
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
タンパク質間相互作用に関与する潜在的に多重特異的なペプチド結合ドメインのアミノ酸選好の計算予測
要約
タンパク質間相互作用(PPI)における多重特異的結合部位のアミノ酸選好性を推定するための、配列の多様化に基づく方法論について説明します。この戦略では、何千もの潜在的なペプチドリガンドが生成され、in silicoでスクリーニングされるため、利用可能な実験方法のいくつかの制限があります。
要約
多くのタンパク質間相互作用には、短いタンパク質セグメントのペプチド結合ドメインへの結合が含まれます。通常、このような相互作用には、可変保存を伴う線形モチーフの認識が必要です。同じリガンド内の高度に保存された領域とより変動しやすい領域の組み合わせは、酵素と細胞シグナル伝達タンパク質の一般的な特性である結合の多特異性に寄与することがよくあります。ペプチド結合ドメインのアミノ酸選好性の特性評価は、タンパク質間相互作用(PPI)のメディエーターの設計にとって重要です。計算法は、高価で面倒な実験手法に代わる効率的な方法であり、後で下流の実験で検証できる可能性のあるメディエーターの設計を可能にします。ここでは、Rosetta分子モデリングパッケージのPepspecアプリケーションを使用して、ペプチド結合ドメインのアミノ酸選好を予測する方法論について説明しました。この方法論は、受容体タンパク質の構造とペプチドリガンドの性質の両方が既知であるか、または推測できる場合に有用です。この方法論は、リガンドからの十分に特徴付けられたアンカーから始まり、アミノ酸残基をランダムに添加することによって拡張されます。このようにして生成されたペプチドの結合親和性は、次にフレキシブルバックボーンペプチドドッキングによって評価され、予測される最高の結合スコアを持つペプチドが選択されます。次に、これらのペプチドを使用してアミノ酸の好みを計算し、オプションでさらなる研究に使用できる位置重みマトリックス(PWM)を計算します。この方法論の適用を説明するために、以前は多重特異性であることが知られていたが、pLxISと呼ばれる短い保存されたモチーフによってグローバルに導かれたヒトインターフェロン調節因子5(IRF5)のサブユニット間の相互作用を使用しました。推定されたアミノ酸選好は、IRF5結合表面に関する以前の知識と一致していました。リン酸化可能なセリン残基が占める位置は、アスパラギン酸とグルタミン酸が高頻度で見られましたが、これはおそらく、それらの負に帯電した側鎖がホスホセリンに類似しているためです。
概要
2つのタンパク質間の相互作用には、多くの場合、アミノ酸の短いセグメントがペプチド結合ドメインに結合することが含まれ、これはタンパク質-ペプチド界面に似ています。このようなタンパク質間相互作用(PPI)に関与する受容体タンパク質は、多くの場合、重なり合っているが異なるリガンド配列の特定のセットを認識する能力を持っています。これは、多特異性1,2として知られています。多重特異性認識は多くの細胞タンパク質の特徴ですが、特に酵素や細胞シグナル伝達タンパク質で顕著です3。多重特異性結合部位と相互作用するタンパク質は、多くの場合、その配列に保存された領域と保存されていない領域の組み合わせを持っています4,5,6。このシナリオでは、より保存された配列モチーフがストリンジェントな分子相互作用に関与しています。逆に、より可変的な配列は、受容体結合部位の何らかの許容性の表面と相互作用します。通常、これらの保存性は低いが機能的に関連性のあるセグメントは、明確な二次構造パターンを欠くループであるか、または天然変性タンパク質に典型的なもののように、さらに動的なコンフォメーションを有する7。
結合部位の潜在的なペプチドリガンドの同定は、通常、対応するPPI8を妨害することができるメディエーターの設計における最初のステップである。しかし、多重特異性結合部位のリガンドのほとんどの配列位置で最も頻度の高い単一のアミノ酸残基を見つけることは、しばしば困難です。それどころか、これらの部位は、その化学的性質に応じて特定のクラスのアミノ酸、例えば、アスパラギン酸またはグルタミン酸などの酸性および負に帯電したアミノ酸、フェニルアラニンのようなかさばる芳香族アミノ酸、または脂肪族アミノ酸アラニン、バリン、ロイシンまたはイソロイシン3のようなより疎水性残基に対して特定の選好を有することがある。いくつかの実験的方法により、指向性進化9、マルチコドンスキャン突然変異誘発10、深部突然変異スキャン11など、タンパク質結合部位のアミノ酸選好に関する洞察を得ることができます。これらの方法はすべて、元のリガンドに突然変異を導入し、受容体タンパク質の機能に対するそれらの影響をさらに分析することに基づく配列多様化のアプローチに従っています(包括的なレビューについては、Bratulic and Badran12 を参照)。しかし、これらの方法では、大規模なシーケンスライブラリの調査が必要になることが多く、煩雑で、コストと時間がかかります。
多重特異性結合部位のアミノ酸選好を推測する計算法は、ウェットラボ法の限界を回避する可能性を秘めています。これらの中で、in silico配列多様化アプローチは、PPI13の構造可塑性を特徴付ける方法として、リガンド配列における幅広いアミノ酸置換のエネルギー的影響を評価する。この方法は、受容体結合部位に結合したペプチドリガンドの構造またはモデルから始まり、その後、リガンド配列に変異を導入します。次に、統計関数とエネルギースコアリング関数を使用して、これらの突然変異が安定性と結合親和性に与える影響を評価します。評価フェーズから得られたベストスコアのリガンド配列のセットは、アミノ酸選好を計算するために使用できます。この戦略は、非常に多くのリガンド配列を効率的に処理できる可能性を秘めています。したがって、ウェットラボアプローチで通常処理できるより限られた数の配列から計算されたものと比較して、アミノ酸選好のより完全で一貫した推論を提供できます。
Rosetta molecular modelingsuite 14 のPepspecアプリケーションは、ペプチド設計モードの主要なステップとして配列の多様化を実行するツールです。このアプリケーションには、結合したペプチドが単一のアミノ酸残基までの長さにまで減少する受容体タンパク質の構造またはモデルが必要であり、これは次のステップのアンカーとして使用されます。次いで、結合したペプチドの配列を(必要に応じて)伸長し、多様化して多数の推定ペプチドリガンドを生成する。次に、これらのペプチドの結合親和性をフレキシブルバックボーンペプチドドッキングによって評価し、予測された結合スコアが最も高いペプチドを選択します。このアプリケーションの主な出力は、設計フェーズの終了時に選択された最良のペプチド候補ですが、このフェーズで受け入れられるはるかに大きなペプチドのセットを使用して、ターゲット結合部位のアミノ酸選好を計算することもできます。アミノ酸選好は、リガンド配列の位置ごとの各アミノ酸残基の頻度として計算され、位置ウェイトマトリックス(PWM)またはより視覚的な配列ロゴとして表されます。
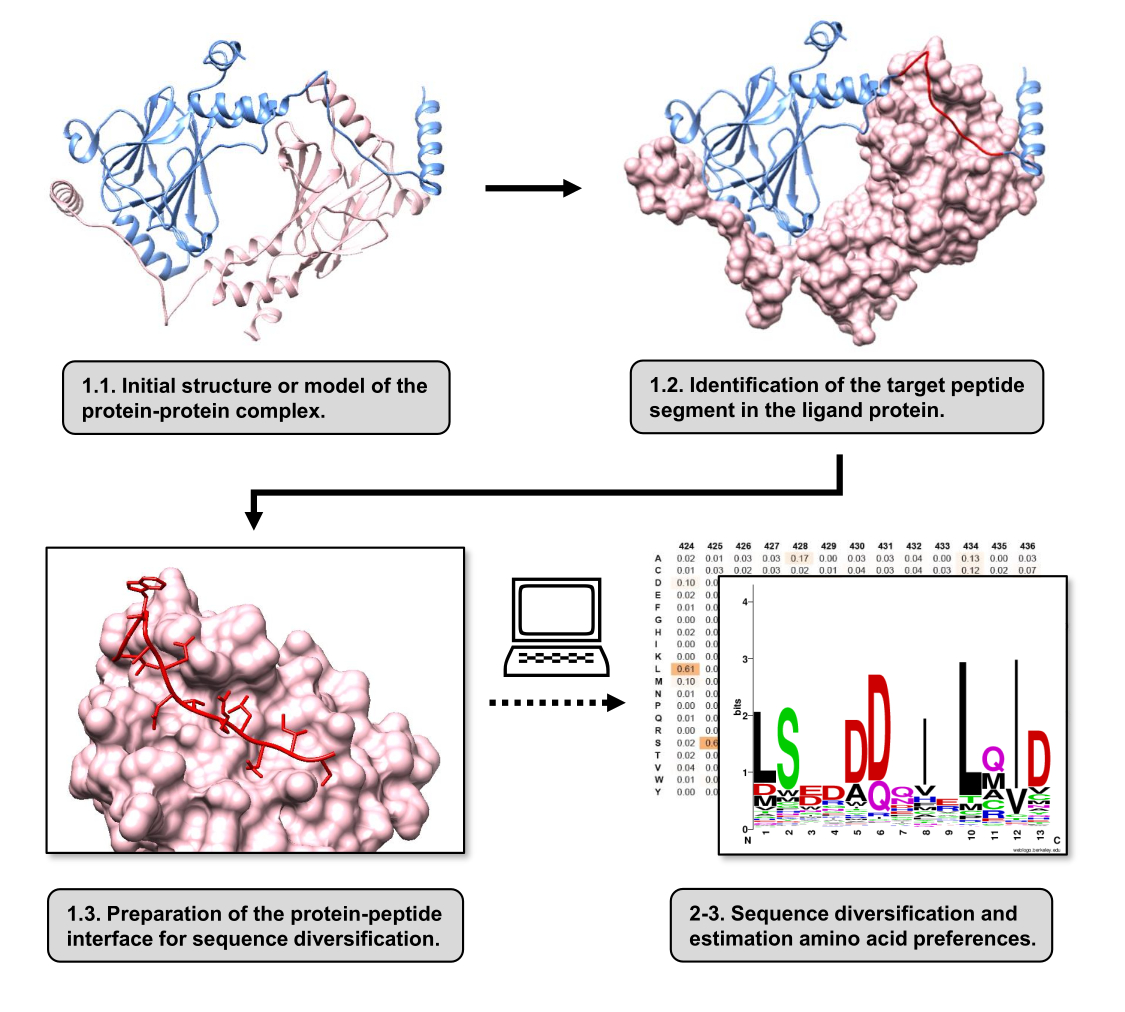
この記事では、PPIに関与する受容体タンパク質の結合表面のアミノ酸選好を推定するプロトコールについて説明します。このプロトコルは、タンパク質-リガンドの線形セグメントが受容体タンパク質に結合することが知られているPPIに焦点を当てているため、シナリオをタンパク質-ペプチドインターフェースとしてモデル化できます。このシナリオでは、リガンドから保存されたモチーフは、通常、受容体結合部位の定義されたポケットと相互作用しますが、PPIに関与するリガンドセグメント全体には、保存領域が少ない場合があります。プロトコルの主な手順をまとめたフローチャートを 図 1 に示します。このプロトコールは、タンパク質-タンパク質複合体の3D構造から始まり、さらにリガンドタンパク質を潜在的に最も相互作用するセグメントに還元し、受容体タンパク質を無傷のままにします。最も相互作用するセグメントは、2つの相互作用タンパク質間のホットスポット残基を同定するために計算アラニン走査突然変異誘発を行うBUDEアラニンスキャンサーバー15を使用することによって推論される。このアプローチでは、配位子からの残基を個々にアラニンで置換し、錯体の自由エネルギーまたは安定性(ΔΔG)の推定変化を使用して、対応する残基のターゲットPPIとの関連性を推測します。最も相互作用の大きいセグメントが推測されると、その受容体タンパク質との複合体を塩基構造としてPepspecに提出し、配列の多様化を行います。

図1:この作業で提案されたプロトコルの主なステップの概要。 番号は、プロトコル セクションのステップ番号と一致します。図は、本文に記載されている例として使用したタンパク質-タンパク質複合体を用いて作成しました。この複合体では、受容体と見なされるタンパク質鎖はピンクで示され、リガンドと見なされる鎖は水色で示され、予測される最もよく相互作用するセグメントは赤で強調表示されます。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
提案されたプロトコルの限界の1つは、タンパク質-ペプチド界面の分離された構造が必要であることです。プロトコルは、代わりに、標的タンパク質−ペプチド界面のモデルから開始してもよいが、特定のモデリングステップは本明細書では説明されていない。さらに、このプロトコルは任意のオペレーティングシステムを実行しているパーソナルコンピューターで実行できますが、Rosettaアプリケーションに関連する手順にはLinux環境が必要です。また、Pepspecでは通常、反復回数が多いため、シーケンスの多様化ステップにはコンピュータークラスターを強くお勧めします。
提案されたプロトコルの適用は、ヒトインターフェロン調節因子(IRF)ファミリーのメンバーであるIRF5の入札表面のアミノ酸選好の推定で示されています。このタンパク質を例として選んだのは、その活性化中に2つのサブユニットが結合して、その構造が十分に特徴付けられている二量体を形成するためです16。IRF二量体では、結合は、一方のサブユニットが結合表面を提供し、もう一方のサブユニットがpLxISと呼ばれる短い保存されたモチーフを含む領域を介して相互作用するタンパク質-ペプチド界面としてモデル化できます17,18。さらに、IRFサブユニットへの結合は多重特異です。したがって、それらはホモ二量体、ヘテロ二量体、およびコアクチベーターとして知られる他の細胞タンパク質との複合体を形成することができる18。
プロトコル
1. タンパク質-ペプチド界面の初期調製
- タンパク質-タンパク質複合体の構造のダウンロード
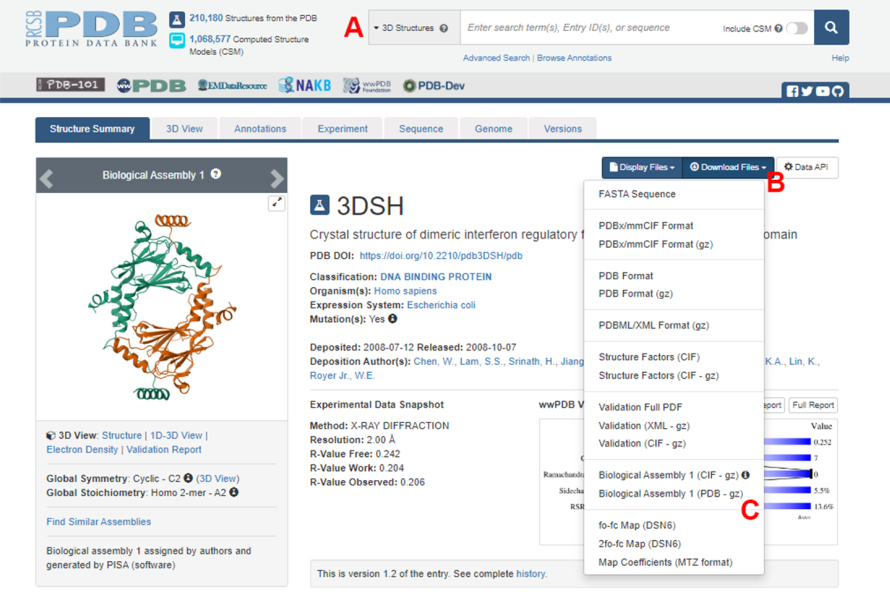
- Protein Data Bank(PDB)のホームページ(https://www.rcsb.org/)に移動し、メインの検索ボックスにタンパク質-タンパク質複合体の構造のPDB IDを入力します(図2A)。この作業で例として使用したIRF5ダイマーの構造のPDB IDは3DSH19です。
- 目的の構造のメインページで、Download Files(図2B)をクリックし、次にBiological Assembly 1(PDB - gz)(図2C)をクリックします。
注:PDBデータベースでは、同一のモノマーによって形成された多くのタンパク質複合体の構造が生物学的集合体として表され、1つのモノマー(非対称ユニット)の構造のみがPDBファイルに格納されます。マルチマーの構造、この場合はIRF5ダイマーは、非対称ユニットの2つのインスタンスを含む生物学的アセンブリとしてダウンロードする必要があります。このプロトコルの次のステップを容易にするために、まず2つのモノマーを分離し、それらに異なる鎖IDを割り当てます。 - ダウンロードしたストラクチャーをUCSF Chimera20 で開き、[ Tools (ツール)] > [Structure Editing (ストラクチャー編集)]> [Change Chain IDs] をクリックします。この例では、生物学的アセンブリの両方のチェーンに A という名前が付けられています。2 番目のチェーン ( #0.2 とラベル付け) の名前を B に変更し、[ OK] をクリックします。
- [お気に入り]>[モデルパネル]をクリックし、2つのチェーンを含むモデルを選択します。[グループ化/グループ解除]ボタンをクリックして、各チェーンを異なるモデルに分割します。次に、2つのモデルを選択し、[コピー/結合]ボタンをクリックします。結合されたモデルの新しい名前を入力し、[Close Source Models] をオンにして [OK] をクリックします。
- Select > Chainをクリックし、ダイマーの各チェーンが異なる文字、つまりAとBで識別されていることを確認します。
- 「File > Save PDB」を使用して、編集した構造を別のPDBファイルに保存します。これは、プロトコルの次のステップで使用されます(ここでは、IRF5_dimer.pdbという名前が使用されました)。

図2:本研究で代表例として使用した構造のProtein Data Bank(PDB)ページ(A)ターゲット構造のPDBアクセッションコードを紹介する検索ボックス。(B)構造をいくつかの形式でダウンロードするためのメニュー。(C)構造が非対称ユニットとして保存されている場合に生物学的アセンブリをダウンロードするオプション(詳細については、ステップ1.1.2を参照)。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
- リガンドタンパク質の標的セグメントの同定
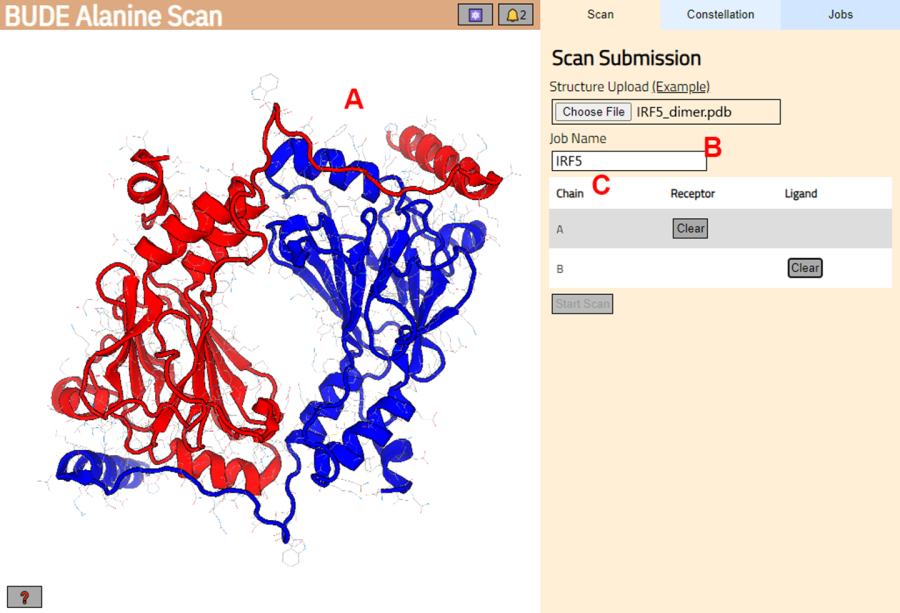
- BUDE Alanine Scanサーバー(https://pragmaticproteindesign.bio.ed.ac.uk/balas/)に移動します。「Structure Upload」の下の「Choose File」ボタンをクリックし、ステップ1.1.6で保存したPDBファイルを選択します。
- 次のページで、構造体が正しくロードされたことを確認し (図 3A)、サーバー内のジョブの名前を入力します (図 3B)。
- 受容体(A)およびリガンド(B)として扱われるPDBからの鎖を設定します(図3C)。次に、「 スキャンの開始 」ボタンをクリックしてジョブを送信します。
- ジョブが終了したら、[ Show Results ]をクリックして結果ページを開きます(図4)。
注:結果ページでは、リガンド構造からの残基は、自由エネルギーの推定変化(ΔΔG)に従って色分けされ、値が高いものは赤で色付けされています。 - 残基リストから、ターゲット結合表面とよりよく相互作用すると予測される残基のストレッチを選択します。これらの残渣が、自由エネルギーの差(ΔΔG)の値が高いほどクラスター化されるようにします。この例では、残基Leu424とSer436の間のセグメントが選択されています( 図4の右側のパネルで赤いボックスで強調表示されています)。
- 配列多様化のためのタンパク質-ペプチド界面の準備
- 手順1.1.6で保存したPDBファイルをChimeraで開き、ターゲットサブユニットの構造に欠落している原子や結合がないことを確認します。
- 元の構造と共結晶化したすべての小分子、イオン、および溶媒を削除します。これを行うには、[ Select > Residues ]をクリックし、標準アミノ酸以外のすべての分子を選択します。次に、「Actions」 >「Atoms/Bonds 」と 「Delete」をクリックします。
- リガンド鎖を、ステップ1.2.5で選択した最も相互作用するセグメントにトリミングします。これを行うには、 Favorites and Sequence をクリックし、次にリガンド(B)と見なされるチェーンをクリックします。 [Sequence ] パネルで、マウスをドラッグして、位置 424 と 436 の間の残基を除くすべての残基を選択します。これらの残基を削除するには、「 Actions」>「Atoms/Bonds 」および 「Delete」をクリックします。
- 「File > Save PDB」を使用して、編集した構造を別のPDBファイルに保存します。これは、プロトコルの次のステップで使用されます(ここでは、IRF5_interface.pdbという名前が使用されました)。

図3:BUDE Alanine Scanサーバーにおける受容体とリガンドの選択 (A)タンパク質-タンパク質複合体のグラフィック表現。(B) サーバー上のジョブの名前を入力するためのテキストボックス。(C)受容体およびリガンドとして考慮される鎖をインタラクティブに選択するパネル(詳細については、ステップ1.2を参照)。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図4:BUDE Alanine Scanサーバーの結果ページ リガンド配列内で最も相互作用しやすいセグメントは、赤いボックスで示されます。左側のパネルでは、予測エネルギー寄与度が高い残基(Leu433)が緑色で強調表示されています。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
2. シーケンスの多様化
注: 次の手順では、 rosetta_main はメインの Rosetta インストール ディレクトリを指し、通常は /opt/rosetta_src__bundle/main/ にあります。ここで 、 はインストールされている Rosetta のバージョンを示します。また、Rosetta アプリケーションはシステム全体でアクセス可能であることを前提としています。そうでない場合は、実行可能ファイルへのフルパスを指定する必要があります。ソースからコンパイルすると、これらの実行可能ファイルは /rosetta_main/source/bin/ ディレクトリにあります。
- アミノ酸側鎖の初期最適化
- 手順 1.3.4 で保存した編集済み構造を、Rosetta アプリケーションからアクセス可能な Linux の場所にコピーします。
- Rosetta の FixBB アプリケーションを使用して、塩基構造のすべてのアミノ酸側鎖の再パックを配列多様化します。この操作では、すべてのアミノ酸側鎖の配向が最適化され、エネルギーが最小限に抑えられ、複合体の安定性が向上します。これを行うには、次のコマンドを実行します。

注 : このコマンドは、元の構造体にちなんで名付けられた PDB ファイルに、数値サフィックス (この例では IRF5_interface_0001.pdb ) を付加して出力します。 - プロトコルの次のステップを容易にするために、次のコマンドを使用して、再パックされた PDB ファイルの名前を _repack サフィックスに変更します。
MV IRF5_interface_0001.pdb IRF5_repack.pdb
- シーケンスの多様化
- Pepspecをデザインモードで実行し、次のコマンドを使用して実際のシーケンス多様化ステップを実行します。

一般的なオプションは次のとおりです。- -s は、入力ファイル (ステップ 2.1.3 で生成された再パックされた PDB ファイル) を示します。
- -o は、出力ファイルに名前を付ける接頭部を示します。
- - database は、メインの Rosetta 3 データベースへのパスを示します。
- -ex1、-ex2、および extrachi_cutoff は rotamer ライブラリオプションです (詳細については Pepspec のドキュメントを参照してください)。
- -overwrite は、前のイテレーションによって生成された既存の出力を上書きするようにアプリケーションに指示します。
- -pepspec:pep_chain は、リガンドと見なされる PDB 鎖を示します (この例では 'b')。
- -pepspec:native_pep_anchor はアンカーとして使用されるアミノ酸残基を示します (この例では、リガンドペプチドの 10 位の Leu 残基)。
- -pepspec:n_peptides は出力するペプチド構造の数を示します。
- -pepspec:no_prepack_prot は、入力ベース構造での再パックをスキップするようにアプリケーションに指示します (これは以前にステップ 2.1 で実行されていたため)。
注:メインのPepspec出力は、デザインフェーズから得られたペプチドのPDBファイルを含むディレクトリであり、出力プレフィックスに.pdbsサフィックスを付けて名前が付けられています(この例ではIRF5.pdbs)。さらに、Pepspec は、配列多様化ステップの一部としてテストされたすべての受け入れられたペプチド配列と、それに対応する Rosetta エネルギースコアを、出力プレフィックスにちなんで名付けられたタブ区切りのテキストファイルに出力します。spec サフィックス (この例では IRF5.spec)。この研究で記述されているプロトコルは、実際のペプチド設計ではなくアミノ酸の好みを推定することを目的としているため、次のステップでは、.pdbsディレクトリ内のPDB構造の代わりにIRF5.specを使用する。
- Pepspecをデザインモードで実行し、次のコマンドを使用して実際のシーケンス多様化ステップを実行します。
3. アミノ酸選好の推定
- PWM の計算
- PWM を生成するには、Rosetta スイートに含まれている gen_pepspec_pwm.py スクリプトを使用します。このスクリプトを実行するには、次のコマンドを使用します。

どこ:- IRF5.spec は、手順 2.2 で生成された Pepspec 出力ファイルです。
- -1 は、シーケンスに追加の N 末端残基がないため、PWM 内の位置が 1 から始まることを示します。
- 0.2 は、スクリプトに対して、Pepspec 出力の上位 20% の最高スコアのペプチドのみを考慮するように指示します (デフォルト値は 0.1 で、10% に対応します)。
- interface_score 、Pepspec 出力ファイルに含まれるさまざまな Rosetta スコアの 1 つであるインターフェイス スコアに基づいてペプチドをランク付けするようにスクリプトに指示します。
注:このスクリプトは、計算されたPWM( .pwm サフィックス付き)用と、PWMの計算に使用されるペプチドのサブセットのシーケンス( .seq サフィックス付き)の2つの出力ファイルを生成します。これらのファイルの名前には、スコアとランキングに使用されたペプチドの割合も含まれています。この例では、これらのファイルの名前はそれぞれ IRF5_interface_score_0.2.pwm と IRF5_interface_score_0.2.seq です。
- PWM を生成するには、Rosetta スイートに含まれている gen_pepspec_pwm.py スクリプトを使用します。このスクリプトを実行するには、次のコマンドを使用します。
- シーケンスロゴの生成
- WebLogoサーバー(https://weblogo.berkeley.edu/logo.cgi)21に移動し、[Upload Sequence Data]の横にある[Choose File]ボタンをクリックします。ステップ3.1.1で生成したペプチド配列(この例ではIRF5_interface_score_0.2.seq)を含むファイルをアップロードします。
- 入力の長さに応じて、ロゴの形式とサイズを選択します。この例では、PDF 形式で 15 cm x 12 cm のサイズを使用しています。 「ロゴを作成」をクリックします。
結果
この記事では、ヒトインターフェロン調節因子として知られる転写因子ファミリーのメンバーであるIRF5の結合表面のアミノ酸選好を予測するためのプロトコルについて説明しました。これらのタンパク質は、自然免疫応答と適応免疫応答の調節因子であり、いくつかの免疫細胞の分化と活性化に関与しています。IRFサブユニットは、高度に可塑性で多重特異な結合?...
ディスカッション
本論文は、in silico配列の多様化に基づいて、潜在的に多重特異的な結合部位のアミノ酸選好を推定するためのプロトコルについて述べる。タンパク質-ペプチド界面のアミノ酸選好を推定するための計算ツールはほとんど開発されていない14,25,26。これらのツールには予測的な性質がありますが、?...
開示事項
著者は何も開示していません。
謝辞
Sistema Nacional de Investigación (SNI) (助成金番号 SNI-043-2023 および SNI-170-2021)、Secretaría Nacional de Ciencia, Tecnología e Innovación (SENACYT)、Instituto para la Formación y Aprovechamiento de Recursos Humanos (IFARHU) による財政支援に感謝の意を表します。著者は、原稿を慎重にレビューしてくれたMiguel Rodríguez博士に感謝します。
資料
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
参考文献
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627 (2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267 (2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33 (2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -. M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999 (2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451 (2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614 (2017).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved