
JoVE 비디오를 활용하시려면 도서관을 통한 기관 구독이 필요합니다. 전체 비디오를 보시려면 로그인하거나 무료 트라이얼을 시작하세요.
Method Article
시험관 내 전사 및 분자 계산을 위한 DNA 테더드 RNA 폴리머라제
요약
우리는 시험관 내 전사 반응을 조절하기 위해 새로운 DNA 테더T7 RNA 폴리머라제의 엔지니어링을 설명합니다. 우리는 단백질 합성 및 특성화를 위한 단계를 논의하고, 개념 증명 전사 적 조절을 검증하며, 분자 컴퓨팅, 진단 및 분자 정보 처리에 대한 응용 분야에 대해 논의합니다.
초록
DNA 나노 기술은 다양한 응용 프로그램에 대한 사용자 처방 모양과 역학으로 핵산의 프로그래밍 가능한 자체 조립을 가능하게합니다. 이 연구는 DNA 나노 기술의 개념이 파지 유래 T7 RNA 폴리머라제(RNAP)의 효소 활성을 프로그래밍하고 확장 가능한 합성 유전자 조절 네트워크를 구축하는 데 사용될 수 있음을 보여줍니다. 첫째, 올리고뉴클레오티드 테더드 T7 RNAP는 N단말 스냅 태그 RNAP의 발현과 SNAP 태그의 후속 화학 결합을 벤질구아닌(BG)-변형 올리고뉴클레오티드와 결합하여 설계된다. 다음으로, 핵산 가닥 변위는 주문형 폴리머라제 전사를 프로그래밍하는 데 사용된다. 또한, 보조 핵산 어셈블리는 DNA 템플릿을 사용하여 DNA 프로그래밍된 T7 RNAP 간의 상호 작용을 조절하기 위해 "인공 전사 인자"로 사용될 수 있다. 이 시험관 내 전사 규제 메커니즘은 디지털 논리, 피드백, 계단식 및 멀티플렉스와 같은 다양한 회로 동작을 구현할 수 있습니다. 이 유전자 규제 아키텍처의 컴포지토리는 설계 추상화, 표준화 및 확장을 용이하게 합니다. 이러한 기능은 생체 감지, 질병 검출 및 데이터 저장과 같은 응용 분야에 대한 시험관 내 유전 장치의 신속한 프로토타이핑을 가능하게 합니다.
서문
DNA 컴퓨팅은 계산을 위한 매체로 디자인된 올리고뉴클레오티드 세트를 사용합니다. 이러한 올리고뉴클레오티드는 사용자 지정 논리에 따라 동적으로 조립하고 특정 핵산 입력에 반응하기 위해 서열로 프로그래밍됩니다. 개념 증명 연구에서, 계산의 출력은 전형적으로 겔 전기전도 또는 형광 판 판독기를 통해 검출될 수 있는 형광표형 올리고뉴클레오티드 세트로 구성됩니다. 지난 30년 동안 다양한 디지털 로직 캐스케이드, 화학 반응 네트워크 및 신경망1,2,3과같이 점점 더 복잡해지는 DNA 전산 회로가 입증되었습니다. 이러한 DNA 회로의 제조를 지원하기 위해, 수학적 모델은 합성 유전자 회로4,5의기능을 예측하는 데 사용되었으며, 직교 DNA 서열 설계6,7,8,9,10을 위해 전산 도구가 개발되었습니다. . 실리콘 기반 컴퓨터에 비해 DNA 컴퓨터의 장점은 생체 분자와 직접 인터페이스하고 전원 공급 장치가없는 상태에서 솔루션에서 작동하는 능력뿐만 아니라 전반적인 컴팩트함과 안정성을 포함합니다. 차세대 시퀀싱이 등장하면서 지난 2년간 DNA 컴퓨터 합성 비용은 무어의 법칙11보다빠른 속도로 감소하고 있습니다. 이러한 DNA 기반 컴퓨터의 응용프로그램은 이제 질병진단(12,13),분자 생물물리학(14)에 전력을 공급하고, 데이터 저장플랫폼(15)과같은 출현하기 시작했다.

그림 1: 발가락 매개 DNA 가닥 변위의 메커니즘. δ 발가락은 부분 이중에 무료, 언바운드 시퀀스입니다. 두 번째 가닥에 보완 도메인(δ*)이 도입되면 자유 δ 도메인은 혼성화를 위한 발판 역할을 하므로 나머지 가닥(ɑ*)이 가닥 마이그레이션이라고 하는 지퍼/압축 해제 가역 반응을 통해 경쟁업체를 천천히 대체할 수 있습니다. δ 길이가 증가함에 따라 전방 반응용 ΔG가 감소하고 변위가 더 쉽게 발생합니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
현재까지 DNA 컴퓨터의 대다수는 발가락 매개 DNA 가닥 변위(TMDSD, 도 1)16으로알려진 동적 DNA 나노 기술 분야에서 잘 확립된 모티프를 활용한다. 이 모티프는 짧은 "발가락"오버행 (즉, 7- 10 뉴클레오티드 (nt)를 표시하는 부분적으로 이중 좌초 DNA (dsDNA) 이중으로 구성된다. 핵산 "입력" 가닥은 발가락을 통해 부분 이중과 상호 작용할 수 있다. 이것은 부분 이중에서 가닥 중 하나의 변위로 이어지며, 이 해방된 가닥은 다운스트림 부분 이중에 대한 입력역할을 할 수 있습니다. 따라서 TMDSD는 신호 계단식 및 정보 처리를 가능하게 합니다. 원칙적으로 직교 TMDSD 모티프는 솔루션에서 독립적으로 작동하여 병렬 정보 처리를 가능하게 합니다. TMDSD 반응에 대한 다양한 변형이 있었다, 발가락 매개 DNA 가닥 교환 (TMDSE)(17),이중 긴 영역을 가진 "누출없는"발가락 홀드(18),서열 불일치 발가락19,및 "손잡이"매개 가닥 변위(20)등이 있었다. 이러한 혁신적인 설계 원칙을 통해 DNA 컴퓨팅 성능을 개선하기 위해 TMDSD의 에너지와 역학을 보다 세밀하게 조정할 수 있습니다.
전사 유전자 회로와 같은 합성 유전자 회로도 계산21,22,23을계산할 수 있다. 이 회로는 특정 조절 DNA 요소에 결합하여 유전자의 전사를 활성화하거나 억압하는 단백질 전사 인자에 의해 조절됩니다. DNA 기반 회로에 비해 전사 회로는 몇 가지 장점이 있습니다. 첫째, 효소 전사는 기존 촉매 DNA 회로보다 훨씬 높은 회전율을 가지므로 입력의 단일 사본당 더 많은 출력 사본을 생성하고 보다 효율적인 신호 증폭 수단을 제공합니다. 또한, 전사 회로는 치료 단백질에 대한 aptamers 또는 메신저 RNA(mRNA) 인코딩과 같은 상이한 기능성 분자를 생성할 수 있으며, 이는 다른 용도에 대해 악용될 수 있는 계산 출력으로. 그러나, 현재 전사 회로의 주요 한계는 확장성의 그들의 부족입니다. 이는 직교 단백질 기반 전사 인자의 매우 제한된 세트가 있기 때문에 새로운 단백질 전사 인자의 de novo 디자인은 기술적으로 도전적이고 시간이 많이 소요됩니다.

도 2: "밧줄"과 "케이지"폴리머라제 복합체의 추상화 및 메커니즘. (A 및 B)올리고뉴클레오티드 테더는 SNAP 태그 반응을 통해 T7 폴리머라아제로 효소로 표시된다. 테더-프로모터가 있는 "가짜" T7 프로모터로 구성된 케이지를 사용하면 테더에 혼성화하고 전사 활동을 차단할 수 있습니다. (C)작업자(a*b*)가존재할 때, 올리고뉴클레오티드테더(ab)에발가락에 결합하고 케이지의 b* 영역을 변위시켜 전사가 발생할 수 있도록 한다. 이 수치는 추와 시27에서수정되었습니다. 약어 : RNAP = RNA 폴리머 라제. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
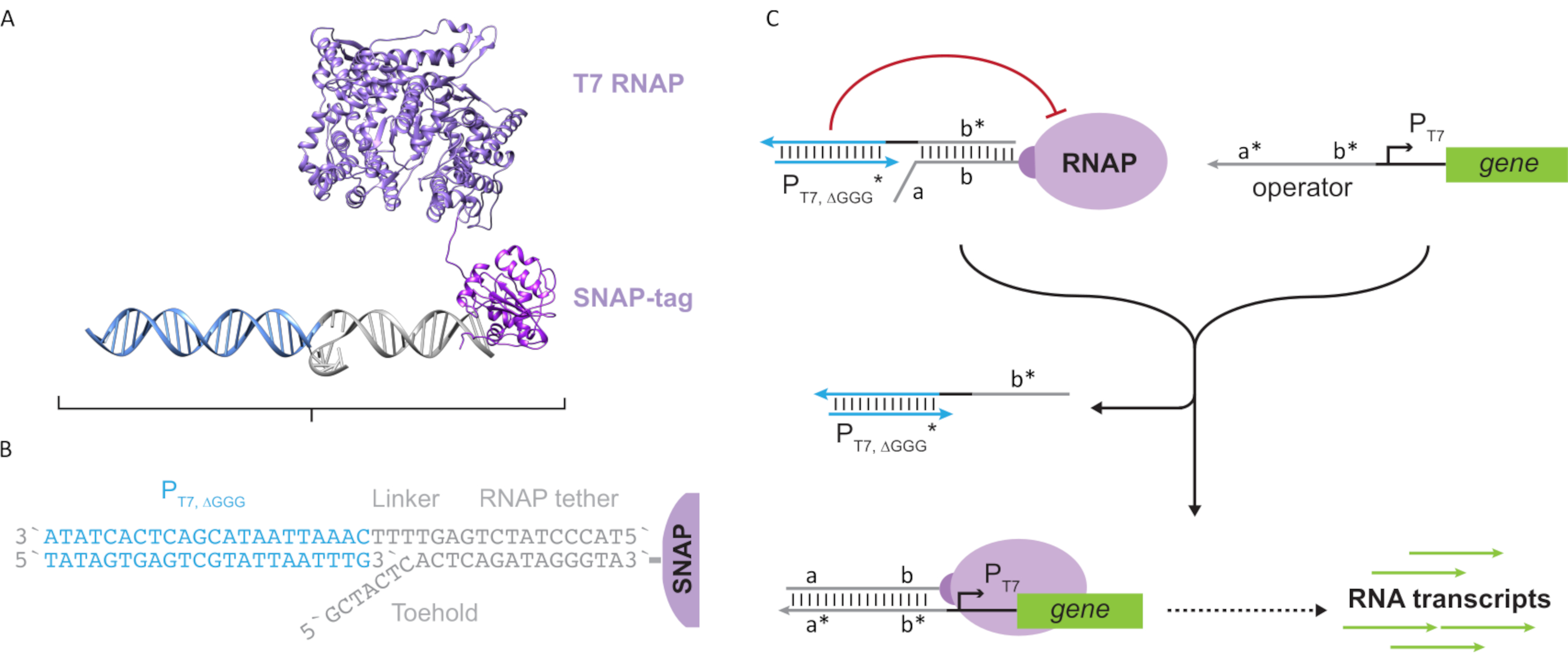{kind=link}
이 논문은 전사 회로의 기능과 DNA 기반 회로의 확장성을 결합한 분자 컴퓨팅을 위한 새로운 빌딩 블록을 소개합니다. 이 빌딩 블록은 단일 좌초 DNA 테더(도2A)와함께 공동으로 부착된 T7 RNAP이다. 이 DNA 테더드 T7 RNAP를 합성하기 위해 폴리머라제는 N 단자 SNAP태그(24)에 융합되고 에슈리치아 대장균에서재조합되었다. SNAP 태그는 BG 기판으로 기능화된 올리고뉴클레오티드로 반응했다. 올리고뉴클레오티드 테더는 DNA 혼성화를 통해 폴리머라제에 근접하여 분자 손님의 위치를 지정할 수 있게 한다. 이러한 손님 중 하나는 "케이지"라고 불리는 경쟁 전사 차단제였으며, 이는 유전자 다운스트림이없는 "가짜"T7 프로모터 DNA 듀플렉스(그림 2B)로구성됩니다. 올리고뉴클레오티드 밧줄을 통해 RNAP에 구속될 때, 케이지는 RNAP 결합을 위한 다른 DNA 템플릿을 능가하여 중합체 활성을 노점으로 하여 RNAP상태를 "OFF"상태(도 2C)로렌더링한다.
"ON" 상태로 중합효소를 활성화하기 위해, 유전자의 T7 프로모터의 상류에 단일 좌초 "연산자" 도메인을 가진 T7 DNA 템플릿이 설계되었다. 연산자 도메인(즉, 도메인 a*b* 도 2C)은TMDSD를 통해 RNAP에서 케이지를 대체하고 RNAP 근위를 유전자의 T7 프로모터에 배치하여 전사를 시동하도록 설계될 수 있다. 대안적으로, DNA 템플릿은 또한 운영자 서열이 "인공 전사 인자"(즉, 도 3A에서TFA 및 TFB 가닥)로 지칭되는 보조 핵산 가닥에 보완된 곳에서 설계되었다. 두 가닥이 반응에 도입되면 운영자 사이트에서 어셈블하여 새로운 의사 연속 도메인 a*b*를만듭니다. 그런 다음 이 도메인은 TMDSD를 통해 케이지를 변위하여전사(그림 3B)를시작할 수 있습니다. 이러한 가닥은 외인성 또는 생산될 수 있다.

그림 3: 3성분 스위치 활성제를 통한 폴리머라제 활성의 선택적 프로그래밍. (A)전사 인자(TFA 및 TFB)가존재할 때, 그들은 프로모터의 연산도메인 상류에 결합하여, 포홀드 중재된 DNA 변위를 통해 케이지를 변위시킬 수 있는 의사 단일 가닥 시퀀스(a*b*)를 형성한다. (B)이 a*b* 도메인은 TMDSD를 통해 케이지를 변위하여 전사를 시작할 수 있습니다. 이 수치는 추와 시27에서수정되었습니다. 약어: TF = 전사 인자; RNAP = RNA 중합체; TMDSD = 발가락 매개 DNA 가닥 변위. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
체외 전사 조절을 위한 핵산 계전사 인자의 사용은 디지털 논리, 피드백 및 신호 계단식과 같은 정교한 회로 동작의 확장 가능한 구현을 가능하게 합니다. 예를 들어, 상류 유전자의 전사체가 다운스트림 유전자를 활성화하는 핵산 서열을 설계함으로써 논리 게이트 캐스케이드를 구축할 수 있다. 이 제안된 기술로 이루어진 계단식 및 멀티플렉스를 활용하는 한 가지 응용 분야는 휴대용 진단 및 분자 데이터 처리를 위한 보다 정교한 분자 컴퓨팅 회로를 개발하는 것입니다. 또한, 분자 컴퓨팅 및 드 노보 RNA 합성 기능을 통합하면 새로운 응용 프로그램이 가능해질 수 있다. 예를 들어, 분자 회로는 사용자 정의 RNA 중 하나 또는 조합을 입력 및 출력 치료 RNA 또는 mRNA로 검출하도록 설계되어 기능성 펩티드 또는 단백질을 치료 시점 의료 응용 분야에 대한 암호화할 수 있다.
Access restricted. Please log in or start a trial to view this content.
프로토콜
1. 버퍼 준비
참고: 단백질 정제 완충제 준비는 어느 날 발생할 수 있습니다. 여기에서 실험을 시작하기 전에 수행되었습니다.
- 50m 트리스(하이드록시메틸)아미노메탄(Tris), 300mM 염화나트륨(NaCl), 5% 글리세롤, 5mM mM β-메르카포에탄올(BME), pH 8을 함유한 리시스/평형 버퍼를 준비한다. 1M Tris 1.5mL, 5M NaCl 1.8mL, 글리세롤 1.5mL, 25.2mL의 탈량(ddH2O)을 50mL 원심분리튜브에 추가하고 사용하기 직전에 14.2M BME의 10.5 μL을 추가합니다.
참고: 트리는 급성 독성을 일으킬 수 있습니다. 따라서 먼지를 호흡하지 말고 피부와 눈접촉을 피하십시오. BME는 독성이 있으며 연기 후드에만 사용해야합니다. 재서스펜션 및 세포 리시스 직전에 BME를 마지막으로 추가하는 것이 중요합니다. 용해 버퍼 수식의 표 1을 참조하십시오. - 50m Tris, 800mM NaCl, 5% 글리세롤, 5mM BME, 20m mM imidazole를 함유한 세척 버퍼(pH 8)를 준비합니다. 1 M Tris의 1.5mL, 5M NaCl의 4.8mL, 글리세롤 1.5mL, ddH2mL 22.2 mL을 50mL 원심분리기 튜브에 넣습니다. 사용하기 직전에 14.2 M BME의 7 μL과 2M 이미다졸2M의 200 μL을 위의 용액의 20mL에 추가하십시오.
참고: 이미다졸로 인한 급성 독성을 방지하려면 개인 보호 장비를 사용하십시오. BME와 imidazole 마지막을 추가하는 것이 중요합니다, 바로 열에서 단백질을 세척하기 전에. 세척 버퍼 수식은 표 2를 참조하십시오. - 50m Tris, 800mM NaCl, 5% 글리세롤, 5mM BME 및 200mM imidazole를 포함하는 용출 버퍼(pH8)를 준비합니다. 1M Tris의 0.5mL, 5M NaCl의 1.6mL, 글리세롤 0.5mL, ddH2O6.4 mL를 15mL 원심분리기 튜브에 추가합니다. 사용하기 직전에 는 위의 용액의 10mL에 14.2 M BME의 3.5 μL과 2M imidazole의 1mL을 추가하십시오.
참고: BME와 이미다졸을 마지막으로 첨가하는 것이 중요하며, 단백질을 컬럼에서 방출하기 직전에 는 이 것을 예의하십시오. 용출 버퍼 수식에 대한 표 3을 참조하십시오. - 100mM Tris, 200mM NaCl, 40m BME 및 2mM 에틸렌디아민트라아세트산(EDTA), 비이온 계면활성제의 0.2%를 포함하는 2배 저장 버퍼(글리세롤로 1:1혼합)를 준비한다. 5m L의 5m L, 5M NaCl의 2mL, ddH2O의 42.56mL, 0.5M EDTA의 200 μL, 비이온 계면활성제의 100 μL을 50mL 원심분리튜브에 추가하여 50mL의 저장 버퍼를 준비한다. 용액이 균일해질 때까지 혼합하고 0.2 μm 주사기 필터를 통해 저장 버퍼를 필터링하고 사용하기 전에 위의 솔루션에 140.8 μL의 BME를 추가합니다.
참고: EDTA로 인한 급성 독성을 방지하려면 먼지를 호흡하지 말고 피부와 눈접촉을 피하십시오. 정제된 단백질을 저장하기 직전에 BME를 마지막으로 추가하고 전체 저장 버퍼 1:1을 글리세롤과 혼합하는 것이 중요합니다. 저장소 버퍼 수식에 대한 표 4를 참조하십시오.
2. 하룻밤 문화 성장: 1일차
- 10mL의 ddH2O로 500 mg의 가나마이신을 용해시켜 1,000x 가나마이신 육수를 준비하십시오.
참고: 카나마이신으로 인한 급성 독성을 방지하기 위해 개인 보호 장비를 사용합니다. - 리소제니 국물의 20mL에 1,000x 가나마이신 재고의 20 μL을 추가합니다. 멸균 파이펫 팁을 사용하여 변형된 BL21 대장균 글리세롤 스톡을 찌른 다음 성장 미디어 국물에 팁을 도입하여 문화를 접종합니다.

그림 4: SNAP T7 RNAP에 대한 플라스미드지도. 플라스미드는 pQE-80L 백본에 락 프레스(lacI) 하에 N단 히스티딘 태그(6x His) 및 SNAP 태그 도메인(SNAP T7 RNAP)이 포함된 T7 RNAP를 인코딩합니다. 다른 특징은 가나마이신 저항을 포함 (KanR) 및 클로람페니콜 저항 (CmR) 유전자. 약어: RNAP = RNA 폴리머라제. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
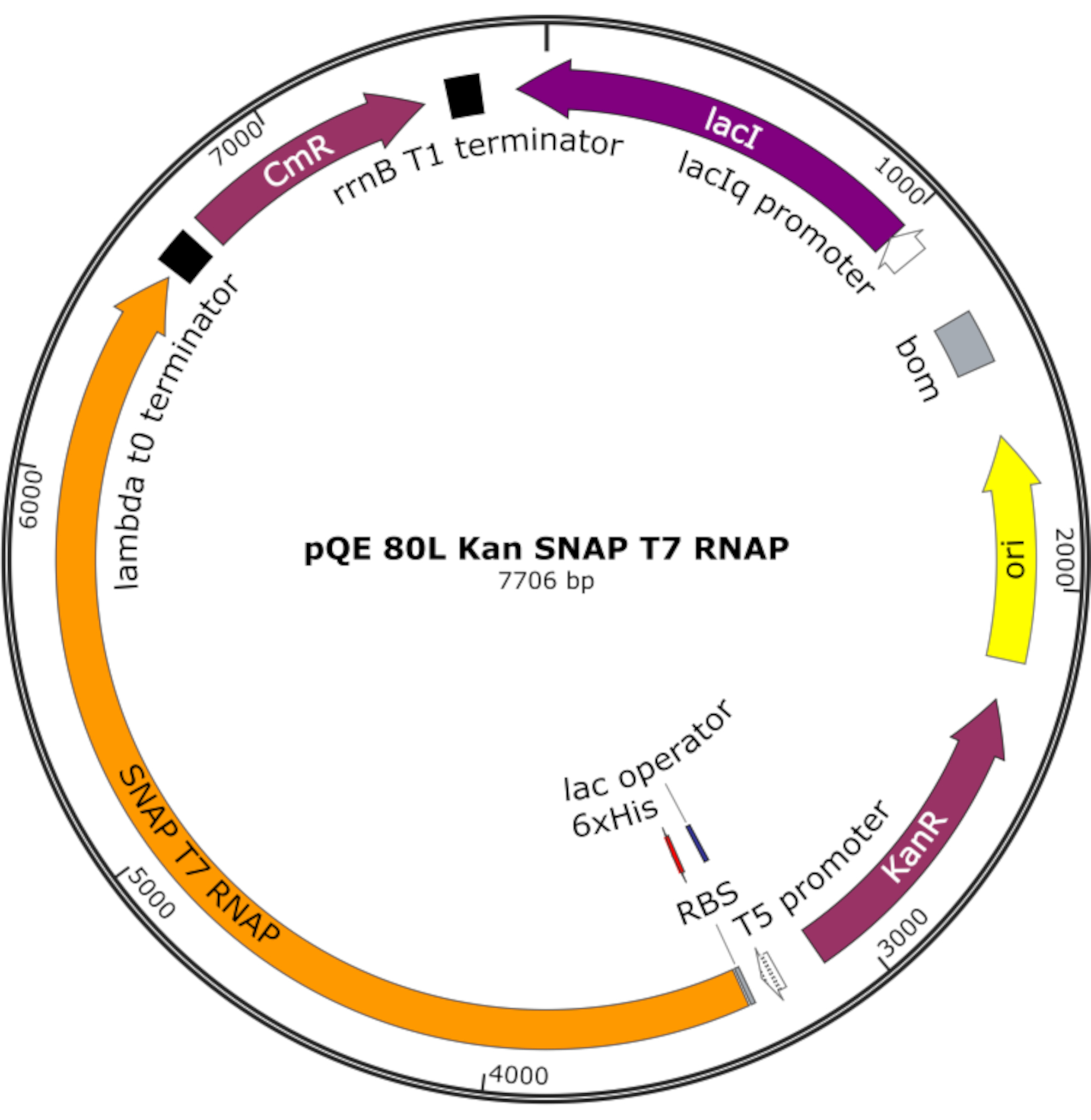{kind=link}
참고: 플라스미드는 N단 히스티딘 태그와 SNAP 태그 도메인(SNAP T7 RNAP)을 포함하는 T7 RNAP뿐만 아니라 pQE-80L 백본(그림4)25하에서가나마이신 저항 유전자를 인코딩한다.
- 다시, 리소겐 국물의 20mL를 포함하는 별도의 배양 플라스크에 1,000x 가나마이신 재고의 20 μL을 추가하고, 제어로 인큐베이션.
- 2개의 샘플(2.2 단계 및 2.3에서) 12-18h에서 37°C에 대해 하룻밤 동안 배양하고, 10 × g에서회전한다.
3. 세포 성장과 유도: 2일차
- 2.4단계에서 야간 성장 배양의 4mL를 함유한 400mL의 리소겐성 육수400mL. 배양 플라스크를 37°C에서 배양하고 10 × g에서 회전합니다.
- 배양이 600nm ~0.5에서 광학 밀도(OD)에 도달하면, 대조군으로서 성장 플라스크로부터 1mL의 시료를 꺼내라. 제어 샘플을 4°C에 저장합니다.
- 배양 100mL당 1M IPTG의 40 μL을 추가하여 이소프로필 β-D-1-티오갈라크토피라노사이드(IPTG)를 사용하여 세포를 유도하여 0.4mM IPTG의 최종 농도를 달성한다. 37°C에서 3시간 동안 시료를 배양하고, 10 × g에서회전한 다음, 유도된 배양을 10분 동안 8,00 ×0 g에서 스핀하여 세포를 펠릿한다. 상체를 제거하고 펠릿을 -20°C에서 추가 로 사용할 때까지 보관하십시오.
참고: IPTG로 인한 급성 독성을 방지하려면 먼지를 호흡하지 말고 피부와 눈의 접촉을 피하십시오. 필요한 경우 여기에서 실험을 일시 중지하고 다음 날 계속할 수 있습니다.
4. 세포 용해, 단백질 정제: 3일차
- 저장된 셀 펠릿을 얼음에 10mL의 리시스 버퍼로 다시 중단하고 부드럽게 소용돌이어 전체 펠릿이 다시 중단되도록 합니다. 그런 다음, 파이펫 1 mL 의 샘플은 얼음에 보관되는 10 1.5 mL 튜브로.
- 30초 동안 50%의 듀티 사이클로 2s용 펄스를 가집니다. 각 샘플 전후에 70% 에탄올과 ddH2O. 초음파 처리 중 및 후 얼음에 모든 샘플을 보관하여 초음파 처리 팁을 청소하십시오.
참고: 열과 화염으로부터 70% 에탄올을 멀리하십시오. - 니켈 충전 된 nitrilotriacetic 산 (Ni-NTA) 정화 스핀 컬럼을 4 °C의 작동 온도로 상화한다. 열을 4°C로 놓고 보관하고 사용 중에 얼음을 보관하십시오.
- 원심분리기는 101mL 샘플에서 4°C에서 20분 동안 × g로 10mL 샘플을 원심분리합니다. 조심스럽게 펠릿을 방해하지 않고 재조합 RNAP를 포함하는 상체를 피펫. 필요한 경우 추가 평형 버퍼를 사용하여 총 볼륨을 ≥ 6mL로 조정합니다.
- Ni-NTA 스핀 열에서 하단 탭을 부드럽게 제거하여 열을 통해 흐를 수 있습니다. 원심분리기 튜브에 기둥을 놓고 얼음 위에 보관하십시오.
참고: 3mL Ni-NTA 스핀 컬럼이 있는 50mL 원심분리기 튜브를 사용합니다. - 700 × g및 4°C에서 2분 동안 기둥을 원심분리하여 저장 버퍼를 제거합니다. 열에 6mL의 평형 버퍼를 추가하여 열을 상화합니다. 버퍼가 수지 침대에 완전히 들어갈 수 있도록 합니다.
- 70 ×0g, 4°C에서 원심분리로 컬럼에서 2분 동안 평형 버퍼를 제거합니다. 준비된 셀 추출기를 열에 추가하기 전에 열을 바닥에 플러그를 놓아 제품을 잃지 않도록 합니다. 그런 다음, 셀 추출물을 컬럼에 넣고 4°C에서 30분 동안 궤도 셰이커 믹서에 섞는다.
- 컬럼에서 하단 플러그를 제거하고 50mL 원심분리기 튜브에 컬럼을 통해 흐름에표시되어 있습니다. 컬럼을 700 × g로 원심분리하여 2분 동안 흐름을 수집합니다.
- 수지 세척을 세척하기 위해 열에 6mL의 워시 버퍼를 추가합니다. 700 × g의 컬럼을 2분 동안 원심분리하여 새로운 원심분리기 튜브에서 분수를 수집하여 세척 1을표시한다. 이 단계를 총 3개의 분리된 분획에 대해 2회 더 반복하고 별도의 원심분리관(세척2및 세척 3)에서분획을 수집합니다.
- 용출 버퍼 3mL을 추가하여 수지에서 태그된 단백질을 엘로우트합니다. 원심분리기는 700 × g에서 2분 동안 원심분리기로 새로운 원심분리기 튜브에서 분수를 수집하여 1호라고표시되어 있다. 이 단계를 총 3개의 분리된 분획에 대해 2회 더 반복하고 분획을 별도의 원심분리기튜브(eluate 2 및 용액 3)로수집합니다.
- 용액을 결합하고 탈염을 수행하여 단백질 용액에서 소금을 제거합니다.
- 파이펫 15 mL 0.05 % w / v 폴리소르 바테 20 이상 100 kDa 원심 필터 장치. 4,000 × g의 원심분리기는 40분 동안 흐르고 유동을 버립니다.
- 코팅된 필터를 사용하여 용출액 1, 2, 3(총 단백질 용출액 + 6mL의 단백질 용출액 + 6mL 의 저장 버퍼)을 ~1,500 μL로 농축하고, 20분 동안 3,220× g에서 필터를 원심분리하고, 강수화를 방지하기 위해 멤브레인을 부드럽게 파이펫 세척한다.
- 저장 버퍼로 샘플을 15mL로 희석합니다. 4.11.2 단계를 두 번 반복하여 저장소 버퍼 1:1,000을 사용하여 버퍼 교환을 수행합니다.
- 280 nm에서 분획의 흡광도를 측정하여 정제 된 단백질을 정량화합니다. 저장 버퍼(4°C에서 2배 스토리지 버퍼)로 분광계를 비우지 마십시오. 결합된 용액의 샘플을 부드럽게 혼합하고 흡광도를 측정합니다.
참고: 단백질 샘플을 1x, 10배 및 50배 희석하여 단백질을 평균화하고 정량화하여 3개의 분리된 판독값을 수행합니다. 저장 버퍼에서 샘플을 희석합니다. - 단백질 샘플을 2배 저장 버퍼를 사용하여 100 μM로 조정합니다. 조정된 샘플을 1:1 부피로 100% 글리세롤으로 희석시킵니다. 결과 단백질 용액을 -80°C에 저장합니다.
5. 나트륨 도데딜 황산염 폴리아크라이알라미드 젤 전기포레시스(SDS-PAGE) 단백질 생성물 분석: 3일차
- 단백질 분석을 위해 SDS-PAGE 젤을 실행합니다. 시료의 9 μL을 4x 리튬 도데딜 황산염(LDS) 단백질 로딩 염료3μL과 혼합합니다. 샘플을 95°C에서 10분 동안 가열합니다.
- 샘플을 4-12% 비스 트리스 SDS-PAGE 젤 설정에 로드합니다. 단백질 사다리를 잘 1에 적재한 다음 샘플(왼쪽에서 오른쪽으로)으로 적재됨) 흐름, 세척 1, 세척 2, 세척 3, 용출 1, 용출 2, 용출 3 및 총 탈염용용.
참고: 표 5에는 SDS-PAGE 젤용 샘플 로딩 테이블이 포함되어 있습니다. - 2-(N-morpholino)에탄에탈포닉산(MES) 버퍼를 200V. 린세클에서 각각 10분 동안 3회 깨끗한 트레이에 함유하고, 젤 매트릭스에서 SDS를 제거하는 부드러운 동요를 한다.
참고: MES로 인한 급성 독성을 피하기 위해 개인 보호 장비를 착용하십시오. - 20mL의 쿠마시 블루로 젤을 염색하고, 부드러운 동요로 실온에서 밤새 젤을 배양합니다. 200mL의 ddH2O로각각 1h에 대해 젤을 두 번 염색하여 궤도 셰이커에 부드럽게 교반합니다.
참고: 젤을 더 긴 기간 동안 세척하거나 물을 자주 교체하면 감도가 향상됩니다. 또한, 과도한 염료를 흡수하기 위해 용기에 접힌 섬세한 작업 닦아 조직을 배치하면 염색 공정을 가속화합니다.
6. 시험관 내 전사를 통해 SNAP T7 RNAP의 기능적 검증
참고: 이 프로토콜은 형광 브로콜리 RNA aptamer에 대한 인코딩 DNA 템플릿을 사용하고 형광 판 판독기에 전사의 역학을 모니터링하기 위해 형광의 사용을 허용합니다.
- SNAP T7 RNAP의 활성과 상업용 소스의 와일드 타입(WT) T7 RNAP 및 버퍼 전용 제어의 활성을 비교하기 위해 3개의 시험관내 전사(IVT) 반응을 설정합니다. 각 반응의 부피를 20 μL로 조정합니다.
- SNAP T7 RNAP IVT 반응을 10x 전사 버퍼2μL, 25m 리보뉴클레오사이드 삼위산염(rNTP) 믹스의 0.4 μL, 500nM DNA 템플릿5μL, 500nM SNAP T7 RNAP 및 10.6 μL의dHDH DD OD DHD OD 의 10.6 μL을 혼합하여 준비한다.
- WT RNAP IVT 반응을 10x 전사 버퍼 2μL, 25m rNTP 믹스의 0.4 μL, 500nM DNA 템플릿 5μL, WT T7 RNAP 2μL, ddH 2O10.6 μL을 혼합하여 WT RNAP IVT 반응을 준비한다.
- 10x 전사 버퍼 2μL, 25m rNTP 믹스의 0.4 μL, 500nM DNA 템플릿 5μL, ddH2O의12.6 μL을 혼합하여 완충 전용 IVT 반응을 준비한다.
참고: RNAP를 마지막으로 추가하여 샘플이 도입될 때까지 얼음 위에 보관합니다. 표 6, 표 7및 표 8에는 IVT 반응 수식이 포함되어 있습니다.
- 470nm의 난초 파장과 512nm의 방출 파장을 사용하여 37°C에서 2시간 간격으로 형광판 판독기의 전사 역학을 모니터링한다.
7. BG 변형 올리고뉴클레오티드 준비: 1일차
- ddH2O에서 3'-amine 변형으로 올리고뉴클레오티드를 1mM의 최종 농도로 용해하십시오. 이 S1에레이블을 지정합니다.
- 1M 나트륨 중탄산염(NaHCO3),100% 디메틸 설산화물(DMSO)의 284 μL, S1의 125μL(올리고뉴클레오티드 스톡), BG-N-하이드록시스쿠니미드(NHS)의 50mM의 66μL을 혼합합니다(BG-GLA-DM) 에스테르(BG-GLA-DM)의 부피를 500 μL로 조정하고 실온에서 100 × g에서하룻밤 동안 배양합니다.
참고: DMSO가 가연성 액체이므로 열과 불꽃으로부터 멀리하십시오. 표 9는 올리고뉴클레오티드에 대한 BG 컨쥬게이션에 대한 반응 공식을 포함한다.
- 1M 나트륨 중탄산염(NaHCO3),100% 디메틸 설산화물(DMSO)의 284 μL, S1의 125μL(올리고뉴클레오티드 스톡), BG-N-하이드록시스쿠니미드(NHS)의 50mM의 66μL을 혼합합니다(BG-GLA-DM) 에스테르(BG-GLA-DM)의 부피를 500 μL로 조정하고 실온에서 100 × g에서하룻밤 동안 배양합니다.
8. BG 올리고뉴클레오티드 컨쥬게이트의 에탄올/아세톤 침전: 2일차
- 원심 분리 단계 7.1.1의 제품. 13,000 × g에서 5 분 동안. 상체를 신선한 튜브로 조심스럽게 옮기고 침전된 BG를 폐기합니다. 반응을 250 μL 알리쿼트로 분할하여 오버플로를 방지하고 두 aliquots에서 다음 단계를 수행합니다.
- 3M 아세테이트(25 μL)의 부피의 1/10을 추가한 다음, 100% 에탄올(625 μL)으로 2.5배 의 부피를 넣습니다. -80°C에서 1h의 인큐베이션을 합니다.
참고: 아세테이트 나트륨(눈, 피부, 소화 및 호흡기에 자극을 일으킬 수 있음)과 에탄올(매우 인화성, 접촉 시 자극을 일으킬 수 있음)을 모두 취급할 때 개인 보호 장비를 사용합니다. 필요한 경우 여기에서 실험을 일시 중지하고 다음 날 계속하십시오. - 원심분리기에 튜브를 놓고 바깥쪽 가장자리를 표시합니다. 4°C에서 30분 동안 17,000 × g의 튜브원심분리기.
참고: 올리고뉴클레오티드 펠릿이 튜브의 표시된 가장자리에 나타납니다. - 펠릿을 방해하지 않고, 상체를 폐기하십시오. 750 μL의 냉각 70% 에탄올로 위로 올려, 4°C에서 10분 동안 17,000 × g로 회전합니다.
- 펠릿을 방해하지 않고, 상체를 폐기하십시오. 100% 아세톤의 750 μL을 얹고, 17,000 × g에서 4°C에서 10분 동안 회전합니다.
참고: 아세톤을 취급할 때는 매우 인화성이 뛰어나고 접촉시 자극을 일으키기 때문에 개인 보호 장비를 사용하십시오. - 튜브 뚜껑을 열고, 증발을 통해 여분의 아세톤을 제거하기 위해 5 분 동안 공기 건조. 올리고뉴클레오티드를 1x Tris-EDTA(TE) 버퍼의 250 μL로 재용해 - 850 μM BG-올리고뉴클레오티드 용액을 생성한다.
- 8.2 ~ 8.6 단계를 반복하고 1x TE 버퍼의 70 μL로 다시 용해하십시오. 이 S2에레이블을 지정합니다.
9. 젤 여과 크로마토그래피를 통한 BG-올리고뉴클레오티드 정화
- 열을 여러 번 적극적으로 반전시켜 행렬을 일시 중단합니다. 상단 캡을 제거하고 열의 하단 끝을 스냅합니다. 1.5mL 원심분리기 튜브에 컬럼을 놓고, 실온에서 1분 동안 1,000g의 원심분리기를 ×. 용출된 버퍼 및 수집 튜브를 폐기합니다.
참고: 진공 형성을 방지하는 것이 중요합니다. 준비된 열을 즉시 사용합니다. - 포장된 컬럼을 깨끗한 1.5mL 원심분리기 튜브에 놓습니다. 1x TE 버퍼 300μL을 열 침대 중앙에 추가하고 1,000 × g의 원심분리기를 2분 동안 추가하여 버퍼 용액을 교환합니다. 다시 한번, 용출 된 버퍼 및 수집 튜브를 폐기합니다.
- 버퍼 교환 컬럼을 깨끗한 1.5mL 원심분리기 튜브에 놓습니다. 최대 75μL의 샘플을 침대 중앙에 발라주세요. 1,000 × g에서 4 분 동안 회전하십시오.
참고: 침대를 방해하거나 열의 측면을 만지지 마십시오. 젤 미디어의 가장 높은 지점은 외부 로터를 가리킨다. - 정제된 핵산을 함유하고 있으므로 수집 관으로부터 용액을 수집합니다. 시료를 정량화하려면 260 nm에서 흡광도를 측정합니다. 이 S3에 레이블을 지정합니다.
참고: 측정에 사용된 경로 길이를 기록하고 맥주-램버트 법칙을 사용하여 농도를 계산합니다.
10. BG 올리고뉴클레오티드 컨쥬게이트의 페이지 분석
- 18% 트리스 보라테-EDTA(TBE)-우레아 페이지 젤을 캐스팅합니다. 4.8 g의 UREA, 4.5 mL 40% 아크릴아미드(19:1), ddH2O2.8mL에서 10x TBE 1mL을 녹인다; 5 μL 테트라메틸레틸레티아민(TEMED)을 넣고 철저히 섞습니다. 10% 암모늄 아황산염(APS)의 100 μL로 반복합니다. 용액을 빈 젤 카세트에 붓고 40 분 동안 중합할 수 있습니다.
참고: 우레아를 취급할 때 적절한 개인 보호 장비를 사용하십시오(눈과 피부에 자극을 일으키기), 아크릴아미드(독성 및 발암성), TEMED(독성, 가연성, 부식성). 표 10에는 18% TBE-UREA 폴리아크라이알라미드 젤에 대한 반응 포뮬러가 포함되어 있습니다. - TBE 버퍼(0.5x)의 500mL를 2분 및 30초 또는 ~70°C까지 전자레인지에 넣고 젤 장치에 붓습니다. 포르마미드(denaturing) 로딩 염료를 95% 포르미미드 + 1mM EDTA 및 브로모페놀 블루를 준비한다. 각 시료와 로딩 염료를 섞고 혼합물을 폴리아크릴아미드 젤에 적재합니다.
참고: 발암성으로 폼아미드를 취급할 때 적절한 개인 보호 장비를 사용하십시오. 표 11에는 샘플 젤 로딩 테이블이 포함되어 있습니다. - 젤을 270 V에서 35분 동안 또는 염료 전면이 끝까지 이동할 때까지 젤을 실행합니다. 젤 상자에 젤을 놓고 이미징 전에 실온에서 15 분 동안 핵산용 시아닌 염료로 얼룩을 놓습니다.
참고: 가연성 으로 시아닌 염료를 취급할 때 적절한 개인 보호 장비를 사용합니다.
11. 올리고뉴클레오티드의 컨쥬게이션을 SNAP T7 RNAP 및 PAGE 분석에
- SNAP T7 RNAP에 BG-올리고뉴클레오티드의 분석 스케일 커플링을 위한 시약을 준비하십시오: ddH2O와 단일 좌초 DNA(ssDNA) 올리고의 9희석을 만들어 5:1에서 1:5까지 의 올리고:RNAP 비율을 생성한다. 단백질 육수를 50 μM로 희석합니다.
참고: 예제 비율은 표 12에서찾을 수 있습니다. 이 비율은 50 μM의 RNAP 농도를 사용하여 계산됩니다. - ssDNA 올리고의 각 희석에 대해, SNAP 버퍼 2μL, BG-올리고뉴클레오티드 4μL, SNAP T7 RNAP4를 포함하는 반응 혼합물의 10 μL을 만듭니다.
참고: 표 13에는 SNAP 태그 라벨링 반응에 대한 반응 수식이 포함되어 있습니다.- 두 개의 더 많은 제어 샘플을 준비: 1) BG-올리고뉴클레오티드를 ddH2O로 대체하여 RNAP 제어; 2) SNAP T7 RNAP을 ddH2O로 대체하여 DNA 대조군(SNAP T7 RNAP의 가장 낮은 올리고뉴클레오티드 농도). 모든 샘플을 실온에서 1시간 동안 배양하고 필요할 때까지 얼음을 유지합니다.
- 각 시료의 2μL을 SNAP 버퍼 4μL에 추가하고 단백질 로딩 염료 2μL을 추가하고 70°C에서 10분 동안 가열하여 111개의 10μL 반응을 설정합니다. 각 샘플의 2 μL을 4-12% 비스 트리스 단백질 젤에 적재하고 35분 동안 200 V에서 얼음에 겔 전기포전을 수행합니다.
참고: 표 14에는 젤 로딩 샘플에 대한 반응 수식이 포함되어 있습니다.- 셰이커에서 3배의 물 교환을 통해 SDS를 씻어내고, 각 세척은 각각 10분 간 지속됩니다. 이미징 전에 15 분 동안 핵산에 대한 시아닌 염료를 가진 얼룩. 1 h에 대 한 쿠마시 블루 얼룩의 20 mL를 사용 하 여 다시 젤을 얼룩. 이미징 전에 ddH2O를 1h(또는 하룻밤)에 대한 염색.
참고: 젤에서, 반응 중 하나는 초과 된 무료 BG-올리고뉴클레오티드의 최소 금액과 함께 가장 테더드 폴리머라아제생성; 이것은 최적의 비율입니다.
- 셰이커에서 3배의 물 교환을 통해 SDS를 씻어내고, 각 세척은 각각 10분 간 지속됩니다. 이미징 전에 15 분 동안 핵산에 대한 시아닌 염료를 가진 얼룩. 1 h에 대 한 쿠마시 블루 얼룩의 20 mL를 사용 하 여 다시 젤을 얼룩. 이미징 전에 ddH2O를 1h(또는 하룻밤)에 대한 염색.
- SNAP T7 RNAP에 BG-올리고뉴클레오티드를 결합하는 전격 스케일에 대한 시약을 준비한다. 분석 척도에서 발견되는 최적의 비율로 결합 반응을 수행합니다.
참고: 사용하지 않을 때는 단백질을 얼음위에 두어 실온에 단백질 노출을 최소화하십시오.
12. 이온 교환 열을 사용하여 올리고뉴클레오티드 테더드 SNAP-T7의 정화
- 여기에 나열된 지침에서 벗어난 경우 튜브 설정에 대한 제조업체의 지침을 따르십시오. 단백질의 등전점보다 pH 높은 정제 완충액을 준비한다.
참고: 본 프로토콜의 예 단백질의 경우, 10mM 나트륨 인산염 버퍼(pH 7)의 정제 완충제가 사용되었다.- 50mM Tris및 0.5M NaCl의 최종 농도를 포함하는 1,000μL의 용출 버퍼를 준비하십시오. 1M 트리의 50 μL, 5M NaCl의 100 μL, ddH2O의 850 μL을 혼합합니다.
참고: 표 15에는 용출 버퍼에 대한 반응 수식이 포함되어 있습니다.
- 50mM Tris및 0.5M NaCl의 최종 농도를 포함하는 1,000μL의 용출 버퍼를 준비하십시오. 1M 트리의 50 μL, 5M NaCl의 100 μL, ddH2O의 850 μL을 혼합합니다.
- 2mL 원심분리기 튜브에 컬럼을 놓고 15분 동안 2,000g× g로 세척하거나 모든 버퍼가 용출될 때까지 세척합니다. 용출된 버퍼를 삭제합니다.
- 3:1 정제 버퍼:샘플 비율로 각 샘플을 희석하고 샘플을 한 번에 400 μL열에 적재합니다. 10분 동안 2,000 × g로 회전하거나 모든 버퍼가 용출될 때까지 회전합니다. 흐름을 수집하고 흐름 스루로 레이블을지정합니다.
- 400 μL의 정제 버퍼를 열 중앙에 추가합니다. 15분 동안 × 2,000 g에서 또는 모든 버퍼가 용출될 때까지 회전합니다. 흐름을 수집하고 세척 1로 라벨을 붙입니다. 2번 더 씻고 3을 씻으시면더 많이 반복하십시오.
- 열 중앙에 용출 버퍼 50μL을 추가합니다. 2,000 × g에서 5분 간 또는 모든 버퍼가 용출될 때까지 회전합니다. 흐름을 수집하고 용액 1로 레이블을 지정합니다. 2를 용해하고 3을 용해하기위해 두 번 더 반복하십시오.
- 풀은 1, 2 및 3 (이 총 용출레이블)을 용출하고, 젤에 대한 각 용출의 작은 부분을 남기고, 260 nm (A260) 및 280 nm (A280)에서 흡광도를 측정합니다. 측정 후 글리세롤을 1:1 비율로 추가하고 추가 사용이 될 때까지 -20°C에 보관하십시오.
- 원심 필터 장치(0.5mL; 30 kDa)를 사용하여 2배 스토리지 버퍼(~1:100)(이 제품에라벨)를 사용하여 총 용출을 버퍼교환합니다. A260/280을 다시 측정합니다. 글리세롤을 1:1 비율로 추가하고 추가 사용이 될 때까지 -20°C에 보관하십시오.
- 각 용출액을 적재: 단백질 사다리와 함께 4-12% 비스 트리S SDS-PAGE 젤에 유동, 세척 1-3, 총 용출 및 제품. 200 V에서 35분 동안 또는 염료 전면이 끝까지 이동될 때까지 실행합니다.
13. 테더드 RNA 폴리머라제 활성의 온디맨드 제어 시연
- 25m Tris, 5mM EDTA 및 25mM 염화 마그네슘(MgCl2)을함유한 5배 아닐링 버퍼를 준비합니다. 각 템플릿(1 μM)의 2.4 μL과 어닐링 버퍼 5μL, ddH2O의 14.2 μL을 혼합하여 1 μM dsDNA 케이지의 25 μL을 형성합니다. 이 용액을 75°C에서 2분 동안 배양합니다. 마찬가지로, 발기인과 말라카이트 녹색 aptamer DNA 템플릿의 감각과 안티 센스 가닥을 음일. 말라카이트 녹색 옥살레이트의 1mM 용액을 준비한다.
참고: 표 16에는 5x 어닐링 버퍼에 대한 반응 공식이 포함되어 있으며, 표 17에는 두 개의 ssDNA 템플릿을 어닐링하기 위한 반응 공식이 포함되어 있습니다. - 500 nM RNAP의 최종 농도에 15 분 동안 실온에서 1:5 어어 비에 dsDNA 케이지와 테더드 된 SNAP T7 RNAP를 배양. 필요할 때까지 얼음을 유지하십시오.
- 플레이트 판독기를 37°C로 예열합니다. 얼음에 25 μL IVT 반응 3회 설정
- 핵산 전사 인자를 가진 케이지 된 SNAP T7RNAP를 포함하는 반응을 설정합니다. 10x IVT 버퍼의 2.5 μL, 25m mM rNTP 믹스의 1 μL, 1m 말라카이트 그린1 μL, RNAP 케이지 혼합물의 2.5 μL, 1 μM 전사 인자 A 및 B 올리고뉴클레오타이드 가닥 각각 2.5 μL, 1mMM 말라키 드 1m 의 3μL을 혼합하십시오.
- 핵산 전사 인자 없이 케이지 된 SNAP T7RNAP를 포함하는 반응을 설정합니다. 10x IVT 버퍼의 2.5 μL, 25mm rNTP 믹스의 1 μL, 1m 말라카이트 그린1μL, RNAP 케이지 혼합물의 2.5 μL, 1m 말라카이트 녹색 aptamer 템플릿의 3 μL을 ddH2O의 15 μL에 혼합한다.
- 버퍼만 포함하는 반응을 설정합니다. 10x IVT 버퍼의 2.5 μL, 25mm rNTP 믹스의 1 μL, 1m 말라카이트 그린의 1 μL, 1m 말라카이트 그린 aptamer 템플릿의 3 μL을 ddH2O의 17.5 μL에 혼합한다.
참고: 표 18에는 시험관 내 전사 반응에 대한 일반적인 참조가 포함되어 있습니다.
- 각 반응을 384웰 플레이트로 옮기. 37°C에서 2시간 동안 형광판 판독기에 말라카이트 녹색 압타머의 전사를 모니터링하고 610nm 발산 및 655nm 방출을 갖는다. 완료되면, 필요할 때까지 얼음에 접시를 유지합니다.
- 전자 레인지 0.5 x TBE 버퍼 2 분 30 s 또는 ~70 °C까지. 20분 동안 280V에서 가열된 0.5배 TBE 버퍼에서 또는 염료 전면이 끝날 때까지 각 우물의 RNA 제품을 12% TBE-Urea 폴리아크릴라미드 젤로 실행한다. 화상 진찰 전에 궤도 셰이커에 10 분 동안 시아닌 염료 핵산 얼룩으로 젤을 얼룩.
참고: 표 19에는 12% TBE-Urea PAGE 젤에 대한 반응 포뮬러가 포함되어 있습니다.
Access restricted. Please log in or start a trial to view this content.
결과

그림 5: SNAP T7 RNAP 발현 및 체외 전사 분석의 SDS-PAGE 분석. (A)SNAP T7 RNAP 단백질 정제 분석, SNAP T7 RNAP 분자량: 119.4kDa. FT = 컬럼에서 의 흐름을 통해, W1 = 용출 분획불을 포함하는 세척 버퍼, 정제 된 제품을 포함하는 E1-3 = 용출 분획, 및 DE = 10 배 희석 총 탈염 용출. 4-1...
Access restricted. Please log in or start a trial to view this content.
토론
이 연구는 N-말단 태그 재조합 T7 RNAP를 BG 기능화 올리고뉴클레오티드와 공동으로 결합하여 T7 RNA 폴리머라제의 활성을 제어하는 DNA 나노 기술에서 영감을 얻은 접근법을 시연하며, 이는 이후 TMDSD 반응을 프로그래밍하는 데 사용되었습니다. 설계에 의하면, SNAP 태그는 폴리머라제의 N-종착체에 위치하였으며, 야생형 T7 RNAP의 C-종점은 단백질 구조 코어 내에 묻혀 DNA템플릿(28)과중?...
Access restricted. Please log in or start a trial to view this content.
공개
저자중 누구도 선언할 경쟁적인 재정적 이해관계는 없습니다.
감사의 말
L.Y.T.C 연구 기금 탐사 (NFRF-E), 캐나다 자연 과학 및 공학 연구 위원회 (NSERC) 디스커버리 그랜트, 캐나다 최초의 연구 우수 기금 (CFREF)의 자금을 받는 디자인 이니셔티브에 의한 토론토 대학의 의학에서 관대 한 지원을 인정합니다.
Access restricted. Please log in or start a trial to view this content.
자료
| Name | Company | Catalog Number | Comments |
| 0.5% polysorbate 20 (TWEEN 20) | BioShop | TWN510.5 | |
| 0.5M ethylenediaminetetraacetic acid (EDTA) | Bio Basic | SD8135 | |
| 10 mM sodium phosphate buffer (pH 7) | Bio Basic | PD0435 | Tablets used to make 10 mM buffer |
| 10% ammonium persulfate (APS) | Sigma Aldrich | A3678-100G | |
| 100 kDa Amicon Ultra-15 Centrifugal Filter Unit | Fisher Scientific | UFC910008 | |
| 100% acetone | Fisher Chemical | A18P4 | |
| 100% ethanol (EtOH) | House Brand | 39752-P016-EAAN | |
| 10x in vitro transcription (IVT) buffer | New England Biolabs | B9012 | |
| 10x Tris-Borate-EDTA (TBE) buffer | Bio Basic | A0026 | |
| 1M Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Sigma Aldrich | I5502-1G | |
| 1M sodium bicarbonate buffer | Sigma Aldrich | S6014-500G | |
| 1M Tris(hydroxymethyl)aminomethane (Tris) | Sigma Aldrich | 648311-1KG | |
| 1X Tris-EDTA (TE) buffer | ThermoFisher | 12090015 | |
| 2M imidazole | Sigma Aldrich | 56750-100G | |
| 2-mercaptoethanol (BME) | Sigma Aldrich | M3148 | |
| 3M sodium acetate | Bio Basic | SRB1611 | |
| 40% acrylamide (19:1) | Bio Basic | A00062 | |
| 4x LDS protein sample loading buffer | Fisher Scientific | NP0007 | |
| 5M sodium chloride (NaCl) | Bio Basic | DB0483 | |
| 5mM dithiothreitol (DTT) | Sigma Aldrich | 43815-1G | |
| 6x gel loading dye | New England Biolabs | B7024S | |
| agarose B powder | Bio Basic | AB0014 | |
| BG-GLA-NHS | New England Biolabs | S9151S | |
| BL21 competent E. coli | Addgene | C2530H | |
| BLUeye prestained protein ladder | FroggaBio | PM007-0500 | |
| bromophenol blue | Bio Basic | BDB0001 | |
| coomassie blue (SimplyBlue SafeStain) | ThermoFisher | LC6060 | |
| cyanine dye (SYBR Gold nucleic acid gel stain) | Fisher Scientific | S11494 | |
| cyanine dye (SYBR Safe nucleic acid gel stain) | Fisher Scientific | S33102 | |
| dry dimethyl sulfoxide (DMSO) | Fisher Scientific | D12345 | |
| formamide | Sigma Aldrich | F9037-100ML | |
| glycerol | Bio Basic | GB0232 | |
| kanamycin sulfate | BioShop | KAN201.5 | |
| lysogeny broth | Sigma Aldrich | L2542-500ML | |
| malachite green oxalate | Sigma Aldrich | 2437-29-8 | |
| N,N,N'N'-Tetramethylethane-1,2-diamine (TEMED) | Sigma Aldrich | T9281-25ML | |
| NuPAGE MES SDS running buffer (20x) | Fisher Scientific | LSNP0002 | |
| NuPAGE Novex 4-12% Bis-Tris gel 1.0 mm 12-well | Life Technologies | NP0322BOX | |
| oligonucleotide (cage antisense) | IDT | N/A | TATAGTGAGTCGTATTAATTTG |
| oligonucleotide (cage sense) | IDT | N/A | TCAGTCACCTATCTGTTTCAAA TTAATACGACTCACTATA |
| oligonucleotide (malachite green aptamer antisense) | IDT | N/A | GGATCCATTCGTTACCTGGCT CTCGCCAGTCGGGATCCTATA GTGAGTCGTATTACAGTTCCAT TATCGCCGTAGTTGGTGTACT |
| oligonucleotide (malachite green aptamer sense) | IDT | N/A | TAATACGACTCACTATAGGATC CCGACTGGCGAGAGCCAGGT AACGAATGGATCC |
| oligonucleotide (Transcription Factor A) | IDT | N/A | AGTACACCAACTACGAGTGAG |
| oligonucleotide (Transcription Factor B) | IDT | N/A | TCAGTCACCTATCTGGCGATAA TGGAACTG |
| oligonucleotide with 3’ Amine modification (tether) | IDT | N/A | GCTACTCACTCAGATAGGTGAC TGA/3AmMO/ |
| Pierce strong ion exchange spin columns | Fisher Scientific | 90008 | |
| plasmid encoding SNAP T7 RNAP and kanamycin resistance genes | Genscript | N/A | custom gene insert |
| protein purification column (HisPur Ni-NTA spin column) | Fisher Scientific | 88226 | |
| rNTP mix | New England Biolabs | N0466S | |
| Roche mini quick DNA spin column | Sigma Aldrich | 11814419001 | |
| Triton X-100 | Sigma Aldrich | T8787-100ML | |
| Ultra Low Range DNA ladder | Fisher Scientific | 10597012 | |
| urea | BioShop | URE001.1 |
참고문헌
- Cherry, K. M., Qian, L. Scaling up molecular pattern recognition with DNA-based winner-take-all neural networks. Nature. 559 (7714), 370-376 (2018).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475 (7356), 368-372 (2011).
- Chen, Y. -J., et al. Programmable chemical controllers made from DNA. Nature Nanotechnology. 8 (10), 755-762 (2013).
- di Bernardo, D., Marucci, L., Menolascina, F., Siciliano, V. Predicting synthetic gene networks. Synthetic Gene Networks: Methods and Protocols. 813, 57-81 (2012).
- Xiang, Y., Dalchau, N., Wang, B. Scaling up genetic circuit design for cellular computing: advances and prospects. Natural Computing. 17 (4), 833-853 (2018).
- Gould, N., Hendy, O., Papamichail, D. Computational tools and algorithms for designing customized synthetic genes. Frontiers in Bioengineering and Biotechnology. 2, (2014).
- MacDonald, J. T., Siciliano, V. Computational sequence design with R2oDNA Designer. Mammalian Synthetic Promoters. 1651, 249-262 (2017).
- Cervantes-Salido, V. M., Jaime, O., Brizuela, C. A., Martínez-Pérez, I. M. Improving the design of sequences for DNA computing: A multiobjective evolutionary approach. Applied Soft Computing. 13 (12), 4594-4607 (2013).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry. 32 (1), 170-173 (2011).
- Fornace, M. E., Porubsky, N. J., Pierce, N. A. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synthetic Biology. 9 (10), 2665-2678 (2020).
- Wetterstrand, K. DNA sequencing costs: Data. Genome.gov. , (2020).
- Lopez, R., Wang, R., Seelig, G. A molecular multi-gene classifier for disease diagnostics. Nature Chemistry. 10 (7), 746-754 (2018).
- Pardee, K., et al. low-cost detection of Zika virus using programmable biomolecular components. Cell. 165 (5), 1255-1266 (2016).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406 (6796), 605-608 (2000).
- Lin, K. N., Volkel, K., Tuck, J. M., Keung, A. J. Dynamic and scalable DNA-based information storage. Nature Communications. 11 (1), 2981(2020).
- Yurke, B., Mills, A. P. Using DNA to power nanostructures. Genetic Programming and Evolvable Machines. 4 (2), 111-122 (2003).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318 (5853), 1121-1125 (2007).
- Wang, B., Thachuk, C., Ellington, A. D., Winfree, E., Soloveichik, D. Effective design principles for leakless strand displacement systems. Proceedings of the National Academy of Sciences. 115 (52), 12182-12191 (2018).
- Machinek, R. R. F., Ouldridge, T. E., Haley, N. E. C., Bath, J., Turberfield, A. J. Programmable energy landscapes for kinetic control of DNA strand displacement. Nature Communications. 5 (1), 5324(2014).
- Cabello-Garcia, J., Bae, W., Stan, G. -B. V., Ouldridge, T. E. Handhold-mediated strand displacement: a nucleic acid-based mechanism for generating far-from-equilibrium assemblies through templated reactions. bioRxiv. , (2020).
- Brophy, J. A. N., Voigt, C. A. Principles of genetic circuit design. Nature Methods. 11 (5), 508-520 (2014).
- Khalil, A. S., et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 150 (3), 647-658 (2012).
- Swank, Z., Laohakunakorn, N., Maerkl, S. J. Cell-free gene-regulatory network engineering with synthetic transcription factors. Proceedings of the National Academy of Sciences. 116 (13), 5892-5901 (2019).
- Howland, S. W., Tsuji, T., Gnjatic, S., Ritter, G., Old, L. J., Wittrup, K. D. Inducing efficient cross-priming using antigen-coated yeast particles. Journal of immunotherapy. 31 (7), 607(2008).
- Abil, Z., Ellefson, J. W., Gollihar, J. D., Watkins, E., Ellington, A. D. Compartmentalized partnered replication for the directed evolution of genetic parts and circuits. Nature Protocols. 12 (12), 2493-2512 (2017).
- Baugh, C., Grate, D., Wilson, C. 2.8 Å crystal structure of the malachite green aptamer11. Journal of Molecular Biology. Doudna, J. A. 301 (1), 117-128 (2000).
- Chou, L. Y. T., Shih, W. M. In vitro transcriptional regulation via nucleic acid-based transcription factors. ACS Synthetic Biology. 8 (11), 2558-2565 (2019).
- Lykke-Andersen, J., Christiansen, J. The C-terminal carboxy group of T7 RNA polymerase ensures efficient magnesium ion-dependent catalysis. Nucleic Acids Research. 26 (24), 5630-5635 (1998).
- Pu, J., Disare, M., Dickinson, B. C. Evolution of C-terminal modification tolerance in full-length and split T7 RNA Polymerase biosensors. Chembiochem. 20 (12), 1547-1553 (2019).
- Gardner, L. P., Mookhtiar, K. A., Coleman, J. E. Initiation, elongation, and processivity of carboxyl-terminal mutants of T7 RNA polymerase. Biochemistry. 36 (10), 2908-2918 (1997).
- Yin, J., Lin, A. J., Golan, D. E., Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nature Protocols. 1 (1), 280-285 (2006).
- Warden-Rothman, R., Caturegli, I., Popik, V., Tsourkas, A. Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Analytical Chemistry. 85 (22), 11090-11097 (2013).
- Zhang, W. -B., Sun, F., Tirrell, D. A., Arnold, F. H. Controlling macromolecular topology with genetically encoded SpyTag-SpyCatcher chemistry. Journal of the American Chemical Society. 135 (37), 13988-13997 (2013).
Access restricted. Please log in or start a trial to view this content.
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유