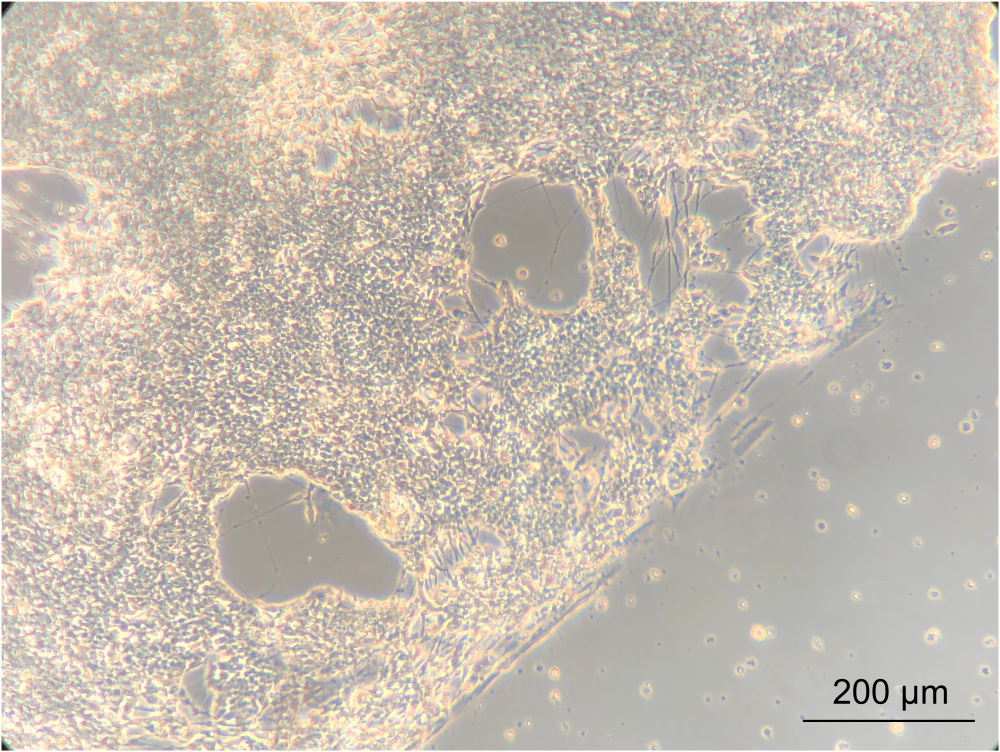
To assess whether culturing the ENs within the device would alter the phenotype of the cells, the gross morphology of the ENs was observed using an inverted phase-contrast microscope. During this step, both the confluency and EN morphology were assessed. Establishment of a confluent neuronal network indicated that the cells had attached well onto the coated device's PC lid. Importantly, this highlights the notion that they grew in co-culture with Caco-2 and L. reuteri. The edge between the confluent neuronal network and the gasket-delineated spiral was clearly apparent (Figure 8).

Figure 8: Enteric neurons after 14 days of culture in the device. On the left side of the image, the neurons have grown to a confluent layer on the spiral. The edge, between the neuronal layer and the space without cells, is the edge of the spiral; magnified 10x, scale bar = 200 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Lids used in the device. Images show top (left) and bottom (right) PC lids. Each side of the PC lid is 6.4 cm. Abbreviation: PC = polycarbonate. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Epithelial chamber gasket on bottom PC lid. Top view of the epithelial chamber gasket placed on the bottom PC lid (left), and bottom view (right) showing the alignment of the epithelial chamber gasket with the inlets and outlets of the bottom PC lid. Each side of the gaskets, as well as the PC lid, measures 6.4 cm. Abbreviation: PC = polycarbonate. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Assembly of the device. (A) Different parts for assembling HuMiX: (1) bottom PC lid; (2) gasket with collagen-coated microporous membrane, which is placed on top of (1); (3) sandwich gasket with a mucin-coated nanoporous membrane in between and placed on top of (2); (4) top PC lid placed on top of (3). Each side of the gaskets and PC lids measures 6.4 cm. (B) All parts from (A) placed together. (C,D) Assembled device-top (left) and side (right) view. B is placed into the clamping system to close the system. (C) Each side of the top clamp measures 8 cm. Abbreviation: PC = polycarbonate. Please click here to view a larger version of this figure.
{kind=link}

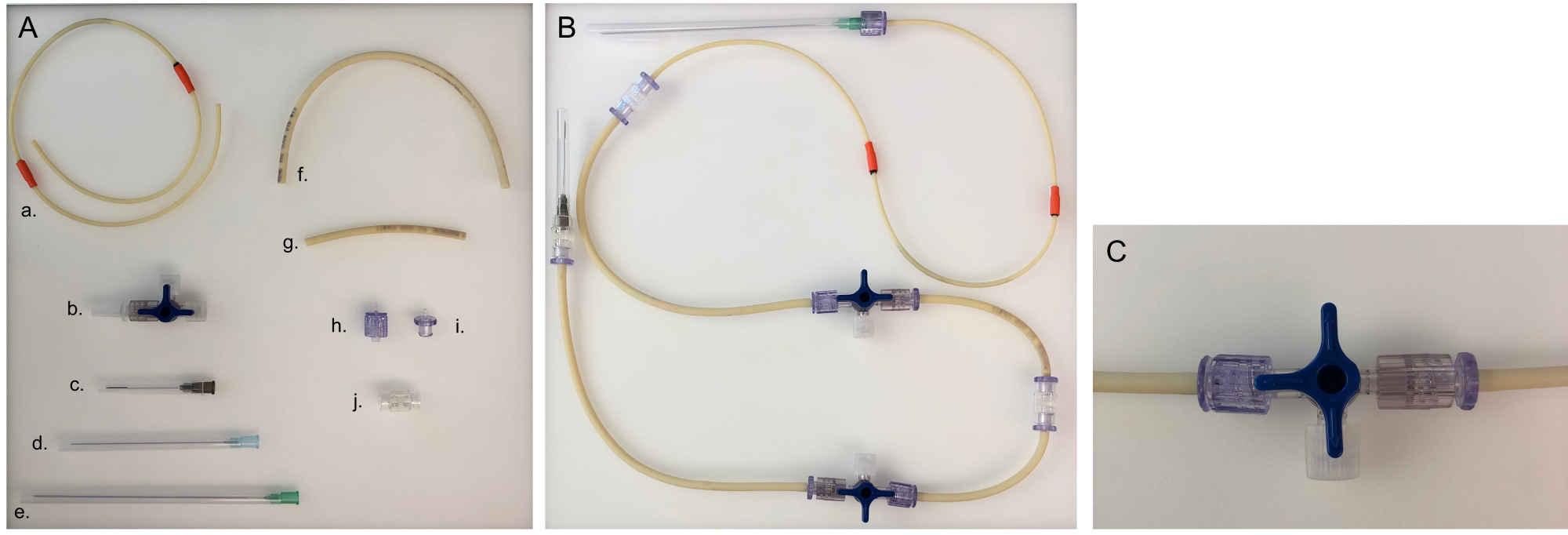
Figure 5: Parts needed for tubing line and assembled tubing line for one chamber. (A) Different parts to build a tubing line: a. pump tubing line; b. three-way stopcock; c. 40 mm needle; d. 80 mm needle; e. 120 mm needle; f. long tubing line (20 cm); g. short tubing line (8 cm); h. male Luer; i. female Luer; j. adaptor. (B) Assembled tubing line for the bacterial or epithelial chamber. For the neuronal chamber, the 120 mm needle would need to be changed to an 80 mm needle. (C) Three-way stopcock valve turned to redirect the medium flow from the device to the 'open connector' and to close the chamber. Please click here to view a larger version of this figure.
{kind=link}
| Day | 0 | 2 | 4 | 6 | 8 | 10 |
| Media Composition | 100% E6 | 100% E6 | 75% E6 | 50% E6 | 25% E6 | 100% N2 |
| + LDN | + LDN | 25% N2 | 50% N2 | 75% N2 | + LDN | |
| + SB | + SB | + LDN | + LDN | + LDN | + SB | |
| + CHIR | + SB | + SB | + SB | + CHIR | ||
| + CHIR | + CHIR | + CHIR | + RA | |||
| + RA | + RA | |||||
| Molecule | [concentration] | |||||
| LDN | 100 nM | |||||
| SB | 10 µM | |||||
| CHIR | 3 µM | |||||
| Retinoic Acid (RA) | 1 µM |
Table 1: Media composition.
| Media | Components (concentrations listed in Table of materials) | Volume (mL) |
| N2 media (50 mL) | DMEM-F12 | 48 |
| N2 Supplement | 0.5 | |
| L-Glutamine | 0.5 | |
| Penicillin/Streptomycin | 0.5 | |
| NEAA | 0.5 | |
| N2B27/ENS Media (50 mL) | Neurobasal | 48 |
| N2 Supplement | 0.5 | |
| L-Glutamine | 0.5 | |
| Penicillin/Streptomycin | 0.5 | |
| B27-A | 0.5 |
Table 2: Media recipes.
| Sterilization Temperature (°C) | 116 |
| Sterilization Time (min) | 20 |
| Dry Time (min) | 10 |
| Pulses | 3 |
| End Temperature (°C) | 99 |
Table 3: HuMiX autoclave run.
| Rotations per minute (rpm) | Average flow rate (µL/min) |
| 0.5 | 13 |
| 2 | 79 |
| 5 | 180 |
Table 4: Flow rates of the peristaltic pump.
Discussion
It is now established that the human gut microbiome influences the host's health and disease. Despite the knowledge suggesting the importance of our microbiome, especially in neurological disorders such as Alzheimer's or Parkinson's disease3,13, it remains largely unknown how the gut microbiome interacts with the enteric nervous system, and subsequently, with the brain.
A representative model to study the interactions between the gut microbiome and the nervous system has thus far been unavailable. Studies regarding the gut-brain axis have traditionally been performed using murine models13. Mice and humans share 85% of their genomic sequences14, but there are significant differences to consider when comparing mice to humans. Regarding the gut, it is important to note that, compared to humans, mice are exclusively herbivores. As a result, their gastrointestinal tract differs in length and characteristics, such as the 'gastric emptying'14. Murine brains also show important differences, whereby the overall structure between mice and humans are different15. Importantly, humans have longer cell cycle times of neural progenitors15. Consequently, it is important to develop representative models that include human-derived cells, including intestinal and neuronal cells5. In this context, the development of more reproducible research viain vitro models reduces the need to use animal models and improves reproducibility.
neuroHuMiX is an advanced version of the previous HuMiX model9. HuMiX is a gut-on-a-chip model allowing proximal and representative co-cultures of epithelial and bacterial cells. Cell-cell communication is possible through the proximal co-culture and diffusion of secreted factors and metabolites via semipermeable membranes. However, to expand the utility of the initial device to study the human gut environment, the introduction of an additional cell type is required. To address this, neuroHuMiX, developed with the introduction of iPSC-derived ENs, enables a proximal co-culture of bacteria, intestinal epithelial cells, and ENs. The resulting in vitro model allows us to address questions regarding the human gut microbiome in relation to the human nervous system. Co-culturing different cell types, especially co-cultures of mammalian cells and bacteria, has several challenges, including the loss of viability, poor adhesion, and overall loss in confluence16. Here, we have demonstrated that within this device, we are able to co-culture three different cell types within the same system while keeping the cell viability high.
A critical step in the protocol is to ensure confluency of the neuronal cells-80%-90% cell confluency and viability-before inoculating into the device. Since it is not possible to assess the cell growth during the run, it is of utmost importance to ensure the cells are confluent and growing well before introducing them in the model. While this may be a limiting factor, the overall viability and confluency observed within the device is generally high.
The device is connected via tubing lines to a peristaltic pump. Each cell chamber has its specific tubing line. The tubing comprises a pump tubing that allows the use of a peristaltic pump for the perfusion of medium, as well as tubing connecting the pump tubing to the device and tubing connecting the device to the outflow/waste bottles. Sampling ports are included before and after the device, to allow the inoculation and sampling of outflow medium. Each chamber can be connected to a different medium, allowing the best culture conditions for each individual cell type. Each chamber can be opened or closed depending on the specific needs for medium supply. In the device, the neuronal chamber stays closed for most of the experiment, while the bacterial and epithelial chambers are open all the time, meaning they get fresh medium throughout the whole experimental run. To make sure the medium is flowing without interruption, it is crucial to not have any air left in the tubings, connectors, or in the device. Therefore, it is important to first let the devices run for a few minutes at the priming step. This often resolves the issue. If not, one of the other lines that are dropping can be closed for a short amount of time by closing the three-way stopcock of the outflow. This redirects the medium to the line with the air bubble, thus resolving the issue by pushing the bubble outward through the tubing.
For any cell culture experiment, the medium is a key component, where each cell type has its respective medium. In a co-culture setup, the medium needs to be compatible not only for the cell type growing in it, but also for the other cell types within the co-culture. This is no different for the device, which poses an additional challenge as we have three different compartments with three different cell types inside-bacterial, epithelial, and neuronal cells. We have, however, shown that by modifying the bacterial media-with the addition of 5% MRS to RPMI 1640 with 10% FBS-all cell types, in particular bacterial and epithelial cells, can be successfully co-cultured within the system. However, in the device, different cell types are co-cultured in proximity, and are hence not in direct contact with one another. Even though this is not fully representative of the direct contact between cells in the human gut, and therefore a limitation, the proximal and representative co-culture condition is a strength for downstream analyses. Soluble factors exchange between the different chambers and cell types; hence, the cells are still interacting with each other. Additionally, the fact that the cell types can be harvested and analyzed separately allows us to study the effect of a healthy and/or diseased microbiome on different cell types (including neuronal cells) and thereby determine/retrieve cell type-specific read-outs. Another limitation is that the morphology of the cells cannot be followed-up during the experimental run, as the device can only be opened and the cells checked at the end of each experiment.
To our knowledge, neuroHuMiX is the first gut-on-a-chip model including ENs. This is a step toward elucidating the communication between the gut microbiota and the enteric nervous system. It is a model allowing investigation of the interplay between a bacterial species, an epithelial layer, and ENs. Its design allows us to study the exchange of soluble factors secreted by the different cell types and their effect on one another. Going forward, it would be important to not only have iPSC-derived ENs, but also iPSC-derived epithelial cells inside the device, to transition the device into a personalized model. Importantly, this personalized model could be used to test pre-, pro-, and synbiotics10,11 and potentially develop personalized screening and therapeutic approaches17. Personalized neuroHuMiX could eventually shed light on the 'dark matter' of the human gut microbiome and its interactions with the nervous system along the gut microbiome-nervous system axis, paving the way for therapeutic assessment and interventions.
We can conclude that being able to have a gut-on-a-chip including the enteric neuronal system is crucial to progressing in the study and understanding of interactions along the gut microbiome-nervous system axis. NeuroHuMiX allows us to study the effects of bacterial species on host cells and provides us with a good basis to improve the model even further in an even more physiologically representative way.
Disclosures
P.W. declares being listed as an inventor on patents PCT/EP2013/056607, PCT/EP2016/062024, PCT/US2017/061602, and PCT/EP2019/081424. P.W., C.S., and L.G. declare being listed as inventors on patent LU503075.
Acknowledgements
The authors would like to thank Dr. Jared Sterneckert for providing us with the cells from the K7 line. We also want to thank the long-standing collaborators Dr. Frederic Zenhausern and Matthew W. Barret from the University of Arizona for their assistance with the engineering aspects. We would also like to acknowledge Dr. Valentina Galata for her help in designing the schematic representation of neuroHuMiX. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement 863664). Figure 1 was partially created with Biorender.com.
Materials
| Name | Company | Catalog Number | Comments |
| 2-Mercaptoethanol | Sigma Aldrich | 10712 | |
| Aeration cannula (length: 1.10 diameter: 30 mm) | VWR (B.Braun) | BRAU4190050 | |
| Agar-agar | Merck Millipore | 1.01614.1000 | |
| Aluminium Crimp | Glasgerätebau Ochs | 102050 | |
| Ascorbic acid | Sigma Aldrich | A4544 | |
| B-27 Supplement Minus Vitamin A (50x) | Gibco | 12587-010 | |
| Bacterial Cell Membrane, pore size: 1 µm | VWR (Whatman) | 515-2084 | |
| Caco-2 cells | DSMZ | ACC169 | |
| Cell Counter & Analyzer CASY | OMNI Life Sceince | ||
| CHIR | Axon Mechem BV | CT99021 | |
| Collagen I, Rat Tail | Invitrogen | A1048301 | |
| Costar 6-well Clear Flat Bottom Ultra-Low Attachment Plates | Corning | 3471 | |
| Difco Lactobacilli MRS Broth | BD Biosciences | 288130 | |
| Discofix 3-way stopcock | B. Braun | BRAU40951111 | |
| DMEM/F12, no glutamine | Thermofisher Scientific | 21331020 | |
| Dulbecco's Phosphate-Buffered Saline, D-PBS | Sigma Aldrich | 14190-169 | |
| Essential 6 Medium | Thermofisher Scientific | A1516401 | |
| Essential 8 Medium | Thermofisher Scientific | A1517001 | |
| Female Luer Lock to Barb Connector | Qosina | 11733 | |
| FGF2 | R&D Systems | 233-FB | |
| Fibronectin | Sigma Aldrich | F1141 | |
| Foetal Bovine Serum, FBS | Thermofisher Scientific | 10500-064 | |
| GDNF | PeproTech | 450-10 | |
| Human Cell Membrane, pore size: 50 nm | Sigma Aldrich (GE Healthcare) | WHA111703 | |
| HuMiX Gasket Collagen | Auer Precision | 216891-003 | |
| HuMiX Gasket Sandwich Bottom | Auer Precision | 216891-002 | |
| HuMiX Gasket Sandwich Top | Auer Precision | 216891-001 | |
| iPSC | Max Planck Institute for Molecular Biomedicine | K7 line | |
| L-Glutamine (200 mM) | Gibco | 25030081 | |
| Laminin from Engelbreth-Holmswarm | Sigma Aldrich | L2020 | |
| LDN193189 | Sigma Aldrich | SML0559 | |
| Limosilactobacillus reuteri | ATCC | 23272 | |
| Live/Dead BacLight Bacterial Viability kit | Thermofisher Scientific | L7012 | |
| Male Luer with Spin Lock to Barb | Qosina | 11735 | |
| Marprene tubing (0.8 mm x 1.6 mm) | Watson-Marlow | 902.0008.J16 | |
| Matrigel hESC-qualified matrix | Corning | 354277 | |
| Mucin, from porcine stomach | Sigma Aldrich | T3924 | |
| N2 Supplement (100x) | Gibco | 17502048 | |
| NEAA | Thermofisher Scientific | 11140050 | |
| Needle (length: 120 mm; diameter: 0.80 mm) | B.Braun (color code: green) | 466 5643 | |
| Needle (length: 40 mm; diameter: 0.70 mm) | Henke Sass Wolf (color code: black) | 4710007040 | |
| Needle (length: 80 mm; diameter: 0.60 mm) | B.Braun (color code: blue) | 466 5635 | |
| Neurobasal Medium | Gibco | 21103049 | |
| PE/Cy7 anti-human CD49d antibody | Biolegend | 304314 | |
| Penicillin-Streptomycin | Sigma Aldrich | P0781 | |
| Peristaltic pump | Watson-Marlow | 205CA | |
| Poly-L-ornithine Hydrobromide | Sigma Aldrich | P3655 | |
| Polycarbonate lids (HuMiX) | University of Arizona | HuMiX 1.0 / 2.0 | |
| Retinoic Acid | Sigma Aldrich | R2625 | |
| RLT Buffer (RNeasy Minikit) | Qiagen | 74104 | |
| RPMI 1640 Medium | Thermofisher Scientific | 72400-021 | |
| SB431542, ALK inhibitor | Abcam | ab120163 | |
| Serum bottles | Glasgerätebau Ochs | 102091 | |
| Syringe | BD Biosciences | 309110 | |
| Trypsin-EDTA solution | Sigma Aldrich | T3924 | |
| Y-27632 Dihydrochloride | R&D Systems | 1254 |
References
- Heintz-Buschart, A., Wilmes, P. Human gut microbiome: function matters. Trends in Microbiology. 26 (7), 563-574 (2018).
- Toor, D., et al. Dysbiosis disrupts gut immune homeostasis and promotes gastric diseases. International Journal of Molecular Sciences. 20 (10), 2432 (2019).
- Braak, H., de Vos, R. A. I., Bohl, J., Del Tredici, K. Gastric α-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neuroscience Letters. 396 (1), 67-72 (2006).
- Schmit, K. J., et al. Dietary fibre deprivation and bacterial curli exposure shift gut microbiome and exacerbate Parkinson's disease-like pathologies in an alpha-synuclein-overexpressing mouse. bioRxiv. , (2022).
- Fritz, J. V., Desai, M. S., Shah, P., Schneider, J. G., Wilmes, P. From meta-omics to causality: experimental models for human microbiome research. Microbiome. 1 (1), 14 (2013).
- Fattahi, F., et al. Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature. 531 (7592), 105-109 (2016).
- Wu, Q., et al. Organ-on-a-chip: Recent breakthroughs and future prospects. BioMedical Engineering Online. 19 (1), 9 (2020).
- May, S., Evans, S., Parry, L. Organoids, organs-on-chips and other systems, and microbiota. Emerging Topics in Life Sciences. 1 (4), 385-400 (2017).
- Shah, P., et al. A microfluidics-based in vitro model of the gastrointestinal human-microbe interface. Nature Communications. 7, 11535 (2016).
- Greenhalgh, K., et al. Integrated in vitro and in silico modeling delineates the molecular effects of a synbiotic regimen on colorectal-cancer-derived cells. Cell Reports. 27 (5), 1621-1632 (2019).
- Mao, J. H., et al. Genetic and metabolic links between the murine microbiome and memory. Microbiome. 8 (1), 53 (2020).
- Moysidou, C. M., Owens, R. M. Advances in modelling the human microbiome-gut-brain axis in vitro. Biochemical Society Transactions. 49 (1), 187-201 (2021).
- Kim, S., et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson's disease. Neuron. 103 (4), 627-641 (2019).
- Hugenholtz, F., de Vos, W. M. Mouse models for human intestinal microbiota research: a critical evaluation. Cellular and Molecular Life Sciences. 75 (1), 149-160 (2018).
- Marshall, J. J., Mason, J. O. Mouse vs man: Organoid models of brain development & disease. Brain Research. 1724, 146427 (2019).
- Goers, L., Freemont, P., Polizzi, K. M. Co-culture systems and technologies: taking synthetic biology to the next level. Journal of the Royal Society Interface. 11 (96), 20140065 (2014).
- Sedrani, C., Wilmes, P. Toward hypothesis-driven, personalized microbiome screening. Cell Reports Methods. 2 (1), 100139 (2022).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved