
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
プログラム可能なインビトロ転写と分子計算のためのDNAテザリングRNAポリメラーゼ
要約
インビトロ転写反応を調節する新規DNAつながされたT7 RNAポリメラーゼの工学について述べています。タンパク質合成と特性評価のステップについて議論し、概念実証の転写制御を検証し、分子コンピューティング、診断、分子情報処理における応用について議論する。
要約
DNAナノテクノロジーは、核酸をプログラム可能な自己集合化をユーザー指定の形状および多様なアプリケーションのためのダイナミクスに可能にします。この研究は、DNAナノテクノロジーの概念を使用して、ファージ由来のT7 RNAポリメラーゼ(RNAP)の酵素活性をプログラムし、スケーラブルな合成遺伝子調節ネットワークを構築できることを実証しています。まず、オリゴヌクレオチドと結合したT7 RNAPは、N末端SNAPタグ付きRNAPの発現と、その後のSNAPタグとベンジルグアニン(BG)修飾オリゴヌクレオチドとの化学的結合を介して設計される。次に、核酸鎖変位を使用して、オンデマンドでポリメラーゼ転写をプログラムします。さらに、補助核酸アセンブリを「人工転写因子」として使用して、DNAプログラムT7 RNAPとDNAテンプレートとの相互作用を調節することができます。このインビトロ転写調節機構は、デジタルロジック、フィードバック、カスケード、多重化などの様々な回路動作を実装できます。この遺伝子調節アーキテクチャの構成性は、設計の抽象化、標準化、およびスケーリングを促進します。これらの機能により、生体センシング、疾患検出、データ保存などのアプリケーションに対するインビトロ遺伝子デバイスの迅速な試作が可能になります。
概要
DNAコンピューティングは、計算の媒体として設計されたオリゴヌクレオチドのセットを使用します。これらのオリゴヌクレオチドは、ユーザー指定のロジックに従って動的に組み立て、特定の核酸入力に応答する配列でプログラムされています。概念実証研究では、計算の出力は、典型的には、ゲル電気泳動または蛍光プレートリーダーを介して検出することができる蛍光標識オリゴヌクレオチドのセットで構成されています。過去30年間で、さまざまなデジタル論理カスケード、化学反応ネットワーク、ニューラルネットワーク1、2、3など、ますます複雑化するDNA計算回路が実証されています。これらのDNA回路の作製を支援するために、合成遺伝子回路4、5の機能性を予測するために数理モデルが使用され、直交DNA配列設計6、7、8、9、10の計算ツールが開発されています。.シリコンベースのコンピュータと比較すると、DNAコンピュータの利点は、生体分子と直接インターフェースし、電源がない場合に溶液中で動作する能力、ならびに全体的なコンパクト性と安定性を備えています。次世代シーケンシングの出現により、DNAコンピュータの合成コストは、ムーアの法則11よりも速い速度で過去20年間減少しています。このようなDNAベースのコンピュータの応用は、現在、疾患診断12、13、分子生物物理学14、およびデータ記憶プラットフォーム15として、現れ始めている。

図1:足のホールド媒介DNA鎖変位のメカニズム δの間のつま先は、部分二重での自由な非バインド シーケンスです。第2の鎖に相補的なドメイン(δ*)が導入されると、フリー δドメインはハイブリダイゼーションの足掛かりとして機能し、ストランド移行と呼ばれる圧縮/解凍可逆反応を通じて競合他社をゆっくりと置き換えることを可能にします( 安打 *)δの長さが長くなるにつれて、前進反応のΔGは減少し、変位はより容易に起こる。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
現在までに、DNAコンピュータの大部分は、足のホールド媒介DNA鎖変位として知られる動的DNAナノテクノロジーの分野において確立されたモチーフを利用している(TMDSD,図1)16。このモチーフは、短い「足掛け」の突出し(すなわち、7-〜10ヌクレオチド(nt))を示す部分的に二本鎖DNA(dsDNA)二重から成る。核酸「入力」鎖は、足止めを通して部分的な二重鎖と相互作用することができる。これにより、部分的なデュプレックスからのストランドの 1 つの変位が起き、この解放されたストランドは下流の部分的なデュプレックスの入力として機能します。このように、TMDSD は信号のカスケードと情報処理を可能にします。基本的に、直交TMDSDモチーフは、ソリューションで独立して動作することができ、並列情報処理を可能にします。TMDSD反応には、足がかりを媒介するDNA鎖交換(TMDSE)17、二重長ドメイン18との「リークレス」足音、配列不一致の足掛かり19、および「ハンドホールド」媒介鎖変位20など多くのバリエーションがあった。これらの革新的な設計原則により、より細かく調整されたTMDSDのエネルギーとダイナミクスがDNAコンピューティングの性能を向上させることができます。
転写遺伝子回路などの合成遺伝子回路は、計算21、22、23も可能である。これらの回路は、タンパク質転写因子によって調節され、特定の調節DNA要素に結合することによって遺伝子の転写を活性化または再抑制する。DNAベースの回路と比較して、転写回路には複数の利点があります。まず、酵素転写は既存の触媒DNA回路よりもはるかに高い回転率を有し、入力の単一コピー当たりの出力のコピーが多く生成され、より効率的な信号増幅手段を提供する。さらに、転写回路は、治療用タンパク質をコードするアプタマーやメッセンジャーRNA(mRNA)などの異なる機能分子を計算出力として生成することができ、異なる用途に利用することができます。しかし、現在の転写回路の大きな制限は、スケーラビリティの欠如です。これは、直交タンパク質ベースの転写因子のセットが非常に限られており、新しいタンパク質転写因子のデノボ設計は技術的に困難で時間がかかるためです。

図2:「テザー」および「ケージ」ポリメラーゼ複合体の抽象化およびメカニズム(AおよびB)のオリゴヌクレオチドテザーは、SNAPタグ反応を通じてT7ポリメラーゼに酵素的に標識されている。テザー補体オーバーハングを有する「フェイク」T7プロモーターからなるケージは、テザーとブロック転写活性にハイブリダイズすることを可能にする。(C)オペレータ(a*b*)が存在すると、オリゴヌクレオチドテザー(ab)上のつま先に結合し、ケージのb*領域を置き換え、転写が起こることを可能にする。この図は、チョウとシー27から変更されています。略語: RNAP = RNA ポリメラーゼこの図の大きなバージョンを表示するには、ここをクリックしてください。
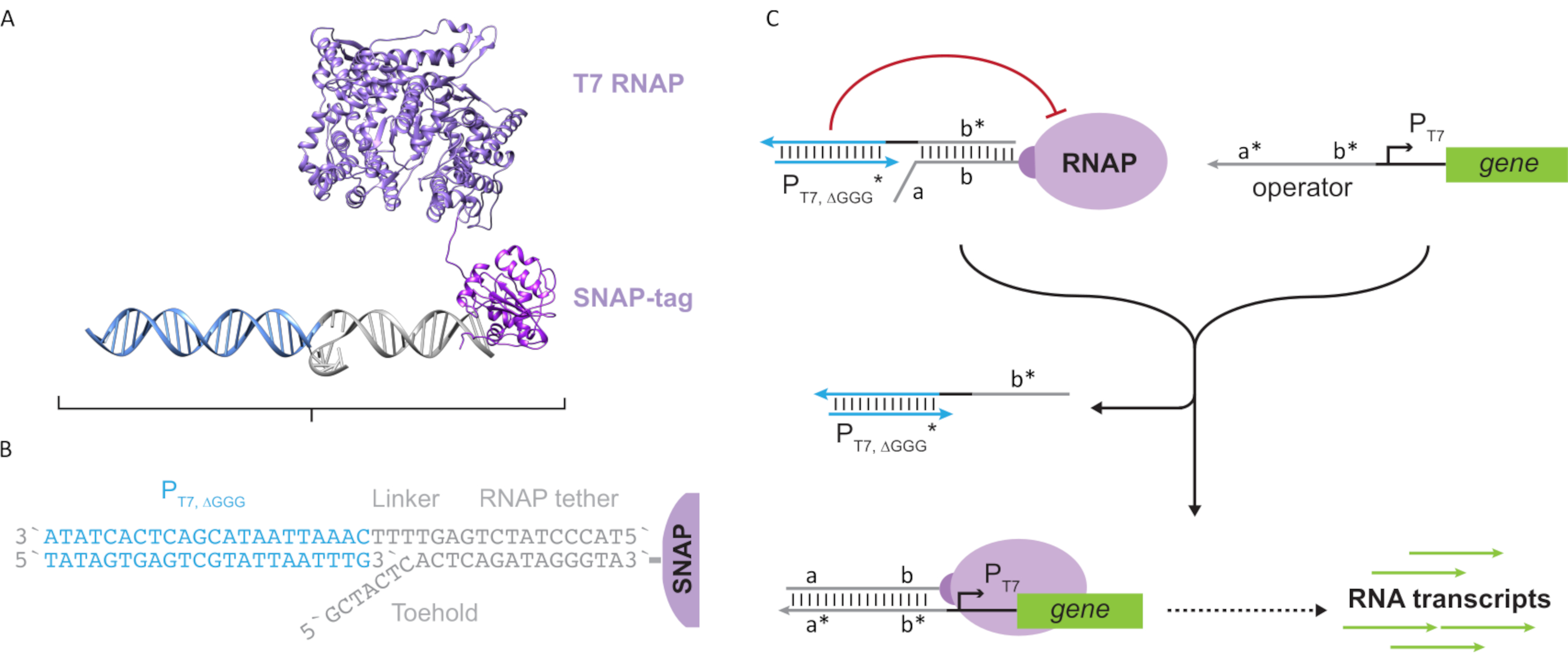{kind=link}
この論文では、転写回路の機能性とDNAベース回路の拡張性を組み合わせた分子計算のための新しいビルディングブロックを紹介する。このビルディングブロックは、T7 RNAP共有結合体である一本鎖DNAテザー(図2A)このDNAテザリングT7 RNAPを合成するために、ポリメラーゼをN末端SNAPタグ24に融合させ、エシェリヒア・コリで組換え発現させた。次いで、SNAPタグを、BG基質と機能するオリゴヌクレオチドと反応させた。オリゴヌクレオチドテザーは、DNAハイブリダイゼーションを介してポリメラーゼに近接した分子ゲストの位置決めを可能にする。そのようなゲストの1つは、下流に遺伝子のない「フェイク」T7プロモーターDNAデュプレックスで構成される「ケージ」と呼ばれる競争的な転写ブロッカーでした(図2B)。オリゴヌクレオチドテザーを介してRNAPに結合すると、ケージはRNAP結合のための他のDNAテンプレートを上回ることによってポリメラーゼ活性を失速させ、RNAPを「オフ」状態でレンダリングする(図2C)。
ポリメラーゼを「ON」状態に活性化させるために、T7遺伝子のT7プロモーターの上流に一本鎖「オペレータ」ドメインを有するT7 DNAテンプレートが設計された。オペレータドメイン(すなわち、ドメインa*b*図2C)は、TMDSDを介してRNAPからケージを置き換え、RNAPを遺伝子のT7プロモーターに近い位置に置き、したがって転写を開始するように設計することができる。あるいは、DNAテンプレートは、オペレータ配列が「人工転写因子」と呼ばれる補助核酸鎖と相補的であるところも設計された(すなわち、図3AのTFAおよびTFB鎖)。両方のストランドが反応に導入されると、それらはオペレータサイトで組み立てられ、新しい擬似連続ドメインa*b*が作成されます。このドメインは、TMDSD を介してケージを置き換えて、転写を開始することができます (図 3B)。これらのストランドは外因性に供給されるか、または製造することができる。

図3:3成分スイッチアクチベータを用いたポリメラーゼ活性の選択的プログラミング (A) 転写因子(TFA およびTFB)が存在する場合、それらはプロモーターのオペレータドメイン上流に結合し、トーホールド媒介DNA変位を通してケージを変位させることができる疑似一本鎖配列(a*b*)を形成する。(B) この a*b* ドメインは、TMDSDを介してケージを置き換えて転写を開始することができます。この図は、チョウとシー27から変更されています。略語: TF = 転写因子;RNAP = RNA ポリメラーゼ;TMDSD = 足掛け媒介DNA鎖変位。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
インビトロ転写制御に核酸ベースの転写因子を使用することで、デジタルロジック、フィードバック、信号カスケードなどの高度な回路挙動をスケーラブルに実装できます。例えば、上流の遺伝子からの転写産物が下流遺伝子を活性化するような核酸配列を設計することによって、ロジックゲートカスケードを構築することができます。この提案された技術によって可能なカスケードと多重化を利用する1つのアプリケーションは、ポータブル診断と分子データ処理のためのより洗練された分子コンピューティング回路の開発です。さらに、分子計算とデノボRNA合成機能の統合により、新たなアプリケーションが可能になります。例えば、分子回路は、ポイント・オブ・ケア・メディカル・アプリケーション用の機能性ペプチドまたはタンパク質をコードする機能性ペプチドまたはタンパク質をコードする、入力および出力治療用RNAまたはmRNAとして、ユーザー定義RNAの1つまたは組み合わせを検出するように設計することができる。
Access restricted. Please log in or start a trial to view this content.
プロトコル
1. バッファの準備
注:タンパク質精製バッファーの調製は、任意の日に発生することができます。ここでは、実験を開始する前に行われました。
- 50 mM トリス(ヒドロキシメチル)アミノメタン(トリス)、300 mM塩化ナトリウム(NaCl)、5%グリセロール、および5 mM β-メルカプトエタノール(BME)、pH 8を含むリシス/平衡バッファーを調製します。1Mトリス1.5mL、5M NaClの1.8mL、グリセロール1.5mL、25.2mLの脱イオン水(ddH2O)を50 mL遠心管に加え、使用直前に10.5μLの10.5 μLを加えます。
注意: トリスは急性毒性を引き起こす可能性があります。したがって、そのほこりを呼吸を避け、皮膚と目の接触を避けてください。BMEは有毒であり、ヒュームフードでのみ使用する必要があります。再懸濁および細胞のリシスの直前に、最後にBMEを追加することが重要です。リシスバッファー式については 、表 1 を参照してください。 - 50 mM トリス、800 mM NaCl、5% グリセロール、5 mM BME、および 20 mM イミダゾールを含む洗浄バッファー (pH 8) を準備します。1 Mトリスの1.5 mL、5 M NaClの4.8 mL、グリセロール1.5mL、および22.2mLのddH2Oを50 mL遠心管に加えます。使用前に、上記の溶液の20 mLに14.2 M BMEの7 μLと2Mイミダゾールの200 μLを加えます。
メモ:イミダゾールによる急性毒性を防ぐために、個人用保護具を使用してください。BMEとイミダゾールを最後に添加することが重要です, ちょうどカラムからタンパク質を洗浄する前に.洗浄バッファーの公式については 、表 2 を参照してください。 - 50 mM トリス、800 mM NaCl、5% グリセロール、5 mM BME、および 200 mM イミダゾールを含む溶出バッファー (pH8) を準備します。1 Mトリスの0.5 mL、5 M NaClの1.6 mL、グリセロールの0.5mL、および6.4mLのddH2Oを15 mL遠心管に加えます。使用前に、14.2 M BMEの3.5 μLと2 Mイミダゾールの1 mLを上記の溶液の10 mLに加えます。
注:BMEとイミダゾールを最後に追加することが重要です, 列からタンパク質を溶出する直前に.溶出バッファー式については 、表 3 を参照してください。 - 100 mMトリス、200 mM NaCl、40 mM BME、および2 mMエチレンジアミネトラ酢酸(EDTA)を含む2xストレージバッファー(グリセロールと1:1を混合する)を用意し、非イオン性界面活性剤の0.2%( 材料表を参照)50 mLの5mLの1 Mトリス、2 mLの5 M NaCl、42.56 mLのddH2O、0.5 M EDTAの200 μL、非イオン性界面活性剤の100 μLを50 mL遠心チューブに加えて調製します。溶液が均質になるまで混ぜ合わせ、0.2 μmのシリンジフィルターでストレージバッファをフィルターし、使用前に上記の溶液に140.8 μLのBMEを追加します。
注:EDTAによる急性毒性を避けるために、ほこりを吸い込むのを避け、皮膚と目の接触を避けてください。BMEを最後に加え、精製されたタンパク質を保存する直前に、保存バッファー全体をグリセロールと1:1混合することが重要です。ストレージ バッファの公式については 、表 4 を参照してください。
2. 一晩の文化の成長:1日目
- 10mLのddH2Oに500mgのカナマイシンを溶解して、1,000mgのカナマイシンストックを調製します。
注:カナマイシンによる急性毒性を防ぐために、個人用保護具を使用してください。 - 1,000xカナマイシンストックの20μLを20mLのリソジニースープに加えます。無菌ピペットチップを使用して、変換されたBL21 大腸菌 グリセロールストックを突き、成長培地のスープにチップを導入して培養液を接種します。

図4:スナップT7 RNAP用プラスミドマップ プラスミドは、pQE-80Lバックボーン上のラックリプレッサー(lacI)の下にN末端ヒスチジンタグ(6x His)とSNAPタグドメイン(SNAP T7 RNAP)を含むT7 RNAPをコード化します。その他の特徴としては、カナマイシン耐性(KanR)およびクロラムフェニコール耐性(CmR)遺伝子が挙げられる。略語: RNAP = RNAポリメラーゼ この図の大きなバージョンを表示するには、ここをクリックしてください。
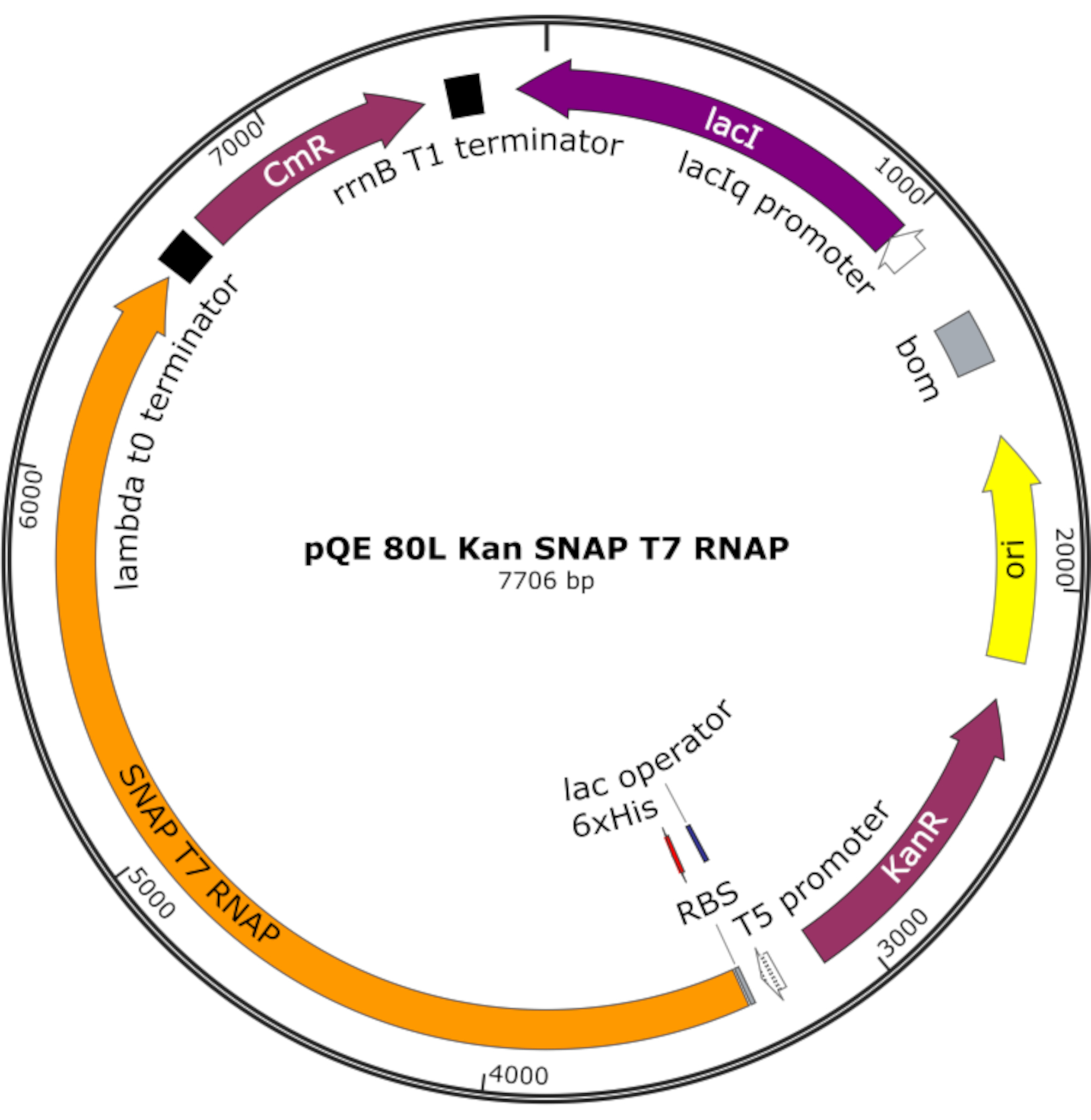{kind=link}
注:プラスミドは、N末端ヒスチジンタグとSNAPタグドメイン(SNAP T7 RNAP)を含むT7 RNAPと、pQE-80Lバックボーン下のカナマイシン耐性遺伝子(図4)25をコードする。
- 再び、20mLのリソジニーブロスを含む別の培養フラスコに1,000xカナマイシンストックの20μLを加え、コントロールとしてインキュベートします。
- 2つのサンプルを(ステップ2.2と2.3から)一晩37°Cで12〜18時間インキュベートし、10×gで回転させる。
3. 細胞の増殖と誘導:2日目
- 400μLのカナマイシンストックを含むリソゲニーブロス400mLを、ステップ2.4から一晩成長培養した4mLを一晩で培養した。培養フラスコを37°Cでインキュベートし、10×gで回転させる。
- 培養液が〜0.5の600nmで光学密度(OD)に達したら、1mLの増殖フラスコをコントロールとして取り出す。4°Cでコントロールサンプルを保存してください。
- 100 mL の培養ごとに 1M IPTG の 40 μL を加えて、イソプロピル β-D-1-チオガラクトピラノシド(IPTG)で細胞を誘導し、最終的な濃度を 0.4 mM IPTG にします。試料を37°Cで3時間インキュベートし、10×gで回転し、8,000×gで細胞をペレット化して10分間培養した。上清を取り出し、ペレットを-20°Cで保管し、さらに使用するまで保管します。
注:IPTGによる急性毒性を避けるために、ほこりを吸い込むのを避け、皮膚と目の接触を避けてください。必要に応じて、ここで実験を一時停止し、翌日に進むことができます。
4. 細胞のリシス、タンパク質精製:3日目
- 貯えた細胞ペレットを氷上に10 mLのリシスバッファーで再懸濁し、ペレット全体が再懸濁されるように静かに旋回します。次いで、10本の1.5mLチューブに1mLのサンプルをピペットし、氷の上に保持する。
- 振幅設定が「1」で各サンプルを超音波処理し、30 sの期間にわたって50%のデューティサイクルで2 sパルスします。各サンプルの前後に、70%エタノールとddH2O.超音波処理中および超音波処理後に氷上のすべてのサンプルを保つ超音波処理の先端をきれいにします。
注:70%エタノールを熱と炎から離してください。 - ニッケル荷電ニトリロトリアセク酸(Ni-NTA)精製スピンカラムを4°Cの作動温度に平衡化する。 4 °Cでカラムを置く/保存し、使用中は氷の上に保管してください。
- 遠心分離100000×gで4°Cで20分間。 組み換えRNAPを含む上清をペレットに邪魔することなく慎重にピペットアウト。必要に応じて、追加の平衡バッファーを使用して、合計ボリュームを6 mL≥に調整します。
- Ni-NTAスピン列から下部タブをそっと取り出して、カラムを通るフローを可能にします。柱を遠心分離管に入れ、氷の上に置きます。
メモ:3 mL Ni-NTAスピンカラムを備えた50 mL遠心管を使用してください。 - 700×gでカラムを遠心し、4°Cで2分間保存バッファーを取り外します。6 mL の平衡バッファーを列に追加して、列を平衡化します。バッファーが樹脂ベッドに完全に入るようにします。
- 700×gで遠心分離し、2分間4°Cでカラムから平衡バッファーを取り除きます。準備したセル抽出物を列に追加する前に、列の下部プラグを置いて製品が失われるのを防ぐ。次に、カラムに細胞抽出物を加え、4°Cで30分間軌道シェーカーミキサーで混ぜます。
- 列から下部プラグを取り外し、流量をラベル付けした50 mL遠心分離管にカラムを入れます。700×gでカラムを2分間遠心して流れを集めます。
- 6 mLの洗浄バッファーをカラムに加え、樹脂を洗浄します。700×gで柱を遠心分離し、2分間、新しい遠心管に分数を集めて洗浄1とラベル付けした。合計3つの別々の分画のためにこのステップをさらに2回繰り返し、別々の遠心管に分数を集める(洗い2と3を洗う)。
- 3 mLの溶出バッファーを加え、樹脂からHisタグ付きタンパク質を溶出します。700×gで列を遠心分離し、2分間、溶出物1とラベル付けされた新しい遠心管に分画を集める。合計3つの別々の分画のためにこのステップをさらに2回繰り返し、分画を別々の遠心管(溶離2と溶出3)に集める。
- 溶出液を組み合わせ、脱塩を行い、タンパク質溶液から塩を除去します。
- ピペット15 mL 0.05 % w/vポリソルベート 20 上の 100 kDa 遠心フィルターユニット。40分間4,000×gで遠心分離機を使用し、フロースルーを廃棄します。
- コーティングされたフィルターを使用して、溶出物1、2、および3(タンパク質溶出物+ 6 mLの合計貯蔵バッファーの9 mL)を約1,500 μLに濃縮し、フィルターを3,220×gで20分間遠心し、沈殿を防ぐために膜を静かにピペット洗浄します。
- サンプルをストレージバッファーで 15 mL に希釈します。ステップ 4.11.2 をさらに 2 回繰り返して、ストレージ バッファ 1:1,000 を使用してバッファ交換を実行します。
- 280nmで画分の吸光度を測定することにより精製タンパク質を定量化する。分光光度計を記憶バッファー(4°Cで2倍の貯蔵バッファー)でブランクにします。溶出物のサンプルを軽く混ぜ合わせ、吸光度を測定します。
注:タンパク質サンプルの1x、10x、および50x希釈値で3つの別々の測定値を実行して、タンパク質を平均化し定量化します。貯蔵バッファーのサンプルを希釈します。 - 2xストレージバッファーを使用して、タンパク質サンプルを100 μMに調整します。調整したサンプルを100%グリセロールで1:1の量で希釈します。得られたタンパク質溶液を-80°Cに保存する。
5. ドデシル硫酸ポリアクリルアミドゲル電気泳動(SDS-PAGE)タンパク質産物の分析:3日目
- タンパク質分析のためにSDS-PAGEゲルを実行します。サンプルの9 μLを、4xリチウムドデシル硫酸塩(LDS)タンパク質ローディング色素の3 μLと混合します。95°Cで10分間加熱します。
- サンプルを 4-12% Bis-Tris SDS-PAGE ゲル設定にロードします。タンパク質のはしごをウェル1にロードし、次にサンプル(左から右へ):フロースルー、ウォッシュ1、ウォッシュ2、ウォッシュ3、溶出1、溶出2、溶出3、および全脱塩溶出。
注: 表 5 に、SDS-PAGE ゲルのサンプルローディングテーブルを示します。 - 2-(N-モルフォリノ)エタネスルホン酸(MES)バッファーで2-(N-モルフォリノ)エタネスルホン酸(MES)バッファーで、200 V.200minでゲルをクリーントレイに3回リンスし、200 mLのddH2Oを使用して、穏やかな攪拌でゲルマトリックスからSDSを除去します。
メモ:MESによる急性毒性を避けるために、個人用保護具を着用してください。 - 20 mLのクマシーブルーでゲルを染色し、穏やかな攪拌で室温でゲルを一晩インキュベートします。200 mLのddH2Oを軌道シェーカーに穏やかに攪拌して、ゲルをそれぞれ1時間2回脱染色します。
注:ゲルを長時間洗うか、頻繁に水を交換すると感度が向上します。さらに、過剰な染料を吸収するために容器に折り畳まれたデリケートな作業ワイプ組織を入れることで、脱染色プロセスが加速します。
6. in vitro転写によるSNAP T7 RNAPの機能検証
注:このプロトコルは、蛍光ブロッコリーRNAアプタマーをコードし、蛍光プレートリーダー上の転写の動態を監視するために蛍光の使用を可能にするDNAテンプレートを使用しています。
- SNAP T7 RNAPの活性を、市販ソースおよびバッファのみの制御から野生型(WT)T7 RNAPと比較するために、3つのin vitro転写(IVT)反応を設定します。各反応の音量を20μLに調整します。
- 10x転写バッファーの2 μL、0.4 μLの25 mMリボヌクレオシド三リン酸(rNTP)ミックス、500 nM DNAテンプレートの5 μL、500 nM SNAP T7 RNAPの2 μL、およびdDH2Oの10.6 μLを混合して、SNAP T7 RNAP IVT反応を調製します。
- 10x転写バッファーの2 μL、25 mM rNTPミックスの0.4 μL、500 nM DNAテンプレートの5 μL、WT T7 RNAPの2 μL、およびdDH2Oの10.6 μLを混合してWT RNAP IVT反応を調製します。
- バッファーのみの IVT 反応を、10x 転写バッファーの 2 μL、25 mM rNTP ミックスの 0.4 μL、500 nM DNA テンプレートの 5 μL、および 12.6 μL の ddH2O を混合して準備します。
注:RNAPを最後に追加し、サンプルを導入するまで氷の上に置いたままにしておきます。表6、表7、表8には、IVT反応式が含まれています。
- 470 nmの励起波長と512 nmの発光波長を使用して、37°Cで2分間、蛍光プレートリーダー上の転写動動態を2分間監視します。
7. BG修飾オリゴヌクレオチドの調製:1日目
- オリゴヌクレオチドをddH2Oの3'アミン修飾で1mMの最終濃度に溶解する。この S1にラベルを付けます。
- 混合1M重炭酸ナトリウム(NaHCO 3)、284 μLの100%ジメチルスルホキシド(DMSO)、S1(オリゴヌクレオチドストック)の125 μL、BG-N-ヒドロキシコキシニミド(NHS)エステル(BG-GLA-NHS)をDMSOで希釈し、50mMの66 μLを混合し、 500 μL にボリュームを調整し、室温で 100 × gで一晩インキュベートします。
注:DMSOは可燃性の液体であるため、熱や炎から離してください。 表9 は、オリゴヌクレオチドに対するBG結合に関する反応式を含む。
- 混合1M重炭酸ナトリウム(NaHCO 3)、284 μLの100%ジメチルスルホキシド(DMSO)、S1(オリゴヌクレオチドストック)の125 μL、BG-N-ヒドロキシコキシニミド(NHS)エステル(BG-GLA-NHS)をDMSOで希釈し、50mMの66 μLを混合し、 500 μL にボリュームを調整し、室温で 100 × gで一晩インキュベートします。
8. BGオリゴヌクレオチド共役のエタノール/アセトン沈殿:2日目
- ステップ7.1.1の製品を遠心分離します。13,000×gで5分間。慎重に新鮮なチューブに上澄み液を移し、任意の沈殿したBGを捨てる.オーバーフローを防ぐために2つの等しい250 μLアリコートに反応を分割し、両方のアリコートで次の手順を実行します。
- 3 M酢酸ナトリウム(25 μL)の体積の1/10分 の1を加えて、100%エタノール(625 μL)に2.5倍の体積を加えます。-80°Cで1時間インキュベートする。
注:酢酸ナトリウム(目、皮膚、消化管、気道に刺激を与える可能性があります)とエタノール(非常に可燃性、接触に刺激を引き起こす)の両方を扱う際には、個人用保護具を使用してください。必要に応じて、ここで実験を一時停止し、翌日に進みます。 - チューブを遠心分離機に配置し、外側のエッジに印を付けます。チューブを4°Cで30分間17,000×gで遠心分離します。
注:オリゴヌクレオチドペレットは、チューブのマークされたエッジに表示されます。 - ペレットを邪魔することなく、上清を捨てます。750 μLの冷やされた70%エタノールを上に上げ、17,000 × g で 4 °C で 10 分間回転します。
- ペレットを邪魔することなく、上清を捨てます。100%アセトンの750 μLで上がり、4°Cで10分間17,000×gでスピンします。
注:アセトンは非常に可燃性で接触に刺激を与えるため、アセトンを取り扱う際には個人用保護具を使用してください。 - チューブ蓋を開けた状態で、空気を5分間乾燥させて、蒸発によって余分なアセトンを取り除きます。1x Tris-EDTA(TE)バッファーの250 μLにオリゴヌクレオチドを再溶解し、約850 μM BGオリゴヌクレオチド溶液を生成した。
- ステップ 8.2 ~ 8.6 を繰り返し、70 μL の 1x TE バッファに再溶解します。この S2にラベルを付けます。
9. ゲル濾過クロマトグラフィーによるBGオリゴヌクレオチドのクリーンアップ
- 列を数回激しく反転させて行列を中断します。上部のキャップを外し、列の下端からスナップします。柱を1.5 mL遠心管に入れ、チューブを1,000×gで遠心し、室温で1分間置きます。溶出したバッファーとコレクション チューブを破棄します。
注:真空の形成を防ぐことが重要です。準備された列をすぐに使用します。 - 詰め込んだ柱をクリーンな1.5 mL遠心チューブに入れます。カラムベッドの中央に300μLの1x TEバッファを加え、遠心分離機を1,000×gで2分間加えて、バッファー溶液を交換します。 もう一度、溶出したバッファーとコレクションチューブを廃棄します。
- バッファー交換カラムをクリーンな1.5 mL遠心チューブに入れます。ベッドの中央に最大75 μLのサンプルを塗布します。1,000×gで4分間スピンします。
注:ベッドを邪魔したり、列の側面に触れないでください。ゲル媒体の最も高い点は、外側のローターを指し示す必要があります。 - それは精製された核酸を含んでいるとして、コレクションチューブから溶出物を収集します。サンプルを定量化するには、260 nmで吸光度を測定します。この S3 にラベルを付けます。
注: 測定で使用されるパスの長さに注意し、ビール・ランバート則を使用して濃度を計算します。
10. BGオリゴヌクレオチドコンジュゲートの変性PAGE解析
- 18%トリスホウ酸EDTA(TBE)-尿素ページゲルをキャストします。4.8 gのUREA、4.5 mLの40%アクリルアミド(19:1)、10x TBEの1mLを2.8 mLのddH2Oで溶解する。5 μL のテトラメチルエチレンアミン (TEMED) を加え、十分に混ぜます。10%過硫酸アンモニウム(APS)の100 μLで繰り返します。溶液を空のゲルカセットに注ぎ、40分間重合させます。
注:尿素(眼や皮膚に刺激を与える)、アクリルアミド(有毒および発がん性)、およびTEMED(有毒、可燃性、腐食性)を取り扱う際には、適切な個人用保護具を使用してください。 表10 は、18%TBE-UREAポリアクリルアミドゲルに対する反応式を含む。 - マイクロ波500mLのTBEバッファー(0.5x)を2分間及び30sまたは〜70°Cまで、ゲル装置に注ぎ込む。95%ホルムアミド+1 mM EDTA及びブロモフェノールブルーを含むホルムアミド(変性)負荷染料を調製する。各サンプルとローディング染料を混合し、ポリアクリルアミドゲルに混合物をロードします。
注意:発がん性があるフォームアミドを取り扱う際には、適切な個人用保護具を使用してください。 表11 は、サンプルゲルローディングテーブルを含む。 - 270 Vで35分間、または染料のフロントが最後まで移動するまでゲルを実行します。ゲルボックスにゲルを入れ、イメージングの前に室温で15分間核酸にシアン色素を染色します。
注:シアニン染料が可燃性であるため、適切な個人用保護具を使用してください。
11. スナップT7 RNAPおよびPAGE分析へのオリゴヌクレオチドの結合
- BGオリゴヌクレオチドとSNAP T7 RNAPへの分析スケール結合用試薬を準備する:ddH2Oで一本鎖DNA(ssDNA)オリゴの9希釈を行い、5:1から1:5までのオリゴ:RNAP比を作成します。タンパク質ストックを50 μMに希釈します。
注: 比率の例は表 12にあります。これらの比率は、50 μMのRNAP濃度を使用して計算されます。 - ssDNAオリゴの希釈ごとに、SNAPバッファーの2 μL、BGオリゴヌクレオチド4 μL、およびSNAP T7 RNAPの4 μLを含む反応混合物の10 μLを作ります。
注: 表 13 に、SNAP タグ標識反応の反応式を示します。- さらに2つの制御サンプルを準備する:1)BGオリゴヌクレオチドをddH2Oに置き換えることによってRNAPコントロール;2)DDH2OでSNAP T7 RNAPを置き換えることによってDNA制御(SNAP T7 RNAPの最も低いオリゴヌクレオチド濃度の場合)。室温で1時間インキュベートし、必要になるまで氷の上に保管してください。
- 各サンプルの2μLを4μLのSNAPバッファーと2μLのタンパク質負荷染料に加え、70°Cで10分間加熱することで、11個の10μL反応を設定します。各サンプルの2μLを4-12%ビストリスタンパク質ゲルに積み込み、200Vの氷上で35分間ゲル電気泳動を行います。
注: 表 14 に、ゲルローディングサンプルの反応式を示します。- シェーカーで3倍の水交換を介してSDSを洗い流し、それぞれ10分持続します。イメージングの前に15分間核酸のシアニン色素で染色する。20 mLのクマシーブルーステインを1時間使用して再びゲルを染色する。ddH2Oを1時間(または一晩)撮影前に脱染色する。
注:ゲルでは、反応の1つは、過剰な遊離BGオリゴヌクレオチドの最小量と一緒に最もつながれたポリメラーゼを生成します。これが最適な比率です。
- シェーカーで3倍の水交換を介してSDSを洗い流し、それぞれ10分持続します。イメージングの前に15分間核酸のシアニン色素で染色する。20 mLのクマシーブルーステインを1時間使用して再びゲルを染色する。ddH2Oを1時間(または一晩)撮影前に脱染色する。
- BGオリゴヌクレオチドをSNAP T7 RNAPに結合する調製スケール用試薬を調製する。分析スケールで見つかった最適な比率でカップリング反応を行います。
注:使用しないときにタンパク質を氷の上に置くことによって、室温へのタンパク質暴露を最小限に抑えます。
イオン交換カラムを用いたオリゴヌクレオチドとつながれたSNAP-T7の精製
- ここに記載されている手順から逸脱している場合は、チューブのセットアップに関する製造元の指示に従ってください。タンパク質の等電点よりもpH高い精製バッファーを調製します。
注:このプロトコルのタンパク質例として、10 mMリン酸ナトリウムバッファー(pH7)の精製バッファーが使用されました。- 50 mMトリスと0.5 M NaClの最終濃度を含む溶出バッファーの1,000 μLを準備します。1 Mトリス50μL、5M NaClの100 μL、ddH2Oの850 μLを混ぜ合わせます。
注: 表 15 に溶出バッファーの反応式が含まれています。
- 50 mMトリスと0.5 M NaClの最終濃度を含む溶出バッファーの1,000 μLを準備します。1 Mトリス50μL、5M NaClの100 μL、ddH2Oの850 μLを混ぜ合わせます。
- 2 mL遠心分離管にカラムを入れ、15分間、または全てのバッファーが溶出するまで2,000×gの精製バッファーで洗浄します。 溶出したバッファを破棄します。
- 精製バッファーで各サンプルを 3:1 の精製バッファー:サンプル比で希釈し、一度に 400 μL のカラムにサンプルをロードします。2,000×gで10分間、またはすべてのバッファーが溶出するまでスピンします。フロースルーを収集し、フロースルーとしてラベルを付けます。
- 400 μL の精製バッファーをカラムの中央に追加します。2,000×gで15分間、またはすべてのバッファが溶出するまで回転します。フロースルーを収集し、それを洗浄1とラベル付けします。2を洗い、3を洗う場合にさらに2回繰り返します。
- 50 μL の溶出バッファーをカラムの中央に追加します。2,000×gで5分間、またはすべてのバッファーが溶出するまでスピンします。フロースルーを収集し、それを溶出1とラベル付けします。2を 2 回繰り返し、3 を溶出します。
- プールは1、2、および3(この 全溶出物にラベルを付ける)、ゲルの各溶出物の小さな分画を残し、260 nm(A260)および280 nm(A280)で吸光度を測定する。測定後、1:1比でグリセロールを加え、さらに使用するまで-20°Cで保存します。
- 2xストレージバッファ(〜1:100)で全溶 出 液をバッファ交換するには、遠心フィルタユニット(0.5 mL;30 kDa)を使用します(この 製品にラベルを付けます)。A260/280をもう一度測定します。グリセロールを1:1比で加え、さらに使用するまで-20°Cで保存します。
- 各溶出物をロード:フロースルー、1-3、全溶出物、および4-12%ビストリSDS-PAGEゲルの製品、およびタンパク質ラダーを含む。200 Vで35分間、または染料フロントが最後まで移動するまで実行します。
13. テザードRNAポリメラーゼ活性のオンデマンド制御の実証
- 25 mMトリス、5 mM EDTA、および25 mM塩化マグネシウム(MgCl2)を含む5xアニール緩衝液を調製する。各テンプレートの2.4 μL(1 μM)を5 μLのアニーリングバッファと14.2 μLのddH2Oと混合し、1 μM dsDNAケージの25 μLを形成します。この溶液を75°Cで2分間インキュベートします。同様に、アニールは、プロモーター及びマラカイトグリーンアプタマーDNAテンプレートのセンス及びアンチセンス鎖をアニールする。マラカイトグリーンシュウ酸塩の1mM溶液を調製します。
注: 表16 は5xアニールバッファの反応式を含み、 表17 には2つのssDNAテンプレートをアニールするための反応式が含まれています。 - テザリングされたSNAP T7 RNAPをdsDNAケージと共に、室温で1:5モル比で15分間、最終濃度の500 nM RNAPにインキュベートします。必要になるまで氷の上に置いておきなさい。
- プレートリーダーを37°Cに予熱します。 氷上に3つの25 μL IVT反応を設定する
- 核酸転写因子を含むケージSNAP T7RNAPを含む反応を設定します。混合 10x IVT バッファーの 2.5 μL、25 mM rNTP ミックスの 1 μL、1 mM マラカイトグリーン 1 μL、RNAP ケージ混合物の 2.5 μL、1 μM 転写因子AとBオリゴヌクレオチドストランドのそれぞれ 2.5 μL、1 mM マラカイトグリーンアプタマーテンプレート 1.5 μL を 10μLd2DD d2DD DH dH d20 μL で混合します。
- 核酸転写因子を含まないケージSNAP T7RNAPを含む反応をセットアップします。10x IVTバッファーの2.5 μL、25 mM rNTPミックスの1 μL、1 mMマラカイトグリーンの1 μL、RNAPケージ混合物の2.5 μL、および1mMマラカイトグリーンアプタマーテンプレートの3 μLを15 μLのddH2Oで混合します。
- バッファーのみを含むリアクションを設定します。10x IVTバッファーの2.5 μL、25 mM rNTPミックスの1 μL、1 mMマラカイトグリーングリーン1μL、1mMマラカイトグリーンアプタマーテンプレートの3 μLを17.5 μLのddH2Oに混ぜます。
注: 表 18 には、インビトロ転写反応の一般的なリファレンスが含まれています。
- 各反応を384ウェルプレートに移します。37°Cで2時間、610 nm励起および655 nmの放出を用いて、蛍光プレートリーダー上のマラカイトグリーンアプタマーの転写を監視する。完成したら、必要になるまでプレートを氷の上に置いておきます。
- マイクロ波0.5x TBEバッファー2分30sまたは〜70°Cまで。 280 Vで20分間、または色素フロントが終わりに達するまで、280 Vで加熱された0.5x TBEバッファー内の変性12%TBE-尿素ポリアクリルアミドゲルで各ウェルのRNA製品を実行します。ゲルをシアニン色素核酸染色で10分間、眼窩シェーカーで染色してからイメージングします。
注: 表19 は変性12%TBE-尿素PAGEゲルのための反応式を含んでいる。
Access restricted. Please log in or start a trial to view this content.
結果

図5:スナップT7 RNAP発現およびインビトロ転写アッセイのSDS-PAGE分析.(A)SNAP T7 RNAPタンパク質精製分析、SNAP T7 RNAP分子量:119.4kDa。FT=カラムからの流れスルーは、W1=不純物を含む洗浄緩衝液の溶出画分、精製物を含むE1-3=溶出画分、およびDE=10倍希釈された全脱塩?...
Access restricted. Please log in or start a trial to view this content.
ディスカッション
本研究は、N末端SNAPタグ組換えT7 RNAPとBG官能オリゴヌクレオチドを共有結合させることによってT7 RNAポリメラーゼの活性を制御するDNAナノテクノロジーに触発されたアプローチを示し、その後TMDSD反応をプログラムするために使用された。設計上、SNAPタグはポリメラーゼのN末語に位置付けられており、野生型T7 RNAPのC末語がタンパク質構造コア内に埋もれ、DNAテンプレート28...
Access restricted. Please log in or start a trial to view this content.
開示事項
著者の誰かによって宣言する競合する財政的利益はありません。
謝辞
L.Y.T.Cは、カナダ自然科学工学研究評議会(NSERC)ディスカバリー・グラント、カナダ第一研究優秀基金(CFREF)から資金を受け取るトロント大学の医学設計イニシアチブに関する研究基金探査(NFRF-E)の新フロンティアからの寛大な支援を認めています。
Access restricted. Please log in or start a trial to view this content.
資料
| Name | Company | Catalog Number | Comments |
| 0.5% polysorbate 20 (TWEEN 20) | BioShop | TWN510.5 | |
| 0.5M ethylenediaminetetraacetic acid (EDTA) | Bio Basic | SD8135 | |
| 10 mM sodium phosphate buffer (pH 7) | Bio Basic | PD0435 | Tablets used to make 10 mM buffer |
| 10% ammonium persulfate (APS) | Sigma Aldrich | A3678-100G | |
| 100 kDa Amicon Ultra-15 Centrifugal Filter Unit | Fisher Scientific | UFC910008 | |
| 100% acetone | Fisher Chemical | A18P4 | |
| 100% ethanol (EtOH) | House Brand | 39752-P016-EAAN | |
| 10x in vitro transcription (IVT) buffer | New England Biolabs | B9012 | |
| 10x Tris-Borate-EDTA (TBE) buffer | Bio Basic | A0026 | |
| 1M Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Sigma Aldrich | I5502-1G | |
| 1M sodium bicarbonate buffer | Sigma Aldrich | S6014-500G | |
| 1M Tris(hydroxymethyl)aminomethane (Tris) | Sigma Aldrich | 648311-1KG | |
| 1X Tris-EDTA (TE) buffer | ThermoFisher | 12090015 | |
| 2M imidazole | Sigma Aldrich | 56750-100G | |
| 2-mercaptoethanol (BME) | Sigma Aldrich | M3148 | |
| 3M sodium acetate | Bio Basic | SRB1611 | |
| 40% acrylamide (19:1) | Bio Basic | A00062 | |
| 4x LDS protein sample loading buffer | Fisher Scientific | NP0007 | |
| 5M sodium chloride (NaCl) | Bio Basic | DB0483 | |
| 5mM dithiothreitol (DTT) | Sigma Aldrich | 43815-1G | |
| 6x gel loading dye | New England Biolabs | B7024S | |
| agarose B powder | Bio Basic | AB0014 | |
| BG-GLA-NHS | New England Biolabs | S9151S | |
| BL21 competent E. coli | Addgene | C2530H | |
| BLUeye prestained protein ladder | FroggaBio | PM007-0500 | |
| bromophenol blue | Bio Basic | BDB0001 | |
| coomassie blue (SimplyBlue SafeStain) | ThermoFisher | LC6060 | |
| cyanine dye (SYBR Gold nucleic acid gel stain) | Fisher Scientific | S11494 | |
| cyanine dye (SYBR Safe nucleic acid gel stain) | Fisher Scientific | S33102 | |
| dry dimethyl sulfoxide (DMSO) | Fisher Scientific | D12345 | |
| formamide | Sigma Aldrich | F9037-100ML | |
| glycerol | Bio Basic | GB0232 | |
| kanamycin sulfate | BioShop | KAN201.5 | |
| lysogeny broth | Sigma Aldrich | L2542-500ML | |
| malachite green oxalate | Sigma Aldrich | 2437-29-8 | |
| N,N,N'N'-Tetramethylethane-1,2-diamine (TEMED) | Sigma Aldrich | T9281-25ML | |
| NuPAGE MES SDS running buffer (20x) | Fisher Scientific | LSNP0002 | |
| NuPAGE Novex 4-12% Bis-Tris gel 1.0 mm 12-well | Life Technologies | NP0322BOX | |
| oligonucleotide (cage antisense) | IDT | N/A | TATAGTGAGTCGTATTAATTTG |
| oligonucleotide (cage sense) | IDT | N/A | TCAGTCACCTATCTGTTTCAAA TTAATACGACTCACTATA |
| oligonucleotide (malachite green aptamer antisense) | IDT | N/A | GGATCCATTCGTTACCTGGCT CTCGCCAGTCGGGATCCTATA GTGAGTCGTATTACAGTTCCAT TATCGCCGTAGTTGGTGTACT |
| oligonucleotide (malachite green aptamer sense) | IDT | N/A | TAATACGACTCACTATAGGATC CCGACTGGCGAGAGCCAGGT AACGAATGGATCC |
| oligonucleotide (Transcription Factor A) | IDT | N/A | AGTACACCAACTACGAGTGAG |
| oligonucleotide (Transcription Factor B) | IDT | N/A | TCAGTCACCTATCTGGCGATAA TGGAACTG |
| oligonucleotide with 3’ Amine modification (tether) | IDT | N/A | GCTACTCACTCAGATAGGTGAC TGA/3AmMO/ |
| Pierce strong ion exchange spin columns | Fisher Scientific | 90008 | |
| plasmid encoding SNAP T7 RNAP and kanamycin resistance genes | Genscript | N/A | custom gene insert |
| protein purification column (HisPur Ni-NTA spin column) | Fisher Scientific | 88226 | |
| rNTP mix | New England Biolabs | N0466S | |
| Roche mini quick DNA spin column | Sigma Aldrich | 11814419001 | |
| Triton X-100 | Sigma Aldrich | T8787-100ML | |
| Ultra Low Range DNA ladder | Fisher Scientific | 10597012 | |
| urea | BioShop | URE001.1 |
参考文献
- Cherry, K. M., Qian, L. Scaling up molecular pattern recognition with DNA-based winner-take-all neural networks. Nature. 559 (7714), 370-376 (2018).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475 (7356), 368-372 (2011).
- Chen, Y. -J., et al. Programmable chemical controllers made from DNA. Nature Nanotechnology. 8 (10), 755-762 (2013).
- di Bernardo, D., Marucci, L., Menolascina, F., Siciliano, V. Predicting synthetic gene networks. Synthetic Gene Networks: Methods and Protocols. 813, 57-81 (2012).
- Xiang, Y., Dalchau, N., Wang, B. Scaling up genetic circuit design for cellular computing: advances and prospects. Natural Computing. 17 (4), 833-853 (2018).
- Gould, N., Hendy, O., Papamichail, D. Computational tools and algorithms for designing customized synthetic genes. Frontiers in Bioengineering and Biotechnology. 2, (2014).
- MacDonald, J. T., Siciliano, V. Computational sequence design with R2oDNA Designer. Mammalian Synthetic Promoters. 1651, 249-262 (2017).
- Cervantes-Salido, V. M., Jaime, O., Brizuela, C. A., Martínez-Pérez, I. M. Improving the design of sequences for DNA computing: A multiobjective evolutionary approach. Applied Soft Computing. 13 (12), 4594-4607 (2013).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry. 32 (1), 170-173 (2011).
- Fornace, M. E., Porubsky, N. J., Pierce, N. A. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synthetic Biology. 9 (10), 2665-2678 (2020).
- Wetterstrand, K. DNA sequencing costs: Data. Genome.gov. , (2020).
- Lopez, R., Wang, R., Seelig, G. A molecular multi-gene classifier for disease diagnostics. Nature Chemistry. 10 (7), 746-754 (2018).
- Pardee, K., et al. low-cost detection of Zika virus using programmable biomolecular components. Cell. 165 (5), 1255-1266 (2016).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406 (6796), 605-608 (2000).
- Lin, K. N., Volkel, K., Tuck, J. M., Keung, A. J. Dynamic and scalable DNA-based information storage. Nature Communications. 11 (1), 2981(2020).
- Yurke, B., Mills, A. P. Using DNA to power nanostructures. Genetic Programming and Evolvable Machines. 4 (2), 111-122 (2003).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318 (5853), 1121-1125 (2007).
- Wang, B., Thachuk, C., Ellington, A. D., Winfree, E., Soloveichik, D. Effective design principles for leakless strand displacement systems. Proceedings of the National Academy of Sciences. 115 (52), 12182-12191 (2018).
- Machinek, R. R. F., Ouldridge, T. E., Haley, N. E. C., Bath, J., Turberfield, A. J. Programmable energy landscapes for kinetic control of DNA strand displacement. Nature Communications. 5 (1), 5324(2014).
- Cabello-Garcia, J., Bae, W., Stan, G. -B. V., Ouldridge, T. E. Handhold-mediated strand displacement: a nucleic acid-based mechanism for generating far-from-equilibrium assemblies through templated reactions. bioRxiv. , (2020).
- Brophy, J. A. N., Voigt, C. A. Principles of genetic circuit design. Nature Methods. 11 (5), 508-520 (2014).
- Khalil, A. S., et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 150 (3), 647-658 (2012).
- Swank, Z., Laohakunakorn, N., Maerkl, S. J. Cell-free gene-regulatory network engineering with synthetic transcription factors. Proceedings of the National Academy of Sciences. 116 (13), 5892-5901 (2019).
- Howland, S. W., Tsuji, T., Gnjatic, S., Ritter, G., Old, L. J., Wittrup, K. D. Inducing efficient cross-priming using antigen-coated yeast particles. Journal of immunotherapy. 31 (7), 607(2008).
- Abil, Z., Ellefson, J. W., Gollihar, J. D., Watkins, E., Ellington, A. D. Compartmentalized partnered replication for the directed evolution of genetic parts and circuits. Nature Protocols. 12 (12), 2493-2512 (2017).
- Baugh, C., Grate, D., Wilson, C. 2.8 Å crystal structure of the malachite green aptamer11. Journal of Molecular Biology. Doudna, J. A. 301 (1), 117-128 (2000).
- Chou, L. Y. T., Shih, W. M. In vitro transcriptional regulation via nucleic acid-based transcription factors. ACS Synthetic Biology. 8 (11), 2558-2565 (2019).
- Lykke-Andersen, J., Christiansen, J. The C-terminal carboxy group of T7 RNA polymerase ensures efficient magnesium ion-dependent catalysis. Nucleic Acids Research. 26 (24), 5630-5635 (1998).
- Pu, J., Disare, M., Dickinson, B. C. Evolution of C-terminal modification tolerance in full-length and split T7 RNA Polymerase biosensors. Chembiochem. 20 (12), 1547-1553 (2019).
- Gardner, L. P., Mookhtiar, K. A., Coleman, J. E. Initiation, elongation, and processivity of carboxyl-terminal mutants of T7 RNA polymerase. Biochemistry. 36 (10), 2908-2918 (1997).
- Yin, J., Lin, A. J., Golan, D. E., Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nature Protocols. 1 (1), 280-285 (2006).
- Warden-Rothman, R., Caturegli, I., Popik, V., Tsourkas, A. Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Analytical Chemistry. 85 (22), 11090-11097 (2013).
- Zhang, W. -B., Sun, F., Tirrell, D. A., Arnold, F. H. Controlling macromolecular topology with genetically encoded SpyTag-SpyCatcher chemistry. Journal of the American Chemical Society. 135 (37), 13988-13997 (2013).
Access restricted. Please log in or start a trial to view this content.
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved