
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
التنبؤ الحسابي لتفضيلات الأحماض الأمينية لمجالات ربط الببتيد متعددة الأنواع المحتملة المشاركة في تفاعلات البروتين والبروتين
In This Article
Summary
وصفنا منهجية تعتمد على تنويع التسلسل لتقدير تفضيلات الأحماض الأمينية لمواقع الارتباط متعددة الأنواع في تفاعلات البروتين والبروتين (PPIs). في هذه الاستراتيجية ، يتم إنشاء الآلاف من روابط الببتيد المحتملة وفحصها في السيليكو ، وبالتالي التغلب على بعض القيود المفروضة على الطرق التجريبية المتاحة.
Abstract
تتضمن العديد من تفاعلات البروتين والبروتين ارتباط شرائح البروتين القصيرة بمجالات ربط الببتيد. عادة ، تتطلب مثل هذه التفاعلات التعرف على الزخارف الخطية مع الحفظ المتغير. غالبا ما يساهم الجمع بين المناطق المحفوظة للغاية والأكثر تنوعا في نفس الروابط في تعدد أنواع الارتباط ، وهي خاصية مشتركة للإنزيمات وبروتينات إشارات الخلايا. يعد توصيف تفضيلات الأحماض الأمينية لمجالات ربط الببتيد أمرا مهما لتصميم وسطاء تفاعلات البروتين والبروتين (PPIs). تعد الطرق الحسابية بديلا فعالا للتقنيات التجريبية المكلفة والمرهقة في كثير من الأحيان ، مما يتيح تصميم وسطاء محتملين يمكن التحقق من صحتها لاحقا في التجارب النهائية. هنا ، وصفنا منهجية باستخدام تطبيق Pepspec لحزمة النمذجة الجزيئية Rosetta للتنبؤ بتفضيلات الأحماض الأمينية لمجالات ربط الببتيد. هذه المنهجية مفيدة عندما تكون بنية بروتين المستقبل وطبيعة ليجند الببتيد معروفة أو يمكن الاستدلال عليها. تبدأ المنهجية بمرساة مميزة جيدا من الليجند ، والتي يتم تمديدها عن طريق إضافة بقايا الأحماض الأمينية بشكل عشوائي. ثم يتم تقييم تقارب الربط للببتيدات المتولدة بهذه الطريقة عن طريق إرساء الببتيد العمود الفقري المرن من أجل اختيار الببتيدات ذات أفضل درجات الربط المتوقعة. ثم تستخدم هذه الببتيدات لحساب تفضيلات الأحماض الأمينية ولحساب مصفوفة وزن الموضع (PWM) التي يمكن استخدامها في مزيد من الدراسات اختياريا. لتوضيح تطبيق هذه المنهجية ، استخدمنا التفاعل بين الوحدات الفرعية للعامل التنظيمي للإنترفيرون البشري 5 (IRF5) ، المعروف سابقا بأنه متعدد الأنواع ولكنه يسترشد عالميا بفكرة قصيرة محفوظة تسمى pLxIS. كانت تفضيلات الأحماض الأمينية المقدرة متوافقة مع المعرفة السابقة حول سطح ربط IRF5. أظهرت المواضع التي تشغلها بقايا سيرين القابلة للفسفورية تواترا عاليا من الأسبارتات والغلوتامات ، على الأرجح لأن سلاسلها الجانبية سالبة الشحنة تشبه الفوسفوسيرين.
Introduction
غالبا ما يتضمن التفاعل بين بروتينين ربط أجزاء قصيرة من الأحماض الأمينية بمجالات ربط الببتيد ، تشبه واجهات البروتين والببتيد. غالبا ما تتمتع بروتينات المستقبلات المشاركة في مثل هذه التفاعلات بين البروتين والبروتين (PPI) بالقدرة على التعرف على مجموعة معينة من تسلسلات الرباط المتداخلة ولكن المتباينة ، وهي خاصية تعرف باسم تعدد الأنواع 1,2. يعد التعرف متعدد الأنواع سمة من سمات العديد من البروتينات الخلوية ، ولكنه ملحوظ بشكل خاص في الإنزيمات وبروتينات إشارات الخلية3. غالبا ما تحتوي البروتينات التي تتفاعل مع مواقع الارتباط متعددة الأنواع على مزيج من المناطق المحفوظة أكثر وأقل في تسلسلها4،5،6. في هذا السيناريو ، تشارك أشكال التسلسل الأكثر حفظا في التفاعلات الجزيئية الصارمة. على العكس من ذلك ، تتفاعل التسلسلات الأكثر تنوعا مع الأسطح المتساهلة بطريقة ما في موقع ربط المستقبلات. عادة ما تكون هذه الأجزاء الأقل حفظا ولكنها لا تزال ذات صلة وظيفية عبارة عن حلقات تفتقر إلى أنماط بنية ثانوية محددة أو لها تطابقات أكثر ديناميكية ، مثل تلك النموذجية للبروتينات المضطربة جوهريا7.
عادة ما يكون تحديد روابط الببتيد المحتملة لمواقع الربط هو الخطوة الأولى في تصميم الوسطاء القادرين على التدخل في PPIsالمقابلة 8. ومع ذلك ، فمن غير المحتمل في كثير من الأحيان العثور على بقايا واحدة من الأحماض الأمينية الأكثر شيوعا في معظم مواضع التسلسل في روابط مواقع الارتباط متعددة الأنواع. بدلا من ذلك ، قد يكون لهذه المواقع تفضيلات خاصة لفئة معينة من الأحماض الأمينية وفقا لخصائصها الكيميائية ، على سبيل المثال ، الأحماض الأمينية الحمضية وسالبة الشحنة مثل الأسبارتات أو الغلوتامات ، والأحماض الأمينية العطرية الضخمة مثل الفينيل ألانين أو المزيد من المخلفات الكارهة للماء مثل الأحماض الأمينية الأليفاتية ألانين ، فالين ، ليوسين أو إيزولوسين3. يمكن أن توفر العديد من الطرق التجريبية رؤى حول تفضيلات الأحماض الأمينية لمواقع ربط البروتين ، بما في ذلك التطور الموجه9 ، ومطفرات المسح متعدد الكودون10 ، والمسح الطفري العميق11. تتبع كل هذه الطرق نهج تنويع التسلسل ، والذي يعتمد على إدخال طفرات إلى الروابط الأصلية وتحليل تأثيرها على وظيفة بروتين المستقبل (انظر Bratulic and Badran12 للحصول على مراجعة شاملة). ومع ذلك ، غالبا ما تتطلب هذه الطرق مسح مكتبات التسلسل الكبيرة ، مما يجعلها أكثر تعقيدا وتكلفة وتستغرق وقتا طويلا.
الطرق الحسابية لاستنتاج تفضيلات الأحماض الأمينية لمواقع الربط متعددة الأنواع لديها القدرة على التحايل على قيود طرق المختبر الرطب. من بين هؤلاء ، يقوم نهج تنويع تسلسل السيليكو بتقييم التأثير النشط لمجموعة واسعة من بدائل الأحماض الأمينية في تسلسل الليجند كطريقة لتوصيف اللدونة الهيكلية ل PPI13. تبدأ هذه الطريقة ببنية أو نموذج ليجند الببتيد المرتبط بموقع ارتباط المستقبلات ، ثم تدخل طفرات إلى تسلسل الليجند. ثم يتم استخدام الوظائف الإحصائية وتسجيل الطاقة لتقييم تأثير هذه الطفرات على الاستقرار وتقارب الارتباط. يمكن بعد ذلك استخدام مجموعة تسلسلات الرباط الأفضل تسجيلا الناتجة عن مرحلة التقييم لحساب تفضيلات الأحماض الأمينية. هذه الاستراتيجية لديها القدرة على معالجة عدد كبير جدا من تسلسلات الليجند بطريقة فعالة. لذلك ، يمكن أن يوفر استدلالا أكثر اكتمالا واتساقا لتفضيلات الأحماض الأمينية مقارنة بتلك المحسوبة من العدد المحدود من التسلسلات التي يمكن معالجتها عادة في مناهج المختبر الرطب.
تطبيق Pepspec لمجموعة النمذجة الجزيئيةRosetta 14 هو أداة تقوم بتنويع التسلسل كخطوة أساسية في وضع تصميم الببتيد. يتطلب هذا التطبيق بنية أو نموذجا لبروتين المستقبل مع ببتيد مرتبط وصولا إلى بقايا حمض أميني واحد في الطول ، والذي يستخدم كمرساة للخطوات التالية. ثم يتم تمديد تسلسل الببتيد المرتبط (إذا لزم الأمر) وتنويعها لتوليد عدد كبير من روابط الببتيد المفترضة. ثم يتم تقييم تقارب الربط لهذه الببتيدات عن طريق إرساء الببتيد العمود الفقري المرن من أجل اختيار تلك التي لديها أفضل درجات الربط المتوقعة. على الرغم من أن الناتج الرئيسي لهذا التطبيق هو أفضل الببتيدات المرشحة المختارة في نهاية مرحلة التصميم ، إلا أنه يمكن أيضا استخدام مجموعة أكبر بكثير من الببتيدات المقبولة خلال هذه المرحلة لحساب تفضيلات الأحماض الأمينية لموقع الربط المستهدف. يتم حساب تفضيلات الأحماض الأمينية على أنها تكرار كل بقايا من الأحماض الأمينية لكل موضع من تسلسل الربيطة ممثلة إما كمصفوفة وزن موضع (PWM) أو كشعار تسلسل مرئي أكثر.
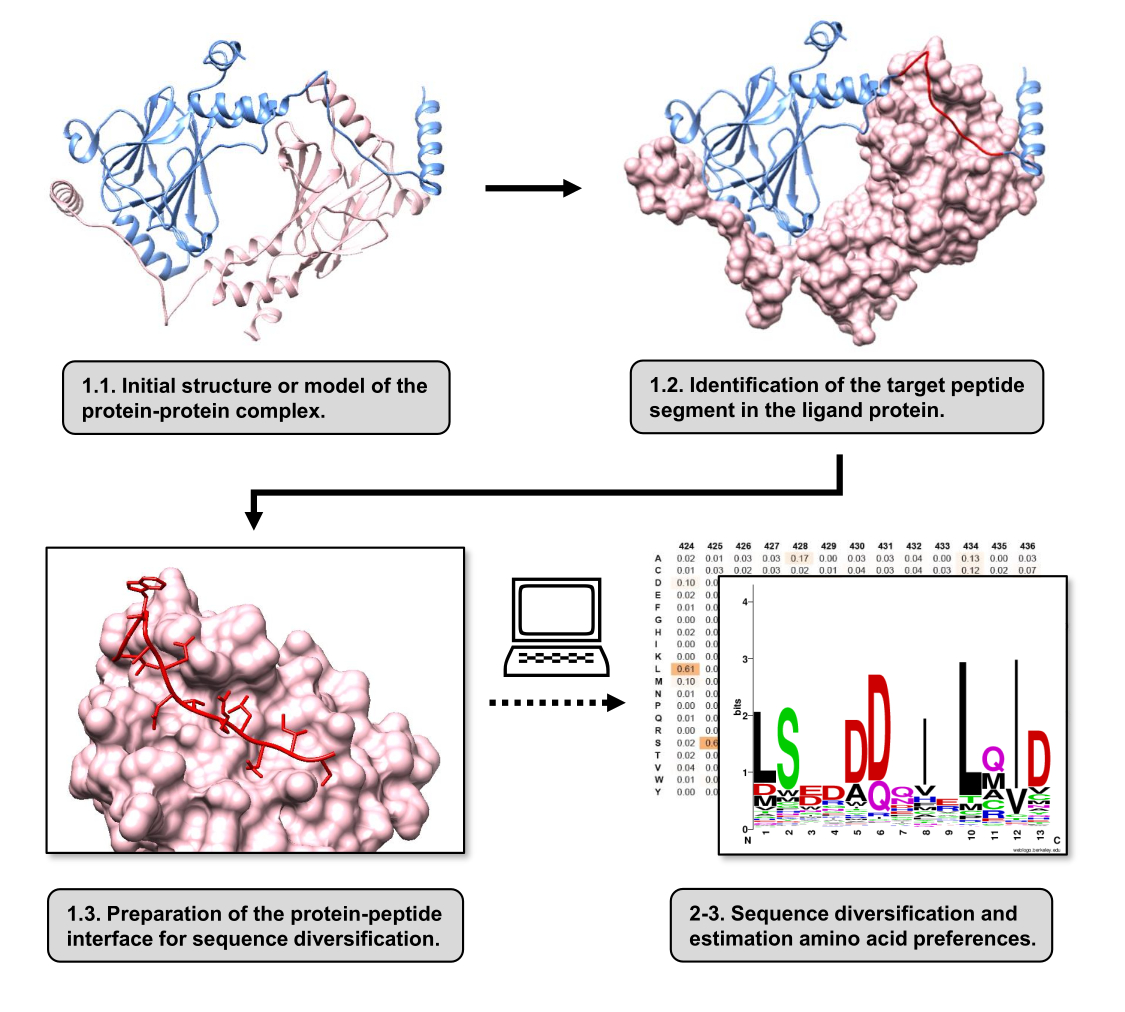
في هذه المقالة ، نصف بروتوكولا لتقدير تفضيلات الأحماض الأمينية لسطح الارتباط لبروتين مستقبلات تشارك في PPI. يركز البروتوكول على PPIs التي يعرف فيها أن الجزء الخطي من بروتين ligand يرتبط ببروتين المستقبل ، لذلك يمكن نمذجة السيناريو كواجهة بروتين-ببتيد. في هذا السيناريو ، تتفاعل الزخارف المحفوظة من الربيطة عادة مع جيوب محددة في موقع ربط المستقبلات ، على الرغم من أن جزء الربيطة بأكمله المتضمن في PPI قد يحتوي على مناطق أقل حفظا. يظهر مخطط انسيابي يلخص الخطوات الرئيسية للبروتوكول في الشكل 1. يبدأ البروتوكول بهيكل 3D لمركب البروتين البروتيني ويقلل من بروتين الليجند إلى الجزء المحتمل الأفضل تفاعلا ، تاركا بروتين المستقبل سليما. يتم الاستدلال على الجزء الأفضل تفاعلا باستخدام خادم BUDE Alanine Scan15 ، الذي يجري طفرات مسح ألانين الحسابية لتحديد بقايا النقاط الساخنة بين البروتينين المتفاعلين. في هذا النهج ، يتم استبدال المخلفات من الربيطة بشكل فردي بالألانين ، ثم يتم استخدام التغير المقدر في الطاقة الحرة أو استقرار المجمع (ΔΔG) لاستنتاج أهمية البقايا المقابلة لمؤشر أسعار المنتجين المستهدف. بمجرد استنتاج الجزء الأفضل تفاعلا ، يتم استخدام معقده مع بروتين المستقبل كهيكل أساسي مقدم إلى Pepspec لإجراء تنويع التسلسل.

الشكل 1: نظرة عامة على الخطوات الرئيسية للبروتوكول المقترح في هذا العمل. تتطابق الأرقام مع أرقام الخطوات في قسم البروتوكول. تم عمل الأرقام باستخدام مركب البروتين والبروتين المستخدم كمثال موصوف في النص. في هذا المركب ، تظهر سلسلة البروتين التي تعتبر مستقبلات باللون الوردي ، بينما تظهر السلسلة التي تعتبر بمثابة الرباط باللون الأزرق الفاتح مع تمييز الجزء الأفضل تفاعلا المتوقع باللون الأحمر. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
أحد قيود البروتوكول المقترح هو شرط وجود بنية تم حلها لواجهة البروتين والببتيد. قد يبدأ البروتوكول بدلا من ذلك بنموذج لواجهة البروتين والببتيد المستهدفة ، على الرغم من أن خطوات النمذجة المحددة غير موصوفة هنا. علاوة على ذلك ، على الرغم من أنه يمكن إجراء البروتوكول على جهاز كمبيوتر شخصي يعمل بأي نظام تشغيل ، إلا أن بيئة Linux مطلوبة للخطوات التي تتضمن تطبيقات Rosetta. يوصى بشدة أيضا باستخدام مجموعة الكمبيوتر لخطوة تنويع التسلسل نظرا للعدد الكبير من التكرارات التي تقوم بها Pepspec عادة.
يتم توضيح تطبيق البروتوكول المقترح من خلال تقدير تفضيلات الأحماض الأمينية لسطح العطاءات ل IRF5 ، وهو عضو في عائلة العامل التنظيمي للإنترفيرون البشري (IRF). اخترنا هذا البروتين كمثال لأنه أثناء تنشيطه ، ترتبط وحدتان فرعيتان لتكوين دايمر يتميز هيكله جيدا16. في ثنائيات IRF ، يمكن نمذجة الربط كواجهة بروتينية بروتينية توفر فيها وحدة فرعية سطح الربط وتتفاعل الوحدة الأخرى عبر منطقة تحتوي على شكل قصير محفوظ يسمى pLxIS17,18. بالإضافة إلى ذلك ، فإن الارتباط بالوحدات الفرعية IRF متعدد الأنواع ؛ لذلك ، يمكن أن تشكل متجانسات ، متغايرة ، ومعقدات مع البروتينات الخلوية الأخرى المعروفة باسم المنشطات18.
Protocol
1. التحضير الأولي لواجهة البروتين الببتيد
- تحميل هيكل مجمع البروتين البروتين
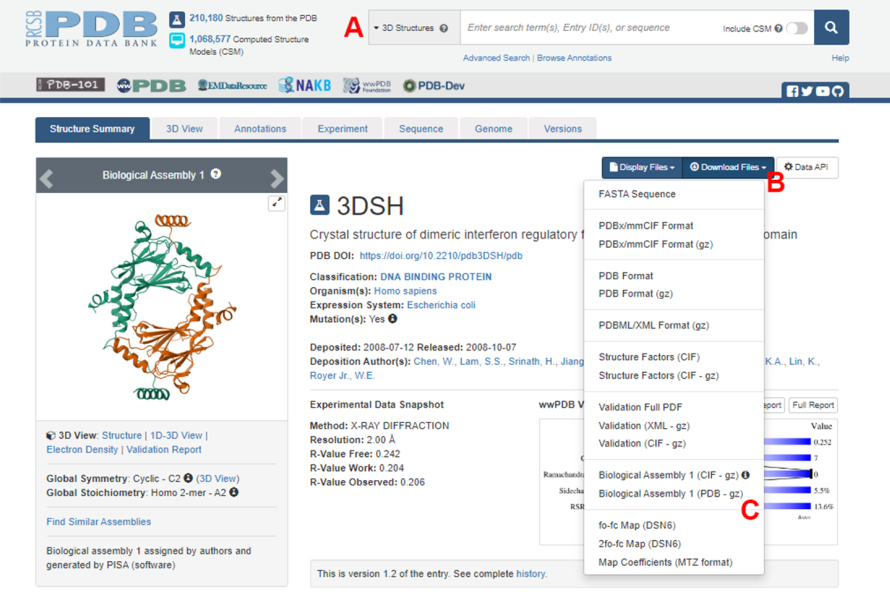
- انتقل إلى الصفحة الرئيسية لبنك بيانات البروتين (PDB) (https://www.rcsb.org/) واكتب معرف PDB لهيكل مركب البروتين والبروتين في مربع البحث الرئيسي (الشكل 2A). معرف PDB لهيكل IRF5 dimer ، المستخدم كمثال في هذا العمل ، هو 3DSH19.
- في الصفحة الرئيسية للهيكل المطلوب ، انقر فوق تنزيل الملفات (الشكل 2 ب) ثم على التجميع البيولوجي 1 (PDB - gz) (الشكل 2C).
ملاحظة: في قاعدة بيانات PDB ، يتم تمثيل هياكل العديد من معقدات البروتين المكونة من مونومرات متطابقة كتجمعات بيولوجية ، حيث يتم تخزين بنية مونومر واحد فقط (وحدة غير متماثلة) في ملف PDB. يجب تنزيل بنية المعدة ، في هذه الحالة ، IRF5 dimer ، كتجميع بيولوجي يحتوي على مثيلين من الوحدة غير المتماثلة. لتسهيل الخطوات التالية من هذا البروتوكول ، يتم فصل المونومرات أولا ، ويتم تعيين معرفات سلسلة مختلفة لهما. - افتح البنية التي تم تنزيلها في UCSF Chimera20 وانقر على أدوات > تحرير الهيكل > تغيير معرفات السلسلة. في هذا المثال ، يتم تسمية كلتا السلسلتين في التجميع البيولوجي A. أعد تسمية السلسلة الثانية (المسماة # 0.2) إلى B وانقر فوق موافق.
- انقر فوق المفضلة > لوحة النموذج ثم حدد النموذج الذي يحتوي على السلسلتين. انقر فوق الزر تجميع / فك تجميع لفصل كل سلسلة في نموذج مختلف. ثم حدد النموذجين وانقر على زر نسخ / دمج . أدخل اسما جديدا للنموذج المدمج، وتحقق من إغلاق نماذج المصدر، وانقر موافق.
- انقر فوق تحديد > السلسلة وتأكد من أن كل سلسلة في dimer يتم تحديدها الآن بحرف مختلف ، وهما A و B.
- استخدم File > Save PDB لحفظ البنية المحررة في ملف PDB مختلف، والذي سيتم استخدامه في الخطوات التالية من البروتوكول (هنا، تم استخدام الاسم IRF5_dimer.pdb ).

الشكل 2: صفحة بنك بيانات البروتين (PDB) للهيكل المستخدم كمثال تمثيلي في هذا العمل. (أ) مربع البحث لتقديم رمز الانضمام PDB للهيكل المستهدف. (ب) قائمة لتنزيل الهيكل بعدة أشكال. (ج) خيارات لتنزيل التجميعات البيولوجية عندما يتم حفظ الهيكل كوحدة غير متماثلة (انظر الخطوة 1.1.2 لمزيد من التفاصيل). يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
- تحديد الشريحة المستهدفة في بروتين الليجند
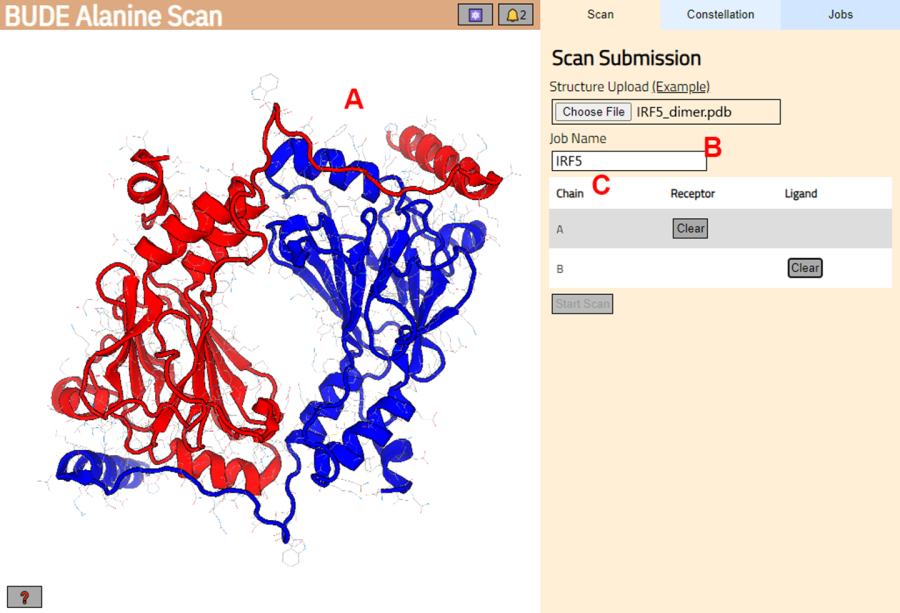
- انتقل إلى خادم BUDE Alanine Scan (https://pragmaticproteindesign.bio.ed.ac.uk/balas/). انقر فوق الزر اختيار ملف ضمن تحميل الهيكل وحدد ملف PDB المحفوظ في الخطوة 1.1.6.
- في الصفحة التالية ، تحقق من تحميل البنية بشكل صحيح (الشكل 3 أ) وأدخل اسما للوظيفة في الخادم (الشكل 3 ب).
- قم بتعيين السلاسل من PDB التي سيتم التعامل معها على أنها مستقبلات (A) و ligand (B) (الشكل 3C). ثم ، انقر فوق الزر "بدء المسح" لإرسال الوظيفة.
- بمجرد الانتهاء من المهمة ، انقر فوق إظهار النتائج لفتح صفحة النتائج (الشكل 4).
ملاحظة: في صفحة النتائج ، يتم تلوين المخلفات من بنية الليجند وفقا لتغيرها المقدر في الطاقة الحرة (ΔΔG) ، ويتم تلوين المخلفات ذات القيم الأعلى باللون الأحمر. - من قائمة المخلفات ، حدد امتداد المخلفات المتوقع أن تتفاعل بشكل أفضل مع سطح الربط المستهدف. تأكد من أن هذه المخلفات تجمع القيم الأعلى للفرق في الطاقة الحرة (ΔΔG). في هذا المثال ، تم تحديد الجزء بين البقايا Leu424 و Ser436 (مظلل بمربع أحمر في اللوحة اليمنى من الشكل 4).
- تحضير واجهة البروتين والببتيد لتنويع التسلسل
- افتح ملف PDB المحفوظ في الخطوة 1.1.6 في Chimera وتحقق من عدم وجود ذرات أو روابط مفقودة في بنية الوحدات الفرعية المستهدفة.
- احذف جميع الجزيئات الصغيرة والأيونات والمذيبات التي تبلورت مع التركيب الأصلي. للقيام بذلك ، انقر فوق تحديد > بقايا ثم حدد جميع الجزيئات بخلاف الأحماض الأمينية القياسية. ثم ، انقر فوق الإجراءات > الذرات / الروابط وحذفها.
- قم بقص سلسلة الليجند إلى الجزء الأفضل تفاعلا الذي تم اختياره في الخطوة 1.2.5. للقيام بذلك ، انقر فوق المفضلة والتسلسل ثم انقر فوق السلسلة التي تعتبر ligand (B). في لوحة التسلسل، اسحب الماوس لتحديد كل البقايا ما عدا تلك الموجودة بين المواضع 424 و436. لحذف هذه المخلفات ، انقر فوق الإجراءات > الذرات / الروابط وحذف.
- استخدم File > Save PDB لحفظ البنية المحررة في ملف PDB مختلف، والذي يتم استخدامه في الخطوات التالية للبروتوكول (هنا، تم استخدام الاسم IRF5_interface.pdb ).

الشكل 3: اختيار المستقبلات والرباط في خادم BUDE Alanine Scan. أ: تمثيل بياني لمركب البروتين والبروتين. (B) مربع نص لإدخال اسم الوظيفة في الخادم. (ج) لوحة لاختيار السلاسل التي سيتم اعتبارها مستقبلات وليجند بشكل تفاعلي (انظر الخطوة 1.2 لمزيد من التفاصيل). يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}

الشكل 4: صفحة نتائج خادم BUDE Alanine Scan. يشار إلى الجزء المحتمل الأفضل تفاعلا في تسلسل الليجند بمربع أحمر. في اللوحة اليسرى ، يتم تمييز البقايا ذات المساهمة الأعلى المتوقعة في الطاقة (Leu433) باللون الأخضر. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
2. تنويع التسلسل
ملاحظة: في الخطوات التالية ، يشير rosetta_main إلى دليل تثبيت Rosetta الرئيسي ، والذي يوجد عادة في / opt / rosetta_src__bundle / main / ، حيث يشير <الإصدار> إلى إصدار Rosetta المثبت. ومن المفترض أيضا أن تكون تطبيقات رشيد متاحة على نطاق المنظومة؛ إذا لم يكن الأمر كذلك ، فيجب توفير المسار الكامل إلى الملفات التنفيذية. عند التحويل البرمجي من المصدر ، توجد هذه الملفات التنفيذية في الدليل /rosetta_main/source/bin/ .
- التحسين الأولي للسلاسل الجانبية للأحماض الأمينية
- انسخ البنية المحررة المحفوظة في الخطوة 1.3.4 إلى موقع Linux يمكن الوصول إليه بواسطة تطبيقات Rosetta.
- استخدم تطبيق FixBB من Rosetta لإجراء إعادة حزم لجميع السلاسل الجانبية للأحماض الأمينية للهيكل الأساسي قبل تنويع التسلسل. في هذه العملية ، يتم تحسين اتجاه جميع السلاسل الجانبية للأحماض الأمينية لتقليل الطاقة وتحسين استقرار المجمع. للقيام بذلك ، قم بتشغيل الأمر التالي:

ملاحظة: ينتج هذا الأمر ملف PDB اسمه بعد البنية الأصلية مع لاحقة رقمية إضافية (IRF5_interface_0001.pdb في هذا المثال). - لتسهيل الخطوة التالية من البروتوكول، أعد تسمية ملف PDB المعاد حزمه باللاحقة _repack باستخدام الأمر التالي:
MV IRF5_interface_0001.pdb IRF5_repack.pdb
- تنويع التسلسل
- قم بتشغيل Pepspec في وضع التصميم لتنفيذ خطوة تنويع التسلسل الفعلية باستخدام الأمر التالي:

فيما يلي خيارات عامة:- -s يشير إلى ملف الإدخال (ملف PDB المعاد تعبئته الذي تم إنشاؤه في الخطوة 2.1.3).
- يشير -o إلى البادئة لتسمية ملفات الإخراج.
- - تشير قاعدة البيانات إلى المسار إلى قاعدة بيانات Rosetta 3 الرئيسية.
- -ex1 و -ex2 و extrachi_cutoff هي خيارات مكتبة rotamer (راجع وثائق Pepspec لمزيد من التفاصيل).
- -الكتابة يخبر التطبيق بالكتابة فوق المخرجات الموجودة مسبقا المحتملة التي تم إنشاؤها بواسطة التكرارات السابقة.
- -pepspec:pep_chain يشير إلى سلاسل PDB التي تعتبر ليجند ('b' في هذا المثال).
- -pepspec:native_pep_anchor يشير إلى بقايا الأحماض الأمينية المستخدمة كمرساة (في هذا المثال ، بقايا Leu في الموضع 10 من ببتيد الرباط).
- -pepspec: n_peptides يشير إلى عدد هياكل الببتيد المراد إخراجها.
- -pepspec:no_prepack_prot يخبر التطبيق بتخطي إعادة التعبئة في بنية قاعدة الإدخال (حيث تم تنفيذ ذلك مسبقا في الخطوة 2.1).
ملاحظة: إخراج Pepspec الرئيسي هو دليل يحتوي على ملفات PDB للببتيدات الناتجة عن مرحلة التصميم ، المسماة باستخدام بادئة الإخراج مع لاحقة .pdbs (IRF5.pdbs في المثال). بالإضافة إلى ذلك ، تقوم Pepspec بإخراج جميع تسلسلات الببتيد المقبولة التي تم اختبارها كجزء من خطوة تنويع التسلسل ودرجات طاقة Rosetta المقابلة لها في ملف نصي محدد بعلامات جدولة سمي على اسم بادئة الإخراج ، مع . لاحقة المواصفات (IRF5.spec في المثال). نظرا لأن البروتوكول الموصوف في هذا العمل يهدف إلى تقدير تفضيلات الأحماض الأمينية بدلا من تصميم الببتيد الفعلي ، فإن الخطوات التالية تستخدم IRF5.spec بدلا من هياكل PDB في دليل .pdbs .
- قم بتشغيل Pepspec في وضع التصميم لتنفيذ خطوة تنويع التسلسل الفعلية باستخدام الأمر التالي:
3. تقدير تفضيلات الأحماض الأمينية
- حوسبة PWM
- لإنشاء PWM ، استخدم البرنامج النصي gen_pepspec_pwm.py المضمن في مجموعة Rosetta. لتشغيل هذا البرنامج النصي، استخدم الأمر التالي:

أين:- IRF5.spec هو ملف إخراج Pepspec الذي تم إنشاؤه في الخطوة 2.2.
- -1 يشير إلى عدم وجود بقايا طرفية N إضافية في التسلسل ، وبالتالي ، فإن المواضع في PWM تستند إلى 1.
- 0.2 يخبر البرنامج النصي أن يأخذ في الاعتبار فقط أفضل 20٪ من الببتيدات الأفضل تسجيلا من إخراج Pepspec (القيمة الافتراضية هي 0.1 ، المقابلة ل 10٪)
- يخبر interface_score البرنامج النصي بترتيب الببتيدات بناء على درجة الواجهة ، وهي واحدة من درجات Rosetta المختلفة المضمنة في ملف إخراج Pepspec.
ملاحظة: يقوم هذا البرنامج النصي بإنشاء ملفين للإخراج ، أحدهما ل PWM المحسوب (مع لاحقة .pwm ) والآخر لتسلسلات المجموعة الفرعية من الببتيدات المستخدمة لحساب PWM (مع لاحقة .seq ). تتضمن أسماء هذه الملفات أيضا النتيجة وجزء الببتيدات المستخدمة في الترتيب. في هذا المثال، يتم تسمية هذه الملفات على التوالي IRF5_interface_score_0.2.pwm و IRF5_interface_score_0.2.seq.
- لإنشاء PWM ، استخدم البرنامج النصي gen_pepspec_pwm.py المضمن في مجموعة Rosetta. لتشغيل هذا البرنامج النصي، استخدم الأمر التالي:
- إنشاء شعار تسلسل
- انتقل إلى خادم WebLogo (https://weblogo.berkeley.edu/logo.cgi)21 وانقر على زر اختيار ملف بجوار تحميل بيانات التسلسل. قم بتحميل الملف بتسلسلات الببتيد التي تم إنشاؤها في الخطوة 3.1.1 (IRF5_interface_score_0.2.seq في هذا المثال).
- اختر التنسيق والحجم المطلوبين للشعار وفقا لطول الإدخال. يستخدم المثال تنسيق PDF وحجم 15 سم × 12 سم. انقر فوق إنشاء شعار.
النتائج
في هذه المقالة ، وصفنا بروتوكولا للتنبؤ بتفضيلات الأحماض الأمينية لسطح الربط ل IRF5 ، وهو عضو في عائلة من عوامل النسخ المعروفة باسم العوامل التنظيمية للإنترفيرون البشري. هذه البروتينات هي منظمات للاستجابات المناعية الفطرية والتكيفية وتشارك في تمايز وتنشيط العديد من الخ?...
Discussion
تصف هذه المقالة بروتوكولا لتقدير تفضيلات الأحماض الأمينية لمواقع الربط متعددة الأنواع المحتملة بناء على تنويع تسلسل السيليكو. تم تطوير عدد قليل من الأدوات الحسابية لتقدير تفضيلات الأحماض الأمينية لواجهات البروتين والببتيد14،25،
Disclosures
المؤلفون ليس لديهم ما يكشفون عنه.
Acknowledgements
ونعرب عن امتناننا للدعم المالي المقدم من النظام الوطني للبحوث (SNI) (أرقام المنح SNI-043-2023 و SNI-170-2021) والأمانة الوطنية للعلوم والتكنولوجيا والابتكار (SENACYT) في بنما ومعهد البحث والتحسين في إعادة تمثيل الإنسان (IFARHU). يود المؤلفون أن يشكروا الدكتور ميغيل رودريغيز على مراجعة المخطوطة بعناية.
Materials
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
References
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627 (2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267 (2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33 (2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -. M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999 (2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451 (2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved