
Bu içeriği görüntülemek için JoVE aboneliği gereklidir. Oturum açın veya ücretsiz deneme sürümünü başlatın.
Method Article
Programlanabilir In vitro Transkripsiyon ve Moleküler Hesaplama için DNA-Bağlı RNA Polimeraz
Bu Makalede
Özet
In vitro transkripsiyon reaksiyonlarını düzenlemek için yeni bir DNA bağlı T7 RNA polimerazının mühendisliğini anlatıyoruz. Protein sentezi ve karakterizasyonu için adımları tartışıyoruz, kavram kanıtı transkripsiyonel düzenlemeyi doğrulıyoruz ve moleküler bilgi işlem, teşhis ve moleküler bilgi işlemedeki uygulamalarını tartışıyoruz.
Özet
DNA nanoteknolojisi, nükleik asitlerin çeşitli uygulamalar için kullanıcı tarafından reçete edilen şekil ve dinamiklere programlanabilir bir şekilde kendi kendine bir şekilde birleştirilebilirliğini sağlar. Bu çalışma, DNA nanoteknolojisinden kavramların, faj türevi T7 RNA polimerazının (RNAP) enzimmatik aktivitesini programlamak ve ölçeklenebilir sentetik gen düzenleyici ağlar oluşturmak için kullanılabileceğini göstermektedir. İlk olarak, oligonükleotid bağlı bir T7 RNAP, N-terminalLY SNAP etiketli bir RNAP ve daha sonra SNAP etiketinin benzilguanine (BG) modifiye edilmiş oligonükleotid ile kimyasal olarak bir bağlantısı ile tasarlanmıştır. Daha sonra, nükleik-asit iplikçik deplasmanı, isteğe bağlı olarak polimeraz transkripsiyonunu programlamak için kullanılır. Ek olarak, yardımcı nükleik asit derlemeleri, DNA programlı T7 RNAP ile DNA şablonları arasındaki etkileşimleri düzenlemek için "yapay transkripsiyon faktörleri" olarak kullanılabilir. Bu in vitro transkripsiyon düzenleyici mekanizması, dijital mantık, geri bildirim, basamaklama ve çoklama gibi çeşitli devre davranışlarını uygulayabilir. Bu gen düzenleyici mimarinin birleştirilmesi tasarım soyutlama, standardizasyon ve ölçeklendirme kolaylaştırır. Bu özellikler, biyo-algılama, hastalık tespiti ve veri depolama gibi uygulamalar için in vitro genetik cihazların hızlı prototiplemesini sağlayacaktır.
Giriş
DNA hesaplama, hesaplama aracı olarak tasarlanmış bir dizi oligonükleotid kullanır. Bu oligonükleotidler, kullanıcı tarafından belirtilen mantığa göre dinamik olarak bir araya gelmek ve belirli nükleik asit girişlerine yanıt vermek için dizilerle programlanmıştır. Kavram kanıtı çalışmalarında, hesaplamanın çıktısı tipik olarak jel elektroforezi veya floresan plaka okuyucuları aracılığıyla tespit edilebilen floresan etiketli bir dizi oligonükleotidden oluşur. Son 30 yılda, çeşitli dijital mantık basamakları, kimyasal reaksiyon ağları ve sinir ağları1,2,3gibi giderek daha karmaşık DNA hesaplama devreleri gösterilmiştir. Bu DNA devrelerinin hazırlanmasına yardımcı olmak için, sentetik gen devrelerinin işlevselliğini tahmin etmek için matematiksel modeller kullanılmıştır4,5ve ortogonal DNA dizi tasarımı 6 , 7,8,9,10 için hesaplamalı araçlar geliştirilmiştir. . Silikon tabanlı bilgisayarlara kıyasla, DNA bilgisayarlarının avantajları arasında doğrudan biyomoleküllerle arayüz kurabilmeleri, güç kaynağı olmadığında çözelti içinde çalışmalarının yanı sıra genel kompaktlıkları ve stabiliteleri yer almaktadır. Yeni nesil sıralamanın ortaya çıkmasıyla, DNA bilgisayarlarını sentezlemenin maliyeti son yirmi yıldır Moore Yasası11'dendaha hızlı bir şekilde azalmaktadır. Bu tür DNA tabanlı bilgisayarların uygulamaları şimdi ortaya çıkmaya başlıyor, hastalık tanısı için12,13, moleküler biyofizik14'e güç sağlamak için ve veri depolama platformlarıolarak 15.

Şekil 1: Toehold aracılı DNA iplikçik yer değiştirme mekanizması. Δ, toehold, kısmi çift yönlü serbest, ilişkisiz bir dizidir. İkinci bir iplikçik üzerinde tamamlayıcı bir etki alanı (δ*) tanıtıldığında, serbest δ etki alanı, hibridizasyon için bir toehold görevi görür ve iplikçik geri kalanının (ɑ*) iplikçik geçişi olarak bilinen bir zipping/unzipping geri dönüşümlü reaksiyon yoluyla rakibini yavaşça yerinden etmesine izin verir. δ uzunluğu arttıkça, ileri reaksiyon için ΔG azalır ve yer değiştirme daha kolay gerçekleşir. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
{kind=link}
Bugüne kadar, DNA bilgisayarlarının çoğunluğu, toehold aracılı DNA iplikçik yer değiştirmesi olarak bilinen dinamik DNA nanoteknolojisi alanında köklü bir motif kullanmaktadır (TMDSD, Şekil 1)16. Bu motif, kısa "toehold" çıkıntılarını (yani, 7 ila 10 nükleotit (nt) gösteren kısmen çift iplikli DNA (dsDNA) dubleksten oluşur. Nükleik asit "giriş" iplikçikler, kısmi dublekslerle toehold yoluyla etkileşime girebilir. Bu, iplikçiklerden birinin kısmi çift yönlüden yer değiştirmesine yol açar ve bu kurtarılmış iplikçik daha sonra aşağı akış kısmi dubleksleri için giriş görevi görebilebilir. Böylece TMDSD sinyal basamaklama ve bilgi işleme sağlar. Prensip olarak, ortogonal TMDSD motifleri çözümde bağımsız olarak çalışarak paralel bilgi işlemeyi mümkün kılar. TMDSD reaksiyonu üzerinde, toehold aracılı DNA iplikçik değişimi (TMDSE)17,çift uzun alan adları 18 , sıra uyumsuz ayak parmakları19ve "handhold" aracılı iplik yer değiştirme20gibi bir dizi varyasyon olmuştur. Bu yenilikçi tasarım ilkeleri, DNA bilgi işlem performansını artırmak için daha ince ayarlanmış TMDSD enerji ve dinamiklerine olanak sağlar.
Transkripsiyonal gen devreleri gibi sentetik gen devreleri de hesaplama yeteneğine sahiptir21,22,23. Bu devreler, belirli düzenleyici DNA elementlerine bağlanarak bir genin transkripsiyonunu aktive eden veya baskılayan protein transkripsiyon faktörleri ile düzenlenir. DNA tabanlı devrelerle karşılaştırıldığında, transkripsiyon devrelerinin çeşitli avantajları vardır. İlk olarak, enzimatik transkripsiyon mevcut katalitik DNA devrelerinden çok daha yüksek bir ciro oranına sahiptir, böylece tek bir giriş kopyası başına daha fazla çıktı kopyası üretir ve daha verimli bir sinyal amplifikasyonu aracı sağlar. Ek olarak, transkripsiyonel devreler, farklı uygulamalar için yararlanılabilen hesaplama çıktıları olarak terapötik proteinler için aptamerler veya haberci RNA (mRNA) kodlaması gibi farklı fonksiyonel moleküller üretebilir. Bununla birlikte, mevcut transkripsiyon devrelerinin önemli bir sınırlaması ölçeklenebilirlik eksikliğidir. Bunun nedeni, çok sınırlı sayıda ortogonal protein bazlı transkripsiyon faktörü olması ve yeni protein transkripsiyon faktörlerinin de novo tasarımının teknik olarak zorlu ve zaman alıcı kalmasıdır.

Şekil 2: "Tether" ve "cage" polimeraz kompleksinin soyutlanması ve mekanizması. (A ve B) Bir oligonükleotid tether, SNAP etiketi reaksiyonu yoluyla bir T7 polimerazine enzymatic olarak etiketlenmiştir. Bağ tamamlayıcı çıkıntlı "sahte" bir T7 promotöründen oluşan bir kafes, bağlamaya melezlemesine ve transkripsiyon aktivitesini engellemesine izin verir. (C) Operatör (a * b *mevcut olduğunda, oligonükleotid bağındaki(ab)toehold'a bağlanır ve kafesin b* bölgesini yerinden çıkararak transkripsiyonun gerçekleşmesini sağlar. Bu rakam Chou ve Shih27'den değiştirilmiştir. Kısaltmalar: RNAP = RNA polimeraz. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
{kind=link}
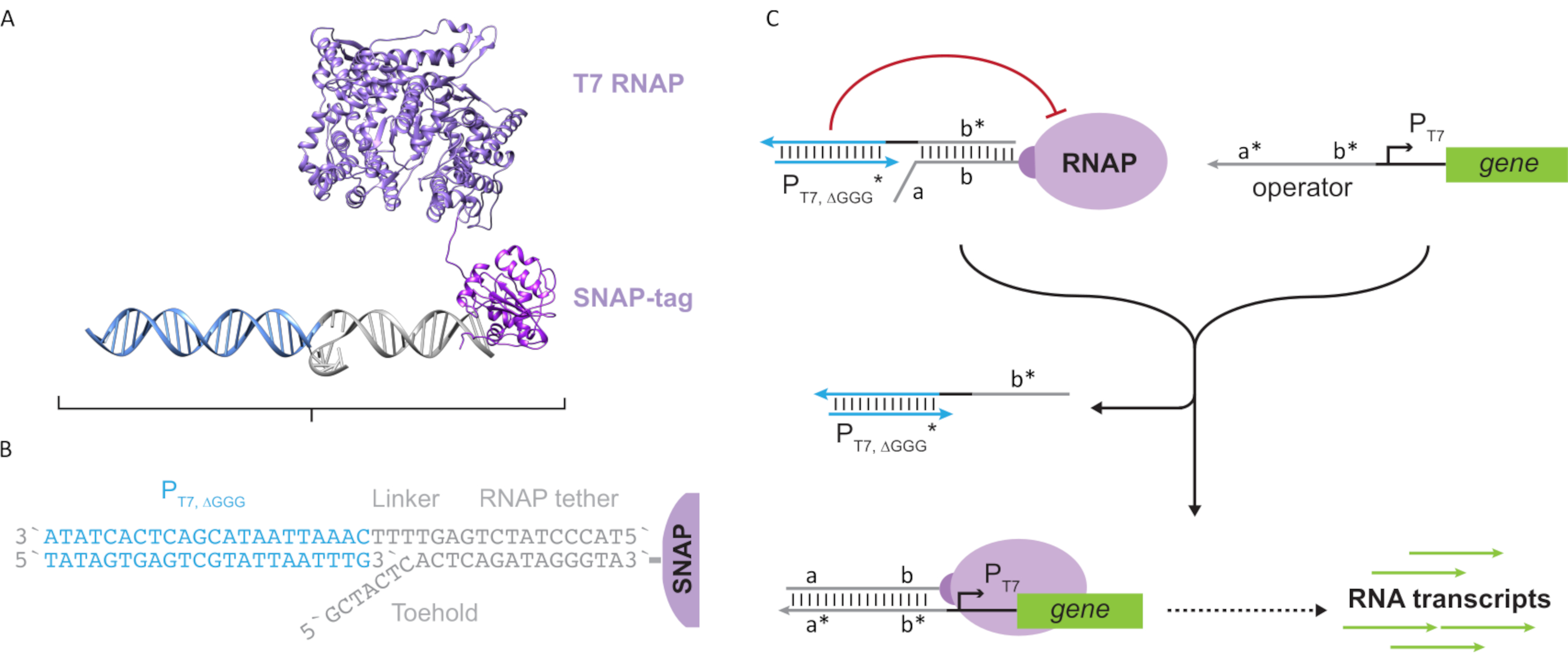
Bu makale, transkripsiyonel devrelerin işlevlerini DNA tabanlı devrelerin ölçeklenebilirliği ile birleştiren moleküler bilgi işlem için yeni bir yapı taşı sunun. Bu yapı taşı, tek iplikli dna tether ile birlikte yaygın olarak bağlanmış bir T7 RNAP'tır (Şekil 2A). Bu DNA bağlı T7 RNAP'ı sentezlemek için, polimeraz bir N-terminal SNAP etiketi24 ile kaynaştırıldı ve Escherichia coliile rekombinant olarak ifade edildi. SNAP etiketi daha sonra BG substratı ile işlevsel hale getirilmiş bir oligonükleotid ile tepkilendi. Oligonükleotid tether, MOLEKÜLER misafirlerin DNA hibridizasyonu yoluyla polimerazın yakınında konumlandırılmasını sağlar. Bu konuklardan biri, "kafes" olarak adlandırılan ve aşağı yönde gen olmayan "sahte" bir T7 promotör DNA dubleksinden oluşan rekabetçi bir transkripsiyon blokerdi (Şekil 2B). Kafes, Oligonükleotid bağıyla RNAP'a bağlandığında, RNAP bağlama için diğer DNA şablonlarını alt ederek polimeraz aktivitesini durdurarak RNAP'ı "KAPASI" durumunda işler (Şekil 2C).
Polimerazı "ON" durumuna etkinleştirmek için, genin T7 promotörünün yukarısındaki tek iplikli "operatör" alan adlarına sahip T7 DNA şablonları tasarlanmıştır. Operatör etki alanı (yani, etki alanı a*b* Şekil 2C),kafesi TMDSD aracılığıyla RNAP'tan yerinden etmek ve RNAP proksimalini genin T7 promotörüne konumlandırmak için tasarlanabilir, böylece transkripsiyon başlatılabilir. Alternatif olarak, OPERATÖR dizisinin "yapay transkripsiyon faktörleri" olarak adlandırılan yardımcı nükleik asit iplikçiklerine tamamlayıcı olduğu DNA şablonları da tasarlanmıştır (yani, Şekil 3A'dakiTFA ve TFB iplikçikleri). Her iki iplikçik de reaksiyona girdiğinde, operatör sahasında bir araya gelecek ve yeni bir sahte bitişik etki alanı a*b*oluşturacaktır. Bu alan adı daha sonra transkripsiyon başlatmak için TMDSD aracılığıyla kafesi yerinden edebilir (Şekil 3B). Bu iplikçikler eksojen olarak temin edilebilir veya üretilebilir.

Şekil 3: Üç bileşenli anahtar aktivatör ile polimeraz aktivitesinin seçici programlanması. (A) Transkripsiyon faktörleri (TFA ve TFB)mevcut olduğunda, organizatörün operatör etki alanına bağlanırlar ve kafesi toehold aracılı DNA yer değiştirme yoluyla yerinden edebilen sahte bir tek telli dizi ( a* b *) oluştururlar. (B) Bu a*b* etki alanı, transkripsiyon başlatmak için TMDSD aracılığıyla kafesi yerinden edebilir. Bu rakam Chou ve Shih27'den değiştirilmiştir. Kısaltmalar: TF = transkripsiyon faktörü; RNAP = RNA polimeraz; TMDSD = toehold aracılı DNA iplikçik yer değiştirmesi. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
{kind=link}
In vitro transkripsiyonal düzenleme için nükleik asit bazlı transkripsiyon faktörlerinin kullanılması, dijital mantık, geri bildirim ve sinyal basamaklama gibi sofistike devre davranışlarının ölçeklenebilir bir şekilde uygulanmasını sağlar. Örneğin, bir yukarı akış geninden gelen transkriptlerin aşağı akış genini aktive edebileceği şekilde nükleik asit dizileri tasarlayarak mantık kapısı basamakları oluşturabilirsiniz. Bu önerilen teknoloji tarafından yetenekli hale getirilebilen basamaklama ve çoklamadan yararlanan bir uygulama, taşınabilir tanılama ve moleküler veri işleme için daha sofistike moleküler bilgi işlem devrelerinin geliştirilmesidir. Buna ek olarak, moleküler bilgi işlem ve de novo RNA sentez yeteneklerini entegre etmek yeni uygulamalara olanak sağlayabilir. Örneğin, bir moleküler devre, kullanıcı tanımlı RNA'ların bir veya bir kombinasyonunu giriş ve çıkış terapötik RNA'lar veya bakım noktası tıbbi uygulamaları için fonksiyonel peptitleri veya proteinleri kodlayan mRNA'lar olarak tespit etmek için tasarlanabilir.
Access restricted. Please log in or start a trial to view this content.
Protokol
1. Tampon hazırlığı
NOT: Protein saflaştırma tampon hazırlığı herhangi bir günde gerçekleşebilir; Burada, deneylere başlamadan önce yapıldı.
- 50 mM tris (hidroksimetil)aminometan (Tris), 300 mM sodyum klorür (NaCl), %5 gliserol ve 5 mM β-mercaptoethanol (BME), pH 8 içeren lizis/denge tamponu hazırlayın. 50 mL santrifüj tüpüne 1,5 mL 1M Tris, 1,8 mL 5M NaCl, 1,5 mL gliserol,25,2mL deiyonize su (ddH 2 O) ekleyin ve kullanımdan hemen önce 10,5 μL 14,2 M BME ekleyin.
NOT: Tris akut toksisiteye neden olabilir; bu nedenle, tozunu solumaktan kaçının ve cilt ve göz temasından kaçının. BME toksiktir ve sadece duman kaputunda kullanılmalıdır. Resüspenzyon ve hücre lizizden hemen önce BME'yi en son eklemek önemlidir. Lizis arabelleği formülü için Tablo 1'e bakın. - 50 mM Tris, 800 mM NaCl, %5 gliserol, 5 mM BME ve 20 mM imidazol içeren yıkama tamponu (pH 8) hazırlayın. 50 mL santrifüj tüpüne 1,5 mL 1 M Tris, 4,8 mL 5 M NaCl, 1,5 mL gliserol ve22,2mL ddH 2 O ekleyin. Kullanmadan hemen önce, yukarıdaki çözeltinin 20 mL'sine 7 μL 14,2 M BME ve 200 μL 2 M imidazol ekleyin.
NOT: Imidazol nedeniyle akut toksisiteyi önlemek için kişisel koruyucu ekipman kullanın. Proteini kolondan yıkamadan hemen önce BME ve imidazol eklemek önemlidir. Yıkama arabelleği formülü için Tablo 2'ye bakın. - 50 mM Tris, 800 mM NaCl, %5 gliserol, 5 mM BME ve 200 mM imidazol içeren elution tamponu (pH8) hazırlayın. 15 mL santrifüj tüpüne 0,5 mL 1 M Tris, 5 M NaCl 1,6 mL, gliserol 0,5 mL ve6,4mL ddH 2 O ekleyin. Kullanmadan hemen önce, yukarıdaki çözeltinin 10 mL'sine 3,5 μL 14,2 M BME ve 1 mL 2 M imidazol ekleyin.
NOT: Proteini sütundan çıkarmadan hemen önce BME ve imidazol eklemek önemlidir. Elution arabellek formülü için Tablo 3'e bakın. - İyonik olmayan bir yüzey aktif maddeden %0,2'si olan 100 mM Tris, 200 mM NaCl, 40 mM BME ve 2 mM etileniaminetetraasetik asit (EDTA) içeren 2x depolama tamponu (gliserol ile 1:1 karıştırılacak şekilde) hazırlayın (Bkz. Malzeme Tablosu). 50 mL santrifüj tüpüne 5 mL 1 M Tris, 2 mL 5 M NaCl, 42,56 mL ddH2O, 200 μL 0,5 M EDTA, 100 μL iyonik olmayan yüzey aktif madde ekleyerek depolama tamponunun 50 mL'lik kısmını hazırlayın. Çözelti homojen olana kadar karıştırın, depolama tamponu 0,2 μm şırıng filtresinden filtreleyin ve kullanmadan önce yukarıdaki çözeltiye 140,8 μL BME ekleyin.
NOT: EDTA'ya bağlı akut toksisiteyi önlemek için tozunu solumaktan kaçının, cilt ve göz temasından kaçının. Bme'yi en son eklemek ve saflaştırılmış proteini depolamadan hemen önce tüm depolama tamponu 1:1'i gliserol ile karıştırmak önemlidir. Depolama arabelleği formülü için Tablo 4'e bakın.
2. Bir gecede kültür büyümesi: 1. Gün
- 10 mL ddH2O'da 500 mg kanamycin eriterek 1.000x kanamycin stoğu hazırlayın.
NOT: Kanamycin nedeniyle akut toksisiteyi önlemek için kişisel koruyucu ekipman kullanın. - 1.000x kanamycin stoğunun 20 μL'sini 20 mL lysogeny suyuna ekleyin. Steril bir pipet ucu kullanarak, dönüştürülmüş bir BL21 E. coli gliserol stoğunu dürtün ve ardından ucu büyüme ortamı suyuna sokarak kültürü aşılayın.

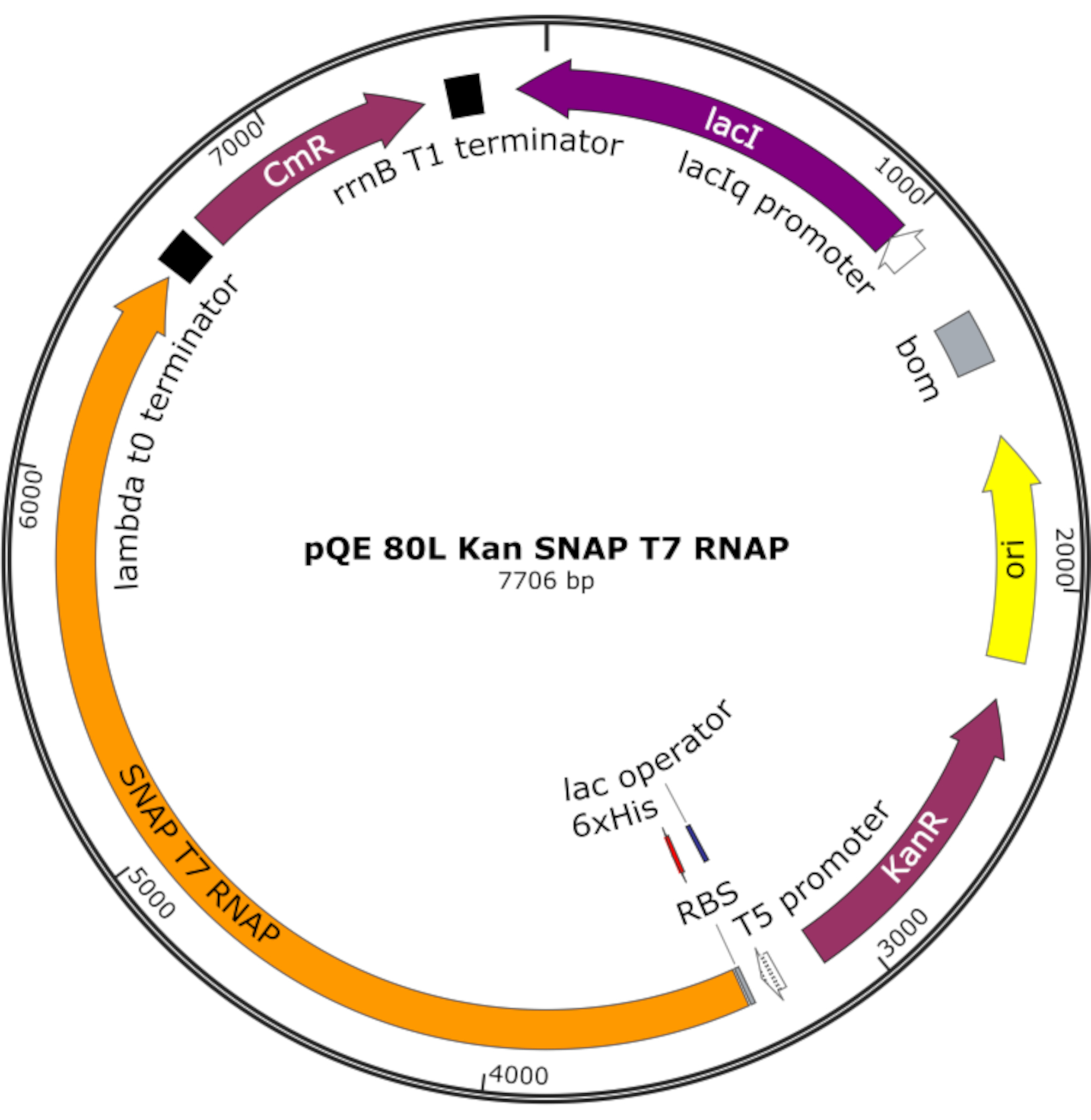
Şekil 4: SNAP T7 RNAP için plazmid harita. Plazmid, pQE-80L omurgadaki bir bağcık represörü (lacI) altında N-terminal histidin etiketi (6x His) ve SNAP-tag etki alanı (SNAP T7 RNAP) içeren bir T7 RNAP'ı kodlar. Diğer özellikler kanamycin direnci (KanR) ve kloramfenikol direnci (CmR) genleridir. Kısaltma: RNAP = RNA polimeraz. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
{kind=link}
NOT: Plazmid, N-terminal histidin etiketi ve SNAP etiketi etki alanı (SNAP T7 RNAP) içeren bir T7 RNAP'ın yanı sıra pQE-80L omurgası altında kanamiş bir direnç geni kodlar (Şekil 4)25.
- Yine, 20 mL lysojeny et suyu içeren ayrı bir kültür şişesine 1.000x kanamycin stoğunun 20 μL'sini ekleyin ve kontrol olarak kuluçkaya yatırın.
- İki numuneyi (adım 2.2 ve 2.3'ten itibaren) 37 °C'de 12-18 saat boyunca gece boyunca kuluçkaya yatırın, 10 × g'dadönerken.
3. Hücre büyümesi ve indüksiyonu: 2. Gün
- Adım 2.4'ten itibaren gecelik büyüme kültürünün 4 mL'si ile 400 μL kanamycin stoğu içeren 400 mL lysogeny et suyu aşıla. Kültür şişelerini 37 °C'de kuluçkaya yatırırken, 10 × g'da döner.
- Kültür ~ 0.5'in 600 nm'sinde bir optik yoğunluğa (OD) ulaştığında, kontrol olarak büyüme şişesinden 1 mL örnek alın. Kontrol numunesini 4 °C'de saklayın.
- 0,4 mM IPTG'lik son konsantrasyonu elde etmek için 100 mL kültür başına 40 μL 1M IPTG ekleyerek izopropil β-D-1-tiyogalactopyranoside (IPTG) ile hücreleri teşvik edin. Numuneyi 37 °C'de 3 saat kuluçkaya yatırın, 10 × g'dadöndürün ve ardından hücreleri peletmek için 10 dakika boyunca 8.000 × g'da indüklenmiş kültürü döndürün. Üst düğmeyi çıkarın ve peletin bir sonraki kullanımına kadar -20 °C'de saklayın.
NOT: IPTG nedeniyle akut toksisiteyi önlemek için tozunu solumaktan kaçının, cilt ve göz temasından kaçının. Gerekirse, denemeyi burada duraklatabilir ve ertesi gün devam edebilirsiniz.
4. Hücre lizisi, protein saflaştırma: Gün 3
- Depolanan hücre peletini buz üzerinde 10 mL lizis tamponu ile yeniden depolayın ve tüm peletin yeniden depolandığından emin olmak için hafifçe döndürün. Daha sonra pipet 1 mL numuneyi buz üzerinde tutulan on adet 1,5 mL tüpe alın.
- Her numuneyi 30 s'lik bir süre boyunca %50 görev döngüsü ile 2 sn boyunca darbeli "1" genlik ayarında sonağlı olarak belirledi. Her numuneden önce ve sonra, sonication ucunu% 70 etanol ve ddH2O ile temizleyin.
NOT: % 70 etanol ısı ve açık ateşten uzak tutun. - Nikel yüklü nitrilotricetic asit (Ni-NTA) saflaştırma spin sütununu 4 °C çalışma sıcaklığına dengelayın. Sütunu 4 °C'ye yerleştirin/saklayın ve kullanım sırasında buzda tutun.
- 15.000 × g'da on adet 1 mL'lik numuneyi 4 °C'de 20 dakika santrifüj edin. Peleti bozmadan rekombinant RNAP içeren süpernatantı dikkatlice pipetle atın. Gerekirse, toplam hacmi 6 mL'ye ≥ ayarlamak için ek denge arabelleği kullanın.
- Sütundan akışa izin vermek için ni-NTA spin sütunundan alt sekmeyi yavaşça kaldırın. Sütunu bir santrifüj tüpüne yerleştirin ve buzda tutun.
NOT: 3 mL Ni-NTA spin sütunları ile 50 mL santrifüj tüpü kullanın. - Depolama tamponunu çıkarmak için sütunu 700 × g ve 4 °C'de 2 dakika santrifüjlayın. Sütuna 6 mL denge arabelleği ekleyerek sütunu dengele. Tamponun reçine yatağına tam olarak girmesine izin verin.
- 700 g ve 4 °C'de 2 dakika × santrifüjleme ile denge tamponunu sütundan çıkarın. Hazırlanan hücre ekstresini sütuna eklemeden önce, herhangi bir ürünün kaybolmaması için sütuna bir alt fiş yerleştirin. Ardından, hücre özünü sütuna ekleyin ve 4 °C'de 30 dakika boyunca bir yörünge çalkalayıcı karıştırıcı üzerinde karıştırın.
- Alt fişi sütundan çıkarın ve sütunu içinden akış etiketli 50 mL santrifüj tüpüne yerleştirin. Akışı toplamak için sütunu 2 dakika boyunca 700 × g'da santrifüjlayın.
- Reçineyi yıkamak için kolona 6 mL yıkama tamponu ekleyin. Fraksiyonu yıkama etiketli yeni bir santrifüj tüpünde toplamak için sütunu 2 dakika boyunca 700 × g'da santrifüjlayın 1. Toplam 3 ayrı fraksiyon için bu adımı iki kez daha tekrarlayın ve fraksiyonları ayrı santrifüj tüplerinde toplayın(2'yi yıkayın ve 3'ü yıkayın).
- His etiketli proteinleri reçineden elute etmek için 3 mL elution tampon ekleyin. Eluat etiketli yeni bir santrifüj tüpünde fraksiyon toplamak için sütunu 2 dakika boyunca 700 × g'da santrifüjlayın 1. Toplam 3 ayrı fraksiyon için bu adımı iki kez daha tekrarlayın ve fraksiyonları ayrı santrifüj tüplerine toplayın(2 eluat ve eluate 3).
- Eluatları birleştirin ve tuzları protein çözeltisinden çıkarmak için tuzdan arındırma işlemi gerçekleştirin.
- Pipet 15 mL 0.05 % w/v polisorbat 20 üzerinde 100 kDa santrifüj filtre ünitesi. 40 dakika boyunca 4.000 × g'da santrifüj ve akış at.
- 1, 2 ve 3 (toplam protein eluat toplamının 9 mL ' si + 6 mL depolama tamponu) için ~1,500 μL ×'ye konsantre etmek için kaplanmış filtreyi kullanın.
- Numuneyi depolama tamponu ile 15 mL'ye seyreltin. 4.11.2 adımlarını iki kez daha yineleyerek depolama arabelleği 1:1.000 kullanarak arabellek değişimi gerçekleştirin.
- Fraksiyonun emiciliğini 280 nm olarak ölçerek saflaştırılmış proteini ölçün. Spektrofotometreyi depolama tamponu ile boş (4 °C'de 2x depolama tamponu). Kombine elüatların örneğini hafifçe karıştırın ve emiciliğini ölçün.
NOT: Protein örneğinin 1x, 10x ve 50x seyreltmelerinde proteinin ortalamasını almak ve ölçmek için üç ayrı okuma gerçekleştirin. Numuneleri depolama tamponunda seyreltin. - Protein örneklerini 2x depolama tamponu kullanarak 100 μM'ye ayarlayın. Ayarlanan numuneyi %100 gliserol ile hacim olarak 1:1 seyreltin. Elde ettiği protein çözeltisini -80 °C'de saklayın.
5. Protein ürününün sodyum dodecyl sülfat-poliakrilamid jel elektroforez (SDS-PAGE) analizi: 3. Gün
- Protein analizi için bir SDS-PAGE jeli çalıştırın. Numunenin 9 μL'sini 3 μL 4x lityum dodecyl sülfat (LDS) protein yükleme boyası ile karıştırın. Numuneleri 95 °C'de 10 dakika ısıtın.
- Numuneleri %4-12 Bis-Tris SDS-PAGE jel kurulumuna yükleyin. Protein merdivenini iyice 1'e yükleyin, ardından örneklerle (soldan sağa): akış, yıkama 1, yıkama 2, yıkama 3, elütion 1, elution 2, elution 3 ve toplam tuzdan arındırılama.
NOT: Tablo 5, SDS-PAGE jel için örnek bir yükleme tablosu içerir. - Yüklü jel örneklerini 2-(N- morfolino) etanesülfonic asit (MES) tamponunda 200 V'ta 35 dakika çalıştırın, jeli her biri 200 mL ddH2O kullanarak 10 dakika boyunca temiz bir tepside üç kez durulayın, jel matrisinden herhangi bir SDS çıkarmak için hafif ajitasyon ile.
NOT: MES nedeniyle akut toksisiteyi önlemek için kişisel koruyucu ekipman giyin. - Jeli 20 mL Coomassie mavisi ile lekelayın ve jel oda sıcaklığında hafif ajitasyonla gece boyunca kuluçkaya yatırın. Bir orbital çalkalayıcı üzerinde nazik ajitasyon ile 200 mL ddH 2 O ile her biri1saat boyunca jeli iki kez lekeden arındırın.
NOT: Jeli daha uzun süre yıkamak veya suyu sık sık değiştirmek hassasiyeti artıracaktır. Ek olarak, fazla boyayı emmek için kabın içine katlanmış hassas görev silme dokusu yerleştirmek leke çözme işlemini hızlandıracaktır.
6. Snap T7 RNAP'ın in vitro transkripsiyon yoluyla fonksiyonel doğrulaması
NOT: Bu protokol, floresan Brokoli RNA aptamerini kodlayan ve floresan plaka okuyucusunda transkripsiyon kinetiğini izlemek için floresan kullanımına izin veren DNA şablonunu kullanır.
- SNAP T7 RNAP'ın aktivitesini ticari bir kaynaktan ve yalnızca tampon kontrolünden vahşi tip (WT) T7 RNAP ile karşılaştırmak için üç in vitro transkripsiyon (IVT) reaksiyonu ayarlayın. Her reaksiyonun hacmini 20 μL'ye ayarlayın.
- 2 μL 10x transkripsiyon tamponunu karıştırarak SNAP T7 RNAP IVT reaksiyonunu hazırlayın, 0,4 μL 25 mM ribonükleozit trifosfat (rNTP) karışımı, 5 μL 500 nM DNA şablonu, 2 μL 500 nM SNAP T7 RNAP ve 10,6 μL ddH2O.
- 2 μL 10x transkripsiyon tamponu, 0,4 μL 25 mM rNTP karışımı, 5 μL 500 nM DNA şablonu, 2 μL WT T7 RNAP ve 10,6 μL ddH2O karıştırarak WT RNAP IVT reaksiyonunu hazırlayın.
- 2 μL 10x transkripsiyon tamponu, 0,4 μL 25 mM rNTP karışımı, 5 μL 500 nM DNA şablonu ve 12,6 μL ddH2O karıştırarak yalnızca tampon IVT reaksiyonunu hazırlayın.
NOT: Numuneleri tanıtımına kadar buzda tutarak RNAP'ı en son ekleyin. Tablo 6, Tablo 7ve Tablo 8 IVT reaksiyon formüllerini içerir.
- 470 nm'lik bir heyecan dalga boyu ve 512 nm emisyon dalga boyu kullanarak, floresan plaka okuyucusunda transkripsiyon kinetiğini 37 °C'de 2 dakika aralıklarla 2 saat boyunca izleyin.
7. BG modifiye oligonükleotidlerin hazırlanması: Gün 1
- Oligonükleotid'i ddH2O'da 3'-amin modifikasyonu ile 1 mM'lik son konsantrasyonda çözün. Bu S1 'i etiketle.
- 25 μL 1 M sodyum bikarbonat (NaHCO3),284 μL% 100 dimetil sülfit (DMSO), 125 μL S1 (oligonuc) karıştırın ve DMSO ile seyreltilmiş BG-N-hidroksysuccinimide (NHS) esterinin (BG-GLA-NHS) 66 μL'si, hacmi 500 μL'ye ayarlayın, ve oda sıcaklığında 100 × g'da gece boyunca kuluçkaya yatırın.
NOT: Yanıcı bir sıvı olduğu için DMSO'yu ısı ve alevden uzak tutun. Tablo 9, Oligonükleotide BG konjugasyonu için reaksiyon formülünü içerir.
- 25 μL 1 M sodyum bikarbonat (NaHCO3),284 μL% 100 dimetil sülfit (DMSO), 125 μL S1 (oligonuc) karıştırın ve DMSO ile seyreltilmiş BG-N-hidroksysuccinimide (NHS) esterinin (BG-GLA-NHS) 66 μL'si, hacmi 500 μL'ye ayarlayın, ve oda sıcaklığında 100 × g'da gece boyunca kuluçkaya yatırın.
8. BG-oligonükleotid konjuge etanol / aseton yağışı: Gün 2
- Adım 7.1.1'in ürününü santrifüj edin. 5 dakika boyunca 13.000 × g'da. Süpernatantı dikkatlice yeni bir tüpe aktarın ve çökemiş BG'leri atın.
- 3 M sodyum asetat(25 μL) hacminin 1/10'unu, ardından %100 etanol (625 μL) hacminin 2,5 katını ekleyin. 1 saat boyunca -80 °C'de kuluçkaya yatırın.
NOT: Hem sodyum asetat (gözlerde, ciltte, sindirimde ve solunum yollarında tahrişe neden olabilir) hem de etanol (aşırı yanıcı, temasta tahrişe neden olabilir) kullanırken kişisel koruyucu ekipman kullanın. Gerekirse, deneyi burada duraklatın ve ertesi gün devam edin. - Tüpleri santrifüje yerleştirin ve dış kenarı işaretleyin. Tüpleri 17.000 × g'da 4 °C'de 30 dakika santrifüj edin.
NOT: Oligonükleotid pelet tüpün işaretli kenarında görünecektir. - Peletin rahatsız etmeden, süpernatant atın. 750 μL soğutulmuş %70 etanol ile üst üste binin ve 4 °C'de 10 dakika boyunca 17.000 × g'da döndürün.
- Peletin rahatsız etmeden, süpernatant atın. %100 aseton 750 μL ile top ve 4 °C'de 10 dakika boyunca 17.000 × g'da döndürün.
NOT: Aseton kullanırken son derece yanıcı olduğu ve temasta tahrişe neden olduğu için kişisel koruyucu ekipman kullanın. - Tüp kapağı açıkken, buharlaşma yoluyla fazla asetonları çıkarmak için hava 5 dakika kurur. ~850 μM BG-oligonükleotid çözeltisi üretmek için oligonükleotid'i 250 μL 1x Tris-EDTA (TE) tamponda yeniden çözün.
- 8,2 ile 8,6 arası adımları yineleyin ve 70 μL 1x TE arabellekte yeniden çözün. Bu S2 'ye etiketle.
9. Jel filtrasyon kromatografisi ile BG-oligonükleotid temizliği
- Sütunları birkaç kez kuvvetli bir şekilde ters çevirerek matrisi askıya alın; üst kapağı çıkarın ve sütunun alt ucunu koparın. Sütunu 1,5 mL santrifüj tüpüne yerleştirin ve tüpü oda sıcaklığında 1 dakika boyunca 1.000 × g'da santrifüjlayın. Zor bulunan tamponu ve toplama tüpünü atın.
NOT: Vakum oluşumunu önlemek önemlidir. Hazırlanan sütunları hemen kullanın. - Paketlenmiş kolonları temiz 1,5 mL santrifüj tüplerine yerleştirin. Sütun yatağının ortasına 300 μL 1x TE arabellek ekleyin ve tampon çözeltisini değiştirmek için 2 dakika boyunca 1.000 × g'da santrifüjleyin. Bir kez daha, zor bulunan tamponu ve toplama tüpünü atın.
- Tamponla değiştir değiştir değiştirmiş kolonları temiz 1,5 mL santrifüj tüplerine yerleştirin. Yatağın ortasına 75 μL'ye kadar numune uygulayın. 4 dakika boyunca 1.000 × g'da döndürün.
NOT: Yatağı rahatsız etmeyin veya sütunun kenarlarına dokunmayın; jel ortamın en yüksek noktası dış rotorayı göstermelidir. - Arıtılmış nükleik asidi içerdiğinden, elüatları toplama tüpünden toplayın. Numuneyi ölçmek için emiciliğini 260 nm olarak ölçün; etiket bu S3.
NOT: Ölçümde kullanılan yol uzunluğuna dikkat edin ve Bira-Lambert yasasını kullanarak konsantrasyonu hesaplayın.
10. BG-oligonükleotid konjugenin SAYFA ANALIZI
- %18 Tris-borat-EDTA (TBE)-Üre PAGE jeli atın. 4,8 g ÜRE, 4,5 mL%40 akrilamid (19:1) ve 1 mL 10x TBE'yi 2,8 mL ddH2O'da çözün; 5 μL tetrametrinediamin (TEMED) ekleyin ve iyice karıştırın. %10 amonyum persülfat (APS) 100 μL ile tekrarlayın. Çözeltiyi boş bir jel kasete dökün ve 40 dakika polimerizasyona izin verin.
NOT: Üre (göz ve ciltte tahrişe neden olur), akrilamid (toksik ve kanserojen) ve TEMED (toksik, yanıcı, aşındırıcı) kullanırken uygun kişisel koruyucu ekipman kullanın. Tablo 10% 18 TBE-ÜRE poliakrilamid jel için reaksiyon formülünü içerir. - Mikrodalga 500 mL TBE tamponu (0,5x) 2 dakika ve 30 s veya ~70 °C'ye kadar ve bir jel aparatına dökün. %95 formamid + 1 mM EDTA ve bromofenol mavisi içeren formamid (denatüre) yükleme boyası hazırlayın. Yükleme boyasını her örnekle karıştırın ve karışımı poliakrilamid jel üzerine yükleyin.
NOT: Kanserojen olduğu için formamid kullanırken uygun kişisel koruyucu ekipman kullanın. Tablo 11 örnek jel yükleme tablosu içerir. - Jeli 270 V'ta 35 dakika boyunca veya boya cephesi sona taşınana kadar çalıştırın. Jelleri bir jel kutusuna yerleştirin ve görüntülemeden önce oda sıcaklığında 15 dakika boyunca nükleik asitler için siyanin boyası ile lekeleyin.
NOT: Siyanin boyasını yanıcı olduğu için kullanırken uygun kişisel koruyucu ekipman kullanın.
11. Oligonükleotit'in SNAP T7 RNAP ve PAGE analizine konjugasyonu
- BG-oligonükleotid'in SNAP T7 RNAP'a analitik ölçekli bağlantısı için reaktifleri hazırlayın: 5:1 ila 1:5 arasında değişen oligo:RNAP oranları oluşturmak için ddH2O ile tek iplikli DNA (ssDNA) oligo'nun 9 seyreltmesini yapın. Protein stoğunu 50 μM'ye seyreltin.
NOT: Örnek oranlar Tablo 12'debulunabilir; bu oranlar 50 μM'lik bir RNAP konsantrasyonu kullanılarak hesaplanır. - SsDNA oligo'nun her seyreltilmesi için, 2 μL SNAP tamponu, 4 μL BG-oligonükleotid ve 4 μL SNAP T7 RNAP içeren reaksiyon karışımından 10 μL yapın.
NOT: Tablo 13, SNAP etiketi etiketleme reaksiyonları için reaksiyon formülleri içerir.- İki kontrol örneği daha hazırlayın: 1) BG-oligonükleotid'i ddH2O ile değiştirerek bir RNAP kontrolü; 2) SNAP T7 RNAP'ı ddH2O ile değiştirerek bir DNA kontrolü (SNAP T7 RNAP'ın en düşük oligonükleotid konsantrasyonu için). Tüm numuneleri oda sıcaklığında 1 saat kuluçkaya yatırın ve ihtiyaç duyulana kadar buzda tutun.
- 4 μL SNAP tampona ve 2 μL protein yükleme boyasına her numuneden 2 μL ekleyerek on bir 10 μL reaksiyon ayarlayın ve 10 dakika boyunca 70 °C'de ısıtın. Her numunenin 2 μL'sini% 4-12 Bis-Tris protein jeline yükleyin ve 35 dakika boyunca 200 V'ta buz üzerinde jel elektroforezi gerçekleştirin.
NOT: Tablo 14 jel yükleme örnekleri için reaksiyon formülleri içerir.- SDS'yi her biri 10 dakika süren bir çalkalayıcı üzerinde 3x su değişimi ile yıkayın. Görüntülemeden önce 15 dakika boyunca nükleik asitler için siyanin boyalı leke. 1 saat boyunca 20 mL Coomassie mavisi leke kullanarak jeli tekrar lekele. Görüntülemeden önce 1 saat (veya gece boyunca) ddH2O ile lekeyi bırakın.
NOT: Jelde, reaksiyonlardan biri en az miktarda fazla ücretsiz BG-oligonükleotid ile birlikte en bağlı polimeraz üretecektir; bu en uygun orandır.
- SDS'yi her biri 10 dakika süren bir çalkalayıcı üzerinde 3x su değişimi ile yıkayın. Görüntülemeden önce 15 dakika boyunca nükleik asitler için siyanin boyalı leke. 1 saat boyunca 20 mL Coomassie mavisi leke kullanarak jeli tekrar lekele. Görüntülemeden önce 1 saat (veya gece boyunca) ddH2O ile lekeyi bırakın.
- BG-oligonükleotid'i SNAP T7 RNAP'a hazırlayan ölçek bağlantısı için reaktifler hazırlayın. Kavrama reaksiyonlarını analitik ölçekte bulunan en uygun oran ile gerçekleştirin.
NOT: Proteini kullanılmadığında buza yerleştirerek proteinin oda sıcaklığına maruz kalmasını en aza indirin.
12. İyon değişim sütunları kullanılarak oligonükleotid bağlı SNAP-T7'nin saflaştırılması
- Burada listelenen talimatlardan sapıyorsa, tüp kurulumu için üreticinin talimatlarını izleyin. Proteinin izoelektrik noktasından daha yüksek pH'a sahip bir arıtma tamponu hazırlayın.
NOT: Bu protokoldeki örnek protein için 10 mM sodyum fosfat tamponu (pH 7) arıtma tamponu kullanılmıştır.- 50 mM Tris ve 0,5 M NaCl son konsantrasyonları içeren 1.000 μL elution tampon hazırlayın. 50 μL 1 M Tris, 100 μL 5 M NaCl ve 850 μL ddH2O karıştırın.
NOT: Tablo 15, elution arabelleği için reaksiyon formülünü içerir.
- 50 mM Tris ve 0,5 M NaCl son konsantrasyonları içeren 1.000 μL elution tampon hazırlayın. 50 μL 1 M Tris, 100 μL 5 M NaCl ve 850 μL ddH2O karıştırın.
- 2 mL santrifüj tüpüne bir sütun yerleştirin ve 15 dakika boyunca veya tüm tampon geçene kadar 2.000 × g'da arıtma tamponu ile yıkayın. Zor arabelleği atın.
- Her numuneyi 3:1 saflaştırma tamponu:numune oranında saflaştırma tamponu ile seyreltin ve numuneyi bir seferde 400 μL sütununa yükleyin. 10 dakika boyunca veya tüm arabellek geçene kadar 2.000 × g'da döndürün. Akış toplayın ve akış olaraketiketleyin.
- Sütunun ortasına 400 μL saflaştırma tamponu ekleyin. 15 dakika boyunca veya tüm arabellek geçene kadar 2.000 g'da 2.000 g'da ×. Akış toplayın ve yıkama 1olarak etiketlenin. Yıkamak için iki kez daha tekrarlayın 2 ve yıkayın 3.
- Sütunun ortasına 50 μL elution tamponu ekleyin. 5 dakika boyunca veya tüm tampon geçene kadar 2.000 × g'da döndürün. Akış toplayın ve eluate 1olarak etiketlenin. 2 eluat için iki kez daha tekrarlayın ve 3'e elüat edin.
- Havuz 1, 2 ve 3'ü elüatlar (bu toplam eluat'ıetiketle), jel için her bir elüatın küçük bir kısmını bırakır ve emiciliği 260 nm (A260) ve 280 nm 'de (A280) ölçer. Ölçümden sonra gliserol'ü 1:1 oranında ekleyin ve bir sonraki kullanıma kadar -20 °C'de saklayın.
- Toplam eluat'ı 2x depolama tamponu (~1:100) (bu ürünüetiketleyin) ile tampon değişimi yapmak için bir santrifüj filtre ünitesi (0,5 mL; 30 kDa) kullanın. A260/280'i tekrar ölçün. Gliserol'u 1:1 oranında ekleyin ve bir sonraki kullanıma kadar -20 °C'de saklayın.
- Her bir elüat yükleyin: akış, 1-3 yıkayın, toplam eluat ve ürün bir protein merdiveni ile birlikte% 4-12 Bis-Tris SDS-PAGE jel. 200 V'ta 35 dakika boyunca veya boya cephesi sona taşınana kadar çalıştırın.
13. Bağlı RNA polimeraz aktivitesinin isteğe bağlı kontrolünün gösterimi
- 25 mM Tris, 5 mM EDTA ve 25 mM magnezyum klorür (MgCl2) içeren 5x tavlama tamponu hazırlayın. Her şablonun 2,4 μL'si (1 μM) 5 μL tavlama tamponu ve 14,2 μL ddH 2 O ile karıştırarak1μM dsDNA kafesinin 25 μL'sini oluşturur. Bu çözeltiyi 2 dakika boyunca 75 °C'de kuluçkaya yatır. Benzer şekilde, organizatör ve malakit yeşil aptamer DNA şablonunun duyu ve antisense iplikçiklerini tavla. 1mM malakit yeşil oksalat çözeltisi hazırlayın.
NOT: Tablo 16, 5x tavlama arabelleği için reaksiyon formülünü içerir, Tablo 17 iki ssDNA şablonlarını tavlama için reaksiyon formülünü içerir. - Bağlı SNAP T7 RNAP'ı dsDNA kafesi ile oda sıcaklığında 15 dakika boyunca 1:5 molar oranında 500 nM RNAP'lık son konsantrasyona inkübte edin. İhtiyaç duyulana kadar buzda tutun.
- Plaka okuyucuyu önceden 37 °C'ye ısıtın. Buz üzerinde üç 25 μL IVT reaksiyon ayarlayın
- Nükleik asit transkripsiyon faktörleri ile kafesli SNAP T7RNAP içeren bir reaksiyon ayarlayın. 2,5 μL 10x IVT tampon, 1 μL 25 mM rNTP karışımı karıştırın, 1 μL 1 mM malakit yeşili, 2,5 μL RNAP kafes karışımı, 1 μM transkripsiyon faktörü A ve B oligonükleotid iplikçiklerinin her biri 2,5 μL ve 10 μL ddH2O'da 3 μL 1 mM malakit yeşil aptamer şablonu.
- Nükleik asit transkripsiyon faktörleri olmadan kafesli SNAP T7RNAP'ı içeren bir reaksiyon ayarlayın. 15 μL ddH 2 O'da 2,5 μL 10x IVT tampon, 1 μL 25 mM rNTP karışımı, 1 μL 1 mM malakit yeşili, 2,5 μL RNAP kafes karışımını ve 3 μL 1 mM malakit yeşil aptamer şablonunun15μL'si karıştırılır.
- Yalnızca arabellek içeren bir reaksiyon ayarlayın. 17,5 μL ddH 2 O'da 2,5 μL 10x IVT tampon, 1 μL25mM rNTP karışımı, 1 μL 1 mM malakit yeşili ve 3 μL 1 mM malakit yeşil aptamer şablonunun karıştırın.
NOT: Tablo 18 in vitro transkripsiyon reaksiyonları için genel bir referans içerir.
- Her reaksiyon 384 kuyu plakasına aktarın. Malakit yeşil aptamerinin 37 °C'de 2 saat boyunca ve 610 nm eksitasyon ve 655 nm emisyon ile floresan plaka okuyucusunda transkripsiyonunu izleyin. Tamamlandığında, plakayı ihtiyaç duyulana kadar buzda tutun.
- Mikrodalga 0,5x TBE tamponu 2 dakika 30 sn veya ~70 °C'ye kadar. Her kuyunun RNA ürünlerini, 280 V'ta 280 V'ta ısıtılmış 0,5x TBE tamponunda 20 dakika boyunca veya boya cephesi sona ulaşana kadar% 12 TBE-Üre poliakrilamid jelinde çalıştırın. Görüntülemeden önce bir orbital çalkalayıcıda 10 dakika boyunca siyanin boyası nükleik asit lekesi ile jeli lekelayın.
NOT: Tablo 19, %12 TBE-Üre PAGE jeli için reaksiyon formülünü içerir.
Access restricted. Please log in or start a trial to view this content.
Sonuçlar

Şekil 5: SNAP T7 RNAP ekspresyonunun SDS-PAGE analizi ve in vitro transkripsiyon tahlil. (A) SNAP T7 RNAP protein saflaştırma analizi, SNAP T7 RNAP moleküler ağırlığı: 119.4kDa. FT = sütundan akış, W1 = safsızlık içeren yıkama tamponunun elüsyon fraksiyonları, saflaştırılmış ürün içeren E1-3 = elution fraksiyonları ...
Access restricted. Please log in or start a trial to view this content.
Tartışmalar
Bu çalışma, N-terminalLY SNAP etiketli bir rekombinant T7 RNAP'ı daha sonra TMDSD reaksiyonlarını programlamak için kullanılan BG fonksiyonelleştirilmiş bir oligonükleotid ile kovalent olarak birleştirerek T7 RNA polimerazının aktivitesini kontrol etmek için DNA nanoteknolojisinden ilham alan bir yaklaşım göstermektedir. Tasarım gereği, SNAP etiketi polimerazın N-terminus'una yerleştirilmiştir, çünkü vahşi tip T7 RNAP'ın C-terminüsü protein yapısı çekirdeğine gömülüdür ve DNA şablon...
Access restricted. Please log in or start a trial to view this content.
Açıklamalar
Yazarların hiçbiri tarafından beyan edilen rakip finansal çıkarlar yoktur.
Teşekkürler
L.Y.T.C, Araştırma Fonu-Keşifte Yeni Sınırlar (NFRF-E), Kanada Doğa Bilimleri ve Mühendisliği Araştırma Konseyi (NSERC) Discovery Grant ve Kanada İlk Araştırma Mükemmellik Fonu'ndan (CFREF) fon alan Toronto Üniversitesi'nin Tasarım Girişimi tarafından cömert destek aldığını kabul ediyor.
Access restricted. Please log in or start a trial to view this content.
Malzemeler
| Name | Company | Catalog Number | Comments |
| 0.5% polysorbate 20 (TWEEN 20) | BioShop | TWN510.5 | |
| 0.5M ethylenediaminetetraacetic acid (EDTA) | Bio Basic | SD8135 | |
| 10 mM sodium phosphate buffer (pH 7) | Bio Basic | PD0435 | Tablets used to make 10 mM buffer |
| 10% ammonium persulfate (APS) | Sigma Aldrich | A3678-100G | |
| 100 kDa Amicon Ultra-15 Centrifugal Filter Unit | Fisher Scientific | UFC910008 | |
| 100% acetone | Fisher Chemical | A18P4 | |
| 100% ethanol (EtOH) | House Brand | 39752-P016-EAAN | |
| 10x in vitro transcription (IVT) buffer | New England Biolabs | B9012 | |
| 10x Tris-Borate-EDTA (TBE) buffer | Bio Basic | A0026 | |
| 1M Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Sigma Aldrich | I5502-1G | |
| 1M sodium bicarbonate buffer | Sigma Aldrich | S6014-500G | |
| 1M Tris(hydroxymethyl)aminomethane (Tris) | Sigma Aldrich | 648311-1KG | |
| 1X Tris-EDTA (TE) buffer | ThermoFisher | 12090015 | |
| 2M imidazole | Sigma Aldrich | 56750-100G | |
| 2-mercaptoethanol (BME) | Sigma Aldrich | M3148 | |
| 3M sodium acetate | Bio Basic | SRB1611 | |
| 40% acrylamide (19:1) | Bio Basic | A00062 | |
| 4x LDS protein sample loading buffer | Fisher Scientific | NP0007 | |
| 5M sodium chloride (NaCl) | Bio Basic | DB0483 | |
| 5mM dithiothreitol (DTT) | Sigma Aldrich | 43815-1G | |
| 6x gel loading dye | New England Biolabs | B7024S | |
| agarose B powder | Bio Basic | AB0014 | |
| BG-GLA-NHS | New England Biolabs | S9151S | |
| BL21 competent E. coli | Addgene | C2530H | |
| BLUeye prestained protein ladder | FroggaBio | PM007-0500 | |
| bromophenol blue | Bio Basic | BDB0001 | |
| coomassie blue (SimplyBlue SafeStain) | ThermoFisher | LC6060 | |
| cyanine dye (SYBR Gold nucleic acid gel stain) | Fisher Scientific | S11494 | |
| cyanine dye (SYBR Safe nucleic acid gel stain) | Fisher Scientific | S33102 | |
| dry dimethyl sulfoxide (DMSO) | Fisher Scientific | D12345 | |
| formamide | Sigma Aldrich | F9037-100ML | |
| glycerol | Bio Basic | GB0232 | |
| kanamycin sulfate | BioShop | KAN201.5 | |
| lysogeny broth | Sigma Aldrich | L2542-500ML | |
| malachite green oxalate | Sigma Aldrich | 2437-29-8 | |
| N,N,N'N'-Tetramethylethane-1,2-diamine (TEMED) | Sigma Aldrich | T9281-25ML | |
| NuPAGE MES SDS running buffer (20x) | Fisher Scientific | LSNP0002 | |
| NuPAGE Novex 4-12% Bis-Tris gel 1.0 mm 12-well | Life Technologies | NP0322BOX | |
| oligonucleotide (cage antisense) | IDT | N/A | TATAGTGAGTCGTATTAATTTG |
| oligonucleotide (cage sense) | IDT | N/A | TCAGTCACCTATCTGTTTCAAA TTAATACGACTCACTATA |
| oligonucleotide (malachite green aptamer antisense) | IDT | N/A | GGATCCATTCGTTACCTGGCT CTCGCCAGTCGGGATCCTATA GTGAGTCGTATTACAGTTCCAT TATCGCCGTAGTTGGTGTACT |
| oligonucleotide (malachite green aptamer sense) | IDT | N/A | TAATACGACTCACTATAGGATC CCGACTGGCGAGAGCCAGGT AACGAATGGATCC |
| oligonucleotide (Transcription Factor A) | IDT | N/A | AGTACACCAACTACGAGTGAG |
| oligonucleotide (Transcription Factor B) | IDT | N/A | TCAGTCACCTATCTGGCGATAA TGGAACTG |
| oligonucleotide with 3’ Amine modification (tether) | IDT | N/A | GCTACTCACTCAGATAGGTGAC TGA/3AmMO/ |
| Pierce strong ion exchange spin columns | Fisher Scientific | 90008 | |
| plasmid encoding SNAP T7 RNAP and kanamycin resistance genes | Genscript | N/A | custom gene insert |
| protein purification column (HisPur Ni-NTA spin column) | Fisher Scientific | 88226 | |
| rNTP mix | New England Biolabs | N0466S | |
| Roche mini quick DNA spin column | Sigma Aldrich | 11814419001 | |
| Triton X-100 | Sigma Aldrich | T8787-100ML | |
| Ultra Low Range DNA ladder | Fisher Scientific | 10597012 | |
| urea | BioShop | URE001.1 |
Referanslar
- Cherry, K. M., Qian, L. Scaling up molecular pattern recognition with DNA-based winner-take-all neural networks. Nature. 559 (7714), 370-376 (2018).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475 (7356), 368-372 (2011).
- Chen, Y. -J., et al. Programmable chemical controllers made from DNA. Nature Nanotechnology. 8 (10), 755-762 (2013).
- di Bernardo, D., Marucci, L., Menolascina, F., Siciliano, V. Predicting synthetic gene networks. Synthetic Gene Networks: Methods and Protocols. 813, 57-81 (2012).
- Xiang, Y., Dalchau, N., Wang, B. Scaling up genetic circuit design for cellular computing: advances and prospects. Natural Computing. 17 (4), 833-853 (2018).
- Gould, N., Hendy, O., Papamichail, D. Computational tools and algorithms for designing customized synthetic genes. Frontiers in Bioengineering and Biotechnology. 2, (2014).
- MacDonald, J. T., Siciliano, V. Computational sequence design with R2oDNA Designer. Mammalian Synthetic Promoters. 1651, 249-262 (2017).
- Cervantes-Salido, V. M., Jaime, O., Brizuela, C. A., Martínez-Pérez, I. M. Improving the design of sequences for DNA computing: A multiobjective evolutionary approach. Applied Soft Computing. 13 (12), 4594-4607 (2013).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry. 32 (1), 170-173 (2011).
- Fornace, M. E., Porubsky, N. J., Pierce, N. A. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synthetic Biology. 9 (10), 2665-2678 (2020).
- Wetterstrand, K. DNA sequencing costs: Data. Genome.gov. , (2020).
- Lopez, R., Wang, R., Seelig, G. A molecular multi-gene classifier for disease diagnostics. Nature Chemistry. 10 (7), 746-754 (2018).
- Pardee, K., et al. low-cost detection of Zika virus using programmable biomolecular components. Cell. 165 (5), 1255-1266 (2016).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406 (6796), 605-608 (2000).
- Lin, K. N., Volkel, K., Tuck, J. M., Keung, A. J. Dynamic and scalable DNA-based information storage. Nature Communications. 11 (1), 2981(2020).
- Yurke, B., Mills, A. P. Using DNA to power nanostructures. Genetic Programming and Evolvable Machines. 4 (2), 111-122 (2003).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318 (5853), 1121-1125 (2007).
- Wang, B., Thachuk, C., Ellington, A. D., Winfree, E., Soloveichik, D. Effective design principles for leakless strand displacement systems. Proceedings of the National Academy of Sciences. 115 (52), 12182-12191 (2018).
- Machinek, R. R. F., Ouldridge, T. E., Haley, N. E. C., Bath, J., Turberfield, A. J. Programmable energy landscapes for kinetic control of DNA strand displacement. Nature Communications. 5 (1), 5324(2014).
- Cabello-Garcia, J., Bae, W., Stan, G. -B. V., Ouldridge, T. E. Handhold-mediated strand displacement: a nucleic acid-based mechanism for generating far-from-equilibrium assemblies through templated reactions. bioRxiv. , (2020).
- Brophy, J. A. N., Voigt, C. A. Principles of genetic circuit design. Nature Methods. 11 (5), 508-520 (2014).
- Khalil, A. S., et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 150 (3), 647-658 (2012).
- Swank, Z., Laohakunakorn, N., Maerkl, S. J. Cell-free gene-regulatory network engineering with synthetic transcription factors. Proceedings of the National Academy of Sciences. 116 (13), 5892-5901 (2019).
- Howland, S. W., Tsuji, T., Gnjatic, S., Ritter, G., Old, L. J., Wittrup, K. D. Inducing efficient cross-priming using antigen-coated yeast particles. Journal of immunotherapy. 31 (7), 607(2008).
- Abil, Z., Ellefson, J. W., Gollihar, J. D., Watkins, E., Ellington, A. D. Compartmentalized partnered replication for the directed evolution of genetic parts and circuits. Nature Protocols. 12 (12), 2493-2512 (2017).
- Baugh, C., Grate, D., Wilson, C. 2.8 Å crystal structure of the malachite green aptamer11. Journal of Molecular Biology. Doudna, J. A. 301 (1), 117-128 (2000).
- Chou, L. Y. T., Shih, W. M. In vitro transcriptional regulation via nucleic acid-based transcription factors. ACS Synthetic Biology. 8 (11), 2558-2565 (2019).
- Lykke-Andersen, J., Christiansen, J. The C-terminal carboxy group of T7 RNA polymerase ensures efficient magnesium ion-dependent catalysis. Nucleic Acids Research. 26 (24), 5630-5635 (1998).
- Pu, J., Disare, M., Dickinson, B. C. Evolution of C-terminal modification tolerance in full-length and split T7 RNA Polymerase biosensors. Chembiochem. 20 (12), 1547-1553 (2019).
- Gardner, L. P., Mookhtiar, K. A., Coleman, J. E. Initiation, elongation, and processivity of carboxyl-terminal mutants of T7 RNA polymerase. Biochemistry. 36 (10), 2908-2918 (1997).
- Yin, J., Lin, A. J., Golan, D. E., Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nature Protocols. 1 (1), 280-285 (2006).
- Warden-Rothman, R., Caturegli, I., Popik, V., Tsourkas, A. Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Analytical Chemistry. 85 (22), 11090-11097 (2013).
- Zhang, W. -B., Sun, F., Tirrell, D. A., Arnold, F. H. Controlling macromolecular topology with genetically encoded SpyTag-SpyCatcher chemistry. Journal of the American Chemical Society. 135 (37), 13988-13997 (2013).
Access restricted. Please log in or start a trial to view this content.
Yeniden Basımlar ve İzinler
Bu JoVE makalesinin metnini veya resimlerini yeniden kullanma izni talebi
Izin talebiThis article has been published
Video Coming Soon
JoVE Hakkında
Telif Hakkı © 2020 MyJove Corporation. Tüm hakları saklıdır