
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
ARN polimerasa atado al ADN para transcripción in vitro programable y computación molecular
En este artículo
Resumen
Describimos la ingeniería de una nueva ARN polimerasa T7 atada al ADN para regular las reacciones de transcripción in vitro. Discutimos los pasos para la síntesis y caracterización de proteínas, validamos la regulación transcripcional de prueba de concepto y discutimos sus aplicaciones en computación molecular, diagnóstico y procesamiento de información molecular.
Resumen
La nanotecnología del ADN permite el autoensamblaje programable de ácidos nucleicos en formas y dinámicas prescritas por el usuario para diversas aplicaciones. Este trabajo demuestra que los conceptos de la nanotecnología del ADN se pueden utilizar para programar la actividad enzimática de la ARN polimerasa T7 derivada de fagos (RNAP) y construir redes reguladoras de genes sintéticos escalables. En primer lugar, un RNAP T7 atado a oligonucleótidos se diseña a través de la expresión de un RNAP marcado con SNAP N-terminal y el posterior acoplamiento químico de la etiqueta SNAP con un oligonucleótido modificado con bencilguanina (BG). A continuación, el desplazamiento de la hebra de ácido nucleico se utiliza para programar la transcripción de la polimerasa bajo demanda. Además, los conjuntos auxiliares de ácidos nucleicos se pueden utilizar como "factores de transcripción artificiales" para regular las interacciones entre el RNAP T7 programado por ADN con sus plantillas de ADN. Este mecanismo regulador de la transcripción in vitro puede implementar una variedad de comportamientos de circuitos como la lógica digital, la retroalimentación, la cascada y la multiplexación. La componibilidad de esta arquitectura reguladora de genes facilita la abstracción, estandarización y escalado del diseño. Estas características permitirán la creación rápida de prototipos de dispositivos genéticos in vitro para aplicaciones como la biodetección, la detección de enfermedades y el almacenamiento de datos.
Introducción
La computación del ADN utiliza un conjunto de oligonucleótidos diseñados como medio para el cálculo. Estos oligonucleótidos están programados con secuencias para ensamblarse dinámicamente de acuerdo con la lógica especificada por el usuario y responder a entradas específicas de ácido nucleico. En los estudios de prueba de concepto, la salida del cálculo generalmente consiste en un conjunto de oligonucleótidos marcados fluorescentemente que se pueden detectar a través de electroforesis en gel o lectores de placas de fluorescencia. En los últimos 30 años, se han demostrado circuitos computacionales de ADN cada vez más complejos, como varias cascadas de lógica digital, redes de reacción química y redes neuronales1,2,3. Para ayudar con la preparación de estos circuitos de ADN, se han utilizado modelos matemáticos para predecir la funcionalidad de los circuitos genéticos sintéticos4,5,y se han desarrollado herramientas computacionales para el diseño de secuencias de ADN ortogonales6,7,8,9,10 . En comparación con las computadoras basadas en silicio, las ventajas de las computadoras de ADN incluyen su capacidad para interactuar directamente con biomoléculas, operar en solución en ausencia de una fuente de alimentación, así como su compacidad y estabilidad generales. Con el advenimiento de la secuenciación de próxima generación, el costo de sintetizar computadoras de ADN ha estado disminuyendo durante las últimas dos décadas a un ritmo más rápido que la Ley11de Moore. Las aplicaciones de tales computadoras basadas en ADN ahora están comenzando a surgir, como para el diagnóstico de enfermedades12,13, para alimentar la biofísica molecular14y como plataformas de almacenamiento de datos15.

Figura 1: Mecanismo de desplazamiento de la cadena de ADN mediada por el dedo del pie. El punto de apoyo, δ, es una secuencia libre y sin encuadernar en un dúplex parcial. Cuando se introduce un dominio complementario (δ*) en una segunda hebra, el dominio de δ libre sirve como punto de apoyo para la hibridación, permitiendo que el resto de la hebra (ɑ*) desplace lentamente a su competidor a través de una reacción reversible de compresión / descompresión conocida como migración de hebras. A medida que aumenta la duración de δ, el ΔG para la reacción hacia adelante disminuye y el desplazamiento ocurre más fácilmente. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Hasta la fecha, la mayoría de las computadoras de ADN utilizan un motivo bien establecido en el campo de la nanotecnología dinámica del ADN conocido como desplazamiento de cadenas de ADN mediadas por dedo del pie (TMDSD, Figura 1)16. Este motivo consiste en un dúplex de ADN parcialmente de doble cadena (dsDNA) que muestra voladizos cortos de "puntera" (es decir, de 7 a 10 nucleótidos (nt)). Las hebras de "entrada" de ácido nucleico pueden interactuar con los dúplex parciales a través del punto de apoyo. Esto conduce al desplazamiento de una de las hebras del dúplex parcial, y esta hebra liberada puede servir como entrada para los dúplex parciales aguas abajo. Por lo tanto, TMDSD permite la señal en cascada y el procesamiento de información. En principio, los motivos ortogonales TMDSD pueden operar de forma independiente en solución, lo que permite el procesamiento paralelo de la información. Ha habido una serie de variaciones en la reacción de TMDSD, como el intercambio de hebras de ADN mediado por el punto de apoyo (TMDSE)17,los puntos de apoyo "sin fugas" con dominios de doble longitud18,los puntos de apoyo no coincidentes con la secuencia19y el desplazamiento de la hebra mediado por el "asidero"20. Estos principios de diseño innovadores permiten una energía y dinámica TMDSD más ajustadas para mejorar el rendimiento de la computación de ADN.
Los circuitos genéticos sintéticos, como los circuitos de genes transcripcionales, también son capaces de calcular21,22,23. Estos circuitos están regulados por factores de transcripción de proteínas, que activan o reprimen la transcripción de un gen uniéndose a elementos reguladores específicos del ADN. En comparación con los circuitos basados en ADN, los circuitos transcripcionales tienen varias ventajas. En primer lugar, la transcripción enzimática tiene una tasa de rotación mucho mayor que los circuitos de ADN catalítico existentes, generando así más copias de salida por copia única de entrada y proporcionando un medio más eficiente de amplificación de la señal. Además, los circuitos transcripcionales pueden producir diferentes moléculas funcionales, como aptámeros o ARN mensajero (ARNm) que codifican para proteínas terapéuticas, como resultados de cálculo, que pueden explotarse para diferentes aplicaciones. Sin embargo, una limitación importante de los circuitos transcripcionales actuales es su falta de escalabilidad. Esto se debe a que hay un conjunto muy limitado de factores de transcripción ortogonales basados en proteínas, y el diseño de novo de nuevos factores de transcripción de proteínas sigue siendo técnicamente desafiante y requiere mucho tiempo.

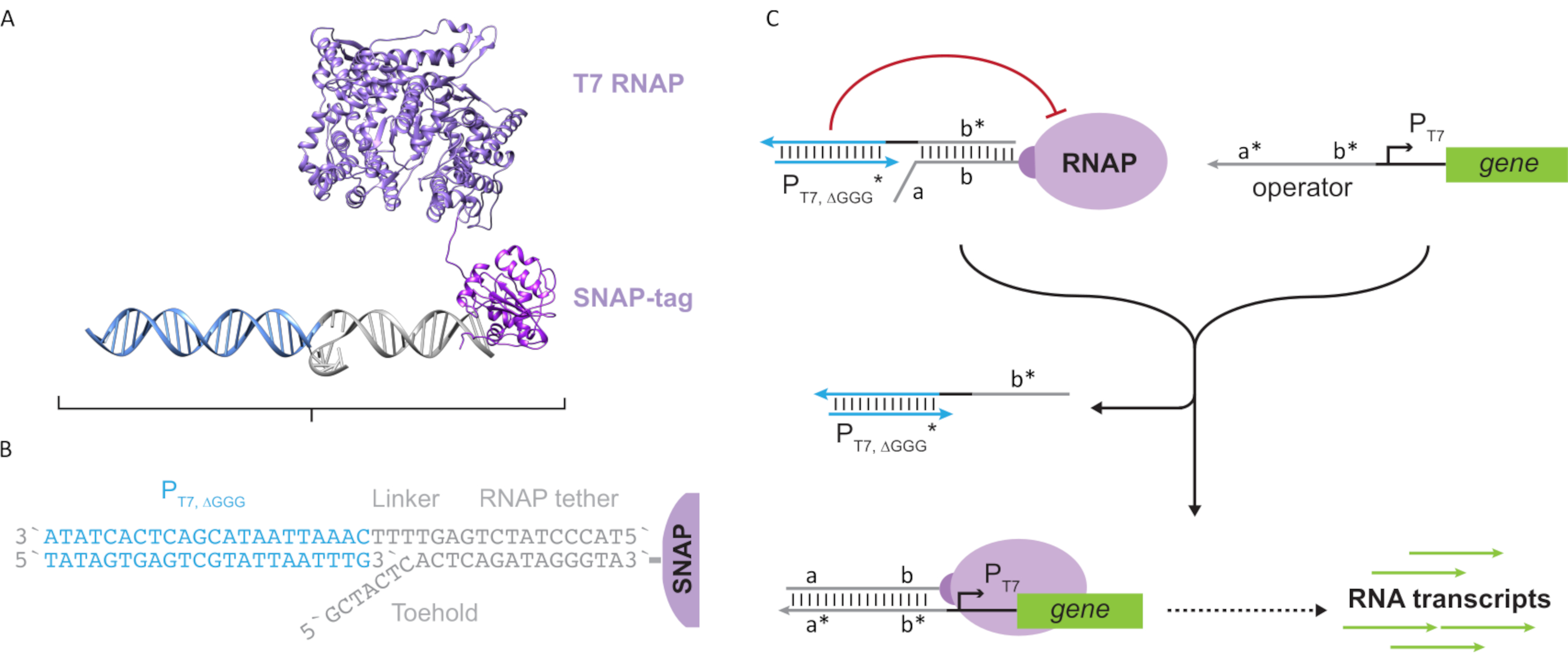
Figura 2: Abstracción y mecanismo del complejo polimerasa "tether" y "cage". (A y B) Una correa de oligonucleótidos se marca enzimáticamente a una polimerasa T7 a través de la reacción SNAP-tag. Una jaula que consiste en un promotor T7 "falso" con un voladizo de complemento de amarre le permite hibridarse con la correa y bloquear la actividad transcripcional. (C) Cuando el operador (a*b*) está presente, se une al punto de apoyo de la correa del oligonucleótido (ab) y desplaza la región b* de la jaula, permitiendo que se produzca la transcripción. Esta figura ha sido modificada de Chou y Shih27. Abreviaturas: RNAP = ARN polimerasa. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Este artículo presenta un nuevo bloque de construcción para la computación molecular que combina las funcionalidades de los circuitos transcripcionales con la escalabilidad de los circuitos basados en ADN. Este bloque de construcción es un RNAP T7 unido covalentemente con una correa de ADN de cadena simple(Figura 2A). Para sintetizar este ARNP T7 atado al ADN, la polimerasa se fusionó con un SNAP-tag24 N-terminal y se expresó recombinantemente en Escherichia coli. La etiqueta SNAP se reaccionó con un oligonucleótido funcionalizado con el sustrato BG. La correa de oligonucleótidos permite el posicionamiento de los huéspedes moleculares en estrecha proximidad a la polimerasa a través de la hibridación del ADN. Uno de esos invitados fue un bloqueador transcripcional competitivo conocido como "jaula", que consiste en un dúplex de ADN promotor T7 "falso" sin gen aguasabajo (Figura 2B). Cuando se une al RNAP a través de su correa de oligonucleótidos, la jaula detiene la actividad de la polimerasa al superar a otras plantillas de ADN para la unión al RNAP, lo que hace que el RNAP esté en un estado "OFF"(Figura 2C).
Para activar la polimerasa a un estado "ON", se diseñaron plantillas de ADN T7 con dominios "operador" monocatenarios aguas arriba del promotor T7 del gen. El dominio operador (es decir, dominio a*b* Figura 2C)puede diseñarse para desplazar la jaula del RNAP a través de TMDSD y posicionar el RNAP proximal al promotor T7 del gen, iniciando así la transcripción. Alternativamente, también se diseñaron plantillas de ADN donde la secuencia del operador era complementaria a las hebras auxiliares de ácido nucleico que se conocen como "factores de transcripción artificiales" (es decir, hebras TFA y TFB en la Figura 3A). Cuando ambas hebras se introducen en la reacción, se ensamblarán en el sitio del operador, creando un nuevo dominio pseudo-contiguo a*b*. Este dominio puede desplazar la jaula a través de TMDSD para iniciar la transcripción(Figura 3B). Estas hebras pueden ser suministradas exógenamente o producidas.

Figura 3: Programación selectiva de la actividad de la polimerasa a través de un activador de interruptor de tres componentes. (A) Cuando los factores de transcripción (TFA y TFB)están presentes, se unen al dominio del operador aguas arriba del promotor, formando una secuencia pseudo de cadena simple (a*b*) capaz de desplazar la jaula a través del desplazamiento del ADN mediado por el punto de apoyo. (B) Este dominio a*b* puede desplazar la jaula a través de TMDSD para iniciar la transcripción. Esta figura ha sido modificada de Chou y Shih27. Abreviaturas: TF = factor de transcripción; RNAP = ARN polimerasa; TMDSD = desplazamiento de la cadena de ADN mediada por el dedo del pie. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
El uso de factores de transcripción basados en ácidos nucleicos para la regulación transcripcional in vitro permite la implementación escalable de comportamientos de circuitos sofisticados como la lógica digital, la retroalimentación y la señal en cascada. Por ejemplo, se pueden construir cascadas de puertas lógicas diseñando secuencias de ácidos nucleicos de tal manera que las transcripciones de un gen aguas arriba activen un gen aguas abajo. Una aplicación que explota la cascada y la multiplexación que esta tecnología propuesta hace capaces es el desarrollo de circuitos de computación molecular más sofisticados para diagnósticos portátiles y procesamiento de datos moleculares. Además, la integración de la computación molecular y las capacidades de síntesis de ARN de novo puede permitir nuevas aplicaciones. Por ejemplo, se puede diseñar un circuito molecular para detectar uno o una combinación de ARN definidos por el usuario como ARN terapéuticos de entrada y salida o ARNm que codifican péptidos o proteínas funcionales para aplicaciones médicas en el punto de atención.
Access restricted. Please log in or start a trial to view this content.
Protocolo
1. Preparación del búfer
NOTA: La preparación del tampón de purificación de proteínas puede ocurrir en cualquier día; aquí, se hizo antes de comenzar los experimentos.
- Preparar tampón de lisis/equilibrio que contenga 50 mM de tris(hidroximetil)aminometano (Tris), 300 mM de cloruro de sodio (NaCl), 5% de glicerol y 5 mM de β-mercaptoetanol (BME), pH 8. Agregue 1.5 mL de 1M Tris, 1.8 mL de 5M NaCl, 1.5 mL de glicerol, 25.2 mL de agua desionizada (ddH2O) en un tubo de centrífuga de 50 mL, y agregue 10.5 μL de 14.2 M BME justo antes de su uso.
NOTA: Tris puede causar toxicidad aguda; por lo tanto, evite respirar su polvo y evite el contacto con la piel y los ojos. BME es tóxico y solo debe usarse en una campana extractora de humos. Es importante agregar BME en último lugar, justo antes de la resuspensión y la lisis celular. Consulte la Tabla 1 para la fórmula del tampón de lisis. - Prepare un tampón de lavado (pH 8) que contenga 50 mM Tris, 800 mM NaCl, 5% de glicerol, 5 mM de BME y 20 mM de imidazol. Agregue 1.5 mL de 1 M Tris, 4.8 mL de 5 M NaCl, 1.5 mL de glicerol y 22.2 mL de ddH2O en un tubo centrífugo de 50 mL. Justo antes de su uso, agregue 7 μL de 14.2 M BME y 200 μL de 2 M de imidazol a 20 mL de la solución anterior.
NOTA: Para prevenir la toxicidad aguda debida al imidazol, use equipo de protección personal. Es importante agregar BME e imidazol por última vez, justo antes de lavar la proteína de la columna. Consulte la Tabla 2 para la fórmula del tampón de lavado. - Prepare el tampón de elución (pH8) que contenga 50 mM Tris, 800 mM NaCl, 5% de glicerol, 5 mM de BME y 200 mM de imidazol. Agregue 0.5 mL de 1 M Tris, 1.6 mL de 5 M NaCl, 0.5 mL de glicerol y 6.4 mL de ddH2O a un tubo centrífugo de 15 mL. Justo antes de su uso, agregue 3.5 μL de 14.2 M BME y 1 mL de imidazol 2 M a 10 mL de la solución anterior.
NOTA: Es importante agregar BME e imidazol por última vez, justo antes de liberar la proteína de la columna. Consulte la Tabla 3 para la fórmula del búfer de elución. - Preparar 2x tampón de almacenamiento (para mezclar 1:1 con glicerol) que contenga 100 mM Tris, 200 mM NaCl, 40 mM BME y 2 mM de ácido etilendiaminotetraacético (EDTA), 0,2% de un tensioactivo no iónico (ver la Tabla de Materiales). Prepare 50 ml del tampón de almacenamiento agregando 5 ml de 1 M Tris, 2 ml de 5 M NaCl, 42,56 ml de ddH2O, 200 μL de 0,5 M EDTA, 100 μL del surfactante no iónico a un tubo centrífugo de 50 ml. Mezclar hasta que la solución sea homogénea, filtrar el tampón de almacenamiento a través de un filtro de jeringa de 0,2 μm y añadir 140,8 μL de BME a la solución anterior antes de su uso.
NOTA: Para evitar la toxicidad aguda debido al EDTA, evite respirar su polvo y evite el contacto con la piel y los ojos. Es importante agregar BME por último y mezclar todo el tampón de almacenamiento 1: 1 con glicerol, justo antes de almacenar la proteína purificada. Consulte la Tabla 4 para ver la fórmula del búfer de almacenamiento.
2. Crecimiento de la cultura durante la noche: Día 1
- Preparar 1.000x de kanamicina disolviendo 500 mg de kanamicina en 10 ml de ddH2O.
NOTA: Use equipo de protección personal para prevenir la toxicidad aguda debida a la kanamicina. - Añadir 20 μL del caldo de kanamicina 1.000x a 20 ml de caldo de lisogenia. Usando una punta de pipeta estéril, pinche un caldo de glicerol BL21 E. coli transformado y luego inocule el cultivo introduciendo la punta en el caldo de medios de crecimiento.

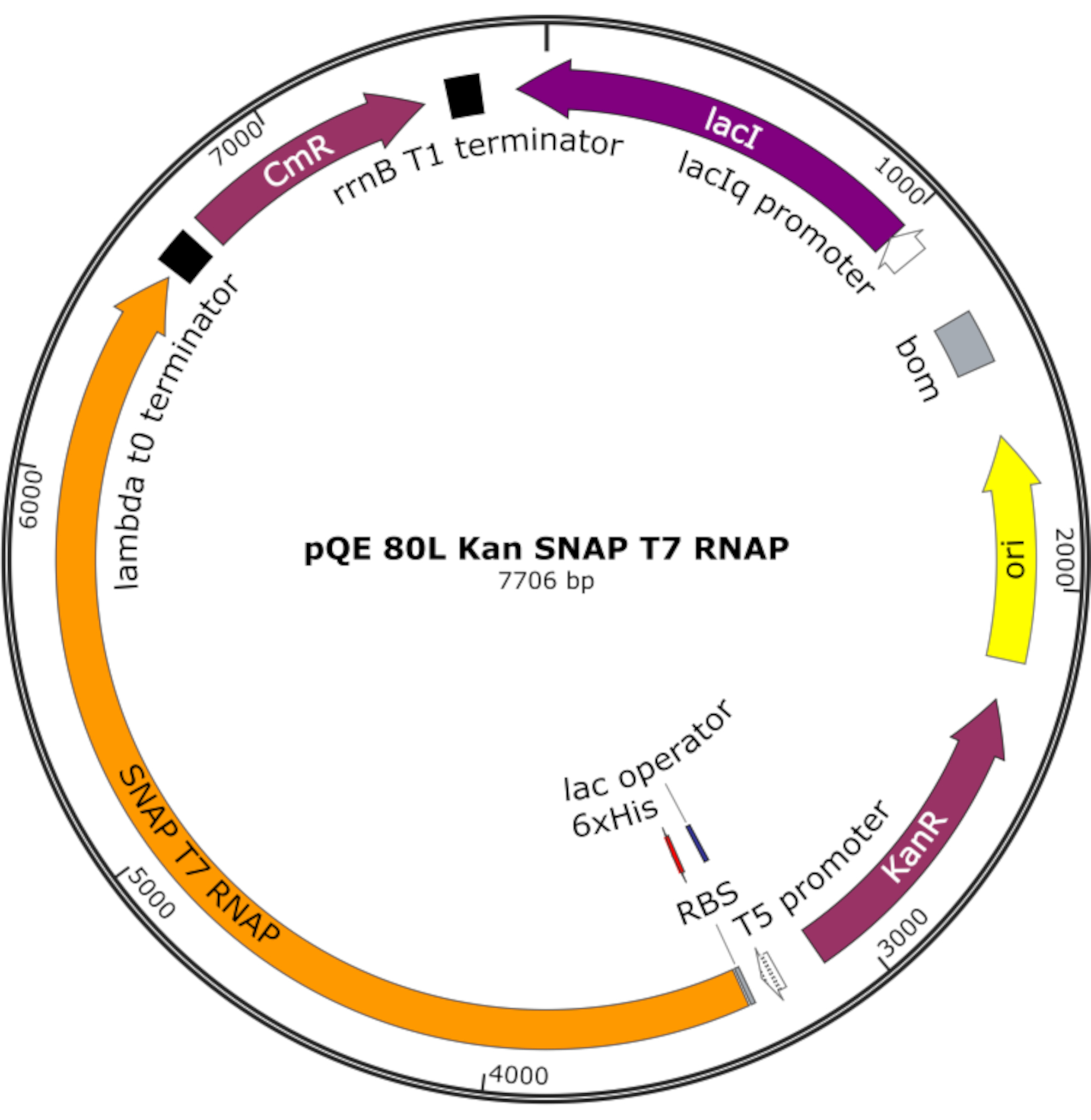
Figura 4: Mapa de plásmidos para SNAP T7 RNAP. El plásmido codifica un RNAP T7 que contiene una etiqueta de histidina N-terminal (6x His) y un dominio snap-tag (SNAP T7 RNAP) bajo un represor lac (lacI) en una columna vertebral pQE-80L. Otras características incluyen la resistencia a la kanamicina (KanR) y los genes de resistencia al cloranfenicol (CmR). Abreviatura: RNAP = ARN polimerasa. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
NOTA: El plásmido codifica un RNAP T7 que contiene una etiqueta de histidina N-terminal y un dominio SNAP-tag (SNAP T7 RNAP), así como un gen de resistencia a la kanamicina bajo una columna vertebral pQE-80L (Figura 4)25.
- Una vez más, agregue 20 μL del stock de kanamicina 1,000x a un matraz de cultivo separado que contenga 20 ml de caldo de lisogenia e incube como control.
- Incubar las dos muestras (de los pasos 2.2 y 2.3) durante la noche durante 12-18 h a 37 °C, mientras se gira a 10 × g.
3. Crecimiento celular e inducción: Día 2
- Inocular 400 ml de caldo de lisogenia que contenga 400 μL de caldo de kanamicina con 4 ml del cultivo de crecimiento durante la noche a partir del paso 2.4. Incubar los matraces de cultivo a 37 °C, girando a 10 × g.
- Una vez que el cultivo haya alcanzado una densidad óptica (OD) a 600 nm de ~0.5, saque 1 ml de muestra del matraz de crecimiento como control. Conservar la muestra de control a 4 °C.
- Inducir las células con isopropil β-D-1-tiogalactopiranósido (IPTG) añadiendo 40 μL de 1M IPTG por 100 mL de cultivo para alcanzar una concentración final de 0,4 mM IPTG. Incubar la muestra durante 3 h a 37 °C, girando a 10 × g,y luego girar el cultivo inducido a 8.000 × g durante 10 min para granular las células. Retire el sobrenadante y guarde el pellet a -20 °C hasta su uso posterior.
NOTA: Para evitar la toxicidad aguda debido a la IPTG, evite respirar su polvo y evite el contacto con la piel y los ojos. Si es necesario, puede pausar el experimento aquí y continuar al día siguiente.
4. Lisis celular, purificación de proteínas: Día 3
- Vuelva a suspender el pellet de célula almacenado con 10 ml de tampón de lisis en hielo y gire suavemente para garantizar que todo el pellet se resuspenda. Luego, pipetee 1 ml de muestra en diez tubos de 1,5 ml que se mantienen en hielo.
- Sonicar cada muestra a un ajuste de amplitud de "1", pulsado durante 2 s con un ciclo de trabajo del 50% durante un período de 30 s. Antes y después de cada muestra, limpie la punta de sonicación con etanol al 70% y ddH2O. Mantenga todas las muestras en hielo durante y después de la sonicación.
NOTA: Mantenga el 70% de etanol lejos del calor y la llama abierta. - Equilibre una columna de espín de purificación de ácido nitrilotriacético cargado de níquel (Ni-NTA) a una temperatura de trabajo de 4 °C. Coloque/guarde la columna a 4 °C y manténgala en hielo durante el uso.
- Centrifugar las diez muestras de 1 ml a 15.000 × g durante 20 min a 4 °C. Pipetee cuidadosamente el sobrenadante que contiene el RNAP recombinante sin perturbar el pellet. Si es necesario, utilice un búfer de equilibrio adicional para ajustar el volumen total a ≥ 6 ml.
- Retire suavemente la pestaña inferior de la columna de giro Ni-NTA para permitir el flujo a través de la columna. Coloque la columna en un tubo de centrífuga y manténgala en hielo.
NOTA: Utilice un tubo centrífugo de 50 ml con las columnas de centrifugado De-NTA de 3 ml. - Centrifugar la columna a 700 × g y 4 °C durante 2 min para eliminar el búfer de almacenamiento. Equilibre la columna agregando 6 ml de búfer de equilibrio a la columna. Permita que el búfer entre completamente en el lecho de resina.
- Retire el tampón de equilibrio de la columna mediante centrifugación a 700 × g y 4 °C durante 2 min. Antes de agregar el extracto de celda preparado a la columna, coloque un tapón inferior en la columna para evitar perder cualquier producto. Luego, agregue el extracto de celda a la columna y mezcle en un mezclador agitador orbital durante 30 minutos a 4 ° C.
- Retire el tapón inferior de la columna y colóquelo en un tubo de centrífuga de 50 ml etiquetado como flujo a travésde . Centrifugar la columna a 700 × g durante 2 min para recoger el flujo.
- Agregue 6 ml de tampón de lavado a la columna para lavar la resina. Centrifugar la columna a 700 × g durante 2 min para recoger la fracción en un nuevo tubo de centrífuga etiquetado como lavado 1. Repita este paso dos veces más para un total de 3 fracciones separadas, y recoja las fracciones en tubos de centrífuga separados(lavar 2 y lavar 3).
- Agregue 3 ml de tampón de elución para eliminar las proteínas marcadas con His de la resina. Centrifugar la columna a 700 × g durante 2 min para recoger la fracción en un nuevo tubo de centrífuga etiquetado como eluido 1. Repita este paso dos veces más para un total de 3 fracciones separadas, y recoja las fracciones en tubos de centrífuga separados (eluar 2 y eluir 3).
- Combine los eluidos y realice la desalinización para eliminar las sales de la solución proteica.
- Pipeta 15 mL de polisorbato 20 p/v 0,05 % sobre una unidad de filtro centrífugo de 100 kDa. Centrifugar a 4.000 × g durante 40 min y desechar el flujo.
- Use el filtro recubierto para concentrar los eluidos 1, 2 y 3 (9 ml del total de eluido de proteína + 6 ml de tampón de almacenamiento) a ~ 1,500 μL. Centrifute el filtro a 3,220 × g durante 20 minutos y lave suavemente la membrana para evitar precipitaciones.
- Diluya la muestra a 15 ml con búfer de almacenamiento. Realice un intercambio de búfer mediante el búfer de almacenamiento 1:1.000 repitiendo el paso 4.11.2 dos veces más.
- Cuantificar la proteína purificada midiendo la absorbancia de la fracción a 280 nm. Deje en blanco el espectrofotómetro con el búfer de almacenamiento (2x búfer de almacenamiento a 4 °C). Mezcle suavemente la muestra de los eluidos combinados y mida su absorbancia.
NOTA: Realice tres lecturas separadas a diluciones de 1x, 10x y 50x de la muestra de proteína para promediar y cuantificar la proteína. Diluya las muestras en el búfer de almacenamiento. - Ajuste las muestras de proteína a 100 μM utilizando un tampón de almacenamiento 2x. Diluir la muestra ajustada 1:1 en volumen con 100% glicerol. Conservar la solución proteica resultante a -80 °C.
5. Análisis de electroforesis en gel de dodecil sulfato de sodio-poliacrilamida (SDS-PAGE) del producto proteico: Día 3
- Ejecute un gel SDS-PAGE para el análisis de proteínas. Mezcle 9 μL de la muestra con 3 μL de 4 colorantes de carga de proteína de dodecil sulfato de litio (LDS). Calentar las muestras a 95 °C durante 10 min.
- Cargue las muestras en una configuración de gel Bis-Tris SDS-PAGE al 4-12%. Cargue la escalera de proteínas en el pozo 1, luego con muestras (de izquierda a derecha): flujo, lavado 1, lavado 2, lavado 3, elución 1, elución 2, elución 3 y elución total desalinizada.
NOTA: La tabla 5 contiene una tabla de carga de muestra para el gel SDS-PAGE. - Ejecute las muestras de gel cargadas en tampón de ácido etanosulfónico (MES) 2-(N-morfolino) durante 35 min a 200 V. Enjuague el gel en una bandeja limpia tres veces durante 10 minutos cada una usando 200 mL de ddH2O, con agitación suave para eliminar cualquier SDS de la matriz de gel.
NOTA: Use equipo de protección personal para evitar toxicidad aguda debido al MES. - Manche el gel con 20 ml de azul Coomassie e incube el gel durante la noche a temperatura ambiente con una agitación suave. Desmante el gel dos veces durante 1 h cada una con 200 ml de ddH2O con agitación suave en un agitador orbital.
NOTA: Lavar el gel durante un período más largo o reemplazar el agua con frecuencia mejorará la sensibilidad. Además, colocar un pañuelo de papel de limpieza de tareas delicadas doblado en el recipiente para absorber el exceso de tinte acelerará el proceso de eliminación de manchas.
6. Verificación funcional del SNAP T7 RNAP mediante transcripción in vitro
NOTA: Este protocolo utiliza la plantilla de ADN, que codifica para el aptámero fluorescente de ARN de brócoli y permite el uso de fluorescencia para monitorear la cinética de la transcripción en un lector de placas de fluorescencia.
- Establecer tres reacciones de transcripción in vitro (IVT) para comparar la actividad de SNAP T7 RNAP con RNAP T7 de tipo salvaje (WT) de una fuente comercial y un control solo tampón. Ajuste el volumen de cada reacción a 20 μL.
- Prepare la reacción SNAP T7 RNAP IVT mezclando 2 μL de tampón de transcripción 10x, 0.4 μL de mezcla de 25 mM de ribonucleósido trifosfato (rNTP), 5 μL de plantilla de ADN 500 nM, 2 μL de 500 nM SNAP T7 RNAP y 10.6 μL de ddH2O.
- Prepare la reacción WT RNAP IVT mezclando 2 μL de tampón de transcripción 10x, 0.4 μL de mezcla rNTP de 25 mM, 5 μL de plantilla de ADN 500 nM, 2 μL de RNAP WT T7 y 10.6 μL de ddH2O.
- Prepare la reacción de IVT solo con tampón mezclando 2 μL de tampón de transcripción 10x, 0.4 μL de mezcla rNTP de 25 mM, 5 μL de plantilla de ADN 500 nM y 12.6 μL de ddH2O.
NOTA: Añadir el RNAP por última vez, manteniendo las muestras en hielo hasta su introducción. La Tabla 6, la Tabla 7y la Tabla 8 contienen las fórmulas de reacción de IVT.
- Monitoree la cinética de transcripción en un lector de placas de fluorescencia durante 2 h a intervalos de 2 minutos a 37 °C utilizando una longitud de onda de excitación de 470 nm y una longitud de onda de emisión de 512 nm.
7. Preparación de oligonucleótidos modificados con BG: Día 1
- Disolver el oligonucleótido con modificación de 3'-amina en ddH2O a una concentración final de 1 mM. Etiquete este S1.
- Mezcle 25 μL de bicarbonato de sodio 1 M (NaHCO3),284 μL de 100% dimetilsulfóxido (DMSO), 125 μL de S1 (stock de oligonucleótidos) y 66 μL de 50 mM del éster BG-N-hidroxisuccinimida (NHS) (BG-GLA-NHS) diluido con DMSO, ajuste el volumen a 500 μL e incube durante la noche a temperatura ambiente a 100 × g.
NOTA: Mantenga el DMSO alejado del calor y la llama, ya que es un líquido combustible. La Tabla 9 contiene la fórmula de reacción para la conjugación de BG con el oligonucleótido.
- Mezcle 25 μL de bicarbonato de sodio 1 M (NaHCO3),284 μL de 100% dimetilsulfóxido (DMSO), 125 μL de S1 (stock de oligonucleótidos) y 66 μL de 50 mM del éster BG-N-hidroxisuccinimida (NHS) (BG-GLA-NHS) diluido con DMSO, ajuste el volumen a 500 μL e incube durante la noche a temperatura ambiente a 100 × g.
8. Precipitación de etanol/acetona del conjugado BG-oligonucleótido: Día 2
- Centrifugar el producto del paso 7.1.1. a 13.000 × g durante 5 min. Transfiera cuidadosamente el sobrenadante a un tubo fresco y deseche cualquier BG precipitado. Divida la reacción en dos alícuotas iguales de 250 μL para evitar el desbordamiento, y realice los siguientes pasos en ambas alícuotas.
- Añadir 1/10del volumen de 3 M de acetato de sodio (25 μL), seguido de 2,5 veces el volumen en etanol al 100% (625 μL). Incubar a -80 °C durante 1 h.
NOTA: Use equipo de protección personal cuando manipule tanto acetato de sodio (puede causar irritación en los ojos, la piel, el tracto digestivo y respiratorio) como el etanol (extremadamente inflamable, causa irritación al contacto). Si es necesario, haga una pausa en el experimento aquí y continúe al día siguiente. - Coloque los tubos en la centrífuga y marque el borde exterior. Centrifugar los tubos a 17.000 × g durante 30 min a 4 °C.
NOTA: El gránulo de oligonucleótido aparecerá en el borde marcado del tubo. - Sin molestar el pellet, deseche el sobrenadante. Rellene con 750 μL de etanol refrigerado al 70% y gire a 17.000 × g durante 10 min a 4 °C.
- Sin molestar el pellet, deseche el sobrenadante. Recargar con 750 μL de acetona al 100% y girar a 17.000 × g durante 10 min a 4 °C.
NOTA: Use equipo de protección personal cuando manipule acetona, ya que es extremadamente inflamable y causa irritación al contacto. - Con la tapa del tubo abierta, seque al aire durante 5 minutos para eliminar cualquier exceso de acetona a través de la evaporación. Vuelva a disolver el oligonucleótido en 250 μL de 1 tampón Tris-EDTA (TE) para producir una solución de ~850 μM de BG-oligonucleótido.
- Repita los pasos 8.2 a 8.6 y vuelva a disolver en 70 μL de 1x búfer TE. Etiquete este S2.
9. Limpieza de oligonucleótidos BG mediante cromatografía de filtración en gel
- Suspenda la matriz invirtiendo vigorosamente las columnas varias veces; retire la tapa superior y cierre la punta inferior de la columna. Coloque la columna en un tubo centrífugo de 1,5 ml y centrífique el tubo a 1.000 × g durante 1 min a temperatura ambiente. Deseche el tampón eluido y el tubo de recolección.
NOTA: Es importante prevenir la formación de vacío. Use columnas preparadas inmediatamente. - Coloque las columnas embaladas en tubos de centrífuga limpios de 1,5 ml. Agregue 300 μL de tampón 1x TE al centro del lecho de columna y centrífique a 1.000 × g durante 2 minutos para intercambiar la solución tampón. Una vez más, deseche el tampón eluido y el tubo de recolección.
- Coloque las columnas intercambiadas por búfer en tubos de centrífuga limpios de 1,5 ml. Aplicar hasta 75 μL de muestra en el centro de la cama. Girar a 1.000 × g durante 4 min.
NOTA: No moleste la cama ni toque los lados de la columna; el punto más alto del medio de gel debe apuntar hacia el rotor exterior. - Recoge el eluido del tubo de recogida, ya que contiene el ácido nucleico purificado. Para cuantificar la muestra, mida su absorbancia a 260 nm; etiquete este S3.
NOTA: Anote la longitud de la trayectoria utilizada en la medición y calcule la concentración utilizando la ley de Beer-Lambert.
10. Análisis PAGE desnaturalización del conjugado BG-oligonucleótido
- Echa un gel TRIS-borate-EDTA (TBE)-Urea PAGE al 18%. Disolver 4,8 g de UREA, 4,5 mL de acrilamida al 40% (19:1) y 1 mL de 10x TBE en 2,8 mL de ddH2O; añadir 5 μL de tetrametilendiamina (TEMED) y mezclar bien. Repetir con 100 μL de persulfato de amonio al 10% (APS). Vierta la solución en un cassette de gel vacío y permita la polimerización durante 40 min.
NOTA: Use el equipo de protección personal adecuado cuando manipule urea (causa irritación en los ojos y la piel), acrilamida (tóxica y cancerígena) y TEMED (tóxica, inflamable, corrosiva). La Tabla 10 contiene la fórmula de reacción para un gel de poliacrilamida TBE-UREA al 18%. - Microondas 500 mL de tampón TBE (0.5x) durante 2 min y 30 s o hasta ~70 °C y vierta en un aparato de gel. Preparar formamida (desnaturalizante) de carga colorante que contenga 95% de formamida + 1 mM de EDTA y azul de bromofenol. Mezcle el tinte de carga con cada muestra y cargue la mezcla en el gel de poliacrilamida.
NOTA: Use el equipo de protección personal adecuado cuando manipule formamida, ya que es cancerígena. La Tabla 11 contiene una tabla de carga de gel de muestra. - Pase el gel a 270 V durante 35 minutos, o hasta que el frente del tinte migre hacia el final. Coloque el gel en una caja de gel y manche con colorante de cianina para ácidos nucleicos durante 15 minutos a temperatura ambiente antes de obtener imágenes.
NOTA: Use el equipo de protección personal adecuado cuando manipule el tinte de cianina, ya que es combustible.
11. Conjugación de oligonucleótidos a SNAP T7 RNAP y análisis PAGE
- Preparar los reactivos para el acoplamiento a escala analítica de BG-oligonucleótido a SNAP T7 RNAP: hacer 9 diluciones de ADN monocatenario (ssDNA) oligo con ddH2O para crear relaciones oligo:RNAP que van desde 5:1 a 1:5. Diluir el stock de proteínas a 50 μM.
NOTA: Ejemplos de ratios se pueden encontrar en la Tabla 12; estas proporciones se calculan utilizando una concentración de RNAP de 50 μM. - Para cada dilución de ssDNA oligo, haga 10 μL de la mezcla de reacción que contenga 2 μL de tampón SNAP, 4 μL de oligonucleótido BG y 4 μL de SNAP T7 RNAP.
NOTA: La Tabla 13 contiene fórmulas de reacción para la reacción de etiquetado de etiquetas SNAP.- Preparar dos muestras de control más: 1) un control de RNAP reemplazando el oligonucleótido BG con ddH2O; 2) un control del ADN mediante la sustitución de SNAP T7 RNAP por ddH2O (para la concentración más baja de oligonucleótidos de SNAP T7 RNAP). Incubar todas las muestras a temperatura ambiente durante 1 h y mantenerlas en hielo hasta que sea necesario.
- Configure once reacciones de 10 μL agregando 2 μL de cada muestra a 4 μL de tampón SNAP y 2 μL de colorante de carga de proteínas, y caliente a 70 °C durante 10 min. Cargue 2 μL de cada muestra en el gel de proteína Bis-Tris al 4-12% y realice la electroforesis en gel en hielo a 200 V durante 35 min.
NOTA: La Tabla 14 contiene fórmulas de reacción para las muestras de carga de gel.- Lave SDS mediante un intercambio de agua 3x en un agitador, cada lavado dura 10 minutos cada uno. Tinción con colorante de cianina para ácidos nucleicos durante 15 minutos antes de la toma de imágenes. Vuelva a manchar el gel con 20 ml de tinción azul de Coomassie durante 1 h. Desmante con ddH2O durante 1 h (o durante la noche) antes de la toma de imágenes.
NOTA: En el gel, una de las reacciones producirá la polimerasa más atada junto con la menor cantidad de exceso de oligonucleótido BG libre; esta es la proporción óptima.
- Lave SDS mediante un intercambio de agua 3x en un agitador, cada lavado dura 10 minutos cada uno. Tinción con colorante de cianina para ácidos nucleicos durante 15 minutos antes de la toma de imágenes. Vuelva a manchar el gel con 20 ml de tinción azul de Coomassie durante 1 h. Desmante con ddH2O durante 1 h (o durante la noche) antes de la toma de imágenes.
- Preparar reactivos para el acoplamiento de escala preparativa BG-oligonucleótido a SNAP T7 RNAP. Realizar la reacción de acoplamiento con la relación óptima que se encuentra en la escala analítica.
NOTA: Minimice la exposición a la proteína a temperatura ambiente colocando la proteína en hielo cuando no esté en uso.
12. Purificación de SNAP-T7 atado a oligonucleótidos utilizando columnas de intercambio iónico
- Siga las instrucciones del fabricante para la configuración del tubo si se desvía de las instrucciones enumeradas aquí. Prepare un tampón de purificación con un pH superior al punto isoeléctrico de la proteína.
NOTA: Para la proteína de ejemplo en este protocolo, se utilizó un tampón de purificación de 10 mM de tampón de fosfato de sodio (pH 7).- Preparar 1.000 μL de tampón de elución que contenga concentraciones finales de 50 mM Tris y 0,5 M NaCl. Mezclar 50 μL de 1 M Tris, 100 μL de 5 M NaCl y 850 μL de ddH2O.
NOTA: La Tabla 15 contiene la fórmula de reacción para el búfer de elución.
- Preparar 1.000 μL de tampón de elución que contenga concentraciones finales de 50 mM Tris y 0,5 M NaCl. Mezclar 50 μL de 1 M Tris, 100 μL de 5 M NaCl y 850 μL de ddH2O.
- Coloque una columna en un tubo de centrífuga de 2 ml y lave con tampón de purificación a 2.000 × g durante 15 minutos, o hasta que se haya eluido todo el tampón. Descarte el búfer eluido.
- Diluya cada muestra con tampón de purificación a una relación tampón de purificación de 3:1:1 y cargue la muestra en la columna de 400 μL a la vez. Girar a 2.000 × g durante 10 min, o hasta que se haya eluido todo el tampón. Recoja el flujo a través y etiquételo como flujo a través.
- Agregue 400 μL de tampón de purificación en el centro de la columna. Girar a 2.000 × g durante 15 min, o hasta que se haya eluido todo el tampón. Recoge el flujo y etiquétalo como lavado 1. Repetir dos veces más para lavar 2 y lavar 3.
- Agregue 50 μL de búfer de elución en el centro de la columna. Girar a 2.000 × g durante 5 min, o hasta que se haya eluido todo el tampón. Recoja el flujo y etiquételo como eluido 1. Repita dos veces más para eluar 2 y eluir 3.
- La piscina eluye 1, 2 y 3 (etiquete este eluido total),dejando una pequeña fracción de cada eluido para el gel, y mida la absorbancia a 260 nm (A260) y 280 nm (A280). Después de la medición, agregue glicerol en una proporción de 1: 1 y guárdelo a -20 ° C hasta su uso posterior.
- Utilice una unidad de filtro centrífugo (0,5 ml; 30 kDa) para intercambiar eluido total con 2x tampón de almacenamiento (~1:100) (etiquete este producto). Vuelva a medir A260/280. Agregue glicerol en una proporción de 1: 1 y guárdelo a -20 ° C hasta su uso posterior.
- Cargue cada eluido: flujo, lavado 1-3, eluido total y producto en un gel Bis-Tris SDS-PAGE al 4-12%, junto con una escalera de proteínas. Correr a 200 V durante 35 min, o hasta que el frente del tinte migre hacia el final.
13. Demostración del control bajo demanda de la actividad de la ARN polimerasa atada
- Prepare un tampón de recocido 5x que contenga 25 mM Tris, 5 mM EDTA y 25 mM de cloruro de magnesio (MgCl2). Mezclar 2,4 μL de cada plantilla (1 μM) con 5 μL de tampón de recocido y 14,2 μL de ddH2O para formar 25 μL de jaula de dsDNA de 1 μM. Incubar esta solución a 75 °C durante 2 min. Del mismo modo, anneal el sentido y las hebras antisentido del promotor y la plantilla de ADN del aptámero verde de malaquita. Prepare una solución de 1 mM de oxalato verde de malaquita.
NOTA: La Tabla 16 contiene la fórmula de reacción para el tampón de recocido 5x, la Tabla 17 contiene la fórmula de reacción para recocir dos plantillas ssDNA. - Incubar el SNAP T7 RNAP atado con la jaula de dsDNA en una proporción molar de 1:5 a temperatura ambiente durante 15 min hasta una concentración final de 500 nM RNAP. Mantener en hielo hasta que sea necesario.
- Precaliente el lector de placas a 37 °C. Configurar tres reacciones de IVT de 25 μL en hielo
- Configure una reacción que contenga el SNAP T7RNAP enjaulado con factores de transcripción de ácidos nucleicos. Mezclar 2,5 μL de 10x tampón IVT, 1 μL de mezcla rNTP de 25 mM, 1 μL de 1 mM verde de malaquita, 2,5 μL de la mezcla RNAP-jaula, 2,5 μL cada una de las hebras de oligonucleótidos A y B de 1 μM de factor de transcripción, y 3 μL de plantilla de aptámero verde de malaquita de 1 mM en 10 μL de ddH2O.
- Configure una reacción que contenga el SNAP T7RNAP enjaulado sin factores de transcripción de ácidos nucleicos. Mezcle 2,5 μL de tampón IVT 10x, 1 μL de mezcla rNTP de 25 mM, 1 μL de 1 mM verde de malaquita, 2,5 μL de la mezcla RNAP-jaula y 3 μL de plantilla de aptámero verde de malaquita 1 mM en 15 μL de ddH2O.
- Configure una reacción que contenga solo búfer. Mezcle 2,5 μL de tampón IVT 10x, 1 μL de mezcla rNTP de 25 mM, 1 μL de 1 mM de verde malaquita y 3 μL de plantilla de aptámero verde de malaquita 1 mM en 17,5 μL de ddH2O.
NOTA: La Tabla 18 contiene una referencia general para las reacciones de transcripción in vitro.
- Transfiera cada reacción a una placa de 384 pocillos. Monitoree la transcripción del aptámero verde de malaquita en un lector de placas de fluorescencia durante 2 h a 37 °C y con excitación de 610 nm y emisión de 655 nm. Una vez terminado, mantenga el plato en hielo hasta que sea necesario.
- Microondas 0.5x TBE buffer durante 2 min 30 s o hasta ~70 °C. Ejecute los productos de ARN de cada pocillo en un gel de poliacrilamida de TBE-Urea al 12% en el tampón TBE calentado 0.5x a 280 V durante 20 min, o hasta que el frente del tinte llegue al final. Tinción del gel con tinción de ácido nucleico colorante cianina durante 10 minutos en un agitador orbitario antes de la toma de imágenes.
NOTA: La Tabla 19 contiene la fórmula de reacción para un gel PAGE deSnaturalizante al 12% de TBE-Urea.
Access restricted. Please log in or start a trial to view this content.
Resultados

Figura 5:Análisis SDS-PAGE de la expresión de SNAP T7 RNAP y ensayo de transcripción in vitro. (A) Análisis de purificación de proteínas SNAP T7 RNAP, snap T7 RNAP peso molecular: 119.4kDa. FT = flujo a través de la columna, W1 = fracciones de elución del tampón de lavado que contiene impurezas, E1-3 = fracciones de elución que cont...
Access restricted. Please log in or start a trial to view this content.
Discusión
Este estudio demuestra un enfoque inspirado en la nanotecnología del ADN para controlar la actividad de la ARN polimerasa T7 mediante el acoplamiento covalente de un RNAP T7 recombinante marcado con SNAP N-terminal con un oligonucleótido funcionalizado de BG, que posteriormente se utilizó para programar reacciones TMDSD. Por diseño, la etiqueta SNAP se colocó en el extremo N de la polimerasa, ya que el extremo C del ARNP T7 de tipo salvaje está enterrado dentro del núcleo de la estructura de la proteína y hace co...
Access restricted. Please log in or start a trial to view this content.
Divulgaciones
No hay intereses financieros en competencia que declarar por ninguno de los autores.
Agradecimientos
L.Y.T.C reconoce el generoso apoyo de New Frontiers in Research Fund-Exploration (NFRF-E), la Beca discovery del Consejo de Investigación de Ciencias Naturales e Ingeniería de Canadá (NSERC) y la Iniciativa de Medicina por Diseño de la Universidad de Toronto, que recibe fondos del Fondo de Excelencia en Investigación de Canadá First (CFREF).
Access restricted. Please log in or start a trial to view this content.
Materiales
| Name | Company | Catalog Number | Comments |
| 0.5% polysorbate 20 (TWEEN 20) | BioShop | TWN510.5 | |
| 0.5M ethylenediaminetetraacetic acid (EDTA) | Bio Basic | SD8135 | |
| 10 mM sodium phosphate buffer (pH 7) | Bio Basic | PD0435 | Tablets used to make 10 mM buffer |
| 10% ammonium persulfate (APS) | Sigma Aldrich | A3678-100G | |
| 100 kDa Amicon Ultra-15 Centrifugal Filter Unit | Fisher Scientific | UFC910008 | |
| 100% acetone | Fisher Chemical | A18P4 | |
| 100% ethanol (EtOH) | House Brand | 39752-P016-EAAN | |
| 10x in vitro transcription (IVT) buffer | New England Biolabs | B9012 | |
| 10x Tris-Borate-EDTA (TBE) buffer | Bio Basic | A0026 | |
| 1M Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Sigma Aldrich | I5502-1G | |
| 1M sodium bicarbonate buffer | Sigma Aldrich | S6014-500G | |
| 1M Tris(hydroxymethyl)aminomethane (Tris) | Sigma Aldrich | 648311-1KG | |
| 1X Tris-EDTA (TE) buffer | ThermoFisher | 12090015 | |
| 2M imidazole | Sigma Aldrich | 56750-100G | |
| 2-mercaptoethanol (BME) | Sigma Aldrich | M3148 | |
| 3M sodium acetate | Bio Basic | SRB1611 | |
| 40% acrylamide (19:1) | Bio Basic | A00062 | |
| 4x LDS protein sample loading buffer | Fisher Scientific | NP0007 | |
| 5M sodium chloride (NaCl) | Bio Basic | DB0483 | |
| 5mM dithiothreitol (DTT) | Sigma Aldrich | 43815-1G | |
| 6x gel loading dye | New England Biolabs | B7024S | |
| agarose B powder | Bio Basic | AB0014 | |
| BG-GLA-NHS | New England Biolabs | S9151S | |
| BL21 competent E. coli | Addgene | C2530H | |
| BLUeye prestained protein ladder | FroggaBio | PM007-0500 | |
| bromophenol blue | Bio Basic | BDB0001 | |
| coomassie blue (SimplyBlue SafeStain) | ThermoFisher | LC6060 | |
| cyanine dye (SYBR Gold nucleic acid gel stain) | Fisher Scientific | S11494 | |
| cyanine dye (SYBR Safe nucleic acid gel stain) | Fisher Scientific | S33102 | |
| dry dimethyl sulfoxide (DMSO) | Fisher Scientific | D12345 | |
| formamide | Sigma Aldrich | F9037-100ML | |
| glycerol | Bio Basic | GB0232 | |
| kanamycin sulfate | BioShop | KAN201.5 | |
| lysogeny broth | Sigma Aldrich | L2542-500ML | |
| malachite green oxalate | Sigma Aldrich | 2437-29-8 | |
| N,N,N'N'-Tetramethylethane-1,2-diamine (TEMED) | Sigma Aldrich | T9281-25ML | |
| NuPAGE MES SDS running buffer (20x) | Fisher Scientific | LSNP0002 | |
| NuPAGE Novex 4-12% Bis-Tris gel 1.0 mm 12-well | Life Technologies | NP0322BOX | |
| oligonucleotide (cage antisense) | IDT | N/A | TATAGTGAGTCGTATTAATTTG |
| oligonucleotide (cage sense) | IDT | N/A | TCAGTCACCTATCTGTTTCAAA TTAATACGACTCACTATA |
| oligonucleotide (malachite green aptamer antisense) | IDT | N/A | GGATCCATTCGTTACCTGGCT CTCGCCAGTCGGGATCCTATA GTGAGTCGTATTACAGTTCCAT TATCGCCGTAGTTGGTGTACT |
| oligonucleotide (malachite green aptamer sense) | IDT | N/A | TAATACGACTCACTATAGGATC CCGACTGGCGAGAGCCAGGT AACGAATGGATCC |
| oligonucleotide (Transcription Factor A) | IDT | N/A | AGTACACCAACTACGAGTGAG |
| oligonucleotide (Transcription Factor B) | IDT | N/A | TCAGTCACCTATCTGGCGATAA TGGAACTG |
| oligonucleotide with 3’ Amine modification (tether) | IDT | N/A | GCTACTCACTCAGATAGGTGAC TGA/3AmMO/ |
| Pierce strong ion exchange spin columns | Fisher Scientific | 90008 | |
| plasmid encoding SNAP T7 RNAP and kanamycin resistance genes | Genscript | N/A | custom gene insert |
| protein purification column (HisPur Ni-NTA spin column) | Fisher Scientific | 88226 | |
| rNTP mix | New England Biolabs | N0466S | |
| Roche mini quick DNA spin column | Sigma Aldrich | 11814419001 | |
| Triton X-100 | Sigma Aldrich | T8787-100ML | |
| Ultra Low Range DNA ladder | Fisher Scientific | 10597012 | |
| urea | BioShop | URE001.1 |
Referencias
- Cherry, K. M., Qian, L. Scaling up molecular pattern recognition with DNA-based winner-take-all neural networks. Nature. 559 (7714), 370-376 (2018).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475 (7356), 368-372 (2011).
- Chen, Y. -J., et al. Programmable chemical controllers made from DNA. Nature Nanotechnology. 8 (10), 755-762 (2013).
- di Bernardo, D., Marucci, L., Menolascina, F., Siciliano, V. Predicting synthetic gene networks. Synthetic Gene Networks: Methods and Protocols. 813, 57-81 (2012).
- Xiang, Y., Dalchau, N., Wang, B. Scaling up genetic circuit design for cellular computing: advances and prospects. Natural Computing. 17 (4), 833-853 (2018).
- Gould, N., Hendy, O., Papamichail, D. Computational tools and algorithms for designing customized synthetic genes. Frontiers in Bioengineering and Biotechnology. 2, (2014).
- MacDonald, J. T., Siciliano, V. Computational sequence design with R2oDNA Designer. Mammalian Synthetic Promoters. 1651, 249-262 (2017).
- Cervantes-Salido, V. M., Jaime, O., Brizuela, C. A., Martínez-Pérez, I. M. Improving the design of sequences for DNA computing: A multiobjective evolutionary approach. Applied Soft Computing. 13 (12), 4594-4607 (2013).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry. 32 (1), 170-173 (2011).
- Fornace, M. E., Porubsky, N. J., Pierce, N. A. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synthetic Biology. 9 (10), 2665-2678 (2020).
- Wetterstrand, K. DNA sequencing costs: Data. Genome.gov. , (2020).
- Lopez, R., Wang, R., Seelig, G. A molecular multi-gene classifier for disease diagnostics. Nature Chemistry. 10 (7), 746-754 (2018).
- Pardee, K., et al. low-cost detection of Zika virus using programmable biomolecular components. Cell. 165 (5), 1255-1266 (2016).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406 (6796), 605-608 (2000).
- Lin, K. N., Volkel, K., Tuck, J. M., Keung, A. J. Dynamic and scalable DNA-based information storage. Nature Communications. 11 (1), 2981(2020).
- Yurke, B., Mills, A. P. Using DNA to power nanostructures. Genetic Programming and Evolvable Machines. 4 (2), 111-122 (2003).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318 (5853), 1121-1125 (2007).
- Wang, B., Thachuk, C., Ellington, A. D., Winfree, E., Soloveichik, D. Effective design principles for leakless strand displacement systems. Proceedings of the National Academy of Sciences. 115 (52), 12182-12191 (2018).
- Machinek, R. R. F., Ouldridge, T. E., Haley, N. E. C., Bath, J., Turberfield, A. J. Programmable energy landscapes for kinetic control of DNA strand displacement. Nature Communications. 5 (1), 5324(2014).
- Cabello-Garcia, J., Bae, W., Stan, G. -B. V., Ouldridge, T. E. Handhold-mediated strand displacement: a nucleic acid-based mechanism for generating far-from-equilibrium assemblies through templated reactions. bioRxiv. , (2020).
- Brophy, J. A. N., Voigt, C. A. Principles of genetic circuit design. Nature Methods. 11 (5), 508-520 (2014).
- Khalil, A. S., et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 150 (3), 647-658 (2012).
- Swank, Z., Laohakunakorn, N., Maerkl, S. J. Cell-free gene-regulatory network engineering with synthetic transcription factors. Proceedings of the National Academy of Sciences. 116 (13), 5892-5901 (2019).
- Howland, S. W., Tsuji, T., Gnjatic, S., Ritter, G., Old, L. J., Wittrup, K. D. Inducing efficient cross-priming using antigen-coated yeast particles. Journal of immunotherapy. 31 (7), 607(2008).
- Abil, Z., Ellefson, J. W., Gollihar, J. D., Watkins, E., Ellington, A. D. Compartmentalized partnered replication for the directed evolution of genetic parts and circuits. Nature Protocols. 12 (12), 2493-2512 (2017).
- Baugh, C., Grate, D., Wilson, C. 2.8 Å crystal structure of the malachite green aptamer11. Journal of Molecular Biology. Doudna, J. A. 301 (1), 117-128 (2000).
- Chou, L. Y. T., Shih, W. M. In vitro transcriptional regulation via nucleic acid-based transcription factors. ACS Synthetic Biology. 8 (11), 2558-2565 (2019).
- Lykke-Andersen, J., Christiansen, J. The C-terminal carboxy group of T7 RNA polymerase ensures efficient magnesium ion-dependent catalysis. Nucleic Acids Research. 26 (24), 5630-5635 (1998).
- Pu, J., Disare, M., Dickinson, B. C. Evolution of C-terminal modification tolerance in full-length and split T7 RNA Polymerase biosensors. Chembiochem. 20 (12), 1547-1553 (2019).
- Gardner, L. P., Mookhtiar, K. A., Coleman, J. E. Initiation, elongation, and processivity of carboxyl-terminal mutants of T7 RNA polymerase. Biochemistry. 36 (10), 2908-2918 (1997).
- Yin, J., Lin, A. J., Golan, D. E., Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nature Protocols. 1 (1), 280-285 (2006).
- Warden-Rothman, R., Caturegli, I., Popik, V., Tsourkas, A. Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Analytical Chemistry. 85 (22), 11090-11097 (2013).
- Zhang, W. -B., Sun, F., Tirrell, D. A., Arnold, F. H. Controlling macromolecular topology with genetically encoded SpyTag-SpyCatcher chemistry. Journal of the American Chemical Society. 135 (37), 13988-13997 (2013).
Access restricted. Please log in or start a trial to view this content.
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados