
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
DNA-Tethered RNA Polymerase für programmierbare In-vitro-Transkription und molekulare Berechnung
In diesem Artikel
Zusammenfassung
Wir beschreiben das Engineering einer neuartigen DNA-gebundenen T7-RNA-Polymerase zur Regulierung von In-vitro-Transkriptionsreaktionen. Wir diskutieren die Schritte für die Proteinsynthese und -charakterisierung, validieren die Proof-of-Concept-Transkriptionsregulation und diskutieren ihre Anwendungen in den Bereichen Molecular Computing, Diagnostik und molekulare Informationsverarbeitung.
Zusammenfassung
Die DNA-Nanotechnologie ermöglicht die programmierbare Selbstorganisation von Nukleinsäuren in vom Benutzer vorgegebene Formen und Dynamiken für verschiedene Anwendungen. Diese Arbeit zeigt, dass Konzepte aus der DNA-Nanotechnologie verwendet werden können, um die enzymatische Aktivität der phagenabgeleiteten T7-RNA-Polymerase (RNAP) zu programmieren und skalierbare synthetische Genregulationsnetzwerke aufzubauen. Zunächst wird ein Oligonukleotid-gebundenes T7 RNAP durch Expression eines N-terminalen SNAP-markierten RNAP und anschließende chemische Kopplung des SNAP-Tags mit einem Benzylguanin (BG)-modifizierten Oligonukleotid hergestellt. Als nächstes wird die Nuklein-Säure-Strangverschiebung verwendet, um die Polymerase-Transkription nach Bedarf zu programmieren. Darüber hinaus können Hilfs-Nukleinsäure-Assemblierungen als "künstliche Transkriptionsfaktoren" verwendet werden, um die Wechselwirkungen zwischen dem DNA-programmierten T7-RNAP und seinen DNA-Templates zu regulieren. Dieser Regulatorische Mechanismus für die In-vitro-Transkription kann eine Vielzahl von Schaltkreisverhalten wie digitale Logik, Feedback, Kaskadierung und Multiplexing implementieren. Die Zusammensetzbarkeit dieser genregulatorischen Architektur erleichtert die Designabstraktion, Standardisierung und Skalierung. Diese Funktionen werden das Rapid Prototyping von in vitro genetischen Geräten für Anwendungen wie Biosensorik, Krankheitserkennung und Datenspeicherung ermöglichen.
Einleitung
DNA-Computing verwendet eine Reihe von entworfenen Oligonukleotiden als Medium für die Berechnung. Diese Oligonukleotide sind mit Sequenzen programmiert, die sich dynamisch nach einer benutzerdefinierten Logik zusammensetzen und auf spezifische Nukleinsäureeinträge reagieren. In Proof-of-Concept-Studien besteht die Ausgabe der Berechnung typischerweise aus einem Satz fluoreszierend markierter Oligonukleotide, die über Gelelektrophorese oder Fluoreszenzplattenleser nachgewiesen werden können. In den letzten 30 Jahren wurden immer komplexere DNA-Rechenschaltungen demonstriert, wie verschiedene digitale Logikkaskaden, chemische Reaktionsnetzwerke und neuronale Netze1,2,3. Um die Herstellung dieser DNA-Schaltkreise zu unterstützen, wurden mathematische Modelle verwendet, um die Funktionalität der synthetischen Genschaltkreise4,5vorherzusagen, und Berechnungswerkzeuge wurden für das orthogonale DNA-Sequenzdesign entwickelt6,7,8,9,10 . Im Vergleich zu siliziumbasierten Computern gehören zu den Vorteilen von DNA-Computern ihre Fähigkeit, direkt mit Biomolekülen zu verbinden, in Lösung zu arbeiten, wenn keine Stromversorgung vorhanden ist, sowie ihre allgemeine Kompaktheit und Stabilität. Mit dem Aufkommen der Next-Generation-Sequenzierung sind die Kosten für die Synthese von DNA-Computern in den letzten zwei Jahrzehnten schneller gesunken als das Mooresche Gesetz11. Anwendungen solcher DNA-basierten Computer beginnen sich nun zu entwickeln, wie z.B. für die Krankheitsdiagnose12,13, für die Stromversorgung der molekularen Biophysik14und als Datenspeicherplattformen15.

Abbildung 1: Mechanismus der Zehengriff-vermittelten DNA-Strangverschiebung. Der Toehold, δ, ist eine freie, ungebundene Sequenz auf einem Teilduplex. Wenn eine komplementäre Domäne (δ*) auf einem zweiten Strang eingeführt wird, dient die freie δ Domäne als Haltepunkt für die Hybridisierung, so dass der Rest des Strangs (ɑ*) seinen Konkurrenten durch eine reversible Reaktion, die als Strangmigration bezeichnet wird, langsam verdrängen kann. Wenn die Länge δ zunimmt, nimmt das ΔG für die Vorwärtsreaktion ab und die Verschiebung erfolgt leichter. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Bis heute verwendet die Mehrheit der DNA-Computer ein etabliertes Motiv auf dem Gebiet der dynamischen DNA-Nanotechnologie, das als Zehengriff-vermittelte DNA-Strangverschiebung (TMDSD, Abbildung 1)16bekannt ist. Dieses Motiv besteht aus einem teilweise doppelsträngigen DNA-Duplex (dsDNA), der kurze "Zehenhalte"-Überhänge (d.h. 7- bis 10 Nukleotide (nt)) aufweist. Nukleinsäure-"Eingangsstränge" können mit den Teilduplexen durch den Zehengriff interagieren. Dies führt zur Verschiebung eines der Stränge aus dem Teilduplex, und dieser freigesetzte Strang kann dann als Eingang für nachgeschaltete Teilduplexe dienen. Somit ermöglicht TMDSD die Signalkaskadierung und Informationsverarbeitung. Prinzipiell können orthogonale TMDSD-Motive in Lösung eigenständig arbeiten und ermöglichen so eine parallele Informationsverarbeitung. Es gab eine Reihe von Variationen der TMDSD-Reaktion, wie z.B. Toehold-vermittelter DNA-Strangaustausch (TMDSE)17, "lecklose" Zehengriffe mit doppelt langen Domänen18, sequenz-mismatched Zehenhalte19und "Handhold"-vermittelte Strangverschiebung20. Diese innovativen Designprinzipien ermöglichen eine feiner abgestimmte TMDSD-Energetik und -Dynamik zur Verbesserung der DNA-Rechenleistung.
Synthetische Genschaltkreise, wie transkriptionelle Genschaltkreise, sind ebenfalls in der Lage,21,22,23zu berechnen. Diese Schaltkreise werden durch Proteintranskriptionsfaktoren reguliert, die die Transkription eines Gens durch Bindung an bestimmte regulatorische DNA-Elemente aktivieren oder unterdrücken. Im Vergleich zu DNA-basierten Schaltkreisen haben Transkriptionsschaltkreise mehrere Vorteile. Erstens hat die enzymatische Transkription eine viel höhere Fluktuationsrate als bestehende katalytische DNA-Schaltkreise, wodurch mehr Kopien der Ausgabe pro einzelner Kopie der Eingabe erzeugt werden und ein effizienteres Mittel zur Signalverstärkung bereitgestellt wird. Darüber hinaus können Transkriptionsschaltkreise verschiedene funktionelle Moleküle wie Aptamere oder Boten-RNA (mRNA), die für therapeutische Proteine kodieren, als Rechenergebnisse erzeugen, die für verschiedene Anwendungen genutzt werden können. Eine wesentliche Einschränkung der aktuellen Transkriptionsschaltungen ist jedoch ihre mangelnde Skalierbarkeit. Dies liegt daran, dass es nur eine sehr begrenzte Anzahl orthogonaler Protein-basierter Transkriptionsfaktoren gibt und das De-novo-Design neuer Proteintranskriptionsfaktoren technisch anspruchsvoll und zeitaufwendig bleibt.

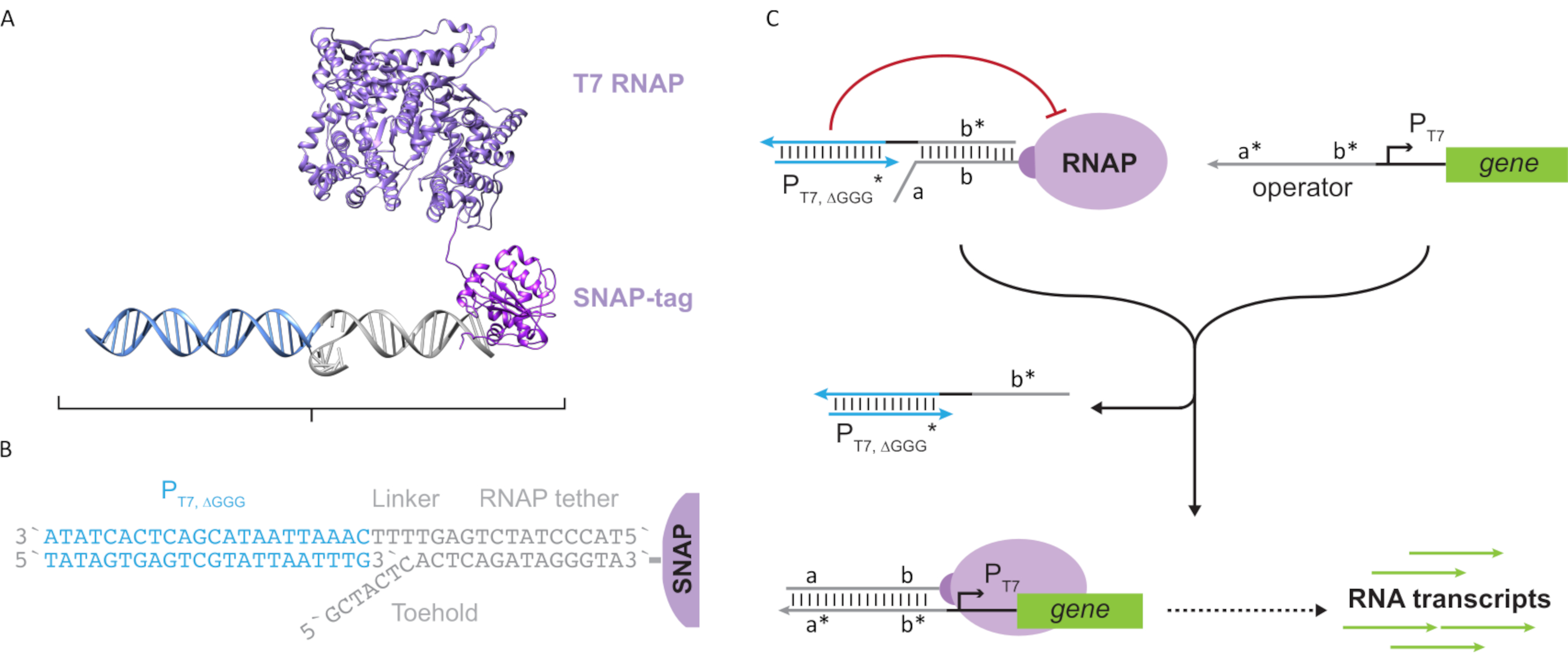
Abbildung 2: Abstraktion und Mechanismus des "Tether"- und "Cage"-Polymerase-Komplexes. (A und B) Ein Oligonukleotid-Tether wird enzymatisch durch die SNAP-Tag-Reaktion zu einer T7-Polymerase markiert. Ein Käfig, der aus einem "künstlichen" T7-Promotor mit einem Tether-Komplement-Überhang besteht, ermöglicht es, mit dem Tether zu hybridisieren und die Transkriptionsaktivität zu blockieren. (C) Wenn der Operator (a*b*) vorhanden ist, bindet er an den Haltepunkt des Oligonukleotid-Tethers (ab) und verschiebt den b*-Bereich des Käfigs, wodurch eine Transkription stattfinden kann. Diese Figur wurde von Chou und Shih27modifiziert. Abkürzungen: RNAP = RNA-Polymerase. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Dieses Papier stellt einen neuartigen Baustein für molekulares Rechnen vor, der die Funktionalitäten von Transkriptionsschaltkreisen mit der Skalierbarkeit von DNA-basierten Schaltkreisen kombiniert. Dieser Baustein ist ein T7 RNAP kovalent gebunden mit einem einzelsträngigen DNA-Tether (Abbildung 2A). Um dieses DNA-tethered T7 RNAP zu synthetisieren, wurde die Polymerase zu einem N-terminalen SNAP-Tag24 fusioniert und rekombinant in Escherichia coliexprimiert. Der SNAP-Tag wurde dann mit einem Oligonukleotid umgesetzt, das mit dem BG-Substrat funktionalisiert wurde. Das Oligonukleotid-Tether ermöglicht die Positionierung molekularer Gäste in unmittelbarer Nähe zur Polymerase mittels DNA-Hybridisierung. Einer dieser Gäste war ein kompetitiver Transkriptionsblocker, der als "Käfig" bezeichnet wird und aus einem "künstlichen" T7-Promotor-DNA-Duplex ohne nachgeschaltetes Gen besteht (Abbildung 2B). Wenn der Käfig über seinen Oligonukleotid-Tether an das RNAP gebunden ist, stoppt er die Polymeraseaktivität, indem er andere DNA-Vorlagen für die RNAP-Bindung überkompetiert, wodurch der RNAP in einen "OFF" -Zustand versetzt wird (Abbildung 2C).
Um die Polymerase in einen "ON"-Zustand zu aktivieren, wurden T7-DNA-Templates mit einzelsträngigen "Operator"-Domänen vor dem T7-Promotor des Gens entworfen. Die Operatordomäne (d.h. Domäne a*b* Abbildung 2C)kann so konstruiert werden, dass sie den Käfig über TMDSD aus dem RNAP verdrängt und die RNAP proximal zum T7-Promotor des Gens positioniert, wodurch die Transkription eingeleitet wird. Alternativ wurden auch DNA-Vorlagen entworfen, bei denen die Operatorsequenz komplementär zu Hilfs-Nukleinsäuresträngen war, die als "künstliche Transkriptionsfaktoren" bezeichnet werden (d. h. TFA- und TFB-Stränge in Abbildung 3A). Wenn beide Stränge in die Reaktion eingeführt werden, werden sie sich am Bedienerstandort zusammensetzen und eine neue pseudo-zusammenhängende Domäne a * b *erzeugen. Diese Domäne kann dann den Käfig über TMDSD verschieben, um die Transkription zu initiieren (Abbildung 3B). Diese Stränge können entweder exogen zugeführt oder hergestellt werden.

Abbildung 3: Selektive Programmierung der Polymeraseaktivität durch einen Drei-Komponenten-Schalteraktivator. (A) Wenn die Transkriptionsfaktoren (TFA und TFB) vorhanden sind, binden sie an die operatordomäne vor dem Promotor und bilden eine pseudo-einzelsträngige Sequenz (a*b*),die in der Lage ist, den Käfig durch Zehengriff-vermittelte DNA-Verschiebung zu verdrängen. (B) Diese a*b*-Domäne kann den Käfig über TMDSD verdrängen, um die Transkription zu initiieren. Diese Figur wurde von Chou und Shih27modifiziert. Abkürzungen: TF = Transkriptionsfaktor; RNAP = RNA-Polymerase; TMDSD = Toehold-vermittelte DNA-Strangverschiebung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Die Verwendung von Nukleinsäure-basierten Transkriptionsfaktoren für die In-vitro-Transkriptionsregulation ermöglicht die skalierbare Implementierung anspruchsvoller Schaltkreisverhaltensweisen wie digitale Logik, Feedback und Signalkaskadierung. Zum Beispiel kann man Logik-Gate-Kaskaden bauen, indem man Nukleinsäuresequenzen so entwirft, dass die Transkripte eines upstream-Gens ein downstreams Gen aktivieren. Eine Anwendung, die die Kaskadierung und das Multiplexing nutzt, die durch diese vorgeschlagene Technologie ermöglicht werden, ist die Entwicklung ausgefeilterer molekularer Computerschaltungen für die tragbare Diagnostik und molekulare Datenverarbeitung. Darüber hinaus kann die Integration der Molekularen Computer- und De-novo-RNA-Synthesefähigkeiten neue Anwendungen ermöglichen. Zum Beispiel kann ein molekularer Schaltkreis entworfen werden, um eine oder eine Kombination von benutzerdefinierten RNAs als therapeutische Input- und Output-RNAs oder mRNAs zu erkennen, die funktionelle Peptide oder Proteine für medizinische Point-of-Care-Anwendungen kodieren.
Access restricted. Please log in or start a trial to view this content.
Protokoll
1. Puffervorbereitung
HINWEIS: Die Vorbereitung des Proteinreinigungspuffers kann an jedem Tag erfolgen. hier wurde es vor Beginn der Experimente gemacht.
- Herstellung eines Lyse-/Gleichgewichtspuffers mit 50 mM Tris(hydroxymethyl)aminomethan (Tris), 300 mM Natriumchlorid (NaCl), 5% Glycerin und 5 mM β-Mercaptoethanol (BME), pH 8. 1,5 mL 1M Tris, 1,8 mL 5M NaCl, 1,5 mL Glycerin, 25,2 mL deionisiertes Wasser (ddH2O) in ein 50-ml-Zentrifugenröhrchen geben und kurz vor Gebrauch 10,5 μL 14,2 M BME hinzufügen.
HINWEIS: Tris kann akute Toxizität verursachen; Vermeiden Sie daher das Einatmen des Staubs und vermeiden Sie Haut- und Augenkontakt. BME ist giftig und sollte nur in einem Abzug verwendet werden. Es ist wichtig, BME zuletzt vor der Resuspension und Zelllyse hinzuzufügen. Siehe Tabelle 1 für die Lysepufferformel. - Waschpuffer (pH 8) mit 50 mM Tris, 800 mM NaCl, 5% Glycerin, 5 mM BME und 20 mM Imidazol herstellen. 1,5 ml 1 M Tris, 4,8 ml 5 M NaCl, 1,5 ml Glycerin und 22,2 mlddH2Oin ein 50 mL Zentrifugenröhrchen geben. Kurz vor der Anwendung 7 μL 14,2 M BME und 200 μL 2 M Imidazol zu 20 mL der oben genannten Lösung hinzufügen.
HINWEIS: Um eine akute Toxizität durch Imidazol zu verhindern, verwenden Sie persönliche Schutzausrüstung. Es ist wichtig, BME und Imidazol zuletzt hinzuzufügen, kurz bevor das Protein aus der Säule gewaschen wird. Siehe Tabelle 2 für die Waschpufferformel. - Elutionspuffer (pH8) mit 50 mM Tris, 800 mM NaCl, 5% Glycerin, 5 mM BME und 200 mM Imidazol herstellen. 0,5 ml 1 M Tris, 1,6 ml 5 M NaCl, 0,5 ml Glycerin und 6,4 mlddH2Oin ein 15 mL Zentrifugenröhrchen geben. Kurz vor der Anwendung 3,5 μL 14,2 M BME und 1 ml 2 M Imidazol zu 10 ml der oben genannten Lösung hinzufügen.
HINWEIS: Es ist wichtig, BME und Imidazol zuletzt hinzuzufügen, kurz bevor das Protein aus der Säule eluiert wird. Siehe Tabelle 3 für die Elutionspufferformel. - Bereiten Sie 2x Speicherpuffer (zu mischen 1:1 mit Glycerin) mit 100 mM Tris, 200 mM NaCl, 40 mM BME und 2 mM Ethylendiamintetraessigsäure (EDTA), 0,2% eines nichtionischen Tensids (siehe Materialtabelle). Bereiten Sie 50 mL des Speicherpuffers vor, indem Sie 5 mL 1 M Tris, 2 mL 5 M NaCl, 42,56 mL ddH2O, 200 μL 0,5 M EDTA, 100 μL des nichtionischen Tensids zu einem 50 mL Zentrifugenröhrchen hinzufügen. Mischen Sie, bis die Lösung homogen ist, filtern Sie den Speicherpuffer durch einen 0,2 μm Spritzenfilter und geben Sie vor der Verwendung 140,8 μL BME in die obige Lösung.
HINWEIS: Um akute Toxizität aufgrund von EDTA zu vermeiden, vermeiden Sie das Einatmen des Staubes und vermeiden Sie Haut- und Augenkontakt. Es ist wichtig, BME zuletzt hinzuzufügen und den gesamten Speicherpuffer 1:1 mit Glycerin zu mischen, kurz bevor das gereinigte Protein gelagert wird. Siehe Tabelle 4 für die Speicherpufferformel.
2. Kulturwachstum über Nacht: Tag 1
- Bereiten Sie 1.000x Kanamycin-Brühe vor, indem Sie 500 mg Kanamycin in 10 mlddH2Oauflösen.
HINWEIS: Verwenden Sie persönliche Schutzausrüstung, um akute Toxizität durch Kanamycin zu verhindern. - Fügen Sie 20 μL der 1.000x Kanamycin-Brühe zu 20 mL Lysogen-Brühe hinzu. Stochern Sie mit einer sterilen Pipettenspitze in einen umgewandelten BL21 E. coli-Glycerinvorrat und impfen Sie dann die Kultur, indem Sie die Spitze in die Wachstumsmedienbrühe einführen.

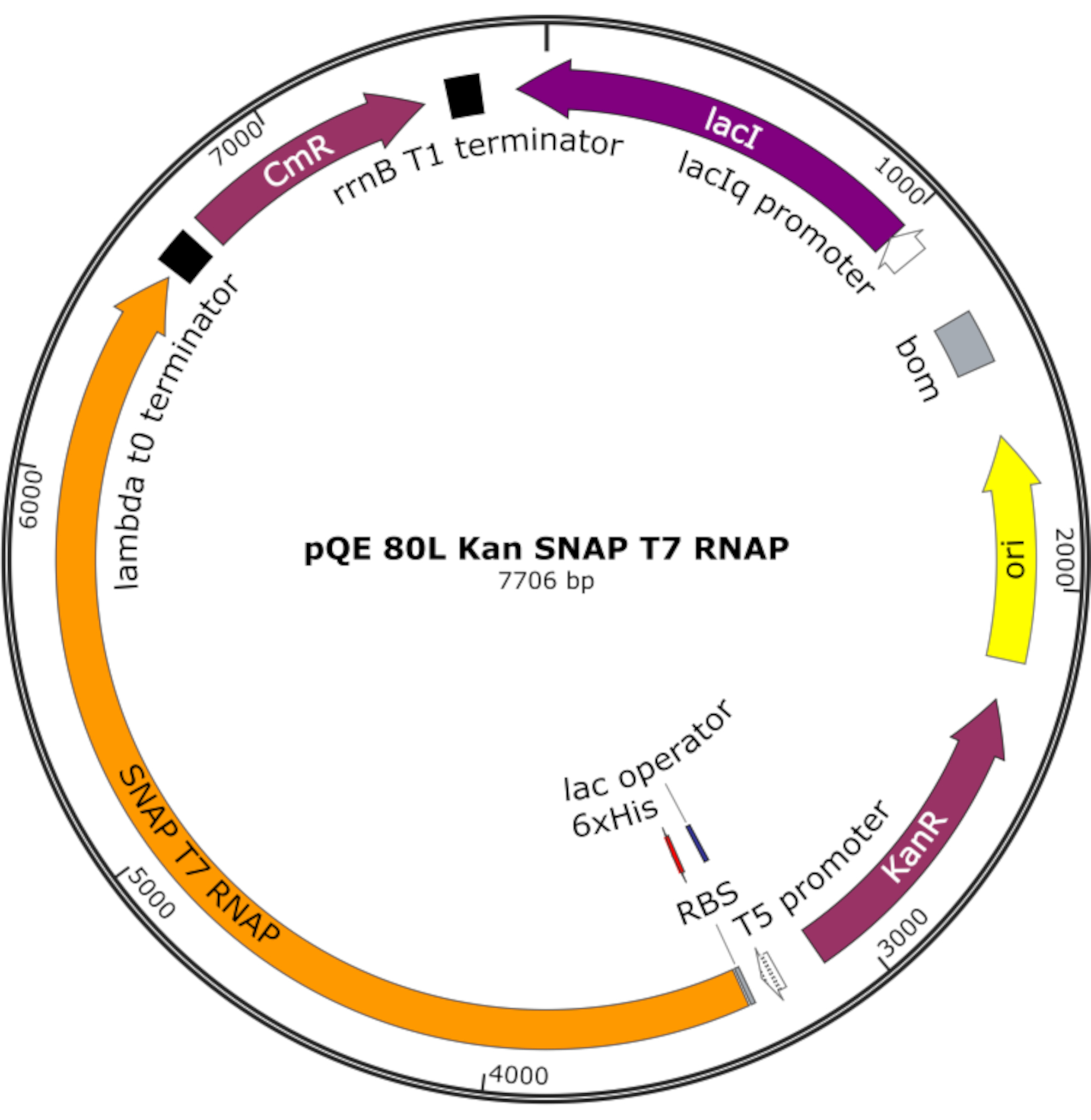
Abbildung 4: Plasmidkarte für SNAP T7 RNAP. Das Plasmid kodiert für ein T7 RNAP, das einen N-terminalen Histidin-Tag (6x His) und eine SNAP-Tag-Domäne (SNAP T7 RNAP) unter einem Lac-Repressor (lacI) auf einem pQE-80L-Backbone enthält. Weitere Merkmale sind die Gene Kanamycin-Resistenz (KanR) und Chloramphenicol-Resistenz (CmR). Abkürzung: RNAP = RNA-Polymerase. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
HINWEIS: Das Plasmid kodiert für ein T7 RNAP, das einen N-terminalen Histidin-Tag und eine SNAP-Tag-Domäne (SNAP T7 RNAP) sowie ein Kanamycin-Resistenzgen unter einem pQE-80L-Backbone enthält (Abbildung 4)25.
- Geben Sie erneut 20 μL des 1.000-fachen Kanamycin-Bestands in einen separaten Kulturkolben mit 20 ml Lysogenbrühe und inkubieren Sie ihn als Kontrolle.
- Inkubieren Sie die beiden Proben (aus den Schritten 2.2 und 2.3) über Nacht für 12-18 h bei 37 °C, während Sie bei 10 × grotieren.
3. Zellwachstum und -induktion: Tag 2
- Impfen Sie 400 ml Lysogenbrühe, die 400 μL Kanamycinvorrat enthält, mit 4 ml der über Nacht angebauten Kultur aus Schritt 2.4. Die Kulturkolben bei 37 °C inkubieren, während sie bei 10 × g gedreht werden.
- Sobald die Kultur eine optische Dichte (OD) bei 600 nm von ~0,5 erreicht hat, nehmen Sie 1 ml Probe aus dem Wachstumskolben als Kontrolle heraus. Lagern Sie die Kontrollprobe bei 4 °C.
- Induzieren Sie die Zellen mit Isopropyl-β-D-1-thiogalactopyranosid (IPTG) durch Zugabe von 40 μL 1M IPTG pro 100 ml Kultur, um eine Endkonzentration von 0,4 mM IPTG zu erreichen. Inkubieren Sie die Probe für 3 h bei 37 °C, rotieren Sie bei 10 × gund spinnen Sie dann die induzierte Kultur bei 8.000 × g für 10 min, um die Zellen zu pelletieren. Entfernen Sie den Überstand und lagern Sie das Pellet bei -20 °C bis zur weiteren Verwendung.
HINWEIS: Um akute Toxizität aufgrund von IPTG zu vermeiden, vermeiden Sie das Einatmen seines Staubes und vermeiden Sie Haut- und Augenkontakt. Bei Bedarf können Sie das Experiment hier pausieren und am nächsten Tag fortsetzen.
4. Zelllyse, Proteinreinigung: Tag 3
- Resuspendieren Sie das gelagerte Zellpellet mit 10 ml Lysepuffer auf Eis und schwenken Sie es vorsichtig, um sicherzustellen, dass das gesamte Pellet resuspendiert wird. Dann pipettieren Sie 1 ml Probe in zehn 1,5 ml Röhrchen, die auf Eis aufbewahrt werden.
- Beschallen Sie jede Probe mit einer Amplitudeneinstellung von "1", gepulst für 2 s mit einem Tastverhältnis von 50% über einen Zeitraum von 30 s. Reinigen Sie die Beschallungsspitze vor und nach jeder Probe mit 70% Ethanol und ddH2O. Bewahren Sie alle Proben während und nach der Beschallung auf Eis auf.
HINWEIS: Halten Sie 70% Ethanol von Hitze und offener Flamme fern. - Eine Nickel-geladene Nitrilotriessigsäure (Ni-NTA)-Reinigungsspinsäule auf eine Arbeitstemperatur von 4 °C ausgleichen. Platzieren/lagern Sie die Säule bei 4 °C und bewahren Sie sie während des Gebrauchs auf Eis auf.
- Zentrifugieren Sie die zehn 1-ml-Proben bei 15.000 × g für 20 min bei 4 °C. Den Überstand, der das rekombinante RNAP enthält, vorsichtig pipettieren, ohne das Pellet zu stören. Verwenden Sie bei Bedarf einen zusätzlichen Gleichgewichtspuffer, um das Gesamtvolumen auf ≥ 6 ml einzustellen.
- Entfernen Sie vorsichtig die untere Lasche aus der Ni-NTA-Spinspalte, um den Durchfluss durch die Säule zu ermöglichen. Legen Sie die Säule in ein Zentrifugenrohr und halten Sie sie auf Eis.
HINWEIS: Verwenden Sie ein 50-ml-Zentrifugenröhrchen mit den 3 ml Ni-NTA-Spinsäulen. - Zentrifugieren Sie die Säule bei 700 × g und 4 °C für 2 min, um den Speicherpuffer zu entfernen. Gleichen Sie die Spalte aus, indem Sie der Spalte 6 ml Äquilibrierungspuffer hinzufügen. Lassen Sie den Puffer vollständig in das Harzbett eindringen.
- Den Gleichgewichtspuffer durch Zentrifugation bei 700 × g und 4 °C für 2 min aus der Säule entfernen. Bevor Sie den vorbereiteten Zellextrakt zur Spalte hinzufügen, legen Sie einen unteren Stecker auf die Säule, um den Verlust eines Produkts zu vermeiden. Dann den Zellextrakt in die Säule geben und auf einem Orbital-Shaker-Mixer für 30 min bei 4 °C mischen.
- Entfernen Sie den unteren Stecker von der Säule und legen Sie die Säule in ein 50 ml Zentrifugenrohr mit der Aufschrift Flow Through. Zentrifugieren Sie die Säule bei 700 × g für 2 min, um den Durchfluss zu sammeln.
- Geben Sie 6 ml Waschpuffer in die Säule, um das Harz zu waschen. Zentrifugieren Sie die Säule bei 700 × g für 2 min, um die Fraktion in einem neuen Zentrifugenröhrchen mit der Bezeichnung Wash 1zu sammeln. Wiederholen Sie diesen Schritt noch zwei weitere Male für insgesamt 3 separate Fraktionen und sammeln Sie die Fraktionen in separaten Zentrifugenröhrchen (Waschen 2 und Waschen 3).
- Fügen Sie 3 ml Elutionspuffer hinzu, um die His-markierten Proteine aus dem Harz zu eluieren. Zentrifugieren Sie die Säule bei 700 × g für 2 min, um die Fraktion in einem neuen Zentrifugenröhrchen mit der Bezeichnung Eluat 1zu sammeln. Wiederholen Sie diesen Schritt zwei weitere Male für insgesamt 3 separate Fraktionen und sammeln Sie die Fraktionen in separaten Zentrifugenröhrchen (Eluat 2 und Eluat 3).
- Kombinieren Sie die Eluate und führen Sie eine Entsalzung durch, um Salze aus der Proteinlösung zu entfernen.
- 15 mL 0,05 % w/v Polysorbat 20 über eine 100 kDa Zentrifugalfiltereinheit pipettieren. Zentrifugiere bei 4.000 × g für 40 min und entsorge den Durchfluss.
- Verwenden Sie den beschichteten Filter, um die Eluate 1, 2 und 3 (9 mL Gesamtproteinelit + 6 ml Speicherpuffer) auf ~ 1.500 μL zu konzentrieren. Zentrifugieren Sie den Filter bei 3.220 × g für 20 minuten und waschen Sie die Membran vorsichtig mit einer Pipette, um eine Ausfällung zu vermeiden.
- Verdünnen Sie die Probe mit Lagerpuffer auf 15 ml. Führen Sie einen Pufferaustausch mit dem Speicherpuffer 1:1.000 durch, indem Sie Schritt 4.11.2 zwei weitere Male wiederholen.
- Quantifizieren Sie das gereinigte Protein, indem Sie die Absorption der Fraktion bei 280 nm messen. Blank das Spektralphotometer mit Speicherpuffer (2x Speicherpuffer bei 4 °C). Mischen Sie die Probe der kombinierten Eluate vorsichtig und messen Sie ihre Absorption.
HINWEIS: Führen Sie drei separate Messwerte bei 1x, 10x und 50x Verdünnungen der Proteinprobe durch, um das Protein zu mittelieren und zu quantifizieren. Verdünnen Sie die Proben im Lagerpuffer. - Stellen Sie die Proteinproben mit 2x Speicherpuffer auf 100 μM ein. Verdünnen Sie die eingestellte Probe 1:1 volumenmäßig mit 100% Glycerin. Die resultierende Proteinlösung wird bei -80 °C gelagert.
5. Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese (SDS-PAGE) Analyse des Proteinprodukts: Tag 3
- Führen Sie ein SDS-PAGE-Gel für die Proteinanalyse aus. Mischen Sie 9 μL der Probe mit 3 μL 4x Lithium-Dodecylsulfat (LDS) Protein-Loading-Farbstoff. Die Proben 10 min bei 95 °C erhitzen.
- Laden Sie die Proben auf ein 4-12% Bis-Tris SDS-PAGE Gel-Setup. Laden Sie die Proteinleiter in Vertiefung 1, dann mit Proben (von links nach rechts): Durchfluss, Waschen 1, Waschen 2, Waschen 3, Elution 1, Elution 2, Elution 3 und gesamte entsalzte Elution.
HINWEIS: Tabelle 5 enthält eine Probenladetabelle für das SDS-PAGE Gel. - Führen Sie die geladenen Gelproben in 2-(N-morpholino)Ethansulfonsäure (MES) Puffer für 35 min bei 200 V. Spülen Sie das Gel in einer sauberen Schale dreimal für jeweils 10 min mit 200 ml ddH2O, mit sanfter Bewegung, um SDS aus der Gelmatrix zu entfernen.
HINWEIS: Tragen Sie persönliche Schutzausrüstung, um akute Toxizität aufgrund des MES zu vermeiden. - Färben Sie das Gel mit 20 ml Coomassie Blue und inkubieren Sie das Gel über Nacht bei Raumtemperatur mit sanfter Bewegung. Entfärben Sie das Gel zweimal für jeweils 1 h mit 200 ml ddH 2 O unter sanfter Bewegung aufeinemOrbitalschüttler.
HINWEIS: Das Gel über einen längeren Zeitraum zu waschen oder das Wasser häufig zu ersetzen, erhöht die Empfindlichkeit. Darüber hinaus beschleunigt das Platzieren eines gefalteten, empfindlichen Wischtuchs in den Behälter, um überschüssigen Farbstoff zu absorbieren, den Entfärbungsprozess.
6. Funktionelle Verifizierung von SNAP T7 RNAP mittels In-vitro-Transkription
HINWEIS: Dieses Protokoll verwendet eine DNA-Vorlage, die für das fluoreszierende Brokkoli-RNA-Aptamer kodiert und die Verwendung von Fluoreszenz zur Überwachung der Kinetik der Transkription auf einem Fluoreszenzplattenleser ermöglicht.
- Richten Sie drei In-vitro-Transkriptionsreaktionen (IVT) ein, um die Aktivität von SNAP T7 RNAP mit Wildtyp (WT) T7 RNAP aus einer kommerziellen Quelle und einer reinen Pufferkontrolle zu vergleichen. Stellen Sie das Volumen jeder Reaktion auf 20 μL ein.
- Bereiten Sie die SNAP T7 RNAP IVT-Reaktion vor, indem Sie 2 μL 10x Transkriptionspuffer, 0,4 μL 25 mM Ribonukleosidtriphosphat (rNTP) Mischung, 5 μL 500 nM DNA-Vorlage, 2 μL 500 nM SNAP T7 RNAP und 10,6 μL ddH2O mischen.
- Bereiten Sie die WT RNAP IVT-Reaktion vor, indem Sie 2 μL 10x Transkriptionspuffer, 0,4 μL 25 mM rNTP-Mix, 5 μL 500 nM DNA-Template, 2 μL WT T7 RNAP und 10,6 μL ddH2O mischen.
- Bereiten Sie die reine Puffer-IVT-Reaktion vor, indem Sie 2 μL 10x Transkriptionspuffer, 0,4 μL 25 mM rNTP-Mix, 5 μL 500 nM DNA-Template und 12,6 μL ddH2O mischen.
HINWEIS: Fügen Sie die RNAP zuletzt hinzu und halten Sie die Proben bis zu ihrer Einführung auf Eis. Tabelle 6, Tabelle 7und Tabelle 8 enthalten die IVT-Reaktionsformeln.
- Überwachung der Transkriptionskinetik auf einem Fluoreszenzplattenleser für 2 h in 2-min-Intervallen bei 37 °C unter Verwendung einer Anregungswellenlänge von 470 nm und einer Emissionswellenlänge von 512 nm.
7. Herstellung von BG-modifizierten Oligonukleotiden: Tag 1
- Das Oligonukleotid mit 3'-Amin-Modifikation inddH2Owird auf eine Endkonzentration von 1 mM gelöst. Beschriften Sie diese S1.
- Mischen Sie 25 μL 1 M Natriumbicarbonat (NaHCO3),284 μL 100% Dimethylsulfoxid (DMSO), 125 μL S1 (Oligonukleotidstock) und 66 μL 50 mM des mit DMSO verdünnten BG-N-Hydroxysuccinimidesters (NHS) (BG-GLA-NHS), stellen Sie das Volumen auf 500 μL ein und inkubieren Sie über Nacht bei Raumtemperatur bei 100 × g.
HINWEIS: Halten Sie DMSO von Hitze und Flammen fern, da es sich um eine brennbare Flüssigkeit handelt. Tabelle 9 enthält die Reaktionsformel für die BG-Konjugation zum Oligonukleotid.
- Mischen Sie 25 μL 1 M Natriumbicarbonat (NaHCO3),284 μL 100% Dimethylsulfoxid (DMSO), 125 μL S1 (Oligonukleotidstock) und 66 μL 50 mM des mit DMSO verdünnten BG-N-Hydroxysuccinimidesters (NHS) (BG-GLA-NHS), stellen Sie das Volumen auf 500 μL ein und inkubieren Sie über Nacht bei Raumtemperatur bei 100 × g.
8. Ethanol/Aceton-Ausfällung von BG-Oligonukleotid-Konjugat: Tag 2
- Zentrifugieren Sie das Produkt von Schritt 7.1.1. bei 13.000 × g für 5 min. Den Überstand vorsichtig in ein frisches Röhrchen überführen und alle ausgefällten BG verwerfen. Teilen Sie die Reaktion in zwei gleiche 250 μL Aliquots auf, um ein Überlaufen zu verhindern, und führen Sie die folgenden Schritte an beiden Aliquots durch.
- Fügen Sie 1/10 des Volumens von 3 M Natriumacetat (25 μL) hinzu, gefolgt vom 2,5-fachen des Volumens in 100% Ethanol (625 μL). Bei -80 °C für 1 h inkubieren.
HINWEIS: Verwenden Sie persönliche Schutzausrüstung, wenn Sie sowohl Natriumacetat (kann Augen, Haut, Verdauungstrakt und Atemwege reizen) als auch Ethanol (hochentzündlich, verursacht Reizungen bei Kontakt) handhaben. Wenn nötig, pausieren Sie das Experiment hier und fahren Sie am nächsten Tag fort. - Legen Sie die Röhrchen in die Zentrifuge und markieren Sie den äußeren Rand. Zentrifugieren Sie die Röhrchen bei 17.000 × g für 30 min bei 4 °C.
HINWEIS: Das Oligonukleotid-Pellet erscheint am markierten Rand des Röhrchens. - Ohne das Pellet zu stören, entsorgen Sie den Überstand. Mit 750 μL gekühltem 70% Ethanol auffüllen und bei 17.000 × g für 10 min bei 4 °C drehen.
- Ohne das Pellet zu stören, entsorgen Sie den Überstand. Mit 750 μL 100% Aceton auffüllen und bei 17.000 × g für 10 min bei 4 °C drehen.
HINWEIS: Verwenden Sie beim Umgang mit Aceton eine persönliche Schutzausrüstung, da es extrem entzündlich ist und bei Kontakt Reizungen verursacht. - Bei geöffnetem Rohrdeckel 5 min an der Luft trocknen, um überschüssiges Aceton durch Verdunstung zu entfernen. Lösen Sie das Oligonukleotid in 250 μL 1x Tris-EDTA (TE) -Puffer wieder auf, um eine ~ 850 μM BG-Oligonukleotidlösung zu erhalten.
- Wiederholen Sie die Schritte 8.2 bis 8.6 und lösen Sie sie in 70 μL 1x TE-Puffer wieder auf. Beschriften Sie diese S2.
9. BG-Oligonukleotid-Reinigung mittels Gelfiltrationschromatographie
- Suspendieren Sie die Matrix, indem Sie die Spalten mehrmals kräftig invertieren; Entfernen Sie die obere Kappe und schnappen Sie die untere Spitze der Spalte ab. Legen Sie die Säule in ein 1,5 ml Zentrifugenröhrchen und zentrifugieren Sie das Röhrchen bei 1.000 × g für 1 min bei Raumtemperatur. Entsorgen Sie den eluierten Puffer und das Auffangrohr.
HINWEIS: Es ist wichtig, Vakuumbildung zu verhindern. Verwenden Sie vorbereitete Spalten sofort. - Legen Sie die verpackten Säulen in saubere 1,5 ml Zentrifugenröhrchen. 300 μL 1x TE-Puffer in die Mitte des Säulenbettes geben und bei 1.000 × g für 2 min zentrifugieren, um die Pufferlösung auszutauschen. Verwerfen Sie erneut den eluierten Puffer und das Auffangrohr.
- Legen Sie die puffergetauschten Säulen in saubere 1,5 ml Zentrifugenröhrchen. Tragen Sie bis zu 75 μL Probe auf die Mitte des Bettes auf. 4 min bei 1.000 × g drehen.
HINWEIS: Stören Sie nicht das Bett oder berühren Sie nicht die Seiten der Säule; der höchste Punkt des Gelmediums sollte in Richtung des äußeren Rotors zeigen. - Sammeln Sie das Eluat aus dem Auffangröhrchen, da es die gereinigte Nukleinsäure enthält. Um die Probe zu quantifizieren, messen Sie ihre Absorption bei 260 nm; beschriften Sie diese S3.
HINWEIS: Notieren Sie sich die bei der Messung verwendete Pfadlänge und berechnen Sie die Konzentration nach dem Beer-Lambert-Gesetz.
10. Denaturierung page Analyse des BG-Oligonukleotidkonjugats
- Gießen Sie ein 18% Tris-Borat-EDTA (FSME)-Harnstoff PAGE Gel. 4,8 g Harnstoff, 4,5 ml 40%iges Acrylamid (19:1) und 1 ml 10x FSME in 2,8 mlddH2O werden gelöst; 5 μL Tetramethylethylendiamin (TEMED) zugeben und gründlich mischen. Wiederholen Sie dies mit 100 μL 10% Ammoniumpersulfat (APS). Gießen Sie die Lösung in eine leere Gelkassette und lassen Sie die Polymerisation für 40 min.
HINWEIS: Verwenden Sie eine geeignete persönliche Schutzausrüstung, wenn Sie mit Harnstoff (verursacht Reizungen von Augen und Haut), Acrylamid (toxisch und krebserregend) und TEMED (giftig, brennbar, ätzend) umgehen. Tabelle 10 enthält die Reaktionsformel für ein 18%iges TBE-UREA Polyacrylamid-Gel. - Mikrowelle 500 ml FSME-Puffer (0,5x) für 2 min und 30 s oder bis ~70 °C und in einen Gelapparat geben. Formamid (Denaturierung) Beladungsfarbstoff mit 95% Formamid + 1 mM EDTA und Bromphenolblau herstellen. Mischen Sie den Beladungsfarbstoff mit jeder Probe und laden Sie die Mischung auf das Polyacrylamid-Gel.
HINWEIS: Verwenden Sie eine geeignete persönliche Schutzausrüstung, wenn Sie mit Formamid umgehen, da es krebserregend ist. Tabelle 11 enthält eine Probengelladetabelle. - Lassen Sie das Gel bei 270 V für 35 min laufen oder bis die Farbstofffront zum Ende wandert. Legen Sie das Gel in eine Gelbox und färben Sie es vor der Bildgebung 15 minuten lang mit Cyaninfarbstoff für Nukleinsäuren bei Raumtemperatur ein.
HINWEIS: Verwenden Sie eine geeignete persönliche Schutzausrüstung, wenn Sie mit Cyaninfarbstoff umgehen, da er brennbar ist.
11. Konjugation von Oligonukleotid zu SNAP T7 RNAP und PAGE Analyse
- Bereiten Sie die Reagenzien für die Kopplung von BG-Oligonukleotid an SNAP T7 RNAP vor: Machen Sie 9 Verdünnungen von einzelsträngigem DNA (ssDNA) -Oligo mit ddH2O, um Oligo: RNAP-Verhältnisse im Bereich von 5: 1 bis 1: 5 zu erzeugen. Verdünnen Sie den Proteinbestand auf 50 μM.
ANMERKUNG: Beispielverhältnisse finden Sie in Tabelle 12; diese Verhältnisse werden unter Verwendung einer RNAP-Konzentration von 50 μM berechnet. - Für jede Verdünnung von ssDNA-Oligo sind 10 μL der Reaktionsmischung herzustellen, die 2 μL SNAP-Puffer, 4 μL BG-Oligonukleotid und 4 μL SNAP T7 RNAP enthält.
HINWEIS: Tabelle 13 enthält Reaktionsformeln für die SNAP-Tag-Etikettierungsreaktion.- Vorbereitung von zwei weiteren Kontrollproben: 1) eine RNAP-Kontrolle durch Ersetzen vonBG-Oligonukleotid durch ddH2O; 2) eine DNA-Kontrolle durch Ersetzen von SNAP T7 RNAP durch ddH2O (für die niedrigste Oligonukleotidkonzentration von SNAP T7 RNAP). Inkubieren Sie alle Proben bei Raumtemperatur für 1 h und bewahren Sie sie bis zum Bedarf auf Eis auf.
- Richten Sie elf 10 μL-Reaktionen ein, indem Sie 2 μL jeder Probe zu 4 μL SNAP-Puffer und 2 μL Proteinbeladungsfarbstoff hinzufügen und 10 Minuten lang bei 70 °C erhitzen. Laden Sie 2 μL jeder Probe auf das 4-12% ige Bis-Tris-Proteingel und führen Sie eine Gelelektrophorese auf Eis bei 200 V für 35 min durch.
ANMERKUNG: Tabelle 14 enthält Reaktionsformeln für die Gelbeladungsproben.- SDB über 3x Wasserwechsel auf einem Shaker abwaschen, wobei jede Wäsche jeweils 10 min dauert. Färben Sie mit Cyaninfarbstoff für Nukleinsäuren für 15 min vor der Bildgebung. Färben Sie das Gel erneut mit 20 ml Coomassie Blue Stain für 1 h. Entfärben Sie mit ddH2O für 1 h (oder über Nacht) vor der Bildgebung.
HINWEIS: Im Gel erzeugt eine der Reaktionen die am stärksten angebundene Polymerase zusammen mit der geringsten Menge an überschüssigem freiem BG-Oligonukleotid; dies ist das optimale Verhältnis.
- SDB über 3x Wasserwechsel auf einem Shaker abwaschen, wobei jede Wäsche jeweils 10 min dauert. Färben Sie mit Cyaninfarbstoff für Nukleinsäuren für 15 min vor der Bildgebung. Färben Sie das Gel erneut mit 20 ml Coomassie Blue Stain für 1 h. Entfärben Sie mit ddH2O für 1 h (oder über Nacht) vor der Bildgebung.
- Bereiten Sie Reagenzien für die präparative Skala vor, die BG-Oligonukleotid an SNAP T7 RNAP koppelt. Führen Sie die Kopplungsreaktion mit dem optimalen Verhältnis durch, das in der analytischen Skala zu finden ist.
HINWEIS: Minimieren Sie die Proteinexposition gegenüber Raumtemperatur, indem Sie das Protein bei Nichtgebrauch auf Eis legen.
12. Reinigung von Oligonukleotid-gebundenem SNAP-T7 mittels Ionenaustauschsäulen
- Befolgen Sie die Anweisungen des Herstellers für die Rohreinrichtung, wenn diese von den hier aufgeführten Anweisungen abweicht. Bereiten Sie einen Reinigungspuffer mit einem pH-Wert vor, der höher ist als der isoelektrische Punkt des Proteins.
HINWEIS: Für das Beispielprotein in diesem Protokoll wurde ein Reinigungspuffer von 10 mM Natriumphosphatpuffer (pH 7) verwendet.- Herstellen von 1.000 μL Elutionspuffer mit Endkonzentrationen von 50 mM Tris und 0,5 M NaCl. Mischen Sie 50 μL 1 M Tris, 100 μL 5 M NaCl und 850 μL ddH2O.
ANMERKUNG: Tabelle 15 enthält die Reaktionsformel für den Elutionspuffer.
- Herstellen von 1.000 μL Elutionspuffer mit Endkonzentrationen von 50 mM Tris und 0,5 M NaCl. Mischen Sie 50 μL 1 M Tris, 100 μL 5 M NaCl und 850 μL ddH2O.
- Legen Sie eine Säule in ein 2-ml-Zentrifugenröhrchen und waschen Sie es mit Reinigungspuffer bei 2.000 × g für 15 min oder bis der gesamte Puffer eluiert wurde. Verwerfen Sie den eluierten Puffer.
- Verdünnen Sie jede Probe mit einem Reinigungspuffer im Verhältnis von 3:1 Reinigungspuffer:Probe und laden Sie die Probe jeweils 400 μL in die Säule. Drehen Sie sich bei 2.000 × g für 10 min oder bis der gesamte Puffer eluiert ist. Sammeln Sie den Durchfluss und kennzeichnen Sie ihn als Durchfluss.
- Fügen Sie 400 μL Reinigungspuffer in die Mitte der Säule hinzu. Drehen Sie bei 2.000 × g für 15 min oder bis der gesamte Puffer eluiert ist. Sammeln Sie den Durchfluss und kennzeichnen Sie ihn als Waschgang 1. Wiederholen Sie noch zweimal für Waschgang 2 und Waschen 3.
- Fügen Sie 50 μL Elutionspuffer in die Mitte der Säule hinzu. Drehen Sie bei 2.000 × g für 5 min oder bis der gesamte Puffer eluiert ist. Sammeln Sie den Durchfluss und beschriften Sie ihn als Eluat 1. Wiederholen Sie noch zweimal für Eluat 2 und Eluat 3.
- Pool eluiert 1, 2 und 3 (markieren Sie dieses Gesamteluat),so dass ein kleiner Bruchteil jedes Eluats für das Gel verbleibt, und messen Sie die Absorption bei 260 nm (A260) und 280 nm (A280). Nach der Messung Glycerin im Verhältnis 1:1 zugeben und bis zur weiteren Verwendung bei -20 °C lagern.
- Verwenden Sie eine Zentrifugalfiltereinheit (0,5 ml; 30 kDa), um das Gesamteluat mit 2x Speicherpuffer (~ 1:100) zu puffern (etikettieren Sie dieses Produkt). Messen Sie erneut A260/280. Glycerin im Verhältnis 1:1 zugeben und bis zur weiteren Verwendung bei -20 °C lagern.
- Laden Sie jedes Eluat: Durchfluss, waschen Sie 1-3, Gesamteluat und Das Produkt in ein 4-12% Bis-Tris SDS-PAGE Gel zusammen mit einer Proteinleiter. Laufen Sie bei 200 V für 35 minuten oder bis die Farbstofffront zum Ende wandert.
13. Nachweis der On-Demand-Kontrolle der Tethered-RNA-Polymerase-Aktivität
- 5x Glühpuffer mit 25 mM Tris, 5 mM EDTA und 25 mM Magnesiumchlorid(MgCl2)herstellen. Mischen Sie 2,4 μL jeder Schablone (1 μM) mit 5 μL Glühpuffer und 14,2 μL ddH2O zu 25 μL 1 μM dsDNA-Käfig. Diese Lösung bei 75 °C für 2 min inkubieren. In ähnlicher Weise glühen die Sinnes- und Antisense-Stränge des Promotors und der malachitgrünen Aptamer-DNA-Schablone. Bereiten Sie eine 1mM-Lösung von Malachit grünem Oxalat vor.
HINWEIS: Tabelle 16 enthält die Reaktionsformel für 5x Glühpuffer, Tabelle 17 enthält die Reaktionsformel für das Glühen von zwei ssDNA-Schablonen. - Inkubieren Sie das angebundene SNAP T7 RNAP mit dem dsDNA-Käfig im Molarenverhältnis 1:5 bei Raumtemperatur für 15 min auf eine Endkonzentration von 500 nM RNAP. Auf Eis halten, bis nötig.
- Plattenleser auf 37 °C vorheizen. Drei 25 μL IVT-Reaktionen auf Eis einrichten
- Richten Sie eine Reaktion ein, die das caged SNAP T7RNAP mit Nukleinsäure-Transkriptionsfaktoren enthält. Mischen Sie 2,5 μL 10x IVT-Puffer, 1 μL 25 mM rNTP-Mix, 1 μL 1 mM Malachitgrün, 2,5 μL der RNAP-Cage-Mischung, je 2,5 μL 1 μM Transkriptionsfaktor A und B Oligonukleotidstränge und 3 μL 1 mM Malachit grüne Aptamer-Schablone in 10 μL ddH2O.
- Richten Sie eine Reaktion ein, die den caged SNAP T7RNAP ohne Nukleinsäure-Transkriptionsfaktoren enthält. Mischen Sie 2,5 μL 10x IVT-Puffer, 1 μL 25 mM rNTP-Mix, 1 μL 1 mM Malachitgrün, 2,5 μL der RNAP-Cage-Mischung und 3 μL 1mM Malachitgrün-Aptamer-Schablone in 15 μL ddH2 O.
- Richten Sie eine Reaktion ein, die nur Puffer enthält. Mischen Sie 2,5 μL 10x IVT-Puffer, 1 μL 25 mM rNTP-Mix, 1 μL 1 mM Malachitgrün und 3 μL 1 mM Malachitgrün-Aptamer-Schablone in 17,5 μL ddH2O.
ANMERKUNG: Tabelle 18 enthält eine allgemeine Referenz für die In-vitro-Transkriptionsreaktionen.
- Übertragen Sie jede Reaktion auf eine 384-Well-Platte. Überwachung der Transkription des Malachitgrünen Aptamers auf einem Fluoreszenzplattenleser für 2 h bei 37 °C und mit 610 nm Anregung und 655 nm Emission. Wenn Sie fertig sind, halten Sie den Teller auf Eis, bis er benötigt wird.
- Mikrowelle 0,5x FSME-Puffer für 2 min 30 s oder bis ~70 °C. Führen Sie die RNA-Produkte jeder Vertiefung in einem denaturierenden 12% igen TBE-Urea-Polyacrylamid-Gel in den erhitzten 0,5-fachen FSME-Puffer bei 280 V für 20 minuten oder bis die Farbstofffront das Ende erreicht. Färben Sie das Gel mit Cyaninfarbstoff-Nukleinsäure-Färbung für 10 min auf einem Orbital-Shaker vor der Bildgebung.
HINWEIS: Tabelle 19 enthält die Reaktionsformel für ein denaturierendes 12%iges TBE-Urea PAGE Gel.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse

Abbildung 5: SDS-PAGE-Analyse der SNAP T7 RNAP-Expression und In-vitro-Transkriptionsassay. (A) SNAP T7 RNAP Proteinreinigungsanalyse, SNAP T7 RNAP Molekulargewicht: 119,4 kDa. FT = Durchfluss aus der Säule, W1 = Elutionsfraktionen des Waschpuffers, der Verunreinigungen enthält, E1-3 = Elutionsfraktionen, die gereinigtes Produkt enthalten, ...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Diese Studie zeigt einen von der DNA-Nanotechnologie inspirierten Ansatz zur Kontrolle der Aktivität der T7-RNA-Polymerase durch kovalente Kopplung eines N-terminalen SNAP-markierten rekombinanten T7 RNAP mit einem BG-funktionalisierten Oligonukleotid, das anschließend zur Programmierung von TMDSD-Reaktionen verwendet wurde. Durch das Design wurde der SNAP-Tag am N-Terminus der Polymerase positioniert, da der C-Terminus des Wildtyps T7 RNAP im Proteinstrukturkern vergraben ist und wichtige Kontakte mit der DNA-Vorlage<...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Es gibt keine konkurrierenden finanziellen Interessen, die von einem der Autoren erklärt werden müssen.
Danksagungen
L.Y.T.C würdigt die großzügige Unterstützung des New Frontiers in Research Fund-Exploration (NFRF-E), des Discovery Grant des Natural Sciences and Engineering Research Council of Canada (NSERC) und der Medicine by Design Initiative der University of Toronto, die vom Canada First Research Excellence Fund (CFREF) finanziert wird.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| 0.5% polysorbate 20 (TWEEN 20) | BioShop | TWN510.5 | |
| 0.5M ethylenediaminetetraacetic acid (EDTA) | Bio Basic | SD8135 | |
| 10 mM sodium phosphate buffer (pH 7) | Bio Basic | PD0435 | Tablets used to make 10 mM buffer |
| 10% ammonium persulfate (APS) | Sigma Aldrich | A3678-100G | |
| 100 kDa Amicon Ultra-15 Centrifugal Filter Unit | Fisher Scientific | UFC910008 | |
| 100% acetone | Fisher Chemical | A18P4 | |
| 100% ethanol (EtOH) | House Brand | 39752-P016-EAAN | |
| 10x in vitro transcription (IVT) buffer | New England Biolabs | B9012 | |
| 10x Tris-Borate-EDTA (TBE) buffer | Bio Basic | A0026 | |
| 1M Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Sigma Aldrich | I5502-1G | |
| 1M sodium bicarbonate buffer | Sigma Aldrich | S6014-500G | |
| 1M Tris(hydroxymethyl)aminomethane (Tris) | Sigma Aldrich | 648311-1KG | |
| 1X Tris-EDTA (TE) buffer | ThermoFisher | 12090015 | |
| 2M imidazole | Sigma Aldrich | 56750-100G | |
| 2-mercaptoethanol (BME) | Sigma Aldrich | M3148 | |
| 3M sodium acetate | Bio Basic | SRB1611 | |
| 40% acrylamide (19:1) | Bio Basic | A00062 | |
| 4x LDS protein sample loading buffer | Fisher Scientific | NP0007 | |
| 5M sodium chloride (NaCl) | Bio Basic | DB0483 | |
| 5mM dithiothreitol (DTT) | Sigma Aldrich | 43815-1G | |
| 6x gel loading dye | New England Biolabs | B7024S | |
| agarose B powder | Bio Basic | AB0014 | |
| BG-GLA-NHS | New England Biolabs | S9151S | |
| BL21 competent E. coli | Addgene | C2530H | |
| BLUeye prestained protein ladder | FroggaBio | PM007-0500 | |
| bromophenol blue | Bio Basic | BDB0001 | |
| coomassie blue (SimplyBlue SafeStain) | ThermoFisher | LC6060 | |
| cyanine dye (SYBR Gold nucleic acid gel stain) | Fisher Scientific | S11494 | |
| cyanine dye (SYBR Safe nucleic acid gel stain) | Fisher Scientific | S33102 | |
| dry dimethyl sulfoxide (DMSO) | Fisher Scientific | D12345 | |
| formamide | Sigma Aldrich | F9037-100ML | |
| glycerol | Bio Basic | GB0232 | |
| kanamycin sulfate | BioShop | KAN201.5 | |
| lysogeny broth | Sigma Aldrich | L2542-500ML | |
| malachite green oxalate | Sigma Aldrich | 2437-29-8 | |
| N,N,N'N'-Tetramethylethane-1,2-diamine (TEMED) | Sigma Aldrich | T9281-25ML | |
| NuPAGE MES SDS running buffer (20x) | Fisher Scientific | LSNP0002 | |
| NuPAGE Novex 4-12% Bis-Tris gel 1.0 mm 12-well | Life Technologies | NP0322BOX | |
| oligonucleotide (cage antisense) | IDT | N/A | TATAGTGAGTCGTATTAATTTG |
| oligonucleotide (cage sense) | IDT | N/A | TCAGTCACCTATCTGTTTCAAA TTAATACGACTCACTATA |
| oligonucleotide (malachite green aptamer antisense) | IDT | N/A | GGATCCATTCGTTACCTGGCT CTCGCCAGTCGGGATCCTATA GTGAGTCGTATTACAGTTCCAT TATCGCCGTAGTTGGTGTACT |
| oligonucleotide (malachite green aptamer sense) | IDT | N/A | TAATACGACTCACTATAGGATC CCGACTGGCGAGAGCCAGGT AACGAATGGATCC |
| oligonucleotide (Transcription Factor A) | IDT | N/A | AGTACACCAACTACGAGTGAG |
| oligonucleotide (Transcription Factor B) | IDT | N/A | TCAGTCACCTATCTGGCGATAA TGGAACTG |
| oligonucleotide with 3’ Amine modification (tether) | IDT | N/A | GCTACTCACTCAGATAGGTGAC TGA/3AmMO/ |
| Pierce strong ion exchange spin columns | Fisher Scientific | 90008 | |
| plasmid encoding SNAP T7 RNAP and kanamycin resistance genes | Genscript | N/A | custom gene insert |
| protein purification column (HisPur Ni-NTA spin column) | Fisher Scientific | 88226 | |
| rNTP mix | New England Biolabs | N0466S | |
| Roche mini quick DNA spin column | Sigma Aldrich | 11814419001 | |
| Triton X-100 | Sigma Aldrich | T8787-100ML | |
| Ultra Low Range DNA ladder | Fisher Scientific | 10597012 | |
| urea | BioShop | URE001.1 |
Referenzen
- Cherry, K. M., Qian, L. Scaling up molecular pattern recognition with DNA-based winner-take-all neural networks. Nature. 559 (7714), 370-376 (2018).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475 (7356), 368-372 (2011).
- Chen, Y. -J., et al. Programmable chemical controllers made from DNA. Nature Nanotechnology. 8 (10), 755-762 (2013).
- di Bernardo, D., Marucci, L., Menolascina, F., Siciliano, V. Predicting synthetic gene networks. Synthetic Gene Networks: Methods and Protocols. 813, 57-81 (2012).
- Xiang, Y., Dalchau, N., Wang, B. Scaling up genetic circuit design for cellular computing: advances and prospects. Natural Computing. 17 (4), 833-853 (2018).
- Gould, N., Hendy, O., Papamichail, D. Computational tools and algorithms for designing customized synthetic genes. Frontiers in Bioengineering and Biotechnology. 2, (2014).
- MacDonald, J. T., Siciliano, V. Computational sequence design with R2oDNA Designer. Mammalian Synthetic Promoters. 1651, 249-262 (2017).
- Cervantes-Salido, V. M., Jaime, O., Brizuela, C. A., Martínez-Pérez, I. M. Improving the design of sequences for DNA computing: A multiobjective evolutionary approach. Applied Soft Computing. 13 (12), 4594-4607 (2013).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry. 32 (1), 170-173 (2011).
- Fornace, M. E., Porubsky, N. J., Pierce, N. A. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synthetic Biology. 9 (10), 2665-2678 (2020).
- Wetterstrand, K. DNA sequencing costs: Data. Genome.gov. , (2020).
- Lopez, R., Wang, R., Seelig, G. A molecular multi-gene classifier for disease diagnostics. Nature Chemistry. 10 (7), 746-754 (2018).
- Pardee, K., et al. low-cost detection of Zika virus using programmable biomolecular components. Cell. 165 (5), 1255-1266 (2016).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406 (6796), 605-608 (2000).
- Lin, K. N., Volkel, K., Tuck, J. M., Keung, A. J. Dynamic and scalable DNA-based information storage. Nature Communications. 11 (1), 2981(2020).
- Yurke, B., Mills, A. P. Using DNA to power nanostructures. Genetic Programming and Evolvable Machines. 4 (2), 111-122 (2003).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318 (5853), 1121-1125 (2007).
- Wang, B., Thachuk, C., Ellington, A. D., Winfree, E., Soloveichik, D. Effective design principles for leakless strand displacement systems. Proceedings of the National Academy of Sciences. 115 (52), 12182-12191 (2018).
- Machinek, R. R. F., Ouldridge, T. E., Haley, N. E. C., Bath, J., Turberfield, A. J. Programmable energy landscapes for kinetic control of DNA strand displacement. Nature Communications. 5 (1), 5324(2014).
- Cabello-Garcia, J., Bae, W., Stan, G. -B. V., Ouldridge, T. E. Handhold-mediated strand displacement: a nucleic acid-based mechanism for generating far-from-equilibrium assemblies through templated reactions. bioRxiv. , (2020).
- Brophy, J. A. N., Voigt, C. A. Principles of genetic circuit design. Nature Methods. 11 (5), 508-520 (2014).
- Khalil, A. S., et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 150 (3), 647-658 (2012).
- Swank, Z., Laohakunakorn, N., Maerkl, S. J. Cell-free gene-regulatory network engineering with synthetic transcription factors. Proceedings of the National Academy of Sciences. 116 (13), 5892-5901 (2019).
- Howland, S. W., Tsuji, T., Gnjatic, S., Ritter, G., Old, L. J., Wittrup, K. D. Inducing efficient cross-priming using antigen-coated yeast particles. Journal of immunotherapy. 31 (7), 607(2008).
- Abil, Z., Ellefson, J. W., Gollihar, J. D., Watkins, E., Ellington, A. D. Compartmentalized partnered replication for the directed evolution of genetic parts and circuits. Nature Protocols. 12 (12), 2493-2512 (2017).
- Baugh, C., Grate, D., Wilson, C. 2.8 Å crystal structure of the malachite green aptamer11. Journal of Molecular Biology. Doudna, J. A. 301 (1), 117-128 (2000).
- Chou, L. Y. T., Shih, W. M. In vitro transcriptional regulation via nucleic acid-based transcription factors. ACS Synthetic Biology. 8 (11), 2558-2565 (2019).
- Lykke-Andersen, J., Christiansen, J. The C-terminal carboxy group of T7 RNA polymerase ensures efficient magnesium ion-dependent catalysis. Nucleic Acids Research. 26 (24), 5630-5635 (1998).
- Pu, J., Disare, M., Dickinson, B. C. Evolution of C-terminal modification tolerance in full-length and split T7 RNA Polymerase biosensors. Chembiochem. 20 (12), 1547-1553 (2019).
- Gardner, L. P., Mookhtiar, K. A., Coleman, J. E. Initiation, elongation, and processivity of carboxyl-terminal mutants of T7 RNA polymerase. Biochemistry. 36 (10), 2908-2918 (1997).
- Yin, J., Lin, A. J., Golan, D. E., Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nature Protocols. 1 (1), 280-285 (2006).
- Warden-Rothman, R., Caturegli, I., Popik, V., Tsourkas, A. Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Analytical Chemistry. 85 (22), 11090-11097 (2013).
- Zhang, W. -B., Sun, F., Tirrell, D. A., Arnold, F. H. Controlling macromolecular topology with genetically encoded SpyTag-SpyCatcher chemistry. Journal of the American Chemical Society. 135 (37), 13988-13997 (2013).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten