
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
الحمض النووي المربوطة الحمض النووي RNA بوليمراز للبرمجة في النسخ المختبري والحوسبة الجزيئية
In This Article
Summary
نحن نصف هندسة رواية الحمض النووي المربوطة T7 الحمض النووي الحمض النووي RNA بوليمرات لتنظيم ردود الفعل في المختبر النسخ. نناقش خطوات تخليق البروتين وتوصيفه، والتحقق من صحة تنظيم النسخ لإثبات المفهوم، ومناقشة تطبيقاته في الحوسبة الجزيئية، والتشخيص، ومعالجة المعلومات الجزيئية.
Abstract
تكنولوجيا النانو الحمض النووي تمكن البرمجة الذاتي التجميع من الأحماض النووية في الأشكال التي يحددها المستخدم وديناميات لتطبيقات متنوعة. هذا العمل يدل على أن المفاهيم من تكنولوجيا النانو الحمض النووي يمكن استخدامها لبرنامج النشاط الأنزيمي من البوليميراز T7 الحمض النووي الريبي المستمدة من phage (RNAP) وبناء شبكات تنظيمية الجينات الاصطناعية قابلة للتطوير. أولا، تم تصميم RNAP T7 المربوطة oligonucleotide عن طريق التعبير عن RNAP N-TERMINALLY SNAP الموسومة والربط الكيميائي اللاحق للSNAP-tag مع oligonucleotide المعدلة بنزيلجوانين (BG). بعد ذلك ، يتم استخدام إزاحة حبلا الحمض النووي لبرنامج نسخ البوليميراز عند الطلب. بالإضافة إلى ذلك، يمكن استخدام تجميعات الحمض النووي المساعدة ك "عوامل نسخ اصطناعية" لتنظيم التفاعلات بين T7 RNAP المبرمجة بالحمض النووي مع قوالب الحمض النووي الخاصة به. يمكن لهذه الآلية التنظيمية للنسخ في المختبر تنفيذ مجموعة متنوعة من سلوكيات الدوائر مثل المنطق الرقمي ، وردود الفعل ، المتتالية ، والتعدد. تسهل قابلية إنشاء هذه البنية التنظيمية الجينية تجريد التصميم والتوحيد القياسي والتحجيم. وستمكن هذه الميزات من النماذج الأولية السريعة للأجهزة الوراثية المختبرية لتطبيقات مثل الاستشعار البيولوجي، والكشف عن الأمراض، وتخزين البيانات.
Introduction
تستخدم حوسبة الحمض النووي مجموعة من أوليغونوكليوتيدات مصممة كوسيلة للحساب. تتم برمجة هذه oligonucleotides مع تسلسل لتجميع ديناميكي وفقا للمنطق المحدد من قبل المستخدم والاستجابة لمدخلات الحمض النووي محددة. في دراسات إثبات المفهوم ، يتكون ناتج الحساب عادة من مجموعة من أوليغونوكليوتيدات الفلورسنت التي يمكن اكتشافها عن طريق الكهروضوئيات الهلامية أو قارئات لوحات الفلورسينس. على مدى السنوات ال 30 الماضية ، وقد ثبت الدوائر الحسابية الحمض النووي معقدة على نحو متزايد ، مثل مختلف الشلالات المنطق الرقمي ، وشبكات التفاعل الكيميائي ، والشبكات العصبية1،2،3. للمساعدة في إعداد هذه الدوائر الحمض النووي، وقد استخدمت نماذج رياضية للتنبؤ وظائف الدوائر الجينية الاصطناعية4،5، وقد وضعت أدوات حسابية لتصميم تسلسل الحمض النووي متعامدة6،7،8،9،10 . بالمقارنة مع أجهزة الكمبيوتر القائمة على السيليكون ، وتشمل مزايا أجهزة الكمبيوتر الحمض النووي قدرتها على التفاعل مباشرة مع الجزيئات الحيوية ، وتعمل في حل في غياب إمدادات الطاقة ، فضلا عن إحكام واستقرارها العام. مع ظهور تسلسل الجيل القادم ، كانت تكلفة تصنيع أجهزة الكمبيوتر الحمض النووي في انخفاض على مدى العقدين الماضيين بمعدل أسرع من قانون مور11. تطبيقات هذه الحواسيب المستندة إلى الحمض النووي بدأت تظهر الآن ، مثل لتشخيص المرض12،13، لتشغيل الفيزياء الحيوية الجزيئية14، وكمنصات لتخزين البيانات15.

الشكل 1:آلية إزاحة حبلا الحمض النووي بوساطة موطئ القدم. موطئ القدم، δ، هو تسلسل حر وغير منضم على مزدوجة جزئية. عندما يتم تقديم مجال تكميلي (δ*) على حبلا ثان ، يعمل نطاق δ الحر كمسند إصبع قدم للتهجين ، مما يسمح لبقية الخيط (Ο*) بإزاحة منافسه ببطء من خلال رد فعل قابل للعكس مضغوط / فك الضغط يعرف باسم هجرة حبلا. مع زيادة طول δ، ينخفض ΔG لرد الفعل الأمامي، ويحدث الإزاحة بسهولة أكبر. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
حتى الآن ، فإن غالبية أجهزة الكمبيوتر الحمض النووي استخدام عزر راسخة في مجال تكنولوجيا النانو الحمض النووي الديناميكي المعروف باسم التشريد حبلا الحمض النووي بوساطة موطئ القدم (TMDSD ، الشكل 1)16. يتكون هذا الشعار من ثنائية جزئيا تقطعت بهم السبل الحمض النووي (dsDNA) المزدوج المزدوج عرض قصيرة "موطئ قدم" يتدلى (أي 7- إلى 10 النيوكليوتيدات (NT)). يمكن أن تتفاعل خيوط "الإدخال" من الحمض النووي مع الدوبلين الجزئي من خلال موطئ القدم. يؤدي هذا إلى إزاحة أحد الخيوط من الدوبلين الجزئي، ويمكن أن يكون هذا الخيط المحرر بمثابة مدخل للدوبلين الجزئيين في المصب. وهكذا، TMDSD تمكن تتالي الإشارات ومعالجة المعلومات. من حيث المبدأ ، يمكن أن تعمل زخارف TMDSD المتعامدة بشكل مستقل في الحل ، مما يتيح معالجة المعلومات الموازية. كان هناك عدد من الاختلافات على رد فعل TMDSD، مثل تبادل حبلا الحمض النووي بوساطة موطئ القدم (TMDSE)17، "تسرب" موطئ قدم مع نطاقات مزدوجة طويلة18،أضواء غير متطابقة تسلسل19،و "اليد" بوساطة حبلاالنزوح 20. تسمح مبادئ التصميم المبتكرة هذه بمزيد من حيوية وديناميكيات TMDSD المضبوطة بدقة لتحسين أداء حوسبة الحمض النووي.
الدوائر الجينية الاصطناعية، مثل الدوائر الجينية النسخية، هي أيضا قادرة على حساب21،22،23. يتم تنظيم هذه الدوائر من خلال عوامل نسخ البروتين ، والتي تنشط أو تقمع نسخ الجين عن طريق الربط بعناصر الحمض النووي التنظيمية المحددة. بالمقارنة مع الدوائر المستندة إلى الحمض النووي ، فإن الدوائر النسخية لها العديد من المزايا. أولا، النسخ الأنزيمي له معدل دوران أعلى بكثير من دوائر الحمض النووي الحفازة الحالية، وبالتالي توليد نسخ أكثر من الإخراج لكل نسخة واحدة من المدخلات وتوفير وسيلة أكثر كفاءة لتضخيم الإشارات. بالإضافة إلى ذلك ، يمكن أن تنتج الدوائر النسخية جزيئات وظيفية مختلفة ، مثل aptamers أو ترميز الحمض النووي الريبي messenger (mRNA) للبروتينات العلاجية ، كخرجات حساب ، والتي يمكن استغلالها لتطبيقات مختلفة. ومع ذلك ، فإن أحد القيود الرئيسية للدوائر النسخية الحالية هو عدم قابليتها للتوسع. وذلك لأن هناك مجموعة محدودة جدا من عوامل النسخ القائمة على البروتين المتعامدة، وتصميم دي نوفو من عوامل نسخ البروتين الجديدة لا تزال صعبة من الناحية الفنية وتستغرق وقتا طويلا.

الشكل 2: التجريد وآلية "الحبل" و "قفص" مجمع البوليميراز. (A و B) يتم وضع علامة على حبل أوليغونوكليوتيد أنزيميا إلى بوليمراز T7 من خلال رد فعل SNAP-tag. قفص يتكون من "فو" T7 المروج مع تراكم حبل تكملة يسمح لها التهجين إلى الحبل ومنع النشاط النسخي. (ج) عندما يكون المشغل(أ * ب *) موجودا ، فإنه يرتبط بمؤشر القدم على حبل oligonucleotide(ab)ويشرد منطقة b * من القفص ، مما يسمح بالنص. تم تعديل هذا الرقم من تشو وشيه27. الاختصارات: RNAP = RNA بوليمراز. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
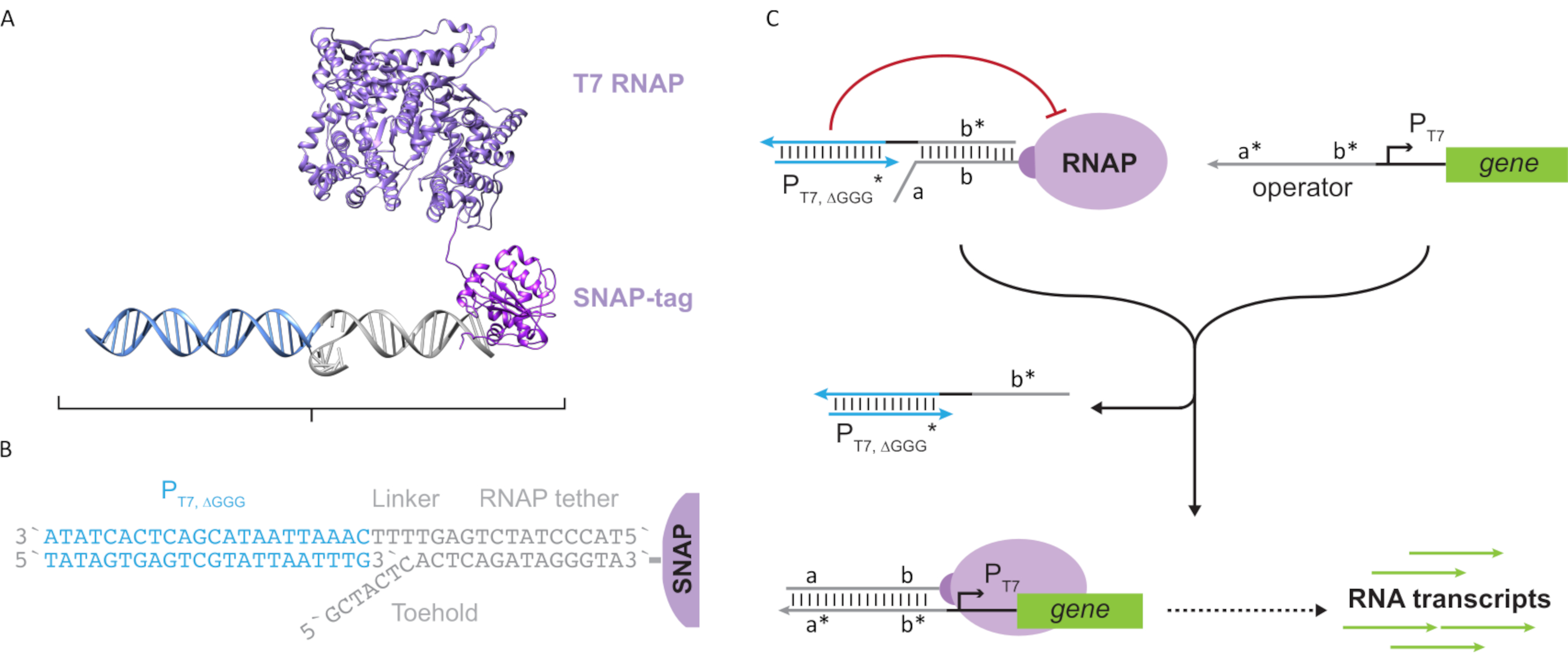
تقدم هذه الورقة لبنة جديدة للحوسبة الجزيئية تجمع بين وظائف الدوائر النسخية وقابلية التوسع في الدوائر القائمة على الحمض النووي. هذه الكتلة البناء هو T7 RNAP المرفقة بشكل مشترك مع حبل الحمض النووي واحد تقطعت بهم السبل (الشكل 2A). لتجميع هذا الحمض النووي المربوطة T7 RNAP، تم دمج البوليميراز إلى N-محطة SNAP-tag24 وأعرب عنه بشكل مركب في الإشريكية القولونية. ثم تفاعلت علامة SNAP مع أوليغونوكليوتيد وظيفية مع ركيزة BG. يسمح حبل أوليغونوكليوتيد بوضع الضيوف الجزيئيين على مقربة من البوليميراز عن طريق تهجين الحمض النووي. وكان أحد هؤلاء الضيوف مانع النسخ التنافسية المشار إليها باسم "قفص"، الذي يتكون من "فو" T7 المروج الحمض النووي المزدوج مع عدم وجود جين المصب(الشكل 2B). عندما ملزمة RNAP عبر الحبل oligonucleotide لها، القفص الأكشاك نشاط البوليميراز عن طريق المنافسة قوالب الحمض النووي الأخرى لربط RNAP، مما يجعل RNAP في حالة "إيقاف" (الشكل 2C).
لتنشيط البوليميراز إلى حالة "ON" ، تم تصميم قوالب الحمض النووي T7 مع نطاقات "المشغل" الوحيدة التي تقطعت بها السبل في المنبع لمروج T7 للجين. يمكن تصميم مجال المشغل (أي المجال أ * ب * الشكل 2C)لإزاحة القفص من RNAP عبر TMDSD ووضع RNAP في مكان قريب من مروج T7 للجين ، وبالتالي بدء النسخ. وبدلا من ذلك، صممت قوالب الحمض النووي أيضا حيث كان تسلسل المشغل مكملا لخيوط الحمض النووي المساعدة التي يشار إليها باسم "عوامل النسخ الاصطناعي" (أي فروع TFA و TFB في الشكل 3A). عندما يتم إدخال كلا الخيوط في رد الفعل ، فإنها سوف تتجمع في موقع المشغل ، وخلق مجال جديد شبه متجاورة أ * ب *. يمكن لهذا المجال ثم إزاحة القفص عبر TMDSD لبدء النسخ(الشكل 3B). ويمكن توفير هذه الخيوط إما بشكل خارجي أو إنتاجها.

الشكل 3:البرمجة الانتقائية لنشاط البوليميراز من خلال منشط تبديل مكون من ثلاثة مكونات. (أ)عندما تكون عوامل النسخ (TFA و TFB)موجودة ، فإنها ترتبط بمجال المشغل في المنبع للمروج ، مما يشكل تسلسلا زائفا تقطعت به السبل(a *b *) قادرا على إزاحة القفص من خلال إزاحة الحمض النووي بوساطة موطئ القدم. (ب) هذا النطاق * ب * يمكن أن تحل محل القفص عبر TMDSD لبدء النسخ. تم تعديل هذا الرقم من تشو وشيه27. المختصرات: TF = عامل النسخ؛ RNAP = RNA بوليمراز; TMDSD = إزاحة حبلا الحمض النووي بوساطة موطئ القدم. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
استخدام عوامل النسخ المستندة إلى الحمض النووي لتنظيم النسخ في المختبر يسمح للتنفيذ القابل للتطوير لسلوكيات الدوائر المتطورة مثل المنطق الرقمي ، وردود الفعل ، وتتالي الإشارات. على سبيل المثال، يمكن للمرء بناء سلاسل بوابة المنطق عن طريق تصميم تسلسل الحمض النووي بحيث النصوص من الجينات المنبع تنشيط الجينات المصب. أحد التطبيقات التي تستغل المتتالية والمتعددة التي يمكن أن تكون قادرة على هذه التكنولوجيا المقترحة هو تطوير دوائر الحوسبة الجزيئية أكثر تطورا للتشخيص المحمول ومعالجة البيانات الجزيئية. وبالإضافة إلى ذلك، يمكن دمج الحوسبة الجزيئية وقدرات التوليف الحمض النووي الريبي دي نوفو تمكين تطبيقات جديدة. على سبيل المثال، يمكن تصميم الدائرة الجزيئية للكشف عن واحد أو مزيج من الرنانات المعرفة من قبل المستخدم كمدخلات وإخراج الرنانات العلاجية أو mRNAs ترميز الببتيدات الوظيفية أو البروتينات للتطبيقات الطبية نقطة الرعاية.
Access restricted. Please log in or start a trial to view this content.
Protocol
1. إعداد المخزن المؤقت
ملاحظة: يمكن أن يحدث إعداد العازلة تنقية البروتين في أي يوم; هنا، تم القيام به قبل بدء التجارب.
- إعداد تحلل / توازن العازلة التي تحتوي على 50 mM تريس (هيدروكسي ميثيل)أمينوميثان (تريس)، 300 mM كلوريد الصوديوم (NaCl)، 5٪ الجلسرين، و 5 مليون β ميركابتوثانول (BME)، PH 8. إضافة 1.5 مل من 1M تريس, 1.8 مل من 5M NaCl, 1.5 مل من الجلسيرول, 25.2 مل من المياه deionized (ddH2O) في أنبوب الطرد المركزي 50 مل, وإضافة 10.5 ميكرولتر من 14.2 M BME قبل الاستخدام.
ملاحظة: يمكن أن يسبب تريس سمية حادة; وبالتالي، تجنب تنفس الغبار، وتجنب الجلد والعين الاتصال. BME سامة وينبغي أن تستخدم فقط في غطاء الدخان. من المهم إضافة BME الماضي، فقط قبل إعادة الإنفاق وتحلل الخلية. راجع الجدول 1 للحصول على صيغة المخزن المؤقت للتحلل. - إعداد عازلة غسل (pH 8) التي تحتوي على 50 متر تريس, 800 م م NaCl, 5٪ الجلسرين, 5 M M BME, و 20 mm imidazole. إضافة 1.5 مل من 1 M تريس, 4.8 مل من 5 M NaCl, 1.5 مل من الجلسيرول, و 22.2 مل من ddH2O في أنبوب الطرد المركزي 50 مل. قبل الاستخدام مباشرة، أضف 7 ميكرولتر من 14.2 M BME و 200 ميكرولتر من 2 M imidazole إلى 20 مل من الحل أعلاه.
ملاحظة: لمنع السمية الحادة بسبب إيميدازول، استخدم معدات الحماية الشخصية. من المهم إضافة BME وimidazole الماضي، فقط قبل غسل البروتين من العمود. راجع الجدول 2 للحصول على تركيبة المخزن المؤقت للغسيل. - إعداد العازلة elution (pH8) التي تحتوي على 50 mM تريس، 800 mM NaCl، 5٪ الجلسرين، 5 M M BME، و 200 mM imidazole. إضافة 0.5 مل من 1 M تريس, 1.6 مل من 5 M NaCl, 0.5 مل من الجلسيرول, و 6.4 مل من ddH2O إلى أنبوب الطرد المركزي 15 مل. قبل الاستخدام مباشرة، أضف 3.5 ميكرولتر من 14.2 M BME و 1 مل من 2 M imidazole إلى 10 مل من الحل أعلاه.
ملاحظة: من المهم إضافة BME وimidazole الماضي، فقط قبل eluting البروتين من العمود. راجع الجدول 3 للحصول على صيغة المخزن المؤقت ل elution. - إعداد مخزن تخزين 2x العازلة (لتكون مختلطة 1:1 مع الجلسرين) التي تحتوي على 100 متر ثلاثي، 200 مليون م م NaCl، 40 M M BME، و 2 M حمض الإيثيلينديامينتتراستيك (EDTA)، 0.2٪ من السطحي غير الأيونية (انظر جدول المواد). إعداد 50 مل من المخزن المؤقت عن طريق إضافة 5 مل من 1 M Tris، و 2 مل من 5 M NaCl، و 42.56 مل من ddH2O، و 200 ميكرولتر من 0.5 M EDTA، و 100 ميكرولتر من السطحي غير الأيونية إلى أنبوب طرد مركزي سعة 50 مل. اخلط حتى يصبح المحلول متجانسا، وتصفية المخزن المؤقت للتخزين من خلال فلتر حقنة 0.2 ميكرومتر، وإضافة 140.8 ميكرولتر من BME إلى الحل أعلاه قبل الاستخدام.
ملاحظة: لتجنب السمية الحادة بسبب EDTA، تجنب تنفس الغبار، وتجنب ملامسة الجلد والعين. من المهم إضافة BME الماضي وخلط المخزن المؤقت تخزين كامل 1:1 مع الجلسرين، فقط قبل تخزين البروتين النقي. راجع الجدول 4 للحصول على صيغة المخزن المؤقت للتخزين.
2. نمو الثقافة بين عشية وضحاها: اليوم الأول
- إعداد 1,000x مخزون كاناميسين عن طريق حل 500 ملغ من الكاناميسين في 10 مل من ddH2O.
ملاحظة: استخدام معدات الحماية الشخصية لمنع السمية الحادة بسبب kanamycin. - إضافة 20 ميكرولتر من المخزون كاناميسين 1000x إلى 20 مل من مرق الليسوجيني. باستخدام طرف ماصة معقمة، كزة تحويل BL21 E. كولاي الجلسرين الأسهم ومن ثم تلقيح الثقافة عن طريق إدخال طرف في مرق وسائل الإعلام النمو.

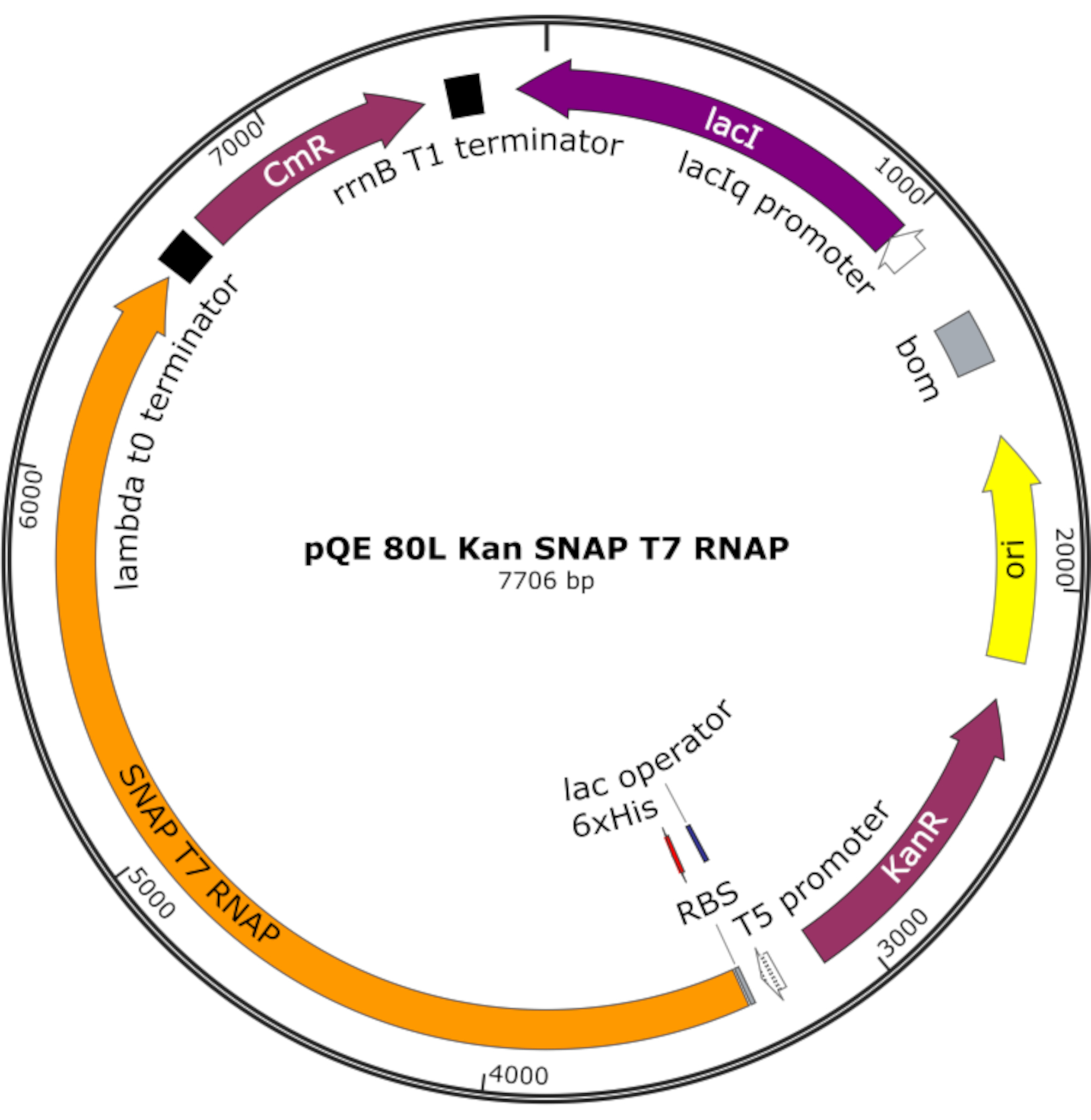
الشكل 4: خريطة بلازميد لSNAP T7 SNAP. يقوم البلازميد بترميز T7 RNAP يحتوي على علامة هيستيدين N-terminal (6x His) ومجال SNAP-tag (SNAP T7 RNAP) تحت مكبوت لاك (lacI) على العمود الفقري pQE-80L. وتشمل الميزات الأخرى مقاومة الكاناميسين (KanR) وجينات مقاومة الكلورامفينيكول (CmR). اختصار: RNAP = RNA بوليمراز. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
ملاحظة: يقوم البلازميد بترميز T7 RNAP يحتوي على علامة هيستيدين N-terminal ومجال SNAP-tag (SNAP T7 RNAP)، بالإضافة إلى جين مقاومة كاناميسين تحت العمود الفقري pQE-80L (الشكل 4)25.
- مرة أخرى، إضافة 20 ميكرولتر من المخزون كاناميسين 1000x إلى قارورة ثقافة منفصلة تحتوي على 20 مل من مرق الليسوجيني، واحتضانه كعنصر تحكم.
- احتضان العينتين (من الخطوتين 2.2 و 2.3) بين عشية وضحاها لمدة 12-18 ساعة في 37 درجة مئوية، في حين تدور في 10 × غرام.
3. نمو الخلية والتعريف: اليوم 2
- تلقيح 400 مل من مرق الليسوجيني الذي يحتوي على 400 ميكرولتر من مخزون الكاناميسين مع 4 مل من ثقافة النمو بين عشية وضحاها من الخطوة 2.4. احتضان قوارير الثقافة عند 37 درجة مئوية ، بينما تدور عند 10 × غرام.
- بمجرد أن تصل الثقافة إلى كثافة بصرية (OD) عند 600 نانومتر من ~ 0.5 ، أخرج 1 مل من العينة من قارورة النمو كعنصر تحكم. تخزين نموذج عنصر التحكم عند 4 °C.
- حث الخلايا مع ايزوبروبيل β-D-1-thiogalactopyranoside (IPTG) بإضافة 40 ميكرولتر من 1M IPTG لكل 100 مل من الثقافة لتحقيق تركيز نهائي من 0.4 M IPTG. احتضان العينة لمدة 3 ساعة في 37 درجة مئوية، بالتناوب في 10 × غرام،ومن ثم تدور الثقافة المستحثة في 8000 × غرام لمدة 10 دقيقة لبيليه الخلايا. إزالة supernatant، وتخزين بيليه في -20 درجة مئوية حتى مزيد من الاستخدام.
ملاحظة: لتجنب السمية الحادة بسبب IPTG، تجنب تنفس غبارها، وتجنب ملامسة الجلد والعين. إذا لزم الأمر، يمكنك إيقاف التجربة مؤقتا هنا والاستمرار في اليوم التالي.
4. تحلل الخلية، تنقية البروتين: اليوم 3
- Resuspend بيليه الخلية المخزنة مع 10 مل من العازلة تحلل على الجليد، ودوامة بلطف لضمان بيليه كامل هو إعادة الإنفاق. ثم، ماصة 1 مل من عينة في عشرة أنابيب 1.5 مل التي يتم الاحتفاظ بها على الجليد.
- Sonicate كل عينة في إعداد السعة من "1"، نبض ل2 ق مع دورة واجب 50٪ على مدى فترة 30 s. قبل وبعد كل عينة، تنظيف طرف سونيكيشن مع الإيثانول 70٪ وddH2O. الحفاظ على جميع العينات على الجليد أثناء وبعد سونيكيشن.
ملاحظة: حافظ على الإيثانول بنسبة 70٪ بعيدا عن الحرارة واللهب المفتوح. - توازن حمض النتريلوترياستيك المشحون بالنيكل (Ni-NTA) عمود دوران تنقية إلى درجة حرارة عمل 4 درجة مئوية. ضع/قم بتخزين العمود عند درجة حرارة 4 درجات مئوية، والحفاظ على الثلج أثناء الاستخدام.
- أجهزة الطرد المركزي العينات العشر 1 مل في 15000 × غرام لمدة 20 دقيقة في 4 درجة مئوية. ماصة بعناية من supernatant التي تحتوي على RNAP المؤتلف دون إزعاج بيليه. إذا لزم الأمر، استخدم المخزن المؤقت الإضافي للتوازن لضبط إجمالي مستوى الصوت إلى ≥ 6 مل.
- قم بإزالة علامة التبويب السفلية برفق من عمود الدوران Ni-NTA للسماح بالتدفق عبر العمود. ضع العمود في أنبوب الطرد المركزي، وأبقه على الجليد.
ملاحظة: استخدم أنبوب طرد مركزي سعة 50 مل مع أعمدة الدوران 3 مل من ني-NTA. - طرد مركزي العمود في 700 × غرام و 4 درجة مئوية لمدة 2 دقيقة لإزالة المخزن المؤقت التخزين. وازن العمود بإضافة 6 مل من المخزن المؤقت للتوازن إلى العمود. السماح للمخزن المؤقت بالدخول الكامل إلى سرير الراتنج.
- إزالة المخزن المؤقت التوازن من العمود عن طريق الطرد المركزي في 700 × غرام و 4 درجة مئوية لمدة 2 دقيقة. قبل إضافة استخراج الخلية المعدة إلى العمود، ضع قابس أسفل على العمود لتجنب فقدان أي منتج. ثم، إضافة استخراج الخلية إلى العمود، وتخلط على خلاط شاكر المداري لمدة 30 دقيقة في 4 درجة مئوية.
- قم بإزالة القابس السفلي من العمود ووضع العمود في أنبوب طرد مركزي سعة 50 مل يسمى التدفق عبر. الطرد المركزي العمود في 700 × غرام لمدة 2 دقيقة لجمع تدفق من خلال.
- إضافة 6 مل من المخزن المؤقت للغسيل إلى العمود لغسل الراتنج. الطرد المركزي العمود في 700 × غرام لمدة 2 دقيقة لجمع الكسر في أنبوب الطرد المركزي الجديد المسمى غسل 1. كرر هذه الخطوة مرتين إضافيتين لما مجموعه 3 كسور منفصلة، وجمع الكسور في أنابيب الطرد المركزي منفصلة(غسل 2 وغسل 3).
- إضافة 3 مل من العازلة elution لe elute البروتينات الموسومة له من الراتنج. الطرد المركزي العمود في 700 × غرام لمدة 2 دقيقة لجمع جزء في أنبوب الطرد المركزي الجديد المسمى eluate 1. كرر هذه الخطوة مرتين إضافيتين لما مجموعه 3 كسور منفصلة، وجمع الكسور في أنابيب الطرد المركزي منفصلة(eluate 2 و eluate 3).
- الجمع بين اليعيل وأداء تحلية لإزالة الأملاح من محلول البروتين.
- Pipette 15 مل من 0.05 ٪ ث / v البوليسوربات 20 على 100 كيلودا وحدة تصفية الطرد المركزي. جهاز الطرد المركزي في 4000 × غرام لمدة 40 دقيقة والتخلص من تدفق من خلال.
- استخدام مرشح المغلفة لتركيز eluates 1 و 2 و 3 (9 مل من مجموع البروتين eluate + 6 مل من المخزن المؤقت) إلى ~ 1500 ميكرولتر. الطرد المركزي مرشح في 3220 × غرام لمدة 20 دقيقة، وبلطف ماصة غسل الغشاء لمنع هطول الأمطار.
- تمييع العينة إلى 15 مل مع المخزن المؤقت للتخزين. تنفيذ تبادل المخزن مؤقت باستخدام المخزن المؤقت التخزين 1:1,000 بتكرار الخطوة 4.11.2 مرتين أكثر.
- قياس البروتين المنقى عن طريق قياس امتصاص الكسر في 280 نانومتر. فارغة مطياف مع المخزن المؤقت التخزين (مخزن تخزين 2x في 4 °C). مزيج بلطف عينة من eluates مجتمعة وقياس امتصاصه.
ملاحظة: إجراء ثلاث قراءات منفصلة في 1x و 10x و 50x تخفيف عينة البروتين إلى متوسط وتحديد كمي البروتين. تمييع العينات في المخزن المؤقت للتخزين. - ضبط عينات البروتين إلى 100 ميكرومتر باستخدام المخزن المؤقت تخزين 2x. تمييع العينة المعدلة 1:1 حسب الحجم مع 100٪ الجلسرين. تخزين محلول البروتين الناتج في -80 درجة مئوية.
5. الصوديوم دودسيل كبريتات البولي أكريلاميد هلام electrophoresis (SDS-PAGE) تحليل منتج البروتين: اليوم 3
- تشغيل هلام SDS-PAGE لتحليل البروتين. مزيج 9 ميكرولتر من العينة مع 3 ميكرولتر من 4x الليثيوم دودسيل كبريتات (LDS) بروتين تحميل صبغة. سخني العينات عند 95 درجة مئوية لمدة 10 دقائق.
- تحميل العينات على 4-12٪ Bis-Tris SDS-PAGE إعداد هلام. تحميل سلم البروتين في بئر 1، ثم مع عينات (من اليسار إلى اليمين): تدفق من خلال، يغسل 1، يغسل 2، يغسل 3، elution 1، elution 2، elution 3، ومجموع elalted elution.
ملاحظة: يحتوي الجدول 5 على جدول تحميل نموذج ل gel SDS-PAGE. - تشغيل عينات هلام محملة في 2-(N-مورفولينو) حمض الإيثانسولفونيك (MES) العازلة لمدة 35 دقيقة في 200 V. شطف الجل في صينية نظيفة ثلاث مرات لمدة 10 دقيقة لكل باستخدام 200 مل من DDH2O، مع التحريض لطيف لإزالة أي SDS من مصفوفة هلام.
ملاحظة: ارتداء معدات الحماية الشخصية لتجنب السمية الحادة بسبب MES. - وصمة عار الجل مع 20 مل من الأزرق Coomassie، واحتضان هلام بين عشية وضحاها في درجة حرارة الغرفة مع التحريض لطيف. إزالة وصمة عار الجل مرتين لمدة 1 ساعة لكل منهما مع 200 مل من DDH2O مع التحريض لطيف على شاكر المدارية.
ملاحظة: غسل الجل لفترة أطول أو استبدال الماء في كثير من الأحيان سيعزز الحساسية. بالإضافة إلى ذلك، فإن وضع أنسجة مسح حساسة مطوية في الحاوية لامتصاص الصبغة الزائدة سيسرع عملية إزالة التلطيخ.
6. التحقق الوظيفي من SNAP T7 RNAP عبر النسخ في المختبر
ملاحظة: يستخدم هذا البروتوكول قالب الحمض النووي، الذي يرمز إلى بيرامر القرنبيط الفلورسنتRNA ويسمح باستخدام الفلورسينس لمراقبة حركية النسخ على قارئ لوحة مضان.
- إعداد ثلاثة ردود فعل النسخ في المختبر (IVT) لمقارنة نشاط SNAP T7 RNAP مع البرية نوع (WT) T7 RNAP من مصدر تجاري وعنصر تحكم المخزن المؤقت فقط. ضبط حجم كل رد فعل إلى 20 ميكرولتر.
- قم بإعداد رد فعل SNAP T7 RNAP IVT عن طريق خلط 2 ميكرولتر من المخزن المؤقت للنسخ 10x، و0.4 ميكرولتر من مزيج ثلاثي الفوسفات ثلاثي الفوسفات (rNTP) 25 mM، و5 ميكرولتر من قالب الحمض النووي 500 nM، و2 ميكرولتر من 500 nM SNAP T7 RNAP، و10.6 ميكرولتر من ddH2O.
- قم بإعداد رد فعل WT RNAP IVT عن طريق خلط 2 ميكرولتر من مخزن النسخ المؤقت 10x، و0.4 ميكرولتر من مزيج rNTP 25 mM، و5 ميكرولتر من قالب الحمض النووي 500 nM، و2 ميكرولتر من WT T7 RNAP، و10.6 ميكرولتر من ddH2O.
- قم بإعداد تفاعل IVT المخزن المؤقت فقط عن طريق خلط 2 ميكرولتر من المخزن المؤقت للنسخ 10x، و0.4 ميكرولتر من مزيج rNTP 25 mM، و5 ميكرولتر من قالب الحمض النووي 500 nM، و12.6 ميكرولتر من ddH2O.
ملاحظة: إضافة RNAP الماضي، وحفظ العينات على الجليد حتى عرضه. يحتوي الجدول 6 والجدول 7والجدول 8 على صيغ رد فعل IVT.
- رصد الحركية النسخ على قارئ لوحة مضان لمدة 2 ساعة في 2 دقيقة فواصل زمنية في 37 درجة مئوية باستخدام الطول الموجي الإثارة من 470 نانومتر وطول موجي انبعاث 512 نانومتر.
7. إعداد أوليغونوكليوتيدات BG المعدلة: اليوم الأول
- حل أوليغونوكليوتيد مع تعديل 3'-أمين في DDH2O إلى تركيز نهائي من 1 mM. تسمية هذا S1.
- مزيج 25 ميكرولتر من بيكربونات الصوديوم 1 م (NaHCO3)،284 ميكرولتر من 100٪ ثنائي ميثيل كبريتوكسي (DMSO)، 125 ميكرولتر من S1 (أوليغونوكلي مخزون الكوديد)، و66 ميكرولتر من 50 مللي متر من إستر BG-N-hydroxysuccinimide (NHS) (BG-GLA-NHS) المخفف مع DMSO، ضبط الحجم إلى 500 ميكرولتر، واحتضان بين عشية وضحاها في درجة حرارة الغرفة في 100 × غرام.
ملاحظة: حافظ على DMSO بعيدا عن الحرارة واللهب لأنه سائل قابل للاحتراق. ويتضمن الجدول 9 صيغة التفاعل لإقران BG بفولق القلة.
- مزيج 25 ميكرولتر من بيكربونات الصوديوم 1 م (NaHCO3)،284 ميكرولتر من 100٪ ثنائي ميثيل كبريتوكسي (DMSO)، 125 ميكرولتر من S1 (أوليغونوكلي مخزون الكوديد)، و66 ميكرولتر من 50 مللي متر من إستر BG-N-hydroxysuccinimide (NHS) (BG-GLA-NHS) المخفف مع DMSO، ضبط الحجم إلى 500 ميكرولتر، واحتضان بين عشية وضحاها في درجة حرارة الغرفة في 100 × غرام.
8. الإيثانول / الأسيتون هطول الأمطار من BG-أوليغونوكليوتيد المصاحبة: اليوم 2
- الطرد المركزي نتاج الخطوة 7.1.1. في 13000 × غرام لمدة 5 دقائق. نقل بعناية supernatant إلى أنبوب جديد وتجاهل أي BG عجلت. تقسيم رد الفعل إلى اثنين من aliquots 250 μL متساوية لمنع تجاوز, وتنفيذ الخطوات التالية على حد سواء aliquots.
- إضافة 1/10th من حجم خلات الصوديوم 3 M (25 ميكرولتر)، تليها 2.5x حجم الإيثانول 100٪ (625 ميكرولتر). حضانة عند -80 درجة مئوية لمدة ساعة واحدة.
ملاحظة: استخدام معدات الحماية الشخصية عند التعامل مع كل من خلات الصوديوم (قد يسبب تهيج في العينين والجلد والجهاز الهضمي والجهاز التنفسي) والإيثانول (قابلة للاشتعال للغاية، ويسبب تهيج عند الاتصال). إذا لزم الأمر، أوقف التجربة هنا و استمر في اليوم التالي. - ضع الأنابيب في جهاز الطرد المركزي، ووضع علامة على الحافة الخارجية. أجهزة الطرد المركزي الأنابيب في 17،000 × غرام لمدة 30 دقيقة في 4 درجة مئوية.
ملاحظة: سوف تظهر بيليه أوليغونوكليوتيد على حافة ملحوظ من الأنبوب. - دون إزعاج بيليه، تجاهل supernatant. أعلى مع 750 ميكرولتر من الإيثانول المبردة 70٪، وتدور في 17،000 × غرام لمدة 10 دقيقة في 4 درجة مئوية.
- دون إزعاج بيليه، تجاهل supernatant. أعلى مع 750 ميكرولتر من الأسيتون 100٪، وتدور في 17،000 × غرام لمدة 10 دقيقة في 4 درجة مئوية.
ملاحظة: استخدام معدات الحماية الشخصية عند التعامل مع الأسيتون كما هو قابل للاشتعال للغاية ويسبب تهيج عند الاتصال. - مع فتح غطاء الأنبوب، يجف الهواء لمدة 5 دقائق لإزالة أي أسيتون زائد من خلال التبخر. إعادة حل أوليغونوكليوتيد في 250 ميكرولتر من 1x تريس-EDTA (TE) العازلة لإنتاج ~ 850 ميكرومتر BG-oligonucleotide الحل.
- كرر الخطوات من 8.2 إلى 8.6، ثم أعد الذوبان في 70 ميكرولتر من المخزن المؤقت 1x TE. تسمية هذا S2.
9. BG-oligonucleotide تنظيف عن طريق الكروماتوغرافيا الترشيح هلام
- تعليق المصفوفة عن طريق عكس الأعمدة بقوة عدة مرات؛ إزالة الغطاء العلوي والقطة قبالة الطرف السفلي من العمود. ضع العمود في أنبوب طرد مركزي سعة 1.5 مل، واطرد الأنبوب عند 1000 × غرام لمدة دقيقة واحدة في درجة حرارة الغرفة. تجاهل المخزن المؤقت eluted وأنبوب التجميع.
ملاحظة: من المهم منع تشكيل فراغ. استخدم الأعمدة المعدة على الفور. - ضع الأعمدة المعبأة في أنابيب طرد مركزي نظيفة سعة 1.5 مل. أضف 300 ميكرولتر من 1x TE العازلة إلى مركز السرير العمود، والطرد المركزي في 1000 × غرام لمدة 2 دقيقة لتبادل الحل العازلة. مرة أخرى، تجاهل المخزن المؤقت eluted وأنبوب جمع.
- ضع أعمدة تبادل المخزن المؤقت في أنابيب طرد مركزي نظيفة سعة 1.5 مل. ضعي ما يصل إلى 75 ميكرولتر من العينة على مركز السرير. تدور في 1000 × غرام لمدة 4 دقائق.
ملاحظة: لا تزعج السرير أو تلمس جانبي العمود؛ أعلى نقطة من وسائل الإعلام هلام يجب أن تشير نحو الدوار الخارجي. - جمع eluate من أنبوب جمع، كما أنه يحتوي على حمض النوى المنقى. لقياس العينة، وقياس امتصاصها في 260 نانومتر؛ تسمية هذا S3.
ملاحظة: لاحظ طول المسار المستخدم في القياس، وحساب التركيز باستخدام قانون بير لامبرت.
10. تحليل الصفحة Denaturing من BG-أوليغونوكليوتيد اقتران
- يلقي 18٪ تريس بورات-EDTA (TBE)-Urea PAGE هلام. حل 4.8 غرام من اليوريا، 4.5 مل من 40٪ أكريلاميد (19:1)، و 1 مل من 10x TBE في 2.8 مل من ddH2O؛ إضافة 5 ميكرولتر رباعي ميثيل إيثيلينديامين (TEMED) وتخلط جيدا. كرر مع 100 ميكرولتر من 10٪ من كبريتات الأمونيوم (APS). صب الحل في كاسيت هلام فارغة والسماح البلمرة لمدة 40 دقيقة.
ملاحظة: استخدام معدات الحماية الشخصية المناسبة عند التعامل مع اليوريا (يسبب تهيج في العينين والجلد)، والاكريلاميد (السامة والمسرطنة)، وTEMED (سامة، قابلة للاشتعال، تآكل). يحتوي الجدول 10 على صيغة التفاعل لجل بولي أكريلاميد TBE-UREA بنسبة 18٪. - ميكروويف 500 مل من TBE العازلة (0.5x) لمدة 2 دقيقة و 30 ق أو حتى ~ 70 درجة مئوية وتصب في جهاز هلام. إعداد فورماميد (denaturing) تحميل صبغة تحتوي على 95٪ فورماميد + 1 mM EDTA والأزرق بروموفينول. اخلط صبغة التحميل مع كل عينة، وحمل الخليط على هلام البولي أكريلاميد.
ملاحظة: استخدام معدات الحماية الشخصية المناسبة عند التعامل مع فورماميد كما هو مسرطن. يحتوي الجدول 11 على جدول تحميل جل عينة. - تشغيل هلام في 270 V لمدة 35 دقيقة، أو حتى الجبهة صبغ يهاجر إلى النهاية. ضع الجل في صندوق هلام وصمة عار مع صبغة السيانين للأحماض النووية لمدة 15 دقيقة في درجة حرارة الغرفة قبل التصوير.
ملاحظة: استخدام معدات الحماية الشخصية المناسبة عند التعامل مع صبغة السيانين كما هو قابل للاحتراق.
11. اقتران أوليغونوكليوتيد إلى SNAP T7 RNAP وتحليل الصفحة
- إعداد الكواشف للاقتران التحليلي على نطاق BG-oligonucleotide إلى SNAP T7 RNAP: جعل 9 تخفيفات من الحمض النووي واحد تقطعت بهم السبل (ssDNA) أوليغو مع ddH2O لخلق نسب oligo:RNAP تتراوح بين 5:1 إلى 1:5. تمييع مخزون البروتين إلى 50 ميكرومتر.
ملاحظة: يمكن العثور على نسب المثال في الجدول 12; يتم حساب هذه النسب باستخدام تركيز RNAP من 50 ميكرومتر. - لكل تخفيف من ssDNA أوليغو، وجعل 10 ميكرولتر من خليط التفاعل التي تحتوي على 2 ميكرولتر من العازلة SNAP، 4 ميكرولتر من BG-oligonucleotide، و 4 ميكرولتر من SNAP T7 RNAP.
ملاحظة: يحتوي الجدول 13 على صيغ رد فعل لرد فعل وضع العلامات SNAP-tag.- إعداد اثنين من عينات التحكم أكثر: 1) التحكم RNAP عن طريق استبدال BG-oligonucleotide مع DDH2O; 2) التحكم في الحمض النووي عن طريق استبدال SNAP T7 RNAP مع DDH2O (لتركيز أوليغونوكليوتيد أدنى من SNAP T7 RNAP). احتضان جميع العينات في درجة حرارة الغرفة لمدة ساعة واحدة، والحفاظ على الجليد حتى الحاجة.
- إعداد أحد عشر 10 ميكرولتر ردود الفعل عن طريق إضافة 2 ميكرولتر من كل عينة إلى 4 ميكرولتر من العازلة SNAP و 2 ميكرولتر من البروتين تحميل صبغة، والحرارة في 70 درجة مئوية لمدة 10 دقيقة. تحميل 2 ميكرولتر من كل عينة على هلام البروتين بيس تريس 4-12٪، وأداء الكهربائي هلام على الجليد في 200 V لمدة 35 دقيقة.
ملاحظة: يحتوي الجدول 14 على صيغ رد فعل لعينات تحميل الجل.- غسل SDS قبالة عن طريق تبادل المياه 3x على شاكر، كل غسل دائم 10 دقيقة لكل منهما. وصمة عار مع صبغة السيانين للأحماض النووية لمدة 15 دقيقة قبل التصوير. وصمة عار الجل مرة أخرى باستخدام 20 مل من وصمة عار زرقاء Coomassie لمدة 1 ساعة. إزالة البقعة مع ddH2O لمدة ساعة واحدة (أو بين عشية وضحاها) قبل التصوير.
ملاحظة: في هلام، واحدة من ردود الفعل سوف تنتج البوليميراز الأكثر المربوطة جنبا إلى جنب مع أقل قدر من فائض الحرة BG-oligonucleotide. هذه هي النسبة المثلى
- غسل SDS قبالة عن طريق تبادل المياه 3x على شاكر، كل غسل دائم 10 دقيقة لكل منهما. وصمة عار مع صبغة السيانين للأحماض النووية لمدة 15 دقيقة قبل التصوير. وصمة عار الجل مرة أخرى باستخدام 20 مل من وصمة عار زرقاء Coomassie لمدة 1 ساعة. إزالة البقعة مع ddH2O لمدة ساعة واحدة (أو بين عشية وضحاها) قبل التصوير.
- إعداد الكواشف للمقياس التحضيري اقتران BG-oligonucleotide إلى SNAP T7 RNAP. قم بإجراء التفاعل مع النسبة المثلى الموجودة في المقياس التحليلي.
ملاحظة: تقليل التعرض للبروتين لدرجة حرارة الغرفة عن طريق وضع البروتين على الجليد عندما لا تكون قيد الاستخدام.
12. تنقية أوليغونوكليوتيد المربوطة SNAP-T7 باستخدام أعمدة تبادل الأيونات
- اتبع إرشادات الشركة المصنعة لإعداد الأنبوب إذا انحرف عن التعليمات المذكورة هنا. إعداد عازلة تنقية مع درجة الحموضة أعلى من نقطة isoelectric من البروتين.
ملاحظة: للحصول على البروتين المثال في هذا البروتوكول، تم استخدام مخزن مؤقت تنقية من 10 mM صوديوم الفوسفات العازلة (pH 7).- إعداد 1000 ميكرولتر من العازلة elution تحتوي على تركيزات النهائي من 50 mM تريس و 0.5 M NaCl. مزيج 50 ميكرولتر من 1 متر تريس، 100 ميكرولتر من 5 م NaCl، و 850 ميكرولتر من ddH2O.
ملاحظة: يحتوي الجدول 15 على صيغة رد الفعل ل المخزن المؤقت elution.
- إعداد 1000 ميكرولتر من العازلة elution تحتوي على تركيزات النهائي من 50 mM تريس و 0.5 M NaCl. مزيج 50 ميكرولتر من 1 متر تريس، 100 ميكرولتر من 5 م NaCl، و 850 ميكرولتر من ddH2O.
- ضع عمودا في أنبوب طرد مركزي سعة 2 مل، واغسله بحاجز تنقية عند 2000 × غرام لمدة 15 دقيقة، أو حتى يتم إعادة تشغيل كل العازلة. تجاهل المخزن المؤقت eluted.
- تمييع كل عينة مع تنقية العازلة في 3:1 تنقية العازلة: نسبة العينة، وتحميل العينة في العمود 400 ميكرولتر في وقت واحد. تدور في 2000 × غرام لمدة 10 دقيقة ، أو حتى تم eluted كل العازلة. جمع التدفق من خلال وتسمية على أنها تدفق من خلال.
- إضافة 400 ميكرولتر من المخزن المؤقت للتنقية في وسط العمود. تدور في 2000 × غرام لمدة 15 دقيقة ، أو حتى تم eluted كل العازلة. جمع تدفق من خلال وتسمية بأنها غسل 1. كرر مرتين أكثر لغسل 2 وغسل 3.
- إضافة 50 ميكرولتر من المخزن المؤقت elution في وسط العمود. تدور في 2000 × غرام لمدة 5 دقائق ، أو حتى تم eluted كل العازلة. جمع تدفق من خلال وتسمية بأنها eluate 1. كرر مرتين أكثر ل eluate 2 و eluate 3.
- تجمع eluates 1, 2, و 3 (تسمية هذا مجموع eluate),ترك جزء صغير من كل eluate للهلام, وقياس امتصاص في 260 نانومتر (A260) و 280 نانومتر (A280). بعد القياس، إضافة الجلسرين بنسبة 1:1 وتخزينها في -20 درجة مئوية حتى مزيد من الاستخدام.
- استخدم وحدة فلتر الطرد المركزي (0.5 مل؛ 30 كيلودا) لإجمالي تبادل المخزن المؤقت مع مخزن تخزين مؤقت 2x (~1:100) (تسمية هذا المنتج). قياس A260/280 مرة أخرى. إضافة الجلسرين بنسبة 1:1 وتخزينها في -20 درجة مئوية حتى مزيد من الاستخدام.
- تحميل كل eluate: تدفق من خلال، وغسل 1-3، eluate مجموع، والمنتج في هلام بيس-تريس SDS-PAGE 4-12٪، جنبا إلى جنب مع سلم البروتين. تشغيل في 200 V لمدة 35 دقيقة، أو حتى الجبهة صبغ يهاجر إلى النهاية.
13. إظهار السيطرة عند الطلب من نشاط البوليميراز الحمض النووي الريبي المربوطة
- إعداد العازلة 5x التلين التي تحتوي على 25 mM تريس، 5 M EDTA، و 25 mM كلوريد المغنيسيوم (MgCl2). امزج 2.4 ميكرولتر لكل قالب (1 ميكرومتر) مع 5 ميكرولتر من العازلة للعلة و14.2 ميكرولتر من ddH2O لتشكيل 25 ميكرولتر من قفص dsDNA 1 ميكرومتر. احتضان هذا الحل في 75 درجة مئوية لمدة 2 دقيقة. وبالمثل، آنال الشعور وخيوط مضادة للمنطق من المروج وmalchite الأخضر aptamer قالب الحمض النووي. إعداد حل 1mM من أوكسالات خضراء ملاخية.
ملاحظة: يحتوي الجدول 16 على صيغة رد الفعل ل 5x عازلة، يحتوي الجدول 17 على صيغة رد الفعل لعلة قوالب ssDNA اثنين. - احتضان المربوطة SNAP T7 RNAP مع قفص dsDNA في نسبة الضرس 1:5 في درجة حرارة الغرفة لمدة 15 دقيقة إلى تركيز نهائي من 500 NM RNAP. ابقي على الثلج حتى الحاجة.
- سخني قارئ اللوحة إلى 37 درجة مئوية. إعداد ثلاثة ردود فعل IVT 25 ميكرولتر على الجليد
- إعداد رد فعل يحتوي على قفص SNAP T7RNAP مع عوامل النسخ حمض النوى. مزيج 2.5 ميكرولتر من 10x المخزن المؤقت IVT، 1 ميكرولتر من مزيج rNTP 25 mM، 1 ميكرولتر من 1 ملاشيتا أخضر، و2.5 ميكرولتر من خليط قفص RNAP، و2.5 ميكرولتر لكل من عامل النسخ 1 ميكرومتر A وB oligonucleotide، و3 ميكرولتر من قالب 1 mm malachite green aptamer في 10 ميكرولتر من DDH2O.
- إعداد رد فعل يحتوي على قفص SNAP T7RNAP دون عوامل نسخ الحمض النووي. مزيج 2.5 ميكرولتر من 10x IVT العازلة، 1 ميكرولتر من 25 mM rNTP مزيج، 1 ميكرولتر من 1 mM malachite الأخضر، 2.5 ميكرولتر من خليط قفص RNAP، و 3 ميكرولتر من 1 mM ملاكيت الأخضر قالب aptamer في 15 ميكروغرام من DDH2O.
- إعداد رد فعل يحتوي على المخزن المؤقت فقط. مزيج 2.5 ميكرولتر من 10x IVT العازلة، 1 ميكرولتر من 25 mM rNTP مزيج، 1 ميكرولتر من 1 ملاشيت الأخضر، و 3 ميكرولتر من 1 ملاخيت الأخضر aptamer قالب في 17.5 ميكرولتر من DDH2O.
ملاحظة: يحتوي الجدول 18 على مرجع عام لتفاعلات النسخ في المختبر.
- نقل كل رد فعل إلى لوحة 384 جيدا. رصد نسخ من aptamer malachite الخضراء على قارئ لوحة مضان لمدة 2 ساعة في 37 درجة مئوية ومع 610 نانومتر الإثارة و 655 نانومتر الانبعاثات. بمجرد الانتهاء، والحفاظ على لوحة على الجليد حتى الحاجة.
- الميكروويف 0.5x TBE العازلة لمدة 2 دقيقة 30 ق أو حتى ~ 70 درجة مئوية. تشغيل منتجات الحمض النووي الريبي من كل بئر في انحطاط 12٪ TBE-اليوريا هلام البولياكريلاميد في المخزن المؤقت TBE 0.5x ساخنة في 280 V لمدة 20 دقيقة، أو حتى الجبهة صبغ يصل إلى النهاية. وصمة عار هلام مع صبغة السيانين وصمة عار حمض النووي لمدة 10 دقيقة على شاكر المداري قبل التصوير.
ملاحظة: يحتوي الجدول 19 على صيغة رد الفعل لجل TBE-Urea PAGE 12٪ من الإزالة.
Access restricted. Please log in or start a trial to view this content.
النتائج

الشكل 5: تحليل SDS-PAGE لتعبير SNAP T7 RNAP ومقايسة النسخ في المختبر. (A) SNAP T7 RNAP تحليل تنقية البروتين، SNAP T7 RNAP الوزن الجزيئي: 119.4kDa. FT = التدفق من خلال العمود، W1 = كسور elution من المخزن المؤقت غسل تحتوي على ال...
Access restricted. Please log in or start a trial to view this content.
Discussion
توضح هذه الدراسة نهجا مستوحى من تكنولوجيا النانو من الحمض النووي للسيطرة على نشاط T7 RNA polymerase من خلال اقتران مركب T7 RNAP المؤتلف N-terminally SNAP الموسومة ب Oligonucleotide الذي يعمل في BG ، والذي تم استخدامه لاحقا لبرنامج ردود فعل TMDSD. حسب التصميم ، تم وضع علامة SNAP في N-terminus من البوليميراز ، حيث يتم دفن C-terminus م...
Access restricted. Please log in or start a trial to view this content.
Disclosures
ولا توجد مصالح مالية متنافسة يعلن عنها أي من أصحاب البلاغ.
Acknowledgements
وتعترف L.Y.T.C بالدعم السخي المقدم من صندوق الحدود الجديدة لاستكشاف البحوث (NFRF-E)، ومجلس أبحاث العلوم الطبيعية والهندسة الكندي (NSERC) منحة ديسكفري، ومبادرة الطب حسب التصميم في جامعة تورنتو، التي تتلقى التمويل من صندوق كندا للتميز البحثي الأول (CFREF).
Access restricted. Please log in or start a trial to view this content.
Materials
| Name | Company | Catalog Number | Comments |
| 0.5% polysorbate 20 (TWEEN 20) | BioShop | TWN510.5 | |
| 0.5M ethylenediaminetetraacetic acid (EDTA) | Bio Basic | SD8135 | |
| 10 mM sodium phosphate buffer (pH 7) | Bio Basic | PD0435 | Tablets used to make 10 mM buffer |
| 10% ammonium persulfate (APS) | Sigma Aldrich | A3678-100G | |
| 100 kDa Amicon Ultra-15 Centrifugal Filter Unit | Fisher Scientific | UFC910008 | |
| 100% acetone | Fisher Chemical | A18P4 | |
| 100% ethanol (EtOH) | House Brand | 39752-P016-EAAN | |
| 10x in vitro transcription (IVT) buffer | New England Biolabs | B9012 | |
| 10x Tris-Borate-EDTA (TBE) buffer | Bio Basic | A0026 | |
| 1M Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Sigma Aldrich | I5502-1G | |
| 1M sodium bicarbonate buffer | Sigma Aldrich | S6014-500G | |
| 1M Tris(hydroxymethyl)aminomethane (Tris) | Sigma Aldrich | 648311-1KG | |
| 1X Tris-EDTA (TE) buffer | ThermoFisher | 12090015 | |
| 2M imidazole | Sigma Aldrich | 56750-100G | |
| 2-mercaptoethanol (BME) | Sigma Aldrich | M3148 | |
| 3M sodium acetate | Bio Basic | SRB1611 | |
| 40% acrylamide (19:1) | Bio Basic | A00062 | |
| 4x LDS protein sample loading buffer | Fisher Scientific | NP0007 | |
| 5M sodium chloride (NaCl) | Bio Basic | DB0483 | |
| 5mM dithiothreitol (DTT) | Sigma Aldrich | 43815-1G | |
| 6x gel loading dye | New England Biolabs | B7024S | |
| agarose B powder | Bio Basic | AB0014 | |
| BG-GLA-NHS | New England Biolabs | S9151S | |
| BL21 competent E. coli | Addgene | C2530H | |
| BLUeye prestained protein ladder | FroggaBio | PM007-0500 | |
| bromophenol blue | Bio Basic | BDB0001 | |
| coomassie blue (SimplyBlue SafeStain) | ThermoFisher | LC6060 | |
| cyanine dye (SYBR Gold nucleic acid gel stain) | Fisher Scientific | S11494 | |
| cyanine dye (SYBR Safe nucleic acid gel stain) | Fisher Scientific | S33102 | |
| dry dimethyl sulfoxide (DMSO) | Fisher Scientific | D12345 | |
| formamide | Sigma Aldrich | F9037-100ML | |
| glycerol | Bio Basic | GB0232 | |
| kanamycin sulfate | BioShop | KAN201.5 | |
| lysogeny broth | Sigma Aldrich | L2542-500ML | |
| malachite green oxalate | Sigma Aldrich | 2437-29-8 | |
| N,N,N'N'-Tetramethylethane-1,2-diamine (TEMED) | Sigma Aldrich | T9281-25ML | |
| NuPAGE MES SDS running buffer (20x) | Fisher Scientific | LSNP0002 | |
| NuPAGE Novex 4-12% Bis-Tris gel 1.0 mm 12-well | Life Technologies | NP0322BOX | |
| oligonucleotide (cage antisense) | IDT | N/A | TATAGTGAGTCGTATTAATTTG |
| oligonucleotide (cage sense) | IDT | N/A | TCAGTCACCTATCTGTTTCAAA TTAATACGACTCACTATA |
| oligonucleotide (malachite green aptamer antisense) | IDT | N/A | GGATCCATTCGTTACCTGGCT CTCGCCAGTCGGGATCCTATA GTGAGTCGTATTACAGTTCCAT TATCGCCGTAGTTGGTGTACT |
| oligonucleotide (malachite green aptamer sense) | IDT | N/A | TAATACGACTCACTATAGGATC CCGACTGGCGAGAGCCAGGT AACGAATGGATCC |
| oligonucleotide (Transcription Factor A) | IDT | N/A | AGTACACCAACTACGAGTGAG |
| oligonucleotide (Transcription Factor B) | IDT | N/A | TCAGTCACCTATCTGGCGATAA TGGAACTG |
| oligonucleotide with 3’ Amine modification (tether) | IDT | N/A | GCTACTCACTCAGATAGGTGAC TGA/3AmMO/ |
| Pierce strong ion exchange spin columns | Fisher Scientific | 90008 | |
| plasmid encoding SNAP T7 RNAP and kanamycin resistance genes | Genscript | N/A | custom gene insert |
| protein purification column (HisPur Ni-NTA spin column) | Fisher Scientific | 88226 | |
| rNTP mix | New England Biolabs | N0466S | |
| Roche mini quick DNA spin column | Sigma Aldrich | 11814419001 | |
| Triton X-100 | Sigma Aldrich | T8787-100ML | |
| Ultra Low Range DNA ladder | Fisher Scientific | 10597012 | |
| urea | BioShop | URE001.1 |
References
- Cherry, K. M., Qian, L. Scaling up molecular pattern recognition with DNA-based winner-take-all neural networks. Nature. 559 (7714), 370-376 (2018).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475 (7356), 368-372 (2011).
- Chen, Y. -J., et al. Programmable chemical controllers made from DNA. Nature Nanotechnology. 8 (10), 755-762 (2013).
- di Bernardo, D., Marucci, L., Menolascina, F., Siciliano, V. Predicting synthetic gene networks. Synthetic Gene Networks: Methods and Protocols. 813, 57-81 (2012).
- Xiang, Y., Dalchau, N., Wang, B. Scaling up genetic circuit design for cellular computing: advances and prospects. Natural Computing. 17 (4), 833-853 (2018).
- Gould, N., Hendy, O., Papamichail, D. Computational tools and algorithms for designing customized synthetic genes. Frontiers in Bioengineering and Biotechnology. 2, (2014).
- MacDonald, J. T., Siciliano, V. Computational sequence design with R2oDNA Designer. Mammalian Synthetic Promoters. 1651, 249-262 (2017).
- Cervantes-Salido, V. M., Jaime, O., Brizuela, C. A., Martínez-Pérez, I. M. Improving the design of sequences for DNA computing: A multiobjective evolutionary approach. Applied Soft Computing. 13 (12), 4594-4607 (2013).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry. 32 (1), 170-173 (2011).
- Fornace, M. E., Porubsky, N. J., Pierce, N. A. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synthetic Biology. 9 (10), 2665-2678 (2020).
- Wetterstrand, K. DNA sequencing costs: Data. Genome.gov. , (2020).
- Lopez, R., Wang, R., Seelig, G. A molecular multi-gene classifier for disease diagnostics. Nature Chemistry. 10 (7), 746-754 (2018).
- Pardee, K., et al. low-cost detection of Zika virus using programmable biomolecular components. Cell. 165 (5), 1255-1266 (2016).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406 (6796), 605-608 (2000).
- Lin, K. N., Volkel, K., Tuck, J. M., Keung, A. J. Dynamic and scalable DNA-based information storage. Nature Communications. 11 (1), 2981(2020).
- Yurke, B., Mills, A. P. Using DNA to power nanostructures. Genetic Programming and Evolvable Machines. 4 (2), 111-122 (2003).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318 (5853), 1121-1125 (2007).
- Wang, B., Thachuk, C., Ellington, A. D., Winfree, E., Soloveichik, D. Effective design principles for leakless strand displacement systems. Proceedings of the National Academy of Sciences. 115 (52), 12182-12191 (2018).
- Machinek, R. R. F., Ouldridge, T. E., Haley, N. E. C., Bath, J., Turberfield, A. J. Programmable energy landscapes for kinetic control of DNA strand displacement. Nature Communications. 5 (1), 5324(2014).
- Cabello-Garcia, J., Bae, W., Stan, G. -B. V., Ouldridge, T. E. Handhold-mediated strand displacement: a nucleic acid-based mechanism for generating far-from-equilibrium assemblies through templated reactions. bioRxiv. , (2020).
- Brophy, J. A. N., Voigt, C. A. Principles of genetic circuit design. Nature Methods. 11 (5), 508-520 (2014).
- Khalil, A. S., et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 150 (3), 647-658 (2012).
- Swank, Z., Laohakunakorn, N., Maerkl, S. J. Cell-free gene-regulatory network engineering with synthetic transcription factors. Proceedings of the National Academy of Sciences. 116 (13), 5892-5901 (2019).
- Howland, S. W., Tsuji, T., Gnjatic, S., Ritter, G., Old, L. J., Wittrup, K. D. Inducing efficient cross-priming using antigen-coated yeast particles. Journal of immunotherapy. 31 (7), 607(2008).
- Abil, Z., Ellefson, J. W., Gollihar, J. D., Watkins, E., Ellington, A. D. Compartmentalized partnered replication for the directed evolution of genetic parts and circuits. Nature Protocols. 12 (12), 2493-2512 (2017).
- Baugh, C., Grate, D., Wilson, C. 2.8 Å crystal structure of the malachite green aptamer11. Journal of Molecular Biology. Doudna, J. A. 301 (1), 117-128 (2000).
- Chou, L. Y. T., Shih, W. M. In vitro transcriptional regulation via nucleic acid-based transcription factors. ACS Synthetic Biology. 8 (11), 2558-2565 (2019).
- Lykke-Andersen, J., Christiansen, J. The C-terminal carboxy group of T7 RNA polymerase ensures efficient magnesium ion-dependent catalysis. Nucleic Acids Research. 26 (24), 5630-5635 (1998).
- Pu, J., Disare, M., Dickinson, B. C. Evolution of C-terminal modification tolerance in full-length and split T7 RNA Polymerase biosensors. Chembiochem. 20 (12), 1547-1553 (2019).
- Gardner, L. P., Mookhtiar, K. A., Coleman, J. E. Initiation, elongation, and processivity of carboxyl-terminal mutants of T7 RNA polymerase. Biochemistry. 36 (10), 2908-2918 (1997).
- Yin, J., Lin, A. J., Golan, D. E., Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nature Protocols. 1 (1), 280-285 (2006).
- Warden-Rothman, R., Caturegli, I., Popik, V., Tsourkas, A. Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Analytical Chemistry. 85 (22), 11090-11097 (2013).
- Zhang, W. -B., Sun, F., Tirrell, D. A., Arnold, F. H. Controlling macromolecular topology with genetically encoded SpyTag-SpyCatcher chemistry. Journal of the American Chemical Society. 135 (37), 13988-13997 (2013).
Access restricted. Please log in or start a trial to view this content.
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved