Method Article
短いビオチン化RNAを用いたRNA結合タンパク質の最適化された定量的プルダウン解析(英語)
要約
ここでは、ヒト細胞からの総タンパク質抽出物、ビオチン化RNAでコーティングされたストレプトアビジンビーズ、および質量分析を使用して、特定のRNA配列のタンパク質相互作用因子を発見、定量、および検証するための最適化された in vitro メソッドを紹介します。
要約
タンパク質-RNA相互作用は、転写レベルおよび転写後レベルで遺伝子発現および細胞機能を調節します。このため、目的のRNAの結合パートナーを特定することは、多くの細胞プロセスの背後にあるメカニズムを明らかにするために依然として非常に重要です。ただし、RNA分子は、一部のRNA結合タンパク質(RBP)、特に非標準的なタンパク質と一過性かつ動的に相互作用する可能性があります。したがって、そのようなRBPを分離および同定するための改善された方法が大いに必要とされています。
既知のRNA配列のタンパク質パートナーを効率的かつ定量的に同定するために、細胞総タンパク質抽出物から始めて、相互作用するすべてのタンパク質のプルダウンと特性評価に基づく方法を開発しました。ストレプトアビジンコーティングビーズにプリロードされたビオチン化RNAを使用して、タンパク質プルダウンを最適化しました。概念実証として、神経変性関連タンパク質TDP-43に結合することが知られている短いRNA配列と、異なるヌクレオチド組成が同じ長さのネガティブコントロールを採用しました。酵母tRNAでビーズをブロックした後、ビオチン化RNA配列をストレプトアビジンビーズにロードし、HEK 293T細胞からの総タンパク質抽出物とともにインキュベートしました。インキュベーションと非特異的結合を除去するためのいくつかの洗浄ステップの後、最も一般的に使用されるタンパク質定量試薬および質量分析用のサンプル調製に適合する高塩溶液で相互作用タンパク質を溶出しました。既知のRNA結合剤を用いて行ったプルダウンにおけるTDP-43の濃縮をネガティブコントロールと比較して質量分析法により定量した。同じ手法を使用して、目的のRNAまたはコントロールの一意の結合剤であると計算的に予測される他のタンパク質の選択的相互作用を検証しました。最後に、適切な抗体によるTDP-43の検出 を介して 、ウェスタンブロットによってプロトコルを検証しました。
このプロトコルにより、生理学的に近い条件で関心のあるRNAのタンパク質パートナーの研究が可能になり、ユニークで予測できないタンパク質-RNA相互作用の発見に役立ちます。
概要
RNA結合タンパク質(RBP)は、mRNAスプライシング、RNA細胞局在、翻訳、修飾、分解などのプロセスに関与しているため、転写および転写後遺伝子の制御における重要なプレーヤーとして浮上しています1,2,3。2つの高分子間のこのような相互作用は、高度に調整され、正確にバランスが取れており、機能ハブとプロセシングハブの形成に不可欠です。これらのハブ内の変異または調節不全は、細かく調節されたタンパク質-RNAネットワークを破壊する可能性があり、癌4,5および神経変性疾患6,7,8を含むさまざまなヒト疾患とますます関連しています。RNA分子とそのタンパク質結合パートナーとの間の相互作用は、安定していて実験的に検証しやすい場合もあれば、非常に動的で一過性で特性評価が難しい場合もあります。
近年、これらの相互作用を理解するための集中的な努力が行われています。最も確立された方法の中で、タンパク質プルダウンアッセイ(PD)は、リボ核タンパク質(RNP)複合体および他のタンパク質-RNA相互作用ネットワークを構成する主要なプレーヤーを解明するためにおそらく最も高く評価され、一般的に使用されるアプローチです3,9,10。PDには、目的のRNA(RIP)11,12またはタンパク質(CLIP)13,14のいずれかの免疫沈降など、幅広い有益な技術が含まれます。これらのRNA-PDプロトコルのいくつかは、タンパク質15の餌として既知のRNAを採用し、ほとんどの場合、ビオチンなどの高親和性タグを利用します。この場合、ビオチン化RNAの相互作用パートナーは、ストレプトアビジンでコーティングされたビーズにRNAを固定することで検出でき、RNPの効率的な単離を可能にします。これらのアプローチの主な制限は、通常、ビオチン化プローブの設計と、標的タンパク質に結合する能力のテストです。この目的のために、標的タンパク質との相互作用のピークに対応する短いRNA領域を高精度で明らかにするので、利用可能な場合、関心のあるタンパク質の公開されたCLIPデータに頼ることは有用であり得る13,16。これらの同じ領域を使用して、PD用のプローブを開発できます。このようなRNAベイトを設計する別の方法は、指数関数的濃縮(SELEX)17によるリガンドの系統的進化であり、包括的なランダム化ライブラリから始まり、一連のPCR駆動の最適化サイクルを介して、in vitro選択を通じてアプタマーの設計を可能にします。ただし、SELEXは複雑で時間がかかり、最終結果は初期ライブラリに大きく依存します。ここに提示されたプロトコルで使用するRNAベイトを選択するために、特定のRNA配列に対する所与のタンパク質の優先的結合を予測するアルゴリズムcatRAPIDの計算能力によってde novoで設計されたRNAベイトを使用することからなるさらに別のアプローチが利用された18、19、20。
ここで紹介するプロトコルは、界面活性剤、変性剤、または高温を使用せずに、生理学的に近い条件で特定のタンパク質パートナーを溶出するように最適化されたRNA-PDのバージョンです。これは、高度に精製されたストレプトアビジンで共有結合でコーティングされたナノ超常磁性ビーズと、餌としての特定の インシリコ 設計ビオチン化RNAの使用に依存しています。このプロトコルは、ビオチン化RNA分子の結合パートナーをネイティブ条件で分離するための迅速かつ効率的な方法を提供し、幅広いダウンストリームアプリケーションの可能性を提供します。このプロトコルを試験するために、タンパク質TAR DNA結合タンパク質43(TDP-43)を高い親和性および特異性で結合するように予め設計された10ヌクレオチド一本鎖RNAアプタマー配列を20使用した。HEK 293T細胞溶解物から始めて、ビオチン化RNAアプタマーの相互作用因子を、高張緩衝液を用いてRNA餌から剥離したサンプルに対して実施した質量分析によって同定した。この分析により、好ましい結合剤としてのTDP-43の同定および定量化が成功したことが確認された。
このプロトコルにより、短いin vitro合成RNAオリゴヌクレオチドのみを使用してタンパク質相互作用因子を同定することができます。さらに、PDプローブ21,22としてインシリコ設計RNAアプタマーを使用することで、大幅に削減されたコストでターゲットに対する特異性が保証されます。
プロトコル
1.一般的な方法と材料
- 選択した哺乳類細胞培養に適した培地を準備し、使用前に37°Cで20分間予熱します。
- 材料の表に記載されているように、必要な材料を事前に準備してください。オートクレーブガラス製品、プラスチック製品、およびバッファーストック。
- 表 1 の説明に従ってバッファーを準備します。成分を最終容量に希釈する前に、濃縮HClまたはNaOHを使用してストック溶液のpHを調整します。
2. 哺乳類細胞株の調製
- 10%ウシ胎児血清(FBS)と100 μg/mLペニシリン/ストレプトマイシン溶液を添加したダルベッコ改変イーグル培地(DMEM)でHEK 293T細胞を増殖させます。5%CO2を供給した加湿インキュベーター内で37°Cでインキュベートします。定期的にセルを分割します。
- 剥離する前に、成長表面を覆うのに十分なリン酸緩衝生理食塩水(PBS)溶液で細胞を洗い流してください。
- PBSを取り外し、トリプシン-EDTA溶液の超薄層を追加します。
- 5%CO2 を備えた加湿インキュベーター内で細胞を37°Cで5分間、または細胞が剥離するまでインキュベートします(顕微鏡観察下では分散しているように見えます)。
- 完全なDMEMを加えてトリプシン-EDTA溶液を10倍に希釈し、不活性化して細胞をカウントします。
- 1.5 x 105 細胞/mLを6ウェルプレートにプレートし、テストする2つのウェル/条件を考慮します。
- 細胞を5%CO2を含む加湿インキュベーター内で37°Cで48時間インキュベートします。
注:細胞株に適した培地とサプリメントの種類については、製造元の説明書を確認してください。また、トリプシン-EDTAの量やインキュベーション時間は細胞株によって異なります。一部の細胞型は、このプロトコルで報告されたものよりも速く/遅く成長します。したがって、播種濃度は事前にテストする必要があります。
3.総タンパク質収穫量
- 細胞が増殖しているウェルから培地を除去します。
- 6ウェルプレートの各ウェルを1 mLのPBSで洗浄します。
- PBS を破棄します。
- プレートを氷上に移動し、ステップ3.5に進むか、乾燥プレートを-80°Cで凍結して溶解を促進します。
- 200 μLの溶解バッファーを各ウェルに加えます。
- セルスクレーパーを使用して、セルを切り離したり壊したりします。
- 2つのウェルに由来する細胞抽出液を同じ1.5 mLチューブに移します。
- タンパク質抽出物を含むチューブを氷上に30分間置きます。
- 細胞ライセートを17,000 x g で4°Cで15分間遠心分離します。
- 各上清を予冷チューブに移します。
注:PD条件ごとに合計10個の6-10 7 セルが推奨されます。細胞溶解とタンパク質の採取は、氷冷バッファーを使用して行う必要があります。プロテアーゼ阻害剤は、タンパク質の分解を防ぐために溶解バッファーに添加する必要があります。
4. タンパク質濃度測定
- 生産者が示すように、ブラッドフォード試薬をdH2Oで5倍に希釈して調製します。
- 試薬1 mLを1 cmキュベットに入れ、サンプルを1 μL加え、転倒混合します。
- 暗所で室温で5〜10分間インキュベートします。
- 595 nmでの吸光度を読み取ります。
- 1.5 mgのタンパク質に相当するタンパク質抽出物の量を計算し、溶解バッファーを使用してすべてのサンプルを最終容量600 μLにします。
- 使用するまでサンプルを氷上に保管してください。
注:バッファー適合性の推奨事項に従って、他のタンパク質濃度決定方法を使用できます。いずれの場合も、溶解バッファーはブランクとして使用する必要があります。多くの試薬はジチオスレイトール(DTT)と互換性がありません。タンパク質定量(DTTを最終濃度1 mMまで)の後にのみDTTまたは他の還元剤を添加することをお勧めします。
5.ビーズの準備
- チューブをフリックして、保存バッファー内のビーズを混合します。
- 100 μLのスラリー媒体/サンプルを計算し、その容量を磁気ラックに入れます。
- ビーズウォッシュ
- 保存液を取り出し、1 mLの溶解バッファー/チューブを加えてビーズを洗浄し、手動で反転させます。
- 磁気ラックを使用してバッファーを取り外します。
- 洗浄手順を繰り返します。
- スラリー培地の初期容量に等しい量の溶解バッファーを加え、チューブをフリックして混合し、サンプルと同じ数の1.5 mLチューブに培地を均一に分注します。
- ビードブロッキング
- 磁気ラックを使用してバッファーを除去し、溶解バッファーで調製した酵母tRNAの0.25 mg/mL溶液600 μLを追加します。
- 回転ホイール上で室温で1時間インキュベートします。
- 磁気ラックを使用してtRNA溶液を除去します。
- 600 μLの溶解バッファーを加え、手動で混合して洗浄します。
- 洗浄ステップを繰り返し、バッファーを廃棄します。
6.ビードローディング
- 最初の100 μLのスラリー培地(現在はブロックビーズ)を含む各チューブについて、600 μLの溶解バッファー中に200 μgのRNAオリゴヌクレオチドを調製します。
- オリゴをビーズに加え、回転させながら室温で1時間インキュベートします。
- 溶液を取り出し、600 μLの溶解バッファーを加え、室温でチューブを5分間回転させてビーズを2回洗浄します。
- バッファーを破棄します。
注意: ビーズをボルテックスしないでください、代わりにフリックします。必要な場合を除いて、ピペッティングステップの数を制限します。可能であれば、カット、1 mLチップを使用してください。ビーズスラリー培地/サンプルの量は、ビーズの結合能力と総タンパク質の開始量に依存します。RNAオリゴがかなりの量の二次構造を有すると予測される場合は、まず80°Cで10分間変性させた後、室温でゆっくりと冷却するか、30°Cで1時間インキュベートしてリフォールドすることをお勧めします。ローディングに必要な量を最適化し、RNAの再利用の可能性を評価するために、ビーズローディング後にRNAオリゴを回収し、残存濃度を決定することをお勧めします。
7. ビーズへのタンパク質結合
注意: 今後は、可能であれば、4°Cで手順を実行してください。
- 600 μLのタンパク質溶液から5%の容量を取り、さらに分析するためにINPUT(IN)として保持します(1.5 mgのタンパク質が600 μLに溶解するため、5%は30 μLおよび75 μgのタンパク質に相当します)。
- 残りのタンパク質混合物をロードしたビーズの各チューブに加え、4°Cで一晩ゆっくりと回転させます。
8.非特異的結合剤の洗浄
- 磁気ラックを使用して、バインドされていない部分を取り外します。容量の5%を保存し、FLOWTHROUGH(FT)としてラベル付けします(非結合容量は約600 μLなので、さらに分析するために30 μLを保管してください)。
- ビーズに1 mLの洗浄バッファー1を加え、4°Cで5分間回転させます。
- バッファーを破棄します。
- 手順 8.2 と 8.3 を繰り返します。
- ビーズに洗浄バッファー2 1 mLを加え、4°Cで5分間回転させます。
- 上澄み液を捨てる。
9. 特定のバインダーの溶出
- 100 μLの溶出バッファー1または溶出バッファー2をビーズに加えます。
- フリックして手動で混合し、室温で5分間インキュベートします。
- チューブをサーモミキサーに入れ、95°Cで5分間激しく振とうします。
- チューブを磁気ラックに入れ、溶出した画分をきれいなチューブに集めます。
- ベンチ遠心分離機でビーズをすばやく回転させ、溶出液の回収率を最大化します。
- さらなる分析のために、総溶出液(EL)容量の5%を節約します(総容量は100 μLなので、5 μLを別のチューブに分けます)。
- 必要に応じて、タンパク質濃度は、溶出バッファーをブランクとして使用することにより、セクション4のように決定できます。
注:タンパク質濃度が決定されるまで、溶出バッファーへのDTTの添加を控えることをお勧めします。タンパク質定量が不要な場合、または還元剤と互換性のあるタンパク質定量キットが利用可能な場合は、最初から1 mM DTTを溶出バッファーに添加できます。このプロトコルでは、溶出バッファー1(1 M NaClを含む)と溶出バッファー2(2 M NaClを含む)の両方をテストしました。イオン強度の増加による標的タンパク質の溶出効率の差は認められませんでしたが、最も適切なバッファーを確立する前に両方の条件をテストすることをお勧めします。溶出バッファー中の塩の存在が多いことがさらなる分析の限界となる場合、溶出バッファー中の界面活性剤の量が非常に少ないため、バッファー交換が可能です。別の方法として、溶出液を所望の塩濃度に達するように希釈することができる。
10. 質量分析によるタンパク質結合剤の同定
- アセトン沈殿
- 溶出したタンパク質を冷(-20°C)アセトンで4倍に希釈して濃縮します。
- チューブをボルテックスし、-20°Cで一晩インキュベートします。
- 17,000 x g で4°Cで30分間スピンします。
- 上清を静かに取り除き、ペレットが完全に乾くまでアセトンを蒸発させます。
- 溶液中のタンパク質消化
- 50 μLの変性バッファーを加えてタンパク質ペレットを溶解します。
- DTTを最終濃度5 mMまで添加し、55°Cで30分間タンパク質を還元します。
- サンプルを室温で冷却し、ヨードアセトアミド(IAA)を10 mMの濃度で15分間添加してタンパク質アルキル化反応を進めます。
- 適切な酵素(トリプシン、LysC)を使用してタンパク質を消化し、サンプルを37°Cで一晩インキュベートします。
- 1 μLの10%トリフルオロ酢酸(TFA)を加えて消化を停止します。
- ペプチドをクリーンアップし、前述のようにカスタム逆相C18マイクロカラムに濃縮します19。
- C18チップからペプチドをバッファーBで溶出します。
- 真空遠心分離機を使用して有機成分を除去し、さらなる分析のためにペプチドを5 μLの0.1%ギ酸に再懸濁します。
注:あるいは、非特異的結合剤を洗浄した直後にタンパク質消化を「オンビーズ」で実行できるため(ステップ8.1〜8.6)、プロトコルが高速になります。ただし、最適な実験タンパク質配列カバレッジを保証するために、溶液消化の標準と比較して「ビーズ上」固定化タンパク質を作動させる酵素の効率をテストすることをお勧めします。
- 液体クロマトグラフィー-タンデム質量分析(LC-MS/MS)
- 分析カラム(C18-固定相)を接続し、運転中は45°Cに保ちます。
- シングルカラム構成で、キャピラリーフィンガータイトフィッティング(20 μm x 550 mm)を介して、LCポンプの6ポートロータリーバルブの出口にカラムを接続します。
- LC 設定を次のように調整します。
- 制御された圧力(980 bar)下でペプチドをバッファーAにロードします。
- 5%-20%のバッファーBグラジエントを300 nL/minで59分間適用し、続いて20%-30%のバッファーBグラジエントを15分間、30%-65%のバッファーBグラジエントを5分間適用します。
- バッファーBの濃度を5分間で最大95%まで増加させ、さらに95%バッファーBで5分間のアイソクラティックステップを追加して、洗浄ステップを追加します。
- 質量分析計をデータ依存取得(DDA)モードで操作して、MSイベントとMSMSイベントを自動的に切り替えます。
- MS イベントと MSMS イベントに対して、自動ゲイン制御 (AGC) ターゲット値 3 x 106 と 1 x 105 を使用して、ループ カウントを 15 に定義します。
- 最大許容イオン蓄積時間を、分解能60 KのMSの場合は20 ms、分解能15 KのMSMSの場合は100 msに設定します。
- 28%の正規化された衝突エネルギーを使用して、20秒の動的排除時間で高衝突解離(HCD)フラグメンテーション実験を実行します。
- ソースパラメータを次のように操作します。
スプレー電圧:1.7 kV
毛細管電圧:275°C
シースも補助ガスも使用しない
注:このプロトコルでは、超高速液体クロマトグラフィー(UHPLC)質量分析(MS)分析は、ハイブリッドトリプル四重極orbitrap装置と組み合わせたLCシングルカラムセットアップを使用して特別に実行されています(材料表)。他のLCMSシステムも使用できますが、パラメータの適応が推奨されます。
- データ分析
- [読み込み] ボタンを使用して、生ファイルをインポートします。
- [ 実験の設定 ] ボタンをクリックして、実験名を定義します。
- グループ固有のパラメータセクションを入力して、識別に関連するすべてのパラメータを指定します。
消化に使用される酵素:トリプシン/ P
胸の谷間を逃した:最大3つ
修正済み修飾: カルバミドメチル化
可変修飾:N-アセチル(タンパク質)、酸化(M) - UniprotKB などのパブリック データベースから入手できる更新された FASTA ファイルをアップロードします。
- 選択したデータベースのソースに応じて正しい解析ルールを指定します。
- タンパク質とペプチドの両方について、親子関係の偽発見率(FDR)値= 1を定義します。
- ラベルフリー定量(LFQ)オプションをラベルフリー定量タブに追加します。
- LFQ 比の最小カウントは 2 に保ちます。
注:ここでは、MaxQuant24 とPerseusソフトウェア25 を使用して、タンパク質の定量とその後の統計分析をそれぞれ実行するデータ分析について説明します。しかしながら、データ分析は、他の任意の市販または無料のバイオインフォマティクスを用いて行うことができる。FDRは、ターゲットデコイデータベースベースのアプローチ26を使用して推定されます。FDRが0.01に等しいペプチドおよびタンパク質は、同定されたペプチドおよびタンパク質が1%の偽陽性を含むと予想されることを意味する。
- 統計解析
- タンパク質グループ.txtファイルをロードして、タンパク質レベルで統計解析を実行します。
- LFQ 値をメイン列として定義します。
- カテゴリ列に基づいて行をフィルタリングすることにより、「逆」と「汚染物質」を削除します。
- カテゴリ注釈行を使用して、さまざまな実験条件をグループ化します。
- データ行列を減らし、以前に定義した各グループの有効な値の数を選択します。
- 実験条件により適した統計的検定を選択します(すなわち、 t検定、複数サンプル検定ANOVA)。
- FDRベースの計算で有意なヒントのカットオフを決定します。通常、0.01 と 0.05 の両方が調整済み p 値のしきい値として受け入れられます。
- 火山プロット表現を使用して、 t検定統計量に基づく差分解析の結果を視覚化します。
- 最終結果テーブルをさらに編集するために、最終マトリックスを.txt形式でエクスポートします。
注:構成フォルダーには、グローバルプロテオミクス実験で一般的な汚染物質と見なされるケラチンなどのタンパク質を含むFASTAファイルが含まれており、出力テーブルでは+のフラグが付けられています。本研究では、2つのサンプル条件を持ち、統計分析に t検定を使用します。
11. ウェスタンブロットによる結果検証
- サンプル調製
- 適切な量の4xサンプルローディングバッファーをIN、FT、およびELの各アリコートに追加します。
- サンプルを95°Cで5分間煮沸します。
- チューブの上部から蒸発したサンプルを回収するための高速スピン。
- SDS-PAGEとゲル転写
- サンプルを4%〜12%の変性ポリアクリルアミドゲルにロードします。
- MES SDSランニングバッファーを使用して、120 Vで1.5時間ゲルを実行します。
- 製造元の指示に従って、半乾式転写カセットを使用してニトロセルロース膜上にゲルを移します。15 V で 10 分の転送をお勧めします。
- 免疫検出
- 穏やかに攪拌しながら、10%ウシ血清アルブミン(BSA)で室温で1時間膜をブロックします。
- 製造元の指示に従って、5%BSAで調製した一次抗体をTBSTに追加します。穏やかに攪拌しながら、4°Cで一晩または室温で1時間放置します。
- 膜をTBSTで3回、毎回5分間洗浄します。
- TBSTで調製した二次抗体を撹拌下で室温で1時間添加する。
- 膜をTBSTで3回、毎回5分間洗浄します。
- ブロットイメージャーを使用して結果を視覚化します。
注:当社のRNAの既知のバインダーであるTDP-43を検出するために、組換えウサギモノクローナル抗体を使用し、メンブレンとともに4°Cで一晩放置しました。 二次抗体として抗ウサギIgG西洋ワサビペルオキシダーゼ(HRP)を用いたが、蛍光二次抗体も同様に機能する。メンブレン上の抗体を可視化するために、メンブレンをクラリティウエスタンECL基質とともに1分間インキュベートしてから、ChemiDocイメージングシステムでイメージングしました。
結果
提案されたプロトコルの有効性を検証するために、ここに提示されたPD実験は、TDP-4320に特異的に結合するようにインシリコで設計されたビオチン化RNAアプタマーを用いて実施された。このRNAは、高い結合親和性(Kd = 90 nM)20でタンパク質標的に結合します。ここで、配列5'-CGGUGUUGCU-3'のこのRNAは、「+RNA」という名称で称される。陰性対照として、+RNAの逆相補配列(ここでは「-RNA」と呼ばれる)を用いた。その配列は5'-AGCAACACCG-3'である。-RNAは、TDP-43(Kd = 1.5 μM)に対して有意に低い結合親和性を示します19。本明細書に記載されるプロトコールの目的のために、これらのRNAオリゴヌクレオチドは、ビオチン分子にコンジュゲートして購入され、ストレプトアビジンビーズへの結合を可能にする。+RNAは、ビオチンと核酸のリン酸基の間に15原子のトリエチレングリコールスペーサーを含む3'末端にビオチン-TEGで購入しました。-RNAは代わりに、その5'末端にビオチンを有し、アミノC6リンカーを介して核酸に結合した。しかし、RNAベイトの設計が堅牢であり、リンカーとRNAの間に構造的または化学的干渉がない限り、ビオチン結合のための他の位置および他のリンカー長を採用することができる。
PD後に+RNAプローブに結合したメインタンパク質の同一性を知ることで、質量分析(MS)とウェスタンブロット(WB)の両方を使用して、溶出液中のTDP-43を同定することにより、プロトコルの検証が可能になりました(図1)。
MS分析は、+RNAまたは-RNAのいずれかを用いて実施した4つのPD反復について実施した(図2)。+RNAと-RNAの相互作用の同定はこのプロトコルの範囲を超えていますが、プロトコルの精度を検証するいくつかの結果が報告されています。注目すべきことに、有意に濃縮されたタンパク質を火山プロットにプロットすると、+RNAから溶出された総タンパク質含有量と濃縮タンパク質は、-RNAから回収されたものよりも有意に高いことが明らかになりました(図2)。これは、同じ長さと構造含量(直鎖状)を有するにもかかわらず、+RNAはより多くの特異的相互作用を確立することができ、それらは高塩で溶出ステップまで保持されることを意味します。代わりに、-RNAは、洗浄ステップ中に破壊される非特異的接触の数が多い可能性があります。予想通り、TDP-43は+RNA20のユニークな相互作用因子として同定されました。+RNAで行った4つのPD反復の平均ラベルフリー定量(LFQ)は31.96 ± 0.56ですが、タンパク質は-RNAの相互作用物質の中で同定されていません。さらに、+RNAのすべてのユニークな相互作用物質の中で、TDP-43は最も豊富に濃縮されたタンパク質であることがわかりました。
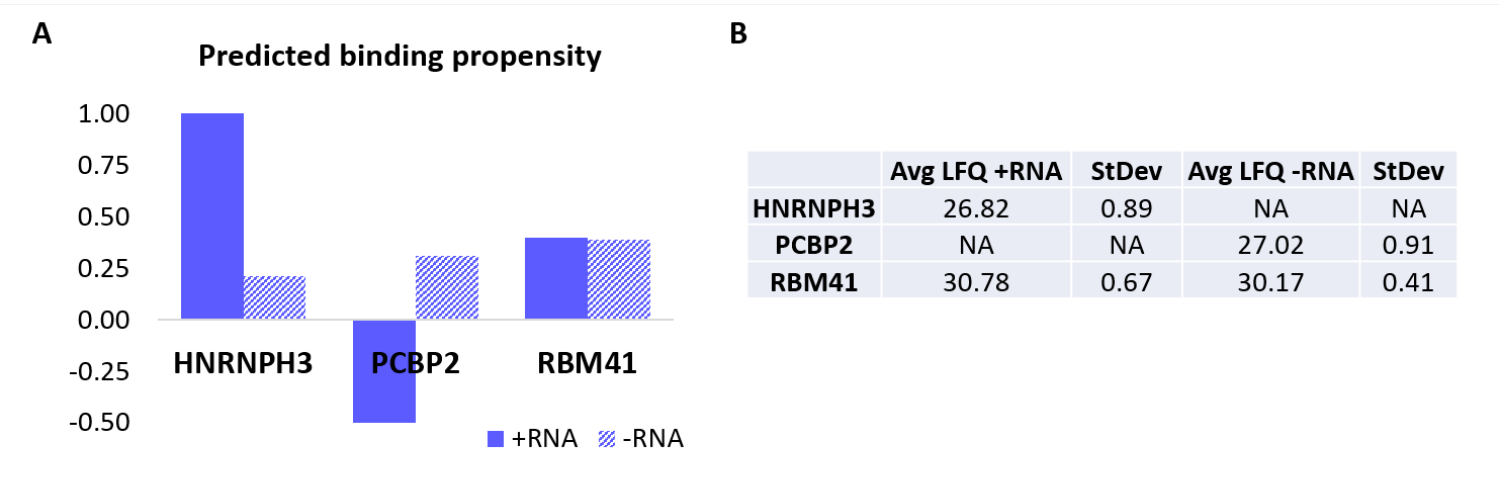
プロトコルをさらに検証するために、社内アルゴリズムcatRAPID18,19を使用して、他のどのタンパク質が+RNAまたは-RNAのいずれかに特異的に結合するかを計算的に予測しました。特に、+RNAおよび-RNAとヒトプロテオームを構成するタンパク質との相互作用スコアは、以前の研究27で定義されているように、catRAPIDの「相互作用傾向」機能を使用して計算されました。高い信頼性でスコアを付けたタンパク質のうち、HNRNPH3は選択的に+RNA(+RNA相互作用スコア= 1.01;-RNA相互作用スコア=0.21)と結合し、PCBP2は-RNAと特異的に相互作用すると予測されました(+RNA相互作用スコア= -0.5;-RNA相互作用スコア= 0.31)(図3A)。さらに、タンパク質RBM41は、両方のRNAオリゴヌクレオチドについて無差別であると予測された(+RNA相互作用スコア= 0.4;-RNA相互作用スコア= 0.39)(図3A)。MS分析では、+RNAと-RNAのPDにそれぞれHNRNPH3とPCBP2が存在することを確認しましたが、RBM41は両方と相互作用していることがわかりました(図3B)。
WBは、結果をさらに確認するためにTDP-43の存在を検出するために使用され、プロトコルの最適化中に使用されました(図4)。ここで説明する手順では、異なるサンプルを異なる段階で収集した。インプットサンプル(IN)は、溶解バッファーで希釈された全タンパク質で構成されていました。フロースルー(FT)は、ビオチン化RNAでプレコーティングされたストレプトアビジンビーズを用いて全タンパク質を一晩インキュベートした後に得られ、RNAに結合しなかったタンパク質の画分を表す。最後に、溶出液(EL)には、FTステップとELステップの間に150 mM塩と0.1%トリトン-Xによる3つの洗浄ステップが最も弱い相互作用が除去されたはずであるため、調査中のRNAを特異的に認識するすべてのタンパク質が含まれていました。
各反復について、同量(5% v/v)のIN、FT、およびELをSDS-PAGE上で並行して実行し、抗TDP-43抗体で染色しました(図4)。+RNAの場合、TDP-43のバンドはINとELで観察され、総タンパク質抽出物の最初から存在するタンパク質は、洗浄ステップ中に+RNAによって保持され、最後に高塩緩衝液でのみ溶出されることを示しています。TDP-43は-RNAのINにも存在していましたが、タンパク質に対応するバンドはFTでも見られ、このRNAがTDP-43に結合しないことを示しています。ELにTDP-43バンドがないことは、この結果を裏付けています。
プロトコルの最適化中、RNA配列に特異的に結合したタンパク質の溶出は、1 M NaClを含む溶出バッファー(EB1)と2 M NaClを含む溶出バッファー(EB2)の両方でプローブされました(図4)。いずれかのEBで得られた溶出液をSDS-PAGE上で比較し、抗TDP-43抗体でブロットした。次いで、得られた画像をImageJ28 で分析し、2つのバッファーで溶出されたTDP-43量の差を定量化した。全体として、有意差は観察されず、これらのアッセイ内では、1 M塩が最も強いタンパク質-RNA相互作用を破壊するのに十分であると結論付けました。
全体として、MSおよびWBについてここで報告された結果は、このプロトコルが特定の方法で特定のRNAのタンパク質相互作用物質を捕捉するのに効率的であり、ダウンストリーム分析と互換性のあるバッファーでの溶出を可能にすることを示しています。

図1:提案されたプロトコルで使用される実験パイプラインのスケッチ 。 (a)ビオチン化RNAオリゴヌクレオチドを溶解バッファー中で適切な濃度で調製する。(B)磁気ストレプトアビジンビーズを洗浄し、酵母tRNAでブロックし、ビオチン化RNAを装填する。(C)培養哺乳動物細胞株由来の総タンパク質抽出物をビーズ-RNA混合物に添加する。(D)非特異的相互作用を除去するために複数回の洗浄が行われる。(E)特定のタンパク質相互作用物質を高張溶液でRNAから分離する。(F)相互作用因子の正体は質量分析によって明らかにされ、特定の症例はウェスタンブロットによって検証されます。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:ラベルフリーMSベースのタンパク質定量のための分析戦略 。 (A)溶出したタンパク質を冷アセトン中で一晩沈殿させる。その後、タンパク質を変性させ、溶液中で消化します。タンパク質分解ペプチドは濃縮され、脱塩されます。(B)ペプチドは、「ショットガンアプローチ」を使用してLC-MS/MS を介して 分析されます。(C) 生データの処理と分析は、それぞれMaxQuantとPerseusソフトウェアを使用して実行されます。(D)統計的に有意に濃縮されたタンパク質が火山プロットに表示されます。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:+RNAおよび-RNAの予測された相互作用傾向と実験的に決定された相互作用との相関。 (A) ネコのRAPID相互作用スコアは、HNRNPH3、PCBP2、およびRBM41と比較して、+RNAに対するHNRNPH3および-RNAに対するPCBP2の優先的な結合を示し、一方、RBM41は両方のRNA配列に無差別に結合すると予測されています。(B)+RNAおよび-RNAで行われたプルダウンからの質量分析によって決定されたラベルフリー定量平均。分析により、HNRNPH3は+RNAのみに結合し、PCBP2は-RNAのみに結合し、RBM41は両方に等しく結合することが確認されています。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図4:選択したRNA配列の相互作用因子におけるTDP-43の有無のウェスタンブロット検証。 WBメンブレンは抗TDP-43抗体で処理されています。IN = 入力;FT =フロースルー;EL(EB1) = 溶出バッファー1による溶出;EL(EB2) = 溶出バッファー2による溶出;記号「+」は、+RNAで実行されたプルダウンに由来するサンプルを示します。記号「-」は、-RNAを用いて実施したプルダウンに由来するサンプルを示す。レーン1にはタンパク質ラダーが含まれています。TDP-43は矢印で示されています。WBは、TDP-43が+RNAインタラクターの間では見出されるが、-RNA相互作用者の間では見出されないことを示している。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
| バッファ名 | 組成 | |||||
| 10x トランスファーバッファ | 250 mMトリス、1.92 Mグリシン、1%SDS、20%メタノール。使用前に10倍に希釈する | ティッカー | ||||
| 20X MES SDS ランニングバッファ | 1 M MES, 1 M トリス, 2% SDS, 20 mM EDTA.pHを7.3に調整します。使用前に20倍に希釈 | |||||
| 4xサンプルローディングバッファ | 0.25 M トリス塩基、0.28 M SDS、40% グリセロール、20% 2-メルカプトエタノール、4 mg/ml ブロムフェノールブルー | |||||
| 溶出バッファー 1 | 20 mM リン酸 pH 7.5、1 M NaCl、0.5 mM EDTA、0.1 % トリトン X-100、1 mM DTT (定量後に添加) | |||||
| 溶出バッファー 2 | 20 mM リン酸 pH 7.5、2 M NaCl、0.5 mM EDTA、0.1 % トリトン X-100、1 mM DTT (定量後に添加) | |||||
| 溶解バッファー | 10 mM トリス塩酸塩 pH 7.4、150 mM NaCl、0.5 mM EDTA、0.1 % Triton X-100、1 mM DTTおよびプロテアーゼ阻害剤 | |||||
| Tween-20を含むトリス緩衝生理食塩水 | 1 M トリス塩酸塩 pH 7.4, 3 M NaCl, 2.0% トゥイーン-20 | |||||
| 洗浄バッファー 1 | 10 mM トリス塩酸塩 pH 7.4、150 mM NaCl、0.5 mM EDTA、0.1 % トリトン TM X-100、1 mM DTT およびプロテアーゼ阻害剤 | |||||
| 洗浄バッファー 2 | 25 mM Hepes pH 8、150 mM NaCl、0.5 mM EDTA、0.1 % Triton X-100、1 mM DTT およびプロテアーゼ阻害剤 | |||||
| バッファ A | 0.1%ギ酸 | さん | ||||
| バッファ B | アセトニトリル60%、ギ酸0.1% | |||||
| 変性バッファー | 8M尿素、50 mMトリス塩酸塩 | |||||
表 1: PD および MS バッファー。 プルダウン実験(PD)または質量分析(MS)に使用されるバッファーの名前と組成。
ディスカッション
この研究では、ビオチン化RNAオリゴヌクレオチドを使用してタンパク質相互作用因子を捕捉するために実行されたPDプロトコルの最適化について報告しています。ここで説明するプロトコルは、実行が簡単で、材料がほとんど必要なく、信頼性の高い結果が得られます。重要なことに、このプロトコルの最も新しい側面は、完全に インシリコで 設計され、タンパク質標的に特異的なRNAベイトの使用と、ストレプトアビジンをビオチンから解離させて高温処理するのではなく、高塩溶液との相互作用を直接破壊することによってRNAベイトに結合したすべてのタンパク質を溶出することです。
このプロトコルは、ビオチンとストレプトアビジン29,30との間の結合の強さを利用する。選択したストレプトアビジンビーズによると、ビオチン化RNAの負荷は、先に進む前にテストおよび定量する必要があります。また、RNAの三次元フォールディングは、ストレプトアビジンへのビオチンの曝露を制限する可能性があるため、ビーズへのローディング効率に影響を与える可能性があります。非ビオチン化tRNAでビーズをブロックすると、ビーズとの非特異的相互作用が制限されるため、結果の清浄度が向上します。ローディングバッファーと溶出バッファーは、ダウンストリームアプリケーションに応じて選択する必要があります。ここでは、大部分の用途に適し、潜在的なタンパク質複合体を保存するために開発された非常に穏やかな条件が提案されました。ただし、この方法は適応性が高いです。ユーザーは任意の細胞株と任意のRNAサイズを選択でき、RNAの折り畳み/折り畳み後にプロトコルを繰り返して、結合特性に対する構造の影響を決定することができます。
このプロトコルの別のオリジナルの態様は、結果の正確性を確実にするための インシリコ 予測ツールの使用である20。どのタンパク質を目的のRNAの相互作用物質として同定すべきかを事前に知ることは、プロトコルの技術的側面を検証するという前例のない利点をもたらします。例えば、単純なWB分析を使用して、MS分析を進める前に、プロトコルのさまざまなステップに由来するサンプル中の既知のタンパク質ターゲットの存在を確認することができますが、これは特殊な機器を必要とし、よりコストがかかります。さらに、社内のタンパク質-RNA予測アルゴリズムである catRAPID20を使用して、標的タンパク質に特異的 なde novo RNAを設計する方法が最近報告されました。最近まで、標的タンパク質のDNA/RNAアプタマーを設計するための唯一の利用可能なパイプラインは、SELEX(指数関数的濃縮によるリガンドの系統的進化)アプローチでした31。 インシリコ 法は、RNAアプタマーのはるかに迅速で費用対効果の高い設計を可能にします。
この方法の主な制限は、ヌクレアーゼフリーのバッファーおよびツールで作業する必要性に関連しています。さらに、de novo設計したRNAと標的タンパク質との結合をPD前にin vitroで確認する必要があると考えられる場合は、タンパク質を産生および精製し、生物物理学的アプローチで結合を決定する必要があります。これは、モノクローナル抗体の産生と共有される制限です。
これらの小さな問題にもかかわらず、ここで紹介するようなRNAとタンパク質の相互作用をマッピングする信頼性の高い方法は、科学者を高分子ネットワークと、癌、心筋症、糖尿病、微生物感染症、遺伝性および神経変性疾患に関与するものなど、多くの生理学的および病理学的メカニズムの複雑な主役を明らかにすることに近づけることができます。
開示事項
著者には、競合する金銭的利益やその他の利益相反はありません。
謝辞
著者らは、タルタリア教授とクオモ博士の研究グループが提供する支援に感謝したいと思います。E.Z.は、マリー・スクロドフスカ・キュリー助成金契約第754490号に基づき、欧州連合のホライズン2020研究およびイノベーションプログラムのMINDEDフェローシップから資金提供を受けました。
資料
| Name | Company | Catalog Number | Comments |
| 6-well tissue culture plates | VWR | 10861-554 | CELLS |
| Cell scrapers | BIOSIGMA | 10153 | CELLS |
| Dulbecco′s Modified Eagle′s Medium (DMEM) | Thermo Fisher Scientfic | 11995065 | CELLS |
| Fetal Bovine Serum, qualified, heat inactivated, Brazil | Thermo Fisher Scientfic | 10500064 | CELLS |
| Phosphate Buffer Saline (PBS, Waltham, MA) | Thermo Fisher Scientfic | 14190169 | CELLS |
| Trypsin (0.25%), phenol red | Thermo Fisher Scientfic | 15050065 | CELLS |
| Anti-rabbit IgG horseradish peroxidase (HRP) | Cellsignal | 7070 | PD |
| Biotinylated RNA | Eurofins | Custom RNA oligonucleotides | PD |
| Bovine serum albumin | Sigma-Aldrich | A9418 | PD |
| Clarity Western ECL Substrate, 500 ml | Biorad | 1705061 | PD |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Merck - Sigma Aldrich | 5056489001 | PD |
| NuPAGE 4 to 12%, Bis-Tris, 1.0 mm, Mini Protein Gel, 10-well | Invitrogen | NP0321BOX | PD |
| Recombinant anti-TDP43 antibody | Abcam | ab109535 | PD |
| Ribonucleic acid, transfer from baker's yeast (S. cerevisiae) | Merck - Sigma Aldrich | R5636-1ML | PD |
| Streptavidin Mag Sepharose | Merck - Sigma Aldrich | GE28-9857-99 | PD |
| Trans-Blot Turbo RTA Mini 0.2 µm PVDF Transfer Kit | Biorad | 1704272 | PD |
| Acetone | Thermo Fisher Scientfic | 022928.K2 | MS |
| C18 cartridge | Thermo Fisher Scientfic | 13-110-018 | MS |
| Dithiothreitol (DTT) | Thermo Fisher Scientfic | 20290 | MS |
| EASY-Spray HPLC Columns | Thermo Scientific | ES902 | MS |
| iodoacetamide (IAA) | Sigma Aldrich S.r.l. | I6125 | MS |
| Lys-C/Trypsin | Promega | V5073 | MS |
| Trifluoroacetic acid (TFA) | Thermo Fisher Scientfic | 28904 | MS |
| Urea | Thermo Fisher Scientfic | J75826.A7 | MS |
| Equipment | |||
| ChemiDoc imaging system | Bio-Rad | CELLS | |
| Dyna Mag -2 , Magnetic rack | Invitrogen | CELLS | |
| Forma Series 3 water jacketed C02 incubator | Thermo Scientific | PD | |
| PROTEAN II xi cell , power supply for PAGE applications | Bio-Rad | PD | |
| Rotating wheel, rotator SB3 | Stuart | PD | |
| Water bath set at 37 °C | VWR | PD | |
| XCell SureLock Mini-Cell electrophoresis system | ThermoFisher Scientific | MS | |
| Easy-nLC 1200 UHPLC | Thermo Scientific | MS | |
| Q exactive Mass Spectrometer | Thermo Scientific | MS | |
| Software | Version | ||
| MaxQuant | 2.0.3.0 | MS | |
| Perseus | 1.6.14.0 | MS |
参考文献
- Gebauer, F., Schwarzl, T., Valcárcel, J., Hentze, M. W. RNA-binding proteins in human genetic disease. Nature Reviews Genetics. 22 (3), 185-198 (2021).
- Hentze, M. W., Castello, A., Schwarzl, T., Preiss, T. A brave new world of RNA-binding proteins. Nature Reviews Molecular Cell Biology. 19 (5), 327-341 (2018).
- Castello, A., et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 149 (6), 1393-1406 (2012).
- Cooper, T. A., Wan, L., Dreyfuss, G. RNA and disease. Cell. 136 (4), 777-793 (2009).
- Qin, H., et al. RNA-binding proteins in tumor progression. Journal of Hematology & Oncology. 13 (1), 90 (2020).
- Nussbacher, J. K., Tabet, R., Yeo, G. W., Lagier-Tourenne, C. Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron. 102 (2), 294-320 (2019).
- Duan, R., Sharma, S., Xia, Q., Garber, K., Jin, P. Towards understanding RNA-mediated neurological disorders. Journal of Genetics and Genomics. 41 (9), 473-484 (2014).
- Maziuk, B., Ballance, H. I., Wolozin, B. Dysregulation of RNA binding protein aggregation in neurodegenerative disorders. Frontiers in Molecular Neuroscience. 10, 89 (2017).
- Zielinski, J., et al. In vivo identification of ribonucleoprotein-RNA interactions. Proceedings of the National Academy of Sciences. 103 (5), 1557-1562 (2006).
- Armaos, A., Zacco, E., Sanchez de Groot, N., Tartaglia, G. G. RNA-protein interactions: Central players in coordination of regulatory networks. BioEssays. 43 (2), 2000118 (2021).
- Weidmann, C. A., Mustoe, A. M., Jariwala, P. B., Calabrese, J. M., Weeks, K. M. Analysis of RNA-protein networks with RNP-MaP defines functional hubs on RNA. Nature Biotechnology. 39 (3), 347-356 (2021).
- Graindorge, A., et al. In-cell identification and measurement of RNA-protein interactions. Nature Communications. 10 (1), 5317 (2019).
- Ule, J., Hwang, H. W., Darnell, R. B. The future of cross-linking and immunoprecipitation (CLIP). Cold Spring Harbor Perspectives in Biology. 10 (8), 032243 (2018).
- Ascano, M., Hafner, M., Cekan, P., Gerstberger, S., Tuschl, T. Identification of RNA-protein interaction networks using PAR-CLIP. Wiley Interdisciplinary Reviews. RNA. 3 (2), 159-177 (2012).
- McHugh, C. A., Russell, P., Guttman, M. Methods for comprehensive experimental identification of RNA-protein interactions. Genome Biology. 15 (1), 203 (2014).
- Sugimoto, Y., et al. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. Genome Biology. 13 (8), (2012).
- Bayat, P., et al. SELEX methods on the road to protein targeting with nucleic acid aptamers. Biochimie. 154, 132-155 (2018).
- Armaos, A., Colantoni, A., Proietti, G., Rupert, J., Tartaglia, G. G. CatRAPID omics v2.0: Going deeper and wider in the prediction of protein-RNA interactions. Nucleic Acids Research. 49, 72-79 (2021).
- Agostini, F., et al. CatRAPID omics: A web server for large-scale prediction of protein-RNA interactions. Bioinformatics. 29 (22), 2928-2930 (2013).
- Zacco, E., et al. Probing TDP-43 condensation using an in silico designed aptamer. Nature Communications. 13 (1), 3306 (2022).
- Leppek, K., Stoecklin, G. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Research. 42 (2), 13 (2014).
- Zhang, Y., Lai, B. S., Juhas, M. Recent advances in aptamer discovery and applications. Molecules. 24 (5), 941 (2019).
- UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Research. 51, 523-531 (2023).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11 (12), 2301-2319 (2016).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13 (9), 731-740 (2016).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2 (8), 1896-1906 (2007).
- Bellucci, M., Agostini, F., Masin, M., Tartaglia, G. G. Predicting protein associations with long noncoding RNAs. Nature Methods. 8 (6), 444-445 (2011).
- Gallo-Oller, G., Ordoñez, R., Dotor, J. A new background subtraction method for Western blot densitometry band quantification through image analysis software. Journal of Immunological Methods. 457, 1-5 (2018).
- Weissinger, R., Heinold, L., Akram, S., Jansen, R. P., Hermesh, O. RNA proximity labeling: A new detection tool for RNA-protein interactions. Molecules. 26 (8), 2270 (2021).
- Hirsch, J. D., et al. Easily reversible desthiobiotin binding to streptavidin, avidin, and other biotin-binding proteins: Uses for protein labeling, detection, and isolation. Analytical Biochemistry. 308 (2), 343-357 (2002).
- Sefah, K., Shangguan, D., Xiong, X., O'Donoghue, M. B., Tan, W. Development of DNA aptamers using cell-SELEX. Nature Protocols. 5 (6), 1169-1185 (2010).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved